

Abstract
Inhibition of postsynaptic glutamate receptors at the Drosophila NMJ initiates a compensatory increase in presynaptic release termed synaptic homeostasis. BMP signaling is necessary for normal synaptic growth and stability. It remains unknown whether BMPs have a specific role during synaptic homeostasis and, if so, whether BMP signaling functions as an instructive retrograde signal that directly modulates presynaptic transmitter release. Here we demonstrate that the BMP receptor (Wit) and ligand (Gbb) are necessary for the rapid induction of synaptic homeostasis. We also provide evidence that both Wit and Gbb have functions during synaptic homeostasis that are separable from NMJ growth. However, further genetic experiments demonstrate that Gbb does not function as an instructive retrograde signal during synaptic homeostasis. Rather our data indicate that Wit and Gbb function via the downstream transcription factor Mad, and that Mad-mediated signaling is continuously required during development to confer competence of motoneurons to express synaptic homeostasis.
Keywords: morphogen, Drosophila, neuromuscular junction, glutamate receptor, axonal transport, dynein, amyotrophic lateral sclerosis, neurodegeneration, homeostasis, synaptic plasticity
INTRODUCTION
Bone Morphogenic Proteins (BMPs) are classical morphogens that are widely expressed in the developing vertebrate and invertebrate nervous systems (Raible, 2006; Teleman et al., 2001). Classical morphogens are defined by their ability to signal at a distance in a concentration-dependent manner (Teleman et al., 2001; Charron and Tessier-Lavigne, 2001). In this way, positional information is conveyed to cells that reside at different positions within a morphogen gradient. The activity of BMP signaling during neuronal fate specification and brain patterning are well established (Chesnutt et al., 2004; Lim et al., 2000; Murali et al., 2005; Rios et al., 2004; Yung et al., 2002). Recently, the BMPs have been shown to have potent activities later in neural development, participating in the mechanisms of axon guidance (Charron and Tessier-Lavigne, 2001), dendrite growth (Withers et al., 2000), synaptic growth (Aberle et al., 2002; Marques et al., 2002; McCabe et al., 2003) and synapse stabilization (Eaton and Davis, 2005). It is generally unknown whether the BMPs participate in these processes as morphogens, signaling at a distance with dose-dependent actions, or whether BMPs function as local, trans-synaptic signaling molecules. This question becomes particularly interesting given recent genetic evidence that BMP signaling may participate in the mechanisms of homeostatic synaptic plasticity (Haghighi et al., 2003).
Homeostatic signaling is believed to regulate cellular excitability throughout the central and peripheral nervous systems (Burrone and Murthy, 2001; Turrigiano and Nelson, 2004; Marder and Goaillard, 2006; Davis, 2006). A form of homeostatic signaling has been documented at the neuromuscular junction of organisms ranging from Drosophila to rodents and human (Petersen et al., 1997; Davis et al., 1998; Paradis et al., 2001; Cull-Candy et al., 1980; Plomp et al., 1992; Sandrock et al., 1997). At the NMJ, decreased postsynaptic neurotransmitter receptor sensitivity leads to a compensatory increase in presynaptic transmitter release that precisely offsets impaired receptor function and restores normal muscle depolarization (Petersen et al., 1997; Davis et al., 1998; Frank et al., 2006). This homeostatic signaling system requires a retrograde signal from muscle to nerve that is able to modulate presynaptic release (Petersen et al., 1997; Davis et al., 1998; Davis, 2006; Frank et al., 2006).
A genetic experiment has provided evidence that the Drosophila type II BMP receptor, Wishful Thinking (Wit), could convey the retrograde signal underlying homeostatic signaling at the Drosophila NMJ (Haghighi et al., 2003). It was shown that expression of a dominant negative glutamate receptor subunit (DN-GluRIIA) in muscle leads to a decrease in the amplitude of spontaneous miniature release events (mEPSP) and a homeostatic increase in presynaptic release. However, when DN-GluRIIA was expressed in muscle in a wit mutant, no homeostatic increase in presynaptic release was observed. Although suggestive, this result is complicated by the fact that the wit mutation also disrupts structural and functional synapse development (Aberle et al., 2002; Marques et al., 2002) as well as synapse stability (Eaton and Davis, 2005). As a result, it remains unclear whether the wit mutation specifically disrupts synaptic homeostasis, or whether this mutation developmentally cripples the NMJ, both structurally and functionally, such that no form of synapse modulation can be expressed (Davis, 2006). Furthermore, it was recently shown that the induction of homeostatic signaling at the Drosophila NMJ is rapid (occurring in 10 minutes), is independent of new protein synthesis, and does not require the presence of the motoneuron cell body (Frank et al., 2006). This would seem to rule out a function for canonical BMP signaling from the NMJ to the motoneuron cell body in the mechanisms responsible for the rapid induction of synaptic homeostasis.
We have addressed the specific functions of BMP signaling during synapse development and homeostatic plasticity by manipulating multiple components of the BMP signaling system. Our experiments provide evidence that BMP signaling is specifically required for homeostatic plasticity, independent of BMP-dependent regulation of synaptic growth or stability. However, our data also argue against a model in which BMPs act as a local, retrograde homeostatic signal to modulate presynaptic release. Rather, we demonstrate that BMPs confer competence for motoneurons to express homeostatic plasticity. Based upon these data we speculate that BMPs may retain many of their morphogen-like signaling properties in the postembryonic nervous system to direct the expression of synaptic plasticity and homeostatic signaling.
RESULTS
Philanthotoxin (PhTx) is a use-dependent glutamate receptor antagonist at the Drosophila NMJ (Frank et al., 2006). Application of sub-blocking concentrations of PhTx to the NMJ initially decreases both mEPSP and EPSP amplitudes by an equivalent amount. This is consistent with the partial blockade of postsynaptic glutamate receptors (Frank et al., 2006). Continued recording in the presence of PhTx demonstrates that EPSP amplitudes gradually increase over the course of 10 minutes without a change in the underlying average mEPSP amplitude. The increase in EPSP amplitude is caused by an increase in presynaptic transmitter release (quantal content) that requires the full functionality of presynaptic CaV2.1 calcium channels (Frank et al., 2006). These data are consistent with the rapid induction of a retrograde, homeostatic signaling system at the NMJ (Frank et al., 2006). Here we use this assay to test the function of BMP signaling in the mechanisms underlying the rapid induction of synaptic homeostasis. Throughout this study, the rapid induction of synaptic homeostasis is achieved by applying sub-blocking concentrations of PhTx to a semi-intact NMJ preparation for 10 minutes (Frank et al., 2006). At this time point we observe a robust, homeostatic increase in presynaptic transmitter release (Frank et al., 2006).
The type II BMP receptor Wit is necessary, presynaptically for the rapid induction of synaptic homeostasis
We first asked whether mutations in the type II BMP receptor wishful thinking (wit) block the rapid induction of synaptic homeostasis following application of PhTx to the NMJ. For the analysis of BMP mutations, we present data both as raw amplitudes (Tables) and as normalized to the same genotype recorded in the absence of PhTx (Figures), as done previously (Frank et al., 2006). This method of data presentation highlights the effects of PhTx application to a given mutant background both in terms of the acute, PhTx dependent change in mEPSP amplitude and the rapid homeostatic modulation of presynaptic release. For example, if we observe that decreased mEPSP amplitude, caused by PhTx application, correlates with increased quantal content compared to the same mutant without PhTx, then we conclude that homeostatic compensation has occurred, even if absolute synaptic strength remains below that observed in wild type (Frank et al., 2006).
In the first set of experiments, we find that application of 6 μM PhTx for 10 minutes to wild type or heterozygous wit mutant animals (wit/+) leads to a decrease in mEPSP amplitude and a homeostatic increase in presynaptic release (Figure 1A, B). We then find that wit null mutants fail to show any compensatory increase in presynaptic release following PhTx application (Figure 1B). Neuronal expression of UAS-wit in the wit mutant background using two independent GAL4 drivers restores the expression of homeostatic compensation demonstrating that Wit is required presynaptically for the rapid induction of synaptic homeostasis (Figure 1D). Importantly, we have confirmed that OK371-GAL4 is specifically expressed in motoneurons (Mahr and Aberle, 2006) and we can, therefore, conclude that wit has a motoneuron-specific activity that is sufficient for the expression of synaptic homeostasis. As an additional experiment, we demonstrate that the known Wit-dependent control of FMRFamide expression in the CNS does not have a role in the expression of synaptic homeostasis (Supplemental Figure 1).
Figure 1. The type-II BMP receptor wishful thinking is required presynaptically for the rapid induction of synaptic homeostasis.
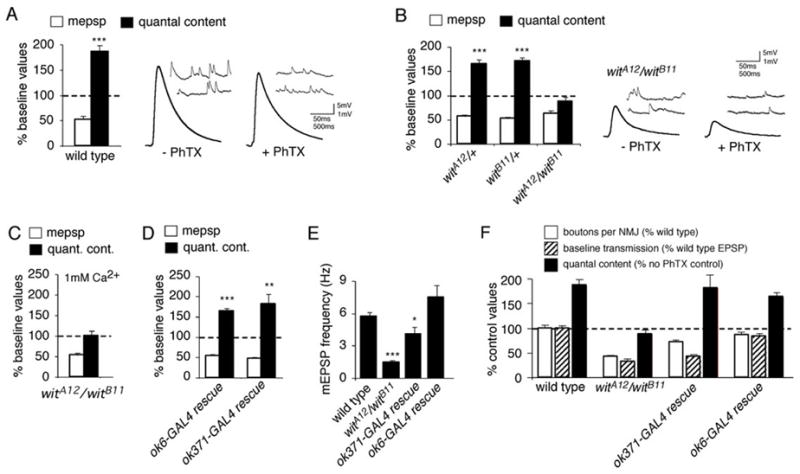
(A) Quantal content (filled bar) and mEPSP amplitude (open bar) are quantified. The dashed line represents normalized wild type baseline values recorded in the absence of PhTx. Bars represent values recorded after 10 minute PhTx application, normalized to wild type in the absence of PhTx. There is a significant decrease in mEPSP amplitude and a significant, compensatory increase in quantal content. Right, representative traces showing mEPSPs (inset) and EPSPs for control and PhTx-treated wild type animals. (B) Data are presented as in (A). Application of PhTx to heterozygous controls (witA12/+ and witB11/+) induces a decrease in mEPSP amplitude and a compensatory increase in quantal content compared to heterozygous controls in the absence of PhTx (p<0.001). No increase in quantal content is observed in the null mutant (witA12/witB11) animals compared to witA12/witB11 animals in the absence of PhTx (p>0.5). Sample traces are shown for the null witA12/witB11 animals with and without PhTx application for 10 minutes. (C) Data are presented as in (A). Synaptic homeostasis remains blocked in witA12/witB11 animals when recordings are conducted in saline containing 1 mM Ca2+ and 10 mM Mg2+. (D) Data are presented as in (A). Expressing UAS-wit using either of the presynaptic GAL4 drivers OK6-GAL4 or OK371-GAL4 in the wit mutant background (witA12/witB11) restores synaptic homeostasis, as demonstrated by a significant increase in quantal content after PhTx challenge (p < 0.001 and p<0.01 respectively). (E) mEPSP frequency in wild type, wit mutant animals, and wit animals in which UAS-wit is expressed presynaptically using OK6-GAL4 or OK371-GAL4. (F) Quantification of data for bouton number (open; percent wild type bouton number), baseline transmission (hatched; percent wild type EPSP amplitude) and quantal content (filled). Values for quantal content are normalized to recordings in the absence of PhTx for a given genotype as in (A). Wit mutant animals (witA12/witB11) have decreased bouton number, decreased EPSP amplitude and no homeostatic increase in quantal content (as shown in B). Presynaptic expression of UAS-wit in the wit mutant using OK371-GAL4 partially restores bouton number (numbers are significantly less than wild type, p<0.01), does not rescue EPSP amplitude, and completely rescues a homeostatic increase in release (p<0.001). Presynaptic expression of UAS-wit in the wit mutant using OK6-GAL4 restores all aspects of synaptic growth and function. Significance is indicated as follows for this figure and all subsequent figures: *p<0.05, **p < 0.01, ***p < 0.001 Student’s t-test.
The wit mutants have a significant decrease in baseline synaptic transmission compared to wild type (Figure 1B; Table 1; Aberle et al., 2002; Marques et al., 2002) and this could be the primary cause of impaired synaptic homeostasis. Therefore, we repeated PhTx application to the wit null mutants and recorded in elevated extracellular calcium saline (1 mM Ca2+, 10 mM Mg2+). Despite enhanced presynaptic release, synaptic homeostasis remained blocked following application of PhTx to the wit mutant (Figure 1C; Table 1). Thus, we conclude that the impaired induction of synaptic homeostasis in the wit mutation is not a direct consequence of decreased quantal release that is observed in the wit mutant NMJ.
Table 1.
Physiological data demonstrating a role for wit in synaptic homeostasis
| Condition | Genotype | PhTX | mEPSP | EPSPa | QC | N |
|---|---|---|---|---|---|---|
| 0.6 mM Ca2+ | w1118 | − | 0.95±0.04 | 39.2±1.5 | 41.8±2.0 | 16 |
| 20 mM Mg2+ | + | 0.49±0.06 | 36.2±1.6 | 78.1±4.9*** | 12 | |
| witA12/+ | − | 0.87±0.06 | 35.2±1.5 | 40.9±1.4 | 6 | |
| + | 0.49±0.02 | 33±1.9 | 67.3±3.4*** | 6 | ||
| witB11/+ | − | 0.93±0.04 | 38.9±1.7 | 42±2.0 | 8 | |
| + | 0.49±0.03 | 34.5±0.6 | 71.8±2.6*** | 8 | ||
| witA12/witB11 | − | 0.64±0.03 | 13.5±1.7 | 21.3±2.7 | 10 | |
| + | 0.40±0.02 | 7.5±0.7** | 18.6±1.8 | 11 | ||
| OK6-Gal4/UAS-wit; | − | 0.91±0.08 | 32.7±1.7 | 37±2.6 | 9 | |
| witA12/witB11 | + | 0.5±0.03 | 29.6±1.1 | 60.5±2.8*** | 9 | |
| OK371-Gal4/UAS-wit; | − | 0.8±0.04 | 16.5±1.8 | 20.8±2.0 | 10 | |
| witA12/witB11 | + | 0.37±0.02 | 13.9±1.8 | 37.6±5.1** | 11 | |
| dLIMK1P1/Y | − | 1.07±0.06 | 43±1.7 | 41.8±3.8 | 9 | |
| + | 0.66±0.02 | 40.7±1.5 | 62.1±2.8*** | 8 | ||
| sax4/Df | − | 0.97±0.13 | 3.0±0.3 | 3.3±0.5 | 6 | |
| 1 mM Ca2+ | witA12/witB11 | − | 0.38±0.03 | 27.7±1.6 | 122.3±12.2 | 11 |
| 10 mM Mg2+ | + | 0.21±0.02 | 18.5±2.2** | 122.9±13.4 | 12 | |
| sax4/Df | − | 0.83±0.07 | 30.4±1.0 | 63.8±3.6 | 11 | |
| + | 0.44±0.05 | 19.2±0.9*** | 63.7±6.8 | 11 |
Values refer to data presented in Figure 1.
Significant changes in average EPSP amplitude and Quantal Content (QC) are determined for each genotype (+/− PhTx) according to:
p<0.05,
p<0.01,
p<0.001. All changes in mEPSP amplitude (+/− PhTx) are statistically significant (p<0.05).
The BMP Ligand Gbb independently specifies synaptic growth and synaptic homeostasis
Glass bottom boat (Gbb) is a BMP ligand for the Wit receptor that is expressed in muscle and within the CNS (Wharton et al., 1999; McCabe et al., 2003). If BMP signaling is required for the rapid induction of synaptic homeostasis, then gbb mutations should also block the rapid induction of synaptic homeostasis. Gbb null mutants are sub-viable (McCabe et al., 2003). Therefore, prior studies examined hypomorphic loss-of-function mutant combinations including a weak gbb loss-of-function condition (gbb1/gbb4) and a severe gbb loss-of-function condition (gbb1/gbb2, UAS-gbb9.9) that is composed of a null mutant allelic combination and leaky expression of UAS-gbb9.9 in the absence of a GAL4 driver (McCabe et al., 2003). We first confirmed that both hypomorphic allelic combinations impair morphological synapse development (McCabe et al., 2003; see below). Unexpectedly, however, both hypomorphic mutant combinations showed robust synaptic homeostasis following a 10-minute PhTx incubation (Figure 2A; Table 2). These data apparently contradict the blockade of homeostasis in the wit mutant.
Figure 2. The BMP ligand Gbb is required for synaptic homeostasis.
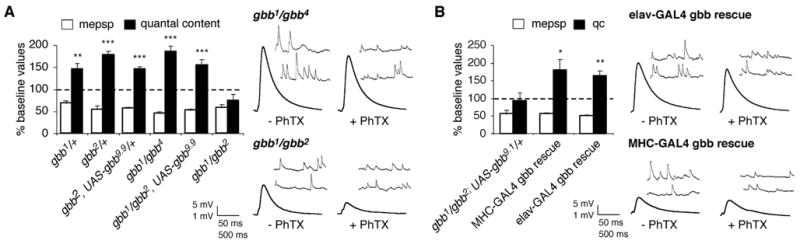
(A) Quantal content (filled bar) and mEPSP amplitude (open bar) are quantified and normalized to amplitudes recorded for each genotype in the absence of PhTx (as in figure 1A). A homeostatic increase in quantal content offsets a significant decrease in mEPSP amplitude in all mutant combinations examined except the null gbb combination gbb1/gbb2 which does not show a homeostatic increase in quantal content in response to PhTx treatment (p > 0.2). Representative traces are shown for indicated genotypes at right. (B) Data are quantified as in (A). Either neuronal-specific (elavC155-GAL4) or muscle-specific (MHC-GAL4) expression of UAS-gbb9.1 in the gbb null mutant background restores a homeostatic increase in quantal content. Representative traces are shown at right.
Table 2.
Physiological data demonstrating a role for gbb in synaptic homeostasis
| Genotype | PhTX | mEPSPb | EPSPa | QC | N |
|---|---|---|---|---|---|
| yw | − | 1.25±0.06 | 35.8±1.5 | 29.3±2.0 | 14 |
| + | 0.64±0.06 | 30.1±2.6* | 48.9±4.0*** | 12 | |
| gbb1/+ | − | 0.84±0.03 | 35.2±1.6 | 41.9±2.1 | 16 |
| + | 0.58±0.05 | 32.1±1.3 | 61±5.3** | 16 | |
| gbb2/+ | − | 1.02±0.09 | 35.2±2.3 | 35.8±3.0 | 7 |
| + | 0.54±0.02 | 34.5±1.7 | 63.5±2.8*** | 5 | |
| gbb2, UAS-gbb9.9/+ | − | 0.91±0.08 | 35.6±1.3 | 41.2±4.1 | 7 |
| + | 0.52±0.02 | 31.1±0.6** | 60.2±2.3*** | 8 | |
| gbb1/gbb4 | − | 1.41±0.13 | 34.4±2.7 | 25.5±2.2 | 11 |
| + | 0.65±0.05 | 29.6±1.4 | 47.2±3.2*** | 9 | |
| gbb1/gbb2, UAS-gbb9.9 | − | 1.27±0.10 | 26.6±1.4 | 22±1.9 | 14 |
| + | 0.67±0.04 | 22.1±1.4* | 34.1±2.6** | 12 | |
| gbb1/gbb2 | − | 1.18±0.10 | 15.8±1.4 | 14.2±1.3 | 14 |
| + | 0.68±0.08 | 6.6±1.2*** | 10.6±2.0 | 11 | |
| gbb1/gbb2; UAS-gbb9.1/+ | − | 1.59±0.21 | 12.9±1.8 | 8.8±1.5 | 9 |
| + | 0.87±0.15 | 5.3±1.0*** | 8.0±2.0 | 9 | |
| gbb1/gbb2; UAS-gbb9.1/ | − | 1.00±0.07 | 8.5±1.3 | 8.3±1.3 | 10 |
| MHC-GAL4 | + | 0.56±0.03 | 8.1±1.1 | 15.0±2.5* | 9 |
| elavC155-GAL4/+; gbb1/gbb2; | − | 1.08±0.11 | 26.4±1.3 | 26.5±2.7 | 10 |
| UAS-gbb9.1/+ | + | 0.55±0.02 | 23.5±2.2 | 43.1±3.8** | 10 |
Values refer to data presented in Figure 2.
Significant changes in average EPSP amplitude and Quantal Content (QC) are determined for each genotype (+/− PhTx) according to:
p<0.05,
p<0.01,
p<0.001. All changes in mEPSP amplitude (+/−PhTx) are statistically significant (p<0.05).
There is a trend toward increased average mEPSP amplitude in the gbb mutants, also observed previously (McCabe et al., 2003), but the data are not statistically significant compared to the appropriate genetic background (yw).
One explanation for the presence of homeostatic compensation in the gbb hypomorphs is that small amounts of Gbb protein fail to support normal synaptic growth, but are sufficient to support normal synaptic homeostasis. To address this possibility we established conditions that allowed us to raise gbb null mutants (gbb1/gbb2) to the third instar stage (see methods). First, we find that synaptic growth is no more severely impaired than that observed in the strong hypomorphic condition (gbb1/gbb2, UAS-gbb9.9) (Figure 3; p>0.3). Importantly, synaptic homeostasis is fully blocked in gbb null mutants (Figure 2A), consistent with the blockade of synaptic homeostasis in wit null mutants. Thus, we conclude that gbb is necessary for synaptic homeostasis, consistent with gbb functioning as the ligand for the Wit receptor in motoneurons.
Figure 3. Impaired synaptic growth in gbb mutants does not correlate with the expression of synaptic homeostasis.

(A–C) Composite images of anti-Synapsin staining at gbb1/gbb2 and gbb1/gbb2, UAS-gbb9.9 as well as wild type synapses. Images represent the NMJ at muscle 6/7. (D) Quantification of bouton number (open; percent wild type bouton number), baseline transmission (hatched; percent wild type EPSP amplitude), and quantal content (filled). Values for quantal content are normalized to control values recorded for each genotype in the absence of PhTx. Bouton number and baseline transmission are significantly impaired in gbb1/gbb2 (p<0.01) and there is no significant homeostatic increase in quantal content (p>0.2). Bouton numbers are significantly decreased in gbb1/gbb4 (p<0.01). Baseline transmission and bouton number are significantly impaired in gbb1/gbb2, UAS-gbb9.9 (p<0.001). Neuronal-specific rescue of gbb (elav-GAL4 gbb rescue) restores synaptic homeostasis and significantly rescues both NMJ growth and baseline neurotransmission (p<0.001). Muscle-specific rescue of gbb (MHC-GAL4 gbb rescue) restores synaptic homeostasis and significantly rescues NMJ growth (p < 0.001) but does not significantly rescue baseline neurotransmission.
The demonstration that gbb null mutations block synaptic homeostasis allows us to test whether expression of UAS-gbb in muscle versus neurons is sufficient to restore synaptic homeostasis to the null mutant background. We expressed the non-leaky UAS-gbb transgene (UAS-gbb9.1) in the gbb null mutant background. The conclusion that UAS-gbb9.1 is not leaky is based on the observation that the presence of UAS-gbb9.1 in the gbb null mutant background (in the absence of a GAL4 driver) does not rescue synaptic function or homeostasis (Figure 2B; Wharton et al., 1999). When we express UAS-gbb9.1 specifically in muscle using the MHC-GAL4 driver in the gbb null mutant background, we restore the rapid induction of synaptic homeostasis (Figure 2B). Similarly, when we express UAS-gbb9.1 specifically in neurons using elav-GAL4 in the gbb null mutant, we find that the rapid induction of synaptic homeostasis is restored (Figure 2B). Since expression of Gbb in neurons is sufficient to rescue normal synaptic homeostasis in the gbb null mutant, these data demonstrate that Gbb need not be released from the muscle to achieve homeostatic compensation. These data argue that Gbb is not the instructive retrograde signal that directly modulates presynaptic release during synaptic homeostasis at the NMJ.
Canonical Mad-mediated signaling is required for synaptic homeostasis
Since the motoneuron cell body is not required for the rapid induction of synaptic homeostasis (Frank et al., 2006), there are two possibilities for how BMP signaling could regulate synaptic homeostasis. First, the BMP receptors could signal locally at the NMJ via a non-canonical pathway involving Lim Kinase (Eaton and Davis, 2005) or other downstream effectors. Alternatively, the BMPs may have a Mad-dependent developmental function in the motoneuron soma that permits the expression of homeostatic plasticity. Mad is a transcription factor that conveys signaling from the BMP receptor to the cell nucleus in the canonical BMP signaling pathway.
To distinguish between these two models we first asked whether Mad-dependent signaling is required for the rapid induction of synaptic homeostasis. As shown previously, the mad null mutants have a deficit in baseline synaptic transmission that is similar to that observed in the wit mutants (Rawson et al., 2002; Supplemental Table 1). Here, we find that mad heterozygous animals show normal synaptic homeostasis while homozygous mad null mutants fail to express synaptic homeostasis in response to PhTx application (Figure 4A). Since Mad is thought to primarily act as a transcription factor (Shi and Massague, 2003), these data suggest that BMP signaling is required at the level of the motoneuron nucleus for normal synaptic homeostasis.
Figure 4. Mad-mediated signaling is required in motoneurons for the expression of synaptic homeostasis.
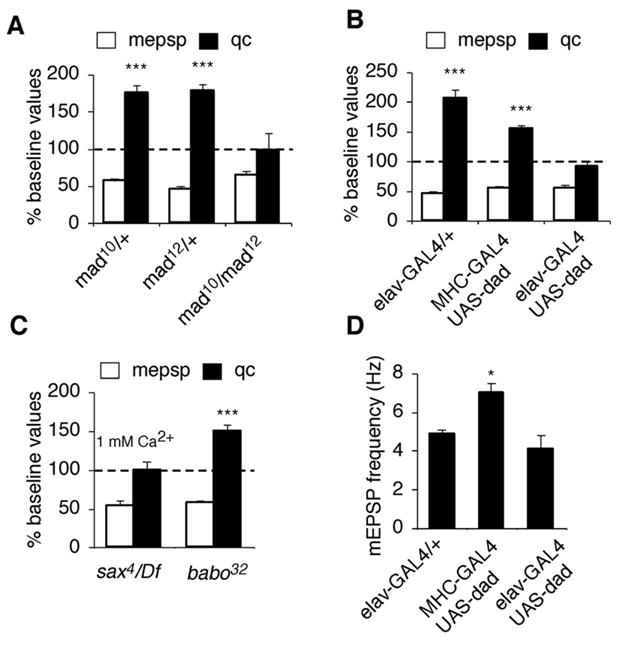
(A) Quantal content (filled bar) and mEPSP amplitude (open bar) are quantified and normalized to amplitudes recorded for each genotype in the absence of PhTx (as in figure 1A). The mad heterozygous animals show a significant decrease in mEPSP amplitude and a significant homeostatic increase in quantal content following PhTx application. The mad null mutant (mad10/mad12) fails to show a homeostatic increase in quantal content in response to decreased mEPSP amplitude. (B) There is no significant increase in quantal content in response to PhTx application in animals that express UAS-dad in neurons using elavC155-GAL4 (p>0.3). A significant, homeostatic increase in quantal content is observed following muscle expression of UAS-dad using MHC-GAL4 (p<0.01). (C) Quantification as in (A) for sax4/Df and babo32/babo32 mutations. Synaptic homeostasis is blocked in the sax4/Df mutants (recorded at elevated calcium as indicated). (D) Quantification of mEPSP frequency.
We next performed experiments to test whether mad is required in muscle versus neurons, with the hypothesis that it functions in the motoneuron downstream of Wit activation. To do so we over-expressed an inhibitory Smad (UAS-dad) in motoneurons. Dad suppresses Mad-mediated signaling by blocking Mad activation and preventing translocation to the cell nucleus (Tsuneizumi et al., 1997; Nakao et al., 1997; Shi and Massague, 2003). We find that UAS-dad expression in neurons blocks synaptic homeostasis whereas expression of UAS-dad in postsynaptic muscle does not (Figure 4B). Together, these data are consistent with the conclusion that Mad-dependent signaling is required in the neuron for the rapid induction of synaptic homeostasis.
To this point we have shown that gbb, wit and mad are necessary for the rapid induction of synaptic homeostasis. To further test whether this branch of the BMP signaling system is specifically required for synaptic homeostasis we also tested mutations in two type 1 BMP receptors, saxophone (sax) and baboon (babo) that can pair with the wit receptor (McCabe et al., 2004; Brummel et al., 1999). The Sax receptor has been shown to function with Wit in the regulation of Mad-mediated NMJ growth and function (McCabe et al., 2004). The Babo receptor is believed to function with Wit to mediate dSmad2 signaling (Brummel et al., 1999; Lee-Hoeflich et al., 2005). Here we demonstrate that the rapid induction of synaptic homeostasis is blocked in the sax4/Df mutant, whereas significant synaptic homeostasis remains in the babo32 mutant (a putative null mutation; Brummel et al., 1999) (Figure 4C). These data are consistent with the conclusion that Mad signaling downstream of the Wit receptor is required for the rapid induction of synaptic homeostasis. Furthermore, the demonstration that Wit is necessary in motoneurons and that neuronal overexpression of UAS-dad also blocks synaptic homeostasis leads us to conclude that Mad signaling is necessary in the motoneuron for normal synaptic homeostasis.
Impaired Retrograde Axonal Transport Blocks the Expression of Synaptic Homeostasis
Our data suggest a model in which the Wit receptor initiates Mad-dependent signaling in the motoneuron nucleus, which is necessary for normal synaptic homeostasis. If this model is correct, then BMP signaling at the NMJ should not be sufficient to achieve synaptic homeostasis if downstream Mad-dependent signaling is prevented from reaching the motoneuron nucleus. It has been previously shown that impaired retrograde axonal transport caused by expression of a dominant-negative p150/glued (UAS-DN-Glued) transgene blocks the accumulation of nuclear P-Mad in Drosophila motoneurons (McCabe et al., 2003; Eaton et al., 2002; Allan et al., 2003). Here we find that neuronal expression of UAS-DN-Glued blocks the rapid induction of synaptic homeostasis (Figure 5A; Supplemental Table 2). Since synaptic homeostasis can be induced in preparations with severed motor axons (Frank et al., 2006) and since local synaptic BMP signaling should be retained in animals expressing UAS-DN-Glued, our data argue that local BMP signaling at the NMJ is not sufficient for the homeostatic modulation of presynaptic transmitter release following the application of sub-blocking concentrations of PhTx. Rather, these data support our model that nuclear BMP signaling is required to specify the competence of motoneurons to express synaptic homeostasis following application of PhTx. However, before this conclusion can be strongly supported, it is necessary to rule out several other mechanisms by which UAS-DN-Glued could indirectly block expression of synaptic homeostasis.
Figure 5. Impaired retrograde axonal transport blocks the rapid induction of synaptic homeostasis.
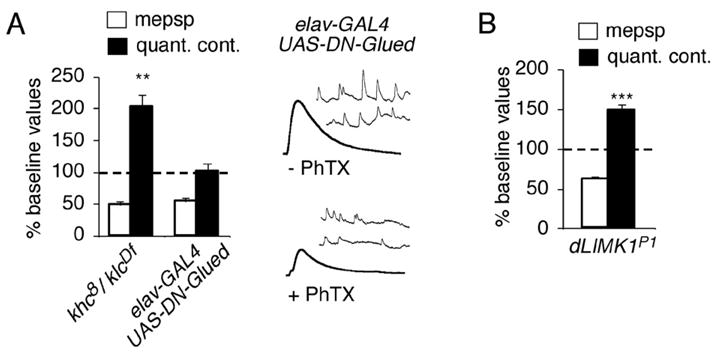
(A) Quantal content (filled bar) and mEPSP amplitude (open bar) are quantified and normalized to amplitudes recorded for each genotype in the absence of PhTx. Neuronal expression of UAS-DN-Glued (elavC155-GAL4/+; UAS-GluedDNΔ84/+) prevents an increase in quantal content in response to PhTx-challenge (p> 0.9). Animals with a double heterozygous combination of mutations in kinesin heavy chain and kinesin light chain (khc8/+; klcDf/+) show a robust homeostatic increase in presynaptic release following PhTx application. (B) Data are quantified as in (A). A robust homeostatic increase in quantal content is observed in a LIM Kinase mutant (DLIMKP1).
First, it was previously shown that UAS-DN-Glued expression not only disrupts retrograde axonal transport, but also destabilizes the NMJ (Eaton et al., 2002). Similarly, it has been shown that impaired BMP signaling disrupts synapse stability (Eaton and Davis, 2005). To test whether NMJ destabilization contributes to the loss of synaptic homeostasis we examined mutations in the Drosophila homolog of LIM Kinase. LIM kinase binds the C-terminal tail of the BMP receptor and mutations in LIM kinase impair synapse stability without altering synaptic growth (Eaton and Davis, 2005). We find that synaptic homeostasis is normal in a LIM kinase mutant previously shown to have impaired synapse stability (Figure 5B; Eaton and Davis, 2005). Thus, impaired synapse stability cannot account for impaired synaptic homeostasis.
Although disrupting dynein/dynactin function primarily alters retrograde axonal transport, it can also influence anterograde transport (Martin et al., 1999). In addition, impaired axonal transport causes the accumulation of protein blockages that could interfere with synaptic homeostasis indirectly by inducing stress-related signaling in the motoneuron (Martin et al., 1999; Cavalli et al., 2005; Byrd and Jin, 2001). Therefore, we examined the induction of synaptic homeostasis in a kinesin mutant combination that is viable to the third instar stage and which has impaired anterograde axonal transport and protein blockages in the motor axon similar in size and severity to that observed when UAS-DN-Glued is expressed neuronally (Martin et al., 1999; data not shown). We find that kinesin mutants show robust homeostatic compensation following a 10-minute incubation in PhTx (Figure 5A). Thus, altered synaptic homeostasis is not a secondary consequence of impaired neuron health, axonal blockage or impaired delivery of synaptic material to the NMJ. We conclude that impaired retrograde axonal transport blocks synaptic homeostasis, most likely due to impaired BMP signaling at the motoneuron soma.
BMP Signaling at the Soma Confers Competence to Express Homeostatic Plasticity
If BMP signaling at the motoneuron soma is required for synaptic homeostasis, it should be possible to restore P-Mad at the soma even in the presence of DN-Glued and rescue synaptic homeostasis. It was shown previously that simultaneous over-expression of UAS-gbb and UAS-DN-Glued in neurons can restore an accumulation of nuclear P-Mad, indicating that BMP signaling can be achieved from UAS-gbb expressed in the CNS without necessitating retrograde axonal transport from peripheral tissues (Allan et al., 2003). Therefore, we over-expressed UAS-Gbb in neurons that also over-express UAS-DN-Glued and find full rescue of synaptic homeostasis (Figure 6A; Supplemental Table 2). These data are consistent with the conclusion that disruption of synaptic homeostasis following neuronal expression of UAS-DN-Glued is a consequence of impaired neuronal BMP signaling. Taken together our data indicate that BMP signaling at the cell soma is required for motoneurons to be competent to express synaptic homeostasis.
Figure 6. Neuronal expression of Gbb in a background of impaired retrograde transport restores synaptic homeostasis but not growth or synaptic efficacy.
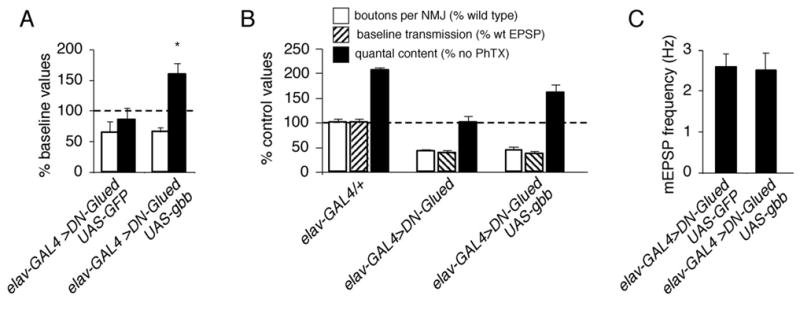
(A). Quantal content (filled bar) and mEPSP amplitude (open bar) are quantified and normalized to amplitudes recorded for each genotype in the absence of PhTx. Animals simultaneously expressing UAS-DN-Glued and UAS-GFP in neurons (elavC155-GAL4/+; UAS-GluedDNΔ84/UAS-CD8-GFP) do not show a homeostatic increase in quantal content compared to controls. However, synaptic homeostasis is restored when UAS-gbb is simultaneously overexpressed with UAS-DN-Glued (elavC155-GAL4/+; UAS-GluedDNΔ84/UAS-gbb9.1). (B) Quantification of bouton number (open; percent wild type bouton number), baseline transmission (hatched; % wild type EPSP amplitude), and quantal content (filled). Values for quantal content are normalized to control values recorded for each genotype in the absence of PhTx. UAS-gbb expression in neurons restores a homeostatic increase in quantal content but does not restore synaptic growth or baseline EPSP amplitudes compared to controls. (C) Quantification of mEPSP frequency.
Independent regulation of homeostasis, baseline neurotransmission and NMJ growth
The BMP signaling system was originally characterized at the Drosophila NMJ as being required for normal NMJ growth and normal baseline transmission. Our data argue that impaired synaptic homeostasis in BMP mutants is not a secondary consequence of impaired synaptic growth or impaired baseline neurotransmission. First, we find two conditions where synaptic growth remains impaired but synaptic homeostasis is intact. For example, synaptic growth in the gbb1/gbb2; UAS-gbb9.9 mutant background is not statistically different from the gbb null (Figure 3; p>.14). However, the presence of the leaky UAS-gbb9.9 transgene restores normal synaptic homeostasis despite impaired growth. A second example is seen when UAS-gbb is co-expressed with UAS-DN-Glued. In this animal, synaptic growth is severely impaired, but synaptic homeostasis is normal (Figure 6B). Thus, synaptic homeostasis can occur at an NMJ with impaired synaptic growth.
A different set of results demonstrates that there is not a correlation between impaired baseline transmission and the expression of synaptic homeostasis. First, when we express UAS-wit in the wit mutant background using OK371-GAL4 we restore synaptic homeostasis without significantly rescuing baseline synaptic function (Figure 1F). Second, when we express UAS-gbb in the gbb null mutant background using MHC-GAL4 we also restore synaptic homeostasis without significantly rescuing baseline synaptic function (Figure 2B, 3D). Third, when UAS-gbb and UAS-DN-Glued are co-expressed in neurons, synaptic homeostasis is restored without rescuing either synaptic growth or baseline transmission (Figure 6B). Together, these three results demonstrate that synaptic homeostasis can be achieved despite impaired baseline transmission. When taken together with the lack of correlation between NMJ growth and synaptic homeostasis, our data suggest that the BMP-dependent mechanisms that specify the expression of synaptic homeostasis can be separated from the mechanisms of BMP-dependent synapse development.
Finally, to test whether the independence of baseline transmission and the expression of synaptic homeostasis generalizes to mutations that directly affect synaptic vesicle fusion, we examined two additional mutations. Baseline synaptic transmission is severely impaired in a csp mutant background (0.6 mM extracellular calcium) (Figure 7A; Supplemental Table 3) consistent with previous studies implicating Csp in synaptic vesicle fusion (Nie et al., 1999). Despite impaired vesicle release we find that synaptic homeostasis is normal in the csp mutant (Figure 7B). Next we examined heterozygous mutations in syntaxin1A, which also have a significant decrease in baseline synaptic transmission (Figure 7C; Supplemental Table 3). Synaptic homeostasis is normal in this mutant background as well (Figure 7C). Together with the BMP mutant data described above, these data demonstrate that synaptic homeostasis can occur in the context of impaired baseline neurotransmission.
Figure 7. Normal synaptic homeostasis in mutations that disrupt synaptic vesicle release.
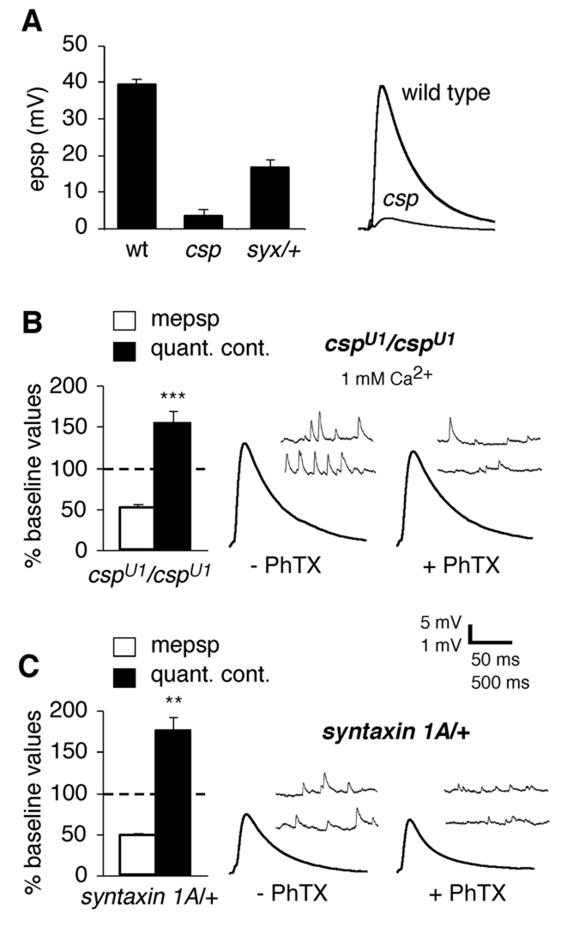
A) Baseline EPSP amplitude is significantly impaired in both cspU1 and syx/+ mutants (wild type amplitudes are repeated from figure 1). Representative EPSP traces are shown at right. B) The cspU1 mutants show normal homeostatic compensation in response to PhTx application, recorded in 1 mM extracellular calcium to increase absolute EPSP amplitude. Representative traces are shown at right. C) The syx/+ mutants show normal synaptic homeostasis in response to PhTx application (normal saline). Representative traces shown at right.
BMP signaling is continuously required for the expression of synaptic homeostasis
Finally, we asked whether BMP signaling is continuously required to support the expression of synaptic homeostasis, or whether BMPs act in a switch like manner, possibly during cell-fate specification, to allow expression of synaptic homeostasis. To examine this question, we inhibited BMP signaling in motoneurons for varying lengths of time during larval development and examined the effect on synaptic homeostasis. First, we demonstrate that expression of UAS-dad with either elavc155-GAL4 or c380-GAL4 (Supplemental Table 1), which initiate expression at different times during embryonic development (Lin and Goodman, 1994; Sanyal et al., 2003), are both sufficient to block PhTx-dependent synaptic homeostasis. In particular, c380-GAL4 initiates expression no earlier than embryonic stage 17 (Sanyal et al., 2003), after motoneuron cell fate specification is completed. This indicates that post-embryonic BMP signaling is required for the expression of synaptic homeostasis.
Next, we refined our analysis by conditionally inhibiting BMP signaling using an inducible GAL4 expression system termed GeneSwitch (Osterwalder et al., 2001; Roman et al., 2001). In the GeneSwitch system, the steroid drug RU486 turns on the GeneSwitch transcription factor elavGS-GAL4. Wild type animals raised on RU486 throughout larval development show normal synaptic homeostasis, baseline transmission and synaptic growth (Figure 8D; Supplemental Table 4 and data not shown). We used this system to drive conditional expression of UAS-dad. Control animals (elavGS-GAL4/+;UAS-dad/+) reared on media lacking RU486 (0 days) have normal synaptic growth (Figure 8A), a slight decrease in baseline transmission (Figure 8B; Supplemental Table 4), normal mEPSP frequency (Figure 8C) and express normal synaptic homeostasis (Figure 8E). By contrast, elavGS-GAL4/+;UAS-dad/+larvae that receive RU486 in their food for the final 2.5 or 4 days of larval development have profound defects in synaptic growth, baseline transmission, mEPSP frequency (Figure 8A–C; Supplemental Table 4) and have severely impaired synaptic homeostasis (Figure 8E). Thus, the GeneSwitch system allows us to express UAS-dad at sufficient levels to impair BMP-dependent synaptic growth, baseline transmission and the BMP-dependent expression of synaptic homeostasis.
Figure 8. Continuous BMP signaling is required to sustain the ability of motoneurons to express homeostatic plasticity.
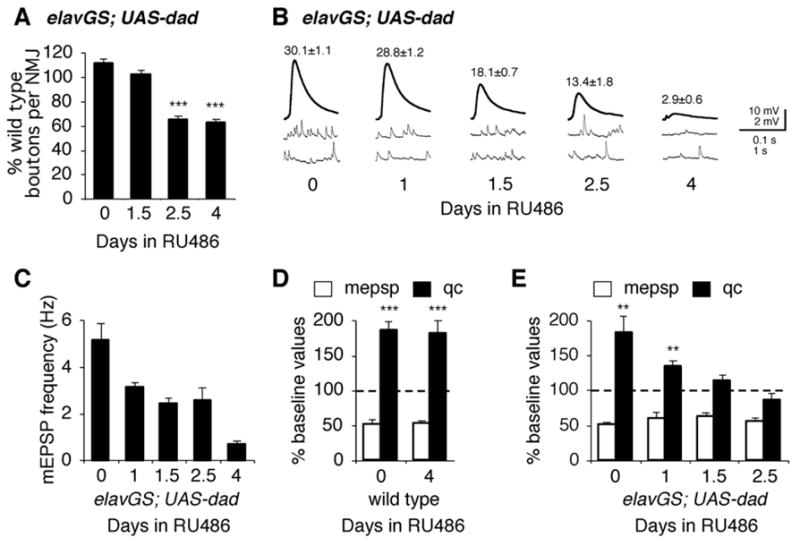
(A) Quantification of bouton number at muscles 6/7 in elavGS-UAS-dad animals (elavGS-GAL4/+; UAS-Dad/+) receiving RU486 administration for different durations of time as indicated. Each data point represents bouton numbers normalized to wild type animals that received identical RU486 administration. Time in RU486 refers to the duration of RU486 exposure prior to the end of larval development. (B) Representative traces and average EPSP amplitudes (numbers above traces) for elavGS-UAS-dad animals raised on RU486 for the indicated durations. RU486 feeding does not have a significant effect on baseline EPSP amplitudes in wild type. (C) Quantification of mEPSP frequency for animals in A–C. (D) Quantal content (filled bar) and mEPSP amplitudes (open bar) are quantified for wild type animals, raised on RU486 for indicated times. Data are normalized to amplitudes recorded for wild type in the absence of PhTx. (E) Data are quantified and presented as in (D) for elavGS-UAS-dad animals raised on RU486 for the indicated durations of time prior to dissection at the end of larval development.
Next we progressively shortened the duration of RU486 administration such that animals spent the final 2.5 days, 1.5 days or 1 day of larval development on food containing RU486. First, there is a clear progressive impairment of bouton number, baseline transmission and homeostatic compensation that corresponds to the duration of RU486 induction of UAS-dad. From this we conclude that Mad-dependent signaling is continuously required during larval development to sustain synapse growth, transmission and homeostatic plasticity. By examining how these three parameters become progressively impaired, we can make an additional conclusion. For example, animals reared on RU486 for 1.5 days of larval development have normal bouton numbers (Figure 8A; p>0.2). However, during this time frame there is a progressive impairment of both baseline transmission and synaptic homeostasis (Figure 8B, E). Clearly, impaired synaptic transmission and impaired homeostatic plasticity are not a secondary consequence of impaired NMJ growth under these conditions. Therefore, for the first time, we can conclude that BMP signaling has an activity at the Drosophila NMJ that is directly relevant to the control of baseline transmission and homeostatic plasticity.
DISCUSSION
The data presented here advance our understanding of BMP signaling at the Drosophila NMJ in several important ways. First, we demonstrate that BMP signaling is essential for the rapid, protein synthesis-independent, induction of synaptic homeostasis previously identified at this NMJ (Frank et al., 2006). Since expression of UAS-wit in motoneurons restores synaptic homeostasis in the wit mutant, and since suppression of Mad-mediated signaling in neurons blocks synaptic homeostasis we conclude that BMP signaling acts upon the motoneuron to enable the rapid induction of synaptic homeostasis. Next, we show that the requirement for BMP signaling during synaptic homeostasis is separable from BMP-dependent support of synaptic growth and baseline neurotransmission. Finally, we dissect the temporal and spatial requirements for BMP signaling. Our data support the conclusion that Mad-mediated signaling is required constitutively, downstream of the Wit receptor, in order to maintain the competence of motoneurons to express homeostatic plasticity. Further, our data argue that Gbb is not the retrograde signal that directly acts upon the presynaptic motoneuron terminal to homeostatically modulate presynaptic release.
Gbb Signaling Confers Competence to Express Homeostatic Plasticity
It has been hypothesized that Gbb could function as a homeostatic retrograde signal at the Drosophila NMJ (McCabe et al., 2003; Keshishian and Kim, 2004). According to this model, Gbb would be released in proportion to the perturbation of postsynaptic muscle excitation in a glutamate receptor mutant and, thereby, instruct the degree of homeostatic compensation expressed by the presynaptic motoneuron terminal (McCabe et al., 2003; Haghighi et al., 2003). In favor of this model, homeostatic compensation observed in a glutamate receptor mutant is blocked by the wit mutation (Haghighi et al., 2003). Here we present two lines of evidence that are consistent with the necessity of BMP signaling for homeostatic compensation. First, we confirm that the rapid induction of homeostatic compensation following application of PhTx is blocked by null mutations in both wit and gbb. Furthermore, we show that muscle-specific rescue of the gbb null mutation is sufficient to restore the rapid induction of homeostatic compensation.
Despite these compelling genetic data, several experiments now argue against the possibility that Gbb functions as an instructive, retrograde signaling that directly modulates presynaptic release during synaptic homeostasis. First, we demonstrate that although muscle-specific rescue of the gbb null mutation is sufficient to restore synaptic homeostasis, so is neuron-specific rescue of the gbb null mutation. Thus, homeostatic compensation can occur even in the absence of muscle-derived Gbb. These data argue against a model in which Gbb functions as the instructive retrograde signal that directly modulates presynaptic release during synaptic homeostasis.
Next, we demonstrate that homeostatic signaling is blocked by expression of DN-Glued in neurons, which disrupts retrograde axonal transport. In this experiment, Gbb signaling at the NMJ should, in theory, persist. Furthermore, we have established that an intact motor axon is not required for the rapid induction of synaptic homeostasis (Frank et al., 2006). Thus, we can conclude that trans-synaptic Gbb signaling from muscle to nerve is not sufficient for the rapid induction of synaptic homeostasis.
Given that Wit and Gbb are necessary for synaptic homeostasis, how do they participate in the process if Gbb is not the instructive retrograde signal? We demonstrate that Mad is necessary for synaptic homeostasis, and we provide evidence that Mad-mediated signaling is required in the motoneuron. In addition, we show that neuronal expression of UAS-Gbb restores homeostatic compensation in the presence of the DN-Glued transgene. These results suggest that the reason DN-Glued disrupts synaptic homeostasis is because it interferes with the retrograde axonal transport of P-Mad downstream of the Wit receptor. This is consistent with the prior demonstration that neuronal expression of Gbb can restore nuclear P-Mad in the presence of UAS-DN-Glued (Allan et al., 2003). Since the induction of synaptic homeostasis does not require the motoneuron soma we conclude that Gbb does not function as an acute, retrograde signal. Rather, Gbb may be a muscle derived signal that acts developmentally to confer the competence of motoneurons to express synaptic homeostasis. Thus, the identity of the homeostatic retrograde signal at the NMJ remains unknown. It remains possible that other TGF-β superfamily signaling molecules could function at the NMJ in this capacity including myoglianin and maverick (Lo and Frasch, 1999; Nguyen et al., 2000), though we have shown that synaptic homeostasis is intact in the baboon receptor mutant.
There are several possible ways in which BMP signaling could confer competence for motoneurons to express homeostatic plasticity. One possibility is that the BMPs control a transcriptional program that is necessary for synaptic homeostasis. For example, BMPs are potent regulators of cell fate during embryonic development (Chizhikov and Millen, 2005). Perhaps the ability of motoneurons to express synaptic homeostasis is related to the maintenance of their cellular or electrical identity. An alternate possibility is that BMPs control the expression of essential presynaptic proteins that are required for synaptic homeostasis. For example, it has been shown in other systems that target-dependent TGF-β signaling can modulate neuronal ion channel expression (Cameron et al., 1998). We recently demonstrated that CaV2.1 calcium channels are required for synaptic homeostasis at the Drosophila NMJ (Frank et al., 2006). However, we consider it unlikely that BMPs control synaptic homeostasis through the regulation of CaV2.1 channel expression because there is not a strong correlation between altered baseline synaptic transmission and the expression of synaptic homeostasis. Furthermore, overexpression of a GFP-tagged CaV2.1 calcium channel (cacophony-GFP) is unable to restore synaptic homeostasis when co-expressed with UAS-dad (data not shown). Finally, BMP signaling could influence the expression of synaptic homeostasis by targeting the rate of spontaneous miniature release. Spontaneous release events that persist in the absence of evoked neurotransmission are sufficient to induce homeostatic compensation at the Drosophila NMJ (Frank et al., 2006). However, we do not find a strong correlation between baseline mEPSP frequency and whether or not a mutant NMJ is able to express synaptic homeostasis. Although the wit mutants show a severe decrease in mEPSP rate compared to wild type, the expression of UAS-dad or UAS-DN-Glued both block synaptic homeostasis without severely impairing baseline mEPSP rate (see Figures 1, 4, 6). Ultimately, continued forward genetic investigation of homeostatic signaling may be required to identify the BMP-dependent mechanisms that control the expression of synaptic homeostasis.
Dissociating BMP-dependent control of synaptic growth, efficacy and plasticity
BMP signaling is required for NMJ growth, baseline neurotransmission and NMJ stability in addition to being required for synaptic homeostasis (Aberle et al., 2002; Marques et al., 2002; McCabe et al., 2003; McCabe et al. 2004; Eaton and Davis, 2005; Haghighi et al., 2003). It is a challenge, therefore, to determine whether BMP signaling has a specific function during synaptic homeostasis versus a more general role during synapse development (Davis, 2006). Here we present several lines of evidence that BMP signaling may have a separable function during synaptic growth versus synaptic homeostasis. First, we demonstrate that synaptic homeostasis can occur at BMP mutant synapses that show severely impaired synaptic growth. For example, the gbb hypomorphic mutant has a decrease in bouton number that is just as severe as the gbb null mutant, but the gbb hypomorphic mutant shows normal homeostatic compensation. As another example, animals in which UAS-gbb and UAS-DN-Glued are co-expressed have a severe decrease in bouton number but normal homeostatic compensation (Figure 6). Thus, we conclude that normal BMP-dependent synaptic growth is not required for the expression of synaptic homeostasis.
We are also able to dissociate BMP-dependent baseline transmission from both synaptic growth and synaptic homeostasis. First, muscle-specific rescue of the gbb null mutation significantly restores synaptic growth and rescues synaptic homeostasis but baseline transmission remains at levels observed in the null mutant (Figure 3). Second, motoneuron-specific rescue of the wit mutation (OK371-GAL4) similarly rescues bouton number and synaptic homeostasis although baseline transmission remains severely impaired (Figure 1). Third, animals in which UAS-gbb and UAS-DN-Glued are co-expressed have a severe decrease in baseline transmission but normal homeostatic compensation (Figure 6). Finally, we have results that show the converse effect. When UAS-dad is expressed for 1.5 days at the end of larval development, both synaptic homeostasis and baseline transmission are significantly impaired, but synaptic bouton numbers remain wild type (Figure 8). From these data we can conclude that impaired synaptic homeostasis is not a secondary consequence of BMP-dependent functional NMJ development. It also appears that there may be distinct effects of BMP signaling on the anatomical versus functional development of the NMJ. One possibility, consistent with BMPs being a classical morphogen, is that different levels of the ligand could initiate specific transcriptional programs with distinct effects on bouton number, baseline transmission and homeostatic plasticity. It is also possible that the site of action of BMP signaling will play an important role in specifying signaling outcome (Baines, 2004).
The relationship between baseline transmission and homeostatic compensation
It was previously speculated that synaptic homeostasis might function, over the course of development, to ensure that the muscle cell is normally depolarized by the NMJ. How can one explain the observation that csp and syx/+ mutations have decreased baseline neurotransmitter release but normal acute synaptic homeostasis in response to PhTx application, or other genotypes explored in this manuscript that show impaired baseline transmission and normal acute synaptic homeostasis? We previously demonstrated that the acute induction of synaptic homeostasis is independent of evoked neurotransmission. Thus, synaptic homeostasis may not function to modulate the absolute amplitude of evoked neurotransmitter release. Rather, synaptic homeostasis might be a rapid system to offset acute perturbations of postsynaptic receptor function. In this case, developmental programs that specify NMJ anatomy and active zone addition would achieve the reproducible development of the NMJ. Alternatively, the mechanisms of acute homeostatic compensation following PhTx application may be separable, either temporally or molecularly, from the other potential mechanisms that monitor and homeostatically control evoked EPSP amplitudes.
Axonal transport, Homeostatic Plasticity and Neurodegenerative Disease
Our data also suggest a possible link between the expression of homeostatic plasticity and the mechanisms of neuromuscular degenerative disease. Genetic mutations that impair retrograde axonal transport have been shown to cause familial amyotrophic lateral sclerosis (Puls et al., 2003). It has also been shown that, in Drosophila and mice, mutations that disrupt dynein-dynactin complex function lead to neuromuscular synapse degeneration (Eaton et al., 2002; Lamonte et al., 2002). It is hypothesized that impaired retrograde axonal transport deprives motoneurons of muscle-derived trophic support leading to motoneuron degeneration (Gauthier et al., 2004; Pun et al., 2006). Here we demonstrate that impaired retrograde axonal transport blocks the expression of homeostatic plasticity at the NMJ. This deficit can be restored by expression of BMPs in the central nervous system, bypassing retrograde axonal transport as the source of BMPs to the motoneuron cell body. It is tempting to speculate that impaired synaptic homeostasis at the NMJ may play a role in the progression of motoneuron disease associated with impaired retrograde axonal transport.
Finally, our data could have relevance to the sustained expression of homeostatic plasticity in regions of the adult nervous system (Davis, 2006; but see Desai et al., 2002). BMPs and downstream signaling proteins such as the Smads continue to be expressed in the adult nervous system (Lopez-Coviella et al, 2006; Sun et al., 2007). In particular, BMPs are secreted into the cerebral spinal fluid at concentrations that are relevant for neuronal signaling (Dattatreyamurty et al., 2001). It is, therefore, interesting to speculate that circulating levels of BMPs might sustain the competence of neurons to express homeostatic plasticity without driving morphological plasticity in the adult nervous system.
EXPERIMENTAL PROCEDURES
Electrophysiology
PhTx treatment and electrophysiology were conducted as previously described (Frank et al., 2006). Unless specified otherwise, recordings were conducted in HL3 saline containing 0.6 mM Ca2+ and 20 mM Mg2+, with a stimulus duration of 3 ms. All recordings were from muscle 6, abdominal segment 3 of third-instar larvae. For PhTx incubations, a semi-intact preparation was used in which a dorsal incision is made with the animal pinned, but not stretched, at the anterior and posterior, and then 200 μL of 6 μM PhTx-433 perfused over the incision (Frank et al., 2006). After 10 minute incubation, the dissection is completed and the animal washed in normal saline. Recordings with Vm < −60 mV were included for analysis. Quantal content was calculated as the ratio of EPSP/mEPSP amplitudes. Average values for mEPSP, EPSP and quantal content were calculated for each recording and then averaged across all recordings for a given genotype. For experiments conducted in higher calcium (Figures 1, 4, 7), quantal content was corrected for nonlinear summation (Martin, 1955).
Immunostaining
For bouton counts, third-instar larval fillets were fixed 2 minutes in Bouin’s solution and stained overnight at 4°C with mouse anti-synapsin antibody. All images and bouton counts are from muscles 6/7, abdominal segment 3. For visualization of FMRFamide in the CNS, a mixture of 4% paraformaldehyde and 7% picric acid in 1x PBS solution was incubated 10 minutes at room temperature, followed by washing in a blocking solution of 1× PBS, 0.3% triton X-100, 1 μg/ml BSA, and 5% goat serum. Anti-PT2 antibody (Jiang et al., 2000; a kind gift of Dr. Paul Taghert, Washington University), and secondary antibody (Goat anti-rabbit alexa-488; Invitrogen) was used at 1:2000 or 1:500 in the same blocking solution at 4°C overnight and 2 hours room temperature, respectively.
Genetics
All stocks were obtained from the Bloomington Stock Collection unless otherwise noted. The following synaptic vesicle mutations were analyzed; sources of fly stocks are indicated in parentheses: cspU1/cspU1 (Konrad Zinsmaier; Zinsmaier et al., 1994) and syntaxin1A06737/+ (Schulze et al., 1995). These and most animals studied were raised at 25 °C. The following BMP mutations were analyzed: witA12/witB11 (Aberle et al., 2002), mad10/mad12 (Sekelsky et al., 1995), gbb1 and gbb2 and gbb4 and UAS-gbb9.9 (Brian McCabe; Wharton et al., 1999), UAS-gbb9.1 (Stephan Thor; Wharton et al., 1999), DLIMKP1 (Eaton and Davis, 2005), sax4/Df(2R)cn7969 (Twombly et al., 1996) and babo32/babo32 (Brummel et al., 1999). The neuronal gbb rescue genotypes analyzed were elavC155-GAL4/+; gbb1/gbb2; UAS-gbb9.1/+, as well as elavC155-GAL4/+; UAS-GluedDNΔ84/+; UAS-gbb9.1/+, as well as elavC155-GAL4/+; UAS-GluedDNΔ84/UAS-CD8-GFP. The muscle gbb rescue genotype analyzed was gbb1/gbb2; MHC-GAL4/UAS-gbb9.1. The following wit rescue genotypes were analyzed: OK6-Gal4/UAS-wit; witA12/witB11 as well as OK371-GAL4/UAS-wit; witA12/witB11. For impairment of anterograde transport, the double heterozygous combination khc8/+; klcDf34ex5/+ (Martin et al., 1999) was analyzed. The following GAL4 lines were used in this study: elavC155-GAL4 (Lin and Goodman, 1994), OK6-GAL4 (Aberle et al., 2002), OK371-GAL4 (Mahr and Aberle, 2006), c380-Gal4 (also referred to as BG380-GAL4; Budnik et al., 1996), MHC-GAL4 (Schuster et al., 1996), elavGS-GAL4 (Haig Keshishian; Osterwalder et al., 2001), c929-GAL4 (Guillermo Marques; Marques et al., 2003). The following UAS lines were used: UAS-dad (Tom Kornberg), UAS-wit (Michael O’Connor; Marques et al., 2002), UAS-GluedDNΔ84 (Rod Murphey; Allen et al., 1999), and UAS-CD8-GFP. Mutant animals were raised on apple plates supplemented with wet yeast paste, and homozygous mutants were selected away from their heterozygous siblings. The w1118 strain controlled for the genetic background of mutations that exist in a w− background. The gbb mutations are in a yw mutant background and, therefore, yw was used as a control. For experiments using GeneSwitch (Osterwalder et al., 2001), 2–3 hour egg lays from parental genotypes elavGS-GAL4 and UAS-dad were conducted on apple juice plates. After the specified time (see text), animals were transferred to apple juice plates containing 25 μg/mL RU486 (a.k.a. mifepristone; Sigma) and topped with a yeast paste made up from 1 g dried yeast and 2 mL 50 μg/mL RU486 in water. RU486 was prepared as a stock solution at 10 mg/ml in ethanol.
Supplementary Material
Acknowledgments
We thank Paul Taghert (Washington University) for anti-PT2 antibody. We thank Guillermo Marques (University of Alabama) for c929-GAL4 flies. We thank Dion Dickman and Edward Pym for their initial work characterizing synaptic homeostasis in the syntaxin1A and csp mutants. We thank Edward Pym and C. Andrew Frank for helpful comments on a previous version of this manuscript. This work was supported by an NSF predoctoral fellowship (C.P.G.) and NIH Grant number NS39313 to GWD.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aberle H, Haghighi AP, Fetter RD, McCabe BD, Magalhaes TR, Goodman CS. wishful thinking encodes a BMP type II receptor that regulates synaptic growth in Drosophila. Neuron. 2002;33:545–558. doi: 10.1016/s0896-6273(02)00589-5. [DOI] [PubMed] [Google Scholar]
- Allen MJ, Shan X, Caruccio P, Froggett SJ, Moffat KG, Murphey RK. Targeted expression of truncated glued disrupts giant fiber synapse formation in Drosophila. J Neurosci. 1999;19:9374–9384. doi: 10.1523/JNEUROSCI.19-21-09374.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allan DW, St Pierre SE, Miguel-Aliaga I, Thor S. Specification of neuropeptide cell identity by the integration of retrograde BMP signaling and a combinatorial transcription factor code. Cell. 2003;113:73–86. doi: 10.1016/s0092-8674(03)00204-6. [DOI] [PubMed] [Google Scholar]
- Baines RA. Synaptic strengthening mediated by bone morphogenetic protein-dependent retrograde signaling in the Drosophila CNS. J Neurosci. 2004;24:6904–6911. doi: 10.1523/JNEUROSCI.1978-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brummel T, Abdollah S, Haerry TE, Shimell MJ, Merriam J, Rafter L, Wrana JL, O’Connor MB. The Drosophila activin receptor baboon signals through dSmad2 and controls cell proliferation but not patterning during larval development. Genes Dev. 1999;13:98–111. doi: 10.1101/gad.13.1.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budnik V, Koh YH, Guan B, Hartmann B, Hough C, Woods D, Gorczyca M. Regulation of synapse structure and function by the Drosophila tumor suppressor gene dlg. Neuron. 1996;17:627–640. doi: 10.1016/s0896-6273(00)80196-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrone J, Murthy VN. Synaptic gain control and homeostasis. Curr Opin Neurobiol. 2003;13:560–567. doi: 10.1016/j.conb.2003.09.007. [DOI] [PubMed] [Google Scholar]
- Byrd DT, Kawasaki M, Walcoff M, Hisamoto N, Matsumoto K, Jin Y. UNC-16, a JNK-signaling scaffold protein, regulates vesicle transport in C. elegans. Neuron. 2001;32:787–800. doi: 10.1016/s0896-6273(01)00532-3. [DOI] [PubMed] [Google Scholar]
- Cameron JS, Lhuillier L, Subramony P, Dryer SE. Developmental regulation of neuronal K+ channels by target-derived TGF beta in vivo and in vitro. Neuron. 1998;21:1045–1053. doi: 10.1016/s0896-6273(00)80622-4. [DOI] [PubMed] [Google Scholar]
- Cavalli V, Kujala P, Klumperman J, Goldstein LS. Sunday Driver links axonal transport to damage signaling. J Cell Biol. 2005;168:775–787. doi: 10.1083/jcb.200410136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charron F, Tessier-Lavigne M. Novel brain wiring functions for classical morphogens: a role as graded positional cues in axon guidance. Development. 2005;132:2251–2262. doi: 10.1242/dev.01830. [DOI] [PubMed] [Google Scholar]
- Chesnutt C, Burrus LW, Brown AM, Niswander L. Coordinate regulation of neural tube patterning and proliferation by TGFbeta and WNT activity. Dev Biol. 2004;274:334–357. doi: 10.1016/j.ydbio.2004.07.019. [DOI] [PubMed] [Google Scholar]
- Chizhikov VV, Millen KJ. Roof plate-dependent patterning of the vertebrate dorsal central nervous system. Dev Biol. 2005;277:287–295. doi: 10.1016/j.ydbio.2004.10.011. [DOI] [PubMed] [Google Scholar]
- Cull-Candy SG, Miledi R, Trautman A, Uchitel OD. On the release of transmitter at normal, myasthenia gravis and myasthenic syndrome affected human endplates. J Physiol. 1980;299:621–638. doi: 10.1113/jphysiol.1980.sp013145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dattatreyamurty B, Roux E, Horbinski C, Kaplan PL, Robak LA, Beck HN, Lein P, Higgins D, Chandrasekaran V. Cerebrospinal fluid contains biologically active bone morphogenetic protein-7. Exp Neurol. 2001;172:273–281. doi: 10.1006/exnr.2001.7728. [DOI] [PubMed] [Google Scholar]
- Davis GW. Homeostatic control of neural activity: from phenomenology to molecular design. Annu Rev Neurosci. 2006;29:307–323. doi: 10.1146/annurev.neuro.28.061604.135751. [DOI] [PubMed] [Google Scholar]
- Davis GW, DiAntonio A, Petersen SA, Goodman CS. Postsynaptic PKA controls quantal size and reveals a retrograde signal that regulates presynaptic transmitter release in Drosophila. Neuron. 1998;20:305–315. doi: 10.1016/s0896-6273(00)80458-4. [DOI] [PubMed] [Google Scholar]
- Desai NS, Cudmore RH, Nelson SB, Turrigiano GG. Critical periods for experience-dependent synaptic scaling in visual cortex. Nat Neurosci. 2002;5:783–789. doi: 10.1038/nn878. [DOI] [PubMed] [Google Scholar]
- Eaton BA, Fetter RD, Davis GW. Dynactin is necessary for synapse stabilization. Neuron. 2002;34:729–741. doi: 10.1016/s0896-6273(02)00721-3. [DOI] [PubMed] [Google Scholar]
- Eaton BA, Davis GW. LIM Kinase1 controls synaptic stability downstream of the type II BMP receptor. Neuron. 2005;47:695–708. doi: 10.1016/j.neuron.2005.08.010. [DOI] [PubMed] [Google Scholar]
- Frank CA, Kennedy MJ, Goold CP, Marek KW, Davis GW. Mechanisms underlying the rapid induction and sustained expression of synaptic homeostasis. Neuron. 2006;52:663–677. doi: 10.1016/j.neuron.2006.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gauthier LR, Charrin BC, Borrell-Pages M, Dompierre JP, Rangone H, Cordelieres FP, De Mey J, MacDonald ME, Lessmann V, Humbert S, Saudou F. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell. 2004;118:127–138. doi: 10.1016/j.cell.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Haghighi AP, McCabe BD, Fetter RD, Palmer JE, Hom S, Goodman CS. Retrograde control of synaptic transmission by postsynaptic CaMKII at the Drosophila neuromuscular junction. Neuron. 2003;39:255–267. doi: 10.1016/s0896-6273(03)00427-6. [DOI] [PubMed] [Google Scholar]
- Jiang N, Kolhekar AS, Jacobs PS, Mains RE, Eipper BA, Taghert PH. PHM is required for normal developmental transitions and for biosynthesis of secretory peptides in Drosophila. Dev Biol. 2000;226:118–136. doi: 10.1006/dbio.2000.9832. [DOI] [PubMed] [Google Scholar]
- Keshishian H, Kim YS. Orchestrating development and function: retrograde BMP signaling in the Drosophila nervous system. Trends Neurosci. 2004;27:143–147. doi: 10.1016/j.tins.2004.01.004. [DOI] [PubMed] [Google Scholar]
- LaMonte BH, Wallace KE, Holloway BA, Shelly SS, Ascano J, Tokito M, Van Winkle T, Howland DS, Holzbauer EL. Disruption of dynein/dynactin inhibits axonal transport in motor neurons causing late-onset progressive degeneration. Neuron. 2002;34:715–727. doi: 10.1016/s0896-6273(02)00696-7. [DOI] [PubMed] [Google Scholar]
- Lee-Hoeflich ST, Zhao X, Mehra A, Attisano L. The Drosophila type II receptor, Wishful thinking, binds BMP and myoglianin to activate multiple TGFβ family signaling pathways. FEBS Lett. 2005;579:4615–4621. doi: 10.1016/j.febslet.2005.06.088. [DOI] [PubMed] [Google Scholar]
- Lim DA, Tramontin AD, Trevejo JM, Herrera DG, Garcia-Verdugo JM, Alvarez-Buylla A. Noggin antagonizes BMP signaling to create a niche for adult neurogenesis. Neuron. 2000;28:713–726. doi: 10.1016/s0896-6273(00)00148-3. [DOI] [PubMed] [Google Scholar]
- Lin DM, Goodman CS. Ectopic and increased expression of Fasciclin II alters motoneuron growth cone guidance. Neuron. 1994;13:507–523. doi: 10.1016/0896-6273(94)90022-1. [DOI] [PubMed] [Google Scholar]
- Lo PC, Frasch M. Sequence and expression of myoglianin, a novel Drosophila gene of the TGF-β superfamily. Mech Dev. 1999;86:171–175. doi: 10.1016/s0925-4773(99)00108-2. [DOI] [PubMed] [Google Scholar]
- Lopez-Coviella I, Mellott TM, Kovacheva VP, Berse B, Slack BE, Zemelko V, Schnitzler A, Blusztajn JK. Developmental pattern of expression of BMP receptors and Smads and activation of Smad1 and Smad5 by BMP9 in mouse basal forebrain. Brain Res. 2006;1088:49–56. doi: 10.1016/j.brainres.2006.02.073. [DOI] [PubMed] [Google Scholar]
- Mahr A, Aberle H. The expression pattern of the Drosophila vesicular glutamate transporter: a marker protein for motoneurons and glutamatergic centers in the brain. Gene Expr Patterns. 2006;6:299–309. doi: 10.1016/j.modgep.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Marder E, Goaillard JM. Variability, compensation and homeostasis in neuron and network function. Nat Rev Neurosci. 2006;7:563–574. doi: 10.1038/nrn1949. [DOI] [PubMed] [Google Scholar]
- Martin AR. A further study of the statistical composition of the end-plate potential. J Physiol. 1955;130:114–122. doi: 10.1113/jphysiol.1955.sp005397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M, Iyadurai SJ, Gassman A, Gindhart JG, Hays TS, Saxton WM. Cytoplasmic dynein, the dynactin complex, and kinesin are interdependent and essential for fast axonal transport. Mol Biol Cell. 1999;10:3717–3728. doi: 10.1091/mbc.10.11.3717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marques G, Bao H, Haerry TE, Shimell MJ, Duchek P, Zhang B, O’Connor MB. The Drosophila BMP type II receptor Wishful Thinking regulates neuromuscular synapse morphology and function. Neuron. 2002;33:529–543. doi: 10.1016/s0896-6273(02)00595-0. [DOI] [PubMed] [Google Scholar]
- Marques G, Haerry TE, Crotty ML, Xue M, Zhang B, O’Connor MB. Retrograde gbb signaling through the Bmp type 2 receptor wishful thinking regulates systemic FMRFa expression in Drosophila. Development. 2003;130:5457–5470. doi: 10.1242/dev.00772. [DOI] [PubMed] [Google Scholar]
- McCabe BD, Marques G, Haghighi AP, Fetter RD, Crotty ML, Haerry TE, Goodman CS, O’Connor MB. The BMP homolog Gbb provides a retrograde signal that regulates synaptic growth at the Drosophila neuromuscular junction. Neuron. 2003;39:241–254. doi: 10.1016/s0896-6273(03)00426-4. [DOI] [PubMed] [Google Scholar]
- McCabe BD, Hom S, Aberle H, Fetter RD, Marques G, Haerry TE, Wan H, O’Connor MB, Goodman CS, Haghighi AP. Highwire regulates presynaptic BMP signaling essential for synaptic growth. Neuron. 2004;41:891–905. doi: 10.1016/s0896-6273(04)00073-x. [DOI] [PubMed] [Google Scholar]
- Murali D, Yoshikawa S, Corrigan RR, Plas DJ, Crair MC, Oliver G, Lyons KM, Mishina Y, Furuta Y. Distinct developmental programs require different levels of Bmp signaling during mouse retinal development. Development. 2005;132:913–923. doi: 10.1242/dev.01673. [DOI] [PubMed] [Google Scholar]
- Nakao A, Afrakhte M, Moren A, Nakayama T, Christian JL, Heuchel R, Itoh S, Kawabata M, Heldin NE, Heldin CH, ten Dijke P. Identification of Smad7, a TGFβ-inducible antagonist of TGF-β signaling. Nature. 1997;389:631–635. doi: 10.1038/39369. [DOI] [PubMed] [Google Scholar]
- Nguyen M, Parker L, Arora K. Identification of maverick, a novel member of the TGF-β superfamily in Drosophila. Mech Dev. 2000;95:201–206. doi: 10.1016/s0925-4773(00)00338-5. [DOI] [PubMed] [Google Scholar]
- Nie Z, Ranjan R, Wenniger JJ, Hong SN, Bronk P, Zinsmaier KE. Overexpression of cysteine-string proteins in Drosophila reveals interactions with syntaxin. J Neurosci. 1999;19:10270–10279. doi: 10.1523/JNEUROSCI.19-23-10270.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osterwalder T, Yoon KS, White BH, Keshishian H. A conditional tissue-specific transgene expression system using inducible GAL4. Proc Natl Acad Sci. 2001;98:12596–12601. doi: 10.1073/pnas.221303298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradis S, Sweeney ST, Davis GW. Homeostatic control of presynaptic release is triggered by postsynaptic membrane depolarization. Neuron. 2001;30:737–749. doi: 10.1016/s0896-6273(01)00326-9. [DOI] [PubMed] [Google Scholar]
- Petersen SA, Fetter RD, Noordermeer JN, Goodman CS, DiAntonio A. Genetic analysis of glutamate receptors in Drosophila reveals a retrograde signal regulating presynaptic transmitter release. Neuron. 1997;19:1237–1248. doi: 10.1016/s0896-6273(00)80415-8. [DOI] [PubMed] [Google Scholar]
- Plomp JJ, van Kempen GT, Molenaar PC. Adaptation of quantal content to decreased postsynaptic sensitivity at single endplates in alpha-bungarotoxin-treated rats. J Physiol. 1992;458:487–499. doi: 10.1113/jphysiol.1992.sp019429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pun S, Santos AF, Saxena S, Xu L, Caroni P. Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nat Neurosci. 2006;9:408–419. doi: 10.1038/nn1653. [DOI] [PubMed] [Google Scholar]
- Puls I, Jonnakuty C, LaMonte BH, Holzbaur EL, Tokito M, Mann E, Floeter MK, Bidus K, Drayna D, Oh SJ, Brown RH, Jr, Ludlow CL, Fischbeck KH. Mutant dynactin in motor neuron disease. Nat Genet. 2003;33:455–456. doi: 10.1038/ng1123. [DOI] [PubMed] [Google Scholar]
- Raible DW. Development of the neural crest: achieving specificity in regulatory pathways. Curr Opin Cell Biol. 2006;18:698–703. doi: 10.1016/j.ceb.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Rawson JM, Lee M, Kennedy EL, Selleck SB. Drosophila neuromuscular synapse assembly and function require the TGF-β type I receptor saxophone and the transcription factor Mad. J Neurobiol. 2002;55:134–150. doi: 10.1002/neu.10189. [DOI] [PubMed] [Google Scholar]
- Rios I, Alvarez-Rodriguez R, Marti E, Pons S. Bmp2 antagonizes sonic hedgehog-mediated proliferation of cerebellar granule neurons through Smad5 signaling. Development. 2004;131:3159–3168. doi: 10.1242/dev.01188. [DOI] [PubMed] [Google Scholar]
- Roman G, Endo K, Zong L, Davis RL. P[Switch], a system for spatial and temporal control of gene expression in Drosophila melanogaster. Proc Natl Acad Sci USA. 2001;98:12602–12607. doi: 10.1073/pnas.221303998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sandrock AW, Dryer SE, Rosen KM, Gozani SN, Kramer R, Theill LE, Fischbach GD. Maintenance of acetylcholine receptor number by neuregulins at the neuromuscular junction in vivo. Science. 1997;276:599–603. doi: 10.1126/science.276.5312.599. [DOI] [PubMed] [Google Scholar]
- Sanyal S, Narayanan R, Consoulas C, Ramaswami M. Evidence for autonomous AP1 function in regulation of Drosophila motor-neuron plasticity. BMC Neurosci. 2003;4:20. doi: 10.1186/1471-2202-4-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Massague J. Mechanisms of TGF-β signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- Schulze KL, Broadie K, Perin MS, Bellen HJ. Genetic and electrophysiological studies of Drosophila syntaxin-1A demonstrate its role in nonneuronal secretion and neurotransmission. Cell. 1995;80:311–320. doi: 10.1016/0092-8674(95)90414-x. [DOI] [PubMed] [Google Scholar]
- Schuster CM, Davis GW, Fetter RD, Goodman CS. Genetic dissection of structural and functional components of synaptic plasticity. I Fasciclin II controls synaptic stabilization and growth. Neuron. 1996;17:641–654. doi: 10.1016/s0896-6273(00)80197-x. [DOI] [PubMed] [Google Scholar]
- Sekelsky JJ, Newfeld SJ, Raftery LA, Chartoff EH, Gelbart WM. Genetic characterization and cloning of mothers against dpp, a gene required for decapentaplegic function in Drosophila melanogaster. Genetics. 1995;139:1347–1358. doi: 10.1093/genetics/139.3.1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun M, Thomas MJ, Herder R, Bofenkamp ML, Selleck SB, O’Connor MB. Presynaptic contributions of chordin to hippocampal plasticity and spatial learning. J Neurosci. 2007;27:7740–7750. doi: 10.1523/JNEUROSCI.1604-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teleman AA, Strigini M, Cohen SM. Shaping morphogen gradients. Cell. 2001;105:559–62. doi: 10.1016/s0092-8674(01)00377-4. [DOI] [PubMed] [Google Scholar]
- Tsuneizumi K, Nakayama T, Kamoshida Y, Kornberg TB, Christian JL, Tabata T. Daughters against dpp modulates dpp organizing activity in Drosophila wing development. Nature. 1997;389:627–631. doi: 10.1038/39362. [DOI] [PubMed] [Google Scholar]
- Turrigiano GG, Nelson SB. Homeostatic plasticity in the developing nervous system. Nat Rev Neurosci. 2004;5:97–107. doi: 10.1038/nrn1327. [DOI] [PubMed] [Google Scholar]
- Twombly V, Blackman RK, Jin H, Graff JM, Padgett RW, Gelbart WM. The TGF-β signaling pathway is essentially for Drosophila oogenesis. Development. 1996;122:1555–1565. doi: 10.1242/dev.122.5.1555. [DOI] [PubMed] [Google Scholar]
- Yung SY, Gokhan S, Jurcsak J, Molero AE, Abrajano JJ, Mehler MF. Differential modulation of BMP signaling promotes the elaboration of cerebral cortical GABAergic neurons or oligodendrocytes from a common sonic hedgehog-responsive ventral forebrain progenitor species. Proc Nat Acad Sci USA. 2002;99:16273–16278. doi: 10.1073/pnas.232586699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wharton KA, Cook JM, Torres-Schumann S, de Castro K, Borod E, Phillips DA. Genetic analysis of the bone morphogenetic protein-related gene, gbb, identifies multiple requirements during Drosophila development. Genetics. 1999;152:629–640. doi: 10.1093/genetics/152.2.629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Withers GS, Higgins D, Charette M, Banker G. Bone morphogenetic protein-7 enhances dendrite growth and receptivity to innervation in cultured hippocampal neurons. Eur J Neuroscience. 2000;12:106–116. doi: 10.1046/j.1460-9568.2000.00889.x. [DOI] [PubMed] [Google Scholar]
- Zinsmaier KE, Eberle KK, Buchner E, Walter N, Benzer S. Paralysis and early death in cysteine string protein mutants of Drosophila. Science. 1994;263:977–980. doi: 10.1126/science.8310297. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.