

Abstract
Cystic fibrosis transmembrane conductance regulator (CFTR) is an adenosine triphosphate (ATP)-gated chloride channel. ATP-induced dimerization of CFTR's two nucleotide-binding domains (NBDs) has been shown to reflect the channel open state, whereas hydrolysis of ATP is associated with channel closure. Pyrophosphate (PPi), like nonhydrolytic ATP analogues, is known to lock open the CFTR channel for tens of seconds when applied with ATP. Here, we demonstrate that PPi by itself opens the CFTR channel in a Mg2+-dependent manner long after ATP is removed from the cytoplasmic side of excised membrane patches. However, the short-lived open state (τ ∼1.5 s) induced by MgPPi suggests that MgPPi alone does not support a stable NBD dimer configuration. Surprisingly, MgPPi elicits long-lasting opening events (τ ∼30 s) when administrated shortly after the closure of ATP-opened channels. These results indicate the presence of two different closed states (C1 and C2) upon channel closure and a state-dependent effect of MgPPi on CFTR gating. The relative amount of channels entering MgPPi-induced long-open bursts during the ATP washout phase decreases over time, indicating a time-dependent dissipation of the closed state (C2) that can be locked open by MgPPi. The stability of the C2 state is enhanced when the channel is initially opened by N6-phenylethyl-ATP, a high affinity ATP analogue, but attenuated by W401G mutation, which likely weakens ATP binding to NBD1, suggesting that an ATP molecule remains bound to the NBD1 site in the C2 state. Taking advantage of the slow opening rate of Y1219G-CFTR, we are able to identify a C2-equivalent state (C2*), which exists before the channel in the C1 state is opened by ATP. This closed state responds to MgPPi much more inefficiently than the C2 state. Finally, we show that MgAMP-PNP exerts its effects on CFTR gating via a similar mechanism as MgPPi. The structural and functional significance of our findings is discussed.
INTRODUCTION
The CFTR, a member of the ATP-binding cassette (ABC) transporter superfamily (Riordan et al. 1989), is a phosphorylation-activated but ATP-gated chloride channel in epithelial cells. Mutations of the CFTR gene resulting in malfunction of this channel cause the lethal genetic disease cystic fibrosis. The CFTR protein incorporates two nucleotide-binding domains (NBDs), NBD1 and NBD2, which serve as the gating machinery to drive the conformational changes during gating transitions (for reviews see Gadsby et al., 2006; Chen and Hwang, 2008). Each NBD holds the Walker A and Walker B motifs that form the major constituents for interactions with ATP (Walker et al., 1982). There is compelling evidence that, similar to other ABC transporters (for review see Higgins and Linton, 2004), once ATP binds to the nucleotide-interacting motifs, the two NBDs of CFTR approach each other to form a head-to-tail dimer with two ATP molecules sandwiched at the dimer interface, and this intramolecular interaction in turn leads to opening of the channel (Vergani et al., 2005; Mense et al., 2006). Because a biochemically stable NBD dimer formation in other ABC transporters is observed only when ATPase activity is abolished (Moody et al., 2002; Smith et al., 2002), it is proposed that ATP hydrolysis causes fast separation of the two NBDs and thus results in closing of the CFTR channel.
The idea that ATP hydrolysis precedes channel closing is further supported by the observations that CFTR mutations whose ATPase activity is abrogated (e.g., K1250A and E1371S) (Ramjeesingh et al., 1999) can remain open for minutes (Gunderson and Kopito, 1995; Zeltwanger et al., 1999; Vergani et al., 2003; Bompadre et al., 2005b), and that channel closure is markedly delayed in the presence of nonhydrolyzable ATP analogue AMP-PNP (Hwang et al., 1994), or of inorganic phosphate analogue orthovanadate, which presumably forms a stable complex with the hydrolytic product ADP (Baukrowitz et al., 1994). Gunderson and Kopito (1994) reported that, similar to AMP-PNP or vanadate, pyrophosphate (PPi), although it fails to open the channel by itself, can “lock” the CFTR channel into a prolonged open state in the presence of ATP. Carson et al. (1995) confirmed and expanded this observation by showing that PPi also strongly potentiates cystic fibrosis–associated mutations ΔF508 and G551S.
Data from several laboratories have suggested that PPi exerts its effect via binding to the ATP-binding site at NBD2. First, several NBD2 mutations, but not corresponding NBD1 mutations (Gunderson and Kopito, 1995; Cotten et al., 1996; Berger et al., 2002), abolish PPi stimulation. Second, PPi fails to enhance the activity of mouse CFTR, and replacing human NBD2 with the equivalent region of mouse CFTR also abolishes the effect of PPi (Lansdell et al., 1998; Scott-Ward et al., 2007). However, the idea that NBD2 determines the PPi sensitivity cannot explain several other findings. For example, PPi, when applied in the absence of ATP, fails to lock open the channel (Gunderson and Kopito, 1994, Carson et al., 1995). Biochemical studies show that PPi paradoxically produces a concentration-dependent increase in 8-N3ATP photolabeling of CFTR (Carson et al., 1995). Furthermore, several results suggest an involvement of NBD1 in PPi's action on CFTR gating. Csanady et al. (2005) reported that deletion of the N-terminal regulatory insertion in NBD1 somewhat weakens PPi's effect. Cai et al. (2006) showed that PPi cannot potentiate G1349D, a mutation at the signature sequence of NBD2, which forms the ATP-binding pocket with the nucleotide-interacting motifs of NBD1. These observations can be nicely explained if one hypothesizes that ATP binding at NBD1 primes the CFTR channel into an activated state that can be locked open by PPi (compare Gunderson and Kopito, 1995). This hypothesis, however appealing, is short of direct evidence.
Because most of the experiments described above were performed when ATP and PPi were applied together, it is difficult to assess whether PPi acts on an open state (compare Hwang et al., 1994) or a closed state (Gunderson and Kopito, 1995). We reasoned that if the open state reflects a stable NBD dimer (Vergani et al., 2005), it seems unlikely that PPi would have access to the nucleotide-binding site buried at the dimer interface in an open-channel configuration. Thus, it is more likely that PPi acts on a closed state. Indeed, in the current study, we provide evidence that once leaving the ATP-induced open state, closed channels respond to PPi in a Mg-dependent manner. We demonstrate that shortly after channel closure, MgPPi alone locks open a closed state (C2), where one ATP molecule is not yet dissociated from the NBD1 site. This result echoes the hypothesis that ATP primes the channel by binding to NBD1.
There are multiple closed states in the gating cycle of CFTR. Here, we are able to differentiate two more closed states (C1 and C2*) based on their different responses to MgPPi. The C1 state exists long after channel closure, and the binding of MgPPi to this state induces relatively short openings. The C2* state is present before the channel is opened from the C1 state by ATP. Although this C2-equivalent state likely has one ATP molecule in the NBD1 site, it has a weaker response to MgPPi compared with the C2 state. Functional and structural implications of our results will be discussed.
MATERIALS AND METHODS
Cell culture and transient expression system
Chinese hamster ovary (CHO) cells were grown at 37°C and 5% CO2 in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. The cDNA constructs of wild-type (WT) or mutant CFTR were cotransfected with pEGFP-C3 (Clontech Laboratories, Inc.) encoding the green fluorescence protein using PolyFect transfection reagent (QIAGEN) according to the manufacturer's instructions. The transfected CHO cells were plated on sterile glass chips in 35-mm tissue culture dishes and incubated at 25°C. Electrophysiological experiments were performed 2–5 d after transfection.
Electrophysiological recordings
Before inside-out patch clamp recordings, glass chips containing CHO cells transfected with various CFTR constructs, W401G, Y1219G, S1347G, E1371S, and WT-CFTR, were transferred to a continuously perfused chamber located on the stage of an inverted microscope (Olympus). Patch clamp pipettes were made from borosilicate capillary glass using a two-stage vertical puller (Narishige). The pipette tips were fire polished with a homemade microforge to ∼1-µm external diameter, resulting in a pipette resistance of 2∼4 MΩ in the bath solution.
CFTR channel currents were recorded at room temperature with an EPC-10 patch clamp amplifier, filtered at 100 Hz with an eight-pole Bessel filter (Warner Instrument Corp.) and captured onto a hard disk at a sampling frequency of 500 Hz. The membrane potential was held at −60 mV. At this membrane potential, downward deflections represent channel openings. However, we inverted the current direction for clearer data presentations.
All inside-out patch clamp experiments were performed with a fast solution exchange device (SF-77B; Warner Instrument Corp.), which can minimize the dead time of solution change to ∼20 ms (Coric et al., 2003). To test the dead time of our solution exchange, we perfused two solutions with different concentrations of NaCl to the patch pipette. Exponential fit of the resulting current changes yielded a time constant of ∼30 ms.
Chemicals and composition of solutions
The pipette solution contained (in mM): 140 NMDG chloride (NMDG-Cl), 2 MgCl2, 5 CaCl2, and 10 HEPES, pH 7.4 with NMDG. Cells were perfused with a bath solution containing (in mM): 145 NaCl, 5 KCl, 2 MgCl2, 1 CaCl2, 5 glucose, 5 HEPES, and 20 sucrose, pH 7.4 with NaOH. After the establishment of an inside-out configuration, the patch was perfused with a standard perfusion solution (i.e., intracellular solution) containing (in mM): 150 NMDG-Cl, 2 MgCl2, 10 EGTA, and 8 Tris, pH 7.4 with NMDG.
MgATP, PPi (tetrasodium salt), and PKA were purchased from Sigma-Aldrich. AMP-PNP was obtained from Roche. N6-(2-phenylethyl)-ATP (P-ATP) was from Biolog Life Science Institute. MgATP, PPi, and AMP-PNP were stored in 250 mM of stock solution at −20°C. 10 mM P-ATP stock solution was stored at −80°C. The [PKA] used in this study was 25 U ml−1. All nucleotides and PPi were diluted in the perfusion solution to the concentrations as indicated in the figures, and the pH was adjusted to 7.4 with NMDG. When NaAMP-PNP or NaPPi was used, the same concentration of MgSO4 was added unless indicated otherwise.
When high concentrations of Mg2+ and NaPPi (>5 mM) were mixed rapidly, precipitations occurred immediately in spite of continuous stirring of the solution. The substances were in a form of amorphous glass-like granules occurring in large number under the microscope. However, when Mg2+ and NaPPi were mixed slowly, the formation of precipitations was much slower, allowing us to test the effects of a wider range of [PPi] (2∼15 mM) on CFTR gating. To minimize a decrease of effective [PPi] due to precipitations, PPi solutions were used no more than 4 h after preparation. In addition, whenever possible, we preferred using PPi concentration <5 mM (e.g., 2 mM) to avoid this problem.
Data analysis and statistics
Steady-state macroscopic current amplitude was measured using the Igor Pro program (version 4.07; Wavemetrics). The baseline current was subtracted before the data were used for presentation. To estimate channel open time, we derived time constants from macroscopic relaxations upon the removal of CFTR ligands. Current relaxations were fitted with single- or double-exponential functions using a Levenberg-Marquardt–based algorithm within the Igor Pro program. Due to the limitation of the program, a current decay trace of two components may not be resolvable by a double-exponential fit when one component has relatively small fractional amplitude (e.g., Figs. 3 B and 4). In this case, single-exponential fitting was used to estimate the time constant of the dominant component.
Figure 3.

MgPPi locks open the closed WT channels. (A) Steady-state macroscopic current activated by 2 mM ATP plus PKA was further increased by a subsequent addition of 10 mM MgPPi. Red line represents single-exponential fit of the current relaxation upon the removal of ATP and MgPPi. (B) MgPPi locks open closed channels shortly after the removal of ATP. Note a 20-s time lapse between the removal of ATP and the application of MgPPi. It should also be noted that the initial part of the current relaxation seems to be faster but was too small to be resolved. Red line marks single-exponential fit of the current decay upon the removal of MgPPi. (C) As the washout time was prolonged to 40 s, fewer channels were locked open by 10 mM MgPPi. Superimposed double-exponential fit (red line) yields τ1 = 1.86 ± 0.34 s and τ2 = 32.46 ± 7.41 s (n = 5).
Figure 4.

Time-dependent reopening of WT-CFTR by 10 mM MgPPi. Mg PPi was applied at different time points after ATP washout. The current amplitude of the slow component (red squares, left y axis) and the fast component (blue squares, right y axis) derived from double-exponential fitting were normalized to ATP current at t = 0. For t = 0 ∼20 s, the fast component was too small to be resolved. Fitting the data points (red curve) by a single-exponential function estimates the lifetime of the C2state to be 27.4 s.
All results are presented as means ± SEM; n represents the number of experiments. Student's t test or paired t test was performed with Sigmaplot (version 8.0; SPSS Science). P < 0.05 was considered significant.
Online supplemental material
Fig. S1 shows that S1347G-CFTR has a weaker response to MgPPi than WT. The observation that MgPPi induces shorter opening events than ATP in E1371S channels is shown in Fig. S2. In Fig. S3, the steady-state current induced by MgPPi can be maintained for minutes once the channel is continuously exposed to MgPPi. The interpretation of these data is presented in Results and Discussion. Figs. S1–S3 are available at http://www.jgp.org/cgi/content/full/jgp.200810186/DC1.
RESULTS
MgPPi supports WT-CFTR gating in the absence of ATP
It is reported that the nonhydrolytic ATP analogue AMP-PNP by itself can slightly increase CFTR channel activity (Aleksandrov et al., 2000; Vergani et al., 2003; see below). We first tested whether PPi has a similar effect in supporting CFTR gating. Macroscopic current was activated by cytoplasmic application of PKA catalytic subunit plus 2 mM ATP in inside-out patches containing hundreds of WT channels (Fig. 1 A). Subsequent withdrawal of ATP resulted in a fast current decay that can be fitted with a single-exponential function with a time constant of 0.42 ± 0.03 s (Fig. 1 B; n = 12). 3 min after ATP removal, patches were exposed to a range of [PPi] supplemented with the same concentration of Mg2+. MgPPi could increase the current in a concentration-dependent manner in the complete absence of ATP (Fig. 1 A). The dose–response relationship for the effect of MgPPi is obtained by normalizing the macroscopic currents induced by different concentrations of MgPPi to that by 2 mM ATP (Fig. 2 B, red line).
Figure 1.

The effect of Mg PPi on WT-CFTR. (A) A recording of WT-CFTR channels in an excised inside-out membrane patch. The channels were activated with 2 mM ATP plus 25 U/ml PKA (not depicted). After phosphorylation-dependent activation, application and removal of ATP result in rapid current rise and decay, respectively. 3 min after washout of ATP, channels were opened by different concentrations of PPi plus Mg2+. 15 mM PPi supplemented with 15 mM MgPPi still seems unable to saturate the macroscopic current. (B) Macroscopic current decay after ATP withdrawal (left) and the ensemble current relaxation after washout of 15 mM MgPPi (right). Channels opened by MgPPi appear to close more slowly than those opened by ATP.
Figure 2.

PPi opens WT-CFTR in a Mg-dependent manner. (A) WT-CFTR channels were exposed to a nucleotide-free solution for 3 min before they were treated with different combinations of [PPi] and [Mg2+]. Raising PPi concentration fails to further increase channel activity if [Mg2+] was not increased simultaneously. (B) Current elicited by PPi at different concentrations was normalized to the original ATP current. (Red line) The PPi solution was supplement with the same concentration of Mg2+. (Blue line) [Mg2+] in the PPi solution was fixed at 2 mM.
For each MgPPi concentration, current traces after MgPPi removal from at least six patches were summed to give ensemble current relaxations (Fig. 1 B). The relaxation time courses can be fitted with a single-exponential function with a similar time constant (τ ∼1.5 s) for all MgPPi concentrations tested, suggesting that the open time for MgPPi-activated channels is longer than that of ATP-opened channels. However, because the channel open time is independent of [MgPPi], a higher concentration of MgPPi should increase the channel open probability mainly by increasing the opening rate. It is noted here that compared with ATP, which maximally activates CFTR at low millimolar concentrations (Zeltwanger et al., 1999), 15 mM MgPPi, the highest concentration tested, was still not able to saturate the current (Fig. 2 B). These results indicate that although MgPPi can increase the open probability of CFTR, its potency is far lower than ATP.
Our observation that MgPPi alone can support CFTR gating contradicts previous reports (Gunderson and Kopito, 1994; Carson et al., 1995) that PPi by itself is unable to open CFTR channels. We suspect that [Mg2+] in the PPi solution may be the culprit because, unlike the current study, it is unclear if additional Mg2+ was added to the PPi solution in those reports. Fig. 2 A shows a continuous current trace demonstrating that PPi's ability to increase channel activity is dependent on Mg2+. The current induced by 10 mM PPi with 10 mM Mg2+ is significantly higher than that with 10 mM PPi and 2 mM Mg2+ (P < 0.01). In fact, when [Mg2+] in 10- and 15-mM PPi solutions was reduced to 2 mM, the ability of PPi in supporting channel gating was dramatically attenuated to that of 2 mM MgPPi (Fig. 2 B, blue line), suggesting that it is MgPPi, not PPi, that activates CFTR. The observed difference is not due to the difference in total salt concentration because the result is essentially the same after adjusting all test solutions to the same salt concentration with NMDG-Cl (not depicted). Unfortunately, the Mg2+ dependence we identified in the current study also prevents us from testing the effect of higher concentrations of MgPPi (e.g., >20 mM) due to the formation of precipitates (see Materials and methods).
Different closed states of WT-CFTR revealed by MgPPi
It has been known for years that PPi, in the presence of ATP, “locks” the channel in an open state with a macroscopic relaxation time constant in tens of seconds (Csanady et al., 2005). In Fig. 3 A, we confirmed these results by showing that adding 10 mM MgPPi to 2 mM ATP further increased the macroscopic WT-CFTR current, and the current decay upon the removal of ATP and MgPPi follows a very slow time course with a time constant of 29.4 ± 4.04 s (n = 5). The difference in the relaxation time constant between MgPPi-opened channels (τ ∼1.5 s in Fig. 1 B) and those opened by ATP plus MgPPi suggests a state-dependent modulation of CFTR gating by MgPPi. It appears that an exposure of the channel to ATP dramatically alters the effect of MgPPi.
To further explore this state-dependent modulation of CFTR gating, we modified our experimental protocol so that MgPPi was applied at different time points after the removal of ATP. Strikingly, we found that when 10 mM MgPPi was applied 20 s after ATP removal, instead of 3 min as in Fig. 1 A, a significant amount of current was elicited (Fig. 3 B; 65 ± 6% of ATP-activated current; n = 4). This PPi effect, similar to that described above (Fig. 2), is Mg2+ dependent (not depicted). After washout of MgPPi, the macrocopic current decayed slowly. Except for the very beginning of the current decay, the time course can be well fitted with a single-exponential function with a time constant of 29.98 ± 6.14 s (Fig. 3 B; n = 4), indicating that many of those closed WT-CFTR channels were locked open by MgPPi alone! These results suggest that there are at least two different closed states (C1 and C2) after ATP removal, and these two states can be differentiated by their distinct responses to MgPPi. When the channel is opened by MgPPi from the C1 state, the open time is ∼1.5 s as depicted in Fig. 1. In contrast, MgPPi locks open the channel with a time constant of ∼30 s when it is in the C2 state.
Transition of C2 to C1
Fig. 3 C shows an experiment where 10 mM MgPPi was applied 40 s after nucleotide withdrawal. Compared with the result shown in Fig. 3 B, 10 mM MgPPi induced a smaller current and its removal resulted in a clear biphasic current decay. The time course can be fitted with a double-exponential function (Fig. 3 C, inset), indicating the presence of two distinct open states. The time constant (32.46 ± 7.41 s; n = 5) for the more stable open state is very similar to that of “locked open” channels shown above. However, the relative current amplitude (27 ± 2% of ATP-induced current; n = 5) attributed to the locked open channels is significantly smaller than that shown in Fig. 3 B when the washout time is 20 s, suggesting a slow dissipation of the C2 closed state. In contrast, the less stable open state with a time constant of 1.86 ± 0.34 s (n = 5), very similar to the time constant for the MgPPi-opened channels after 3 min of ATP removal (Fig. 1 A), made a distinguishable appearance in this condition (compare with Fig. 3 B). Therefore, the longer the time lapse after ATP washout, the more channels were accumulated in the C1 state upon which MgPPi acted to induce less stable openings.
To further elaborate this time-dependent alteration of C1 and C2 state distribution, we applied 10 mM MgPPi after WT channel, initially opened by ATP, was subsequently washed with nucleotide-free solution for different lengths of time. Except for the data obtained with a washout time of <20 s (see Material and methods for details), the current relaxation after MgPPi removal was fitted with a double-exponential function giving the fractional amplitude of slow and fast components. Fig. 4 plots the waiting time versus the two fractions of current induced by MgPPi relative to the original current in the presence of PKA and ATP. The left-hand y axis represents the ratio of the current attributable to lock-open bursts at time t to that activated by ATP at time zero (red squares). This ratio decreases monotonically as the washout time was prolonged. In contrast, when the current resulted from short openings was normalized to ATP-induced current (Fig. 4, right-hand y axis, blue squares), the ratio gradually increased and reached a plateau after 60 s of washout time.
Overall, these data support the idea of a two-step channel closure scheme:
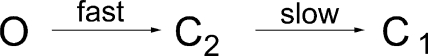 |
(SCHEME 1) |
The first step, O→C2, is fast presumably because of a rapid hydrolysis of ATP and subsequent dissociation of the hydrolytic products. The second step, C2→C1, is slow as MgPPi can lock open a significant number of channels even 20 s after washout of ATP. It should be noted here that this scheme may be oversimplified because there are likely other closed states in the channel closure process that we fail to identify due to the limitation of the current experimental design.
Assuming that MgPPi can efficiently lock open all channels in the C2 state, in theory the lifetime of the C2 state can be approximated by exponential fitting of the slow component of the current induced by MgPPi (Fig. 4, red line). Unfortunately, because of the slow current rise upon the application of MgPPi (probably due to a low binding affinity of MgPPi), many channels in the C2 state already dissipate without being locked open during the rising phase of MgPPi-induced current increase. Therefore, the time constant of 27.4 s derived from a single-exponential fit of data points (red squares) in Fig. 4 can only be considered a rough estimate of the lifetime of the C2 state.
Possible biochemical basis of the stable C2 closed state
What makes C1 and C2 different closed states? We noticed that although a long washout of ATP finally results in a complete transition of the C2 state to the C1 state, reapplication of ATP can once again bring channels back to the O state and subsequently the C2 state (not depicted). Thus, the channel seems to have an ability to remember the gating history for tens of seconds and if so, this long-lasting “memory” likely comes from a prior exposure of the channels to ATP.
It has been shown that although the NBD2 of CFTR has a high nucleotide turnover rate, presumably due to its ATPase activity, ATP can be “trapped” in NBD1 for a long time (Szabo et al., 1999; Aleksandrov et al., 2001, 2002; Basso et al., 2003). Using these biochemical data, Vergani et al. (2003) proposed that once ATP binds to the NBD1 site, it is occluded there; therefore, during a gating cycle, when the channel closes, the ATP molecule remains bound at the NBD1 site. We wondered if the C2 state described above may represent this elusive state, defined based on biochemical data. If the C2 state indeed has a bound ATP in NBD1, the effect of MgPPi on the C2 state should meet two key predictions. First, with the NBD1 site occupied by ATP, MgPPi likely binds to NBD2 to lock open the channel. Reports from several different groups have already suggested that is the case (see Introduction). Thus, when MgPPi and ATP (or other nucleotides such as ADP) are applied together, they may compete for the NBD2 site. Second, the binding affinity of ATP or ATP analogue in NBD1 may affect the lifetime of the C2 state.
To test whether MgPPi and ATP, when applied together, compete for a common binding site, we prepared solutions containing 2 mM MgPPi with various concentrations of ATP. In Fig. 5 A, WT-CFTR channels in inside-out patches were first activated by PKA and 2 mM ATP. After the macroscopic current reached the steady state, subsequent application of 10 mM ATP plus 2 mM MgPPi further increased the current. Washout of this mixed solution resulted in a bi-exponential current decay with a fast component reflecting ATP-induced open state (τ ∼400 ms) and a slow component (τ ∼30 s) representing channels in the lock-open state (compare Vergani et al., 2003). After the current reached the baseline, PKA and 2 mM ATP were applied again to ensure full phosphorylation of the channels. Once the channels were fully activated, 2 mM ATP and 2 mM MgPPi were applied. Note that not only is the steady-state current with 2 mM ATP plus 2 mM MgPPi higher than that with 10 mM ATP plus 2 mM MgPPi, the current increase is also faster. In addition, the biphasic current decay after the removal of 2 mM ATP plus 2 mM MgPPi shows a noticeably smaller faction of the fast phase, indicating that more channels enter the lock-open state (see the arrows in Fig. 5 A). Thus, although MgPPi only locks open CFTR after the channel is primed by ATP, a higher [ATP] actually exerts an inhibitory effect. As summarized in Fig. 5 B (red symbols), the fraction of the slow component decreased as the concentration of ATP was elevated. On the other hand, when [ATP] was fixed at 2 mM, an increased concentration of MgPPi led to a larger fraction of the slow component (Fig. 5 B, blue symbols). These results are consistent with the idea that ATP and MgPPi compete for a common binding site.
Figure 5.

ATP and MgPPi compete for a common binding site. (A) Steady-state macroscopic current of WT-CFTR activated by 2 mM ATP and PKA was further increased by the application of 2 mM MgPPi plus 2 mM ATP (right) or 2 mM MgPPi plus 10 mM ATP (left). With lower [ATP] in the PPi solution, the current increase proceeds more rapidly (right). Fitting the current relaxation yields two time constants after the removal of 2 mM ATP plus 2 mM PPi (τ1 = 0.55 ± 0.06 s and τ2 = 26.9 ± 1.56 s; n = 12) or after 2 mM MgPPi plus 10 mM ATP (τ1 = 0.34 ± 0.03 s and τ2 = 30.4 ± 3.1 s; n = 4). Arrows indicate the end of the fast component during current decay. (B) The fractional amplitude of the slow component under different combinations of [MgPPi] and [ATP]. Raising [ATP] (red line, lower x axis) or reducing [PPi] (blue line, upper x axis) decreases the fractional amplitude of the slow component. (C) Effects of PPi on Y1219G-CFTR. 2 mM ATP plus PKA activated a small amount of current due to a reduced apparent affinity for ATP by the mutation. Upon application of 2 mM PPi plus 2 mM ATP, the current was greatly enhanced. The current decays monotonically with τ = 30.7 ± 4.5 s (n = 5).
It seems counterintuitive that not all channels in the membrane patches are locked open in the presence of equal concentrations of ATP and MgPPi when one considers the fact that the lock-open state (τ ∼30 s) is >50-fold more stable than the regular open state (τ ∼400 ms). This phenomenon, however, can be readily explained by a proposition that ATP out-competes MgPPi in opening the channel. For example, when MgPPi and ATP are both at 2 mM, the slow component has a time constant of τs = 26.9 ± 1.56 s (n = 12), whereas the time constant of the fast component (τf) is 0.55 ± 0.06 s. From the fractional amplitude of the slow component (70.2 ± 3.7%), we estimated that only 1 in every ∼20 openings results in a lock-open event. This conclusion is probably not surprising as results from Zhou et al. (2006) suggested that the ring–ring stacking interaction (Lewis et al., 2005; but compare Lewis et al., 2004 and Thibodeau et al., 2005), which MgPPi is lacking, between the side chain of aromatic residues in NBDs (W401 in NBD1 or Y1219 in NBD2) and the adenine ring of ATP may play an important role in nucleotide binding. In fact, we have already shown above that MgPPi has a lower potency than ATP to open channels from the C1 state.
If NBD2 is indeed the common binding site for ATP and MgPPi, one expects that removal of the side chain of Y1219 should diminish ATP's advantage in competing for the binding pocket. Fig. 5 C shows that 2 mM MgPPi, when added to 2 mM ATP solution, dramatically increased the Y1219G-CFTR macroscopic current. Unlike the data for WT-CFTR (Fig. 5 A), the current relaxation upon the removal of ATP and MgPPi follows a monotonic decay with a time constant of τ = 30.7 ± 4.5 s (n = 5), indicating that almost all Y1219G-CFTR channels have been locked open under this experimental condition. As a control, when a similar experiment was performed for W401G, ATP still out-competes MgPPi as demonstrated by a large fraction of the fast component during current relaxation (not depicted).
Although Scheme 1 depicts the first step of channel closure as a single step, as described above, this is likely an oversimplification. If this step indeed involves ATP hydrolysis and dissociation of the hydrolytic products as we speculated above, there must exist other closed state configurations. For example, one can imagine one closed state with ADP remaining bound and the other closed state with an empty binding pocket. These two possible states can be differentiated by adding MgPPi in the presence of ADP. If ADP inhibits the effect of MgPPi as ATP, one can argue that the closed state where MgPPi acts should have an unoccupied NBD2. In Fig. 6 A (n = 5), we show that a direct solution switch from 2 mM ATP to 2 mM MgPPi (a deadtime of ∼20 ms) resulted in fast channel closure, followed by a reopening of the channels. However, when the MgPPi solution was mixed with 5 mM ADP, the second-phase current rise was mostly abolished (summarized in Fig. 6 B), indicating that ADP indeed inhibits the effect of MgPPi. It is known that ADP inhibits the ATP-dependent WT-CFTR activity (Anderson and Welsh, 1992; Gunderson and Kopito, 1994; Winter et al., 1994; Schultz et al., 1995) mainly by competing for NBD2 of CFTR (Bompadre et al., 2005a). Our results thus suggest that regardless of whether the NBD2 binding site is occupied by ATP that leads to channel opening, or by ADP that keeps the channel in a closed state with a bound ADP at the NBD2 site, MgPPi fails to exert its effects. We therefore propose that NBD2 is vacant when the channel is in the C2 state.
Figure 6.

ADP inhibits the reopening of WT-CFTR by MgPPi. (A) WT channels were first opened by ATP. Once current reached steady state, a direct switch of the solution to MgPPi (dead time, ∼30 ms) resulted in a rapid decrease of the current, followed by an increase of the current. In the same patch, the effect of MgPPi was dramatically reduced when applied together with 5 mM ADP. (B) Summary of data presented in A. Steady-state current induced by MgPPi or MgPPi plus ADP was normalized to original ATP-dependent current. IMgPPi/IATP = 0.53 ± 0.036 (n = 5); IMgPPi-ADP/IATP = 0.065 ± 0.014 (n = 5; P < 0.01).
Lifetime of the C2 state is affected by the nucleotide biding affinity in NBD1
Although the NBD2 site is vacant in the C2 state, one ATP molecule may remain tightly bound at NBD1 to account for the long-lasting memory assumed by the C2 state. We hypothesize that it is the dissociation of ATP at NBD1 that is responsible for the C2→C1 transition. This hypothesis predicts that a high affinity ATP analogue in NBD1 should slow down the dissipation of the C2 state. To test this prediction, we used P-ATP, which is 50-fold more potent than ATP (Zhou et al., 2005), to activate WT-CFTR. After macroscopic current was first elicited by the nucleotide at a saturating concentration (i.e., 2 mM ATP or 50 µM P-ATP), changing the perfusion solution directly to 2 mM MgPPi alone caused a rapid current decline due to channel closure through ATP hydrolysis and a subsequent current rise from the lock-open channels by MgPPi (Fig. 7 A). We measured the ratio of the peak current induced by MgPPi (IPPi) to the original nucleotide-activated current (IN). It should be noted that because the open probabilities of WT-CFTR in the presence of ATP or P-ATP are 0.45 and 0.65, respectively (Zeltwanger et al., 1999; Zhou et al., 2006), a fair comparison of the two ratios (IPPi /IATP and IPPi /IPATP) requires normalization of the open probability. We recalibrated the ratio using the following strategy. First, we obtained the current level (IN) reflecting all channels being in the open state by dividing IATP (or IP-ATP) with the open probability (0.45 for IATP, and 0.65 for IP-ATP). Then, we divided the current generated by MgPPi (i.e., IPPi) with IN (the left-hand y axis of Fig. 7 C). This type of analysis more accurately portrays the actual number of channels entering the lock-open state under different conditions. As summarized in Fig. 7 C, when the channels were first opened by P-ATP, 2 mM MgPPi locks open 49.3 ± 5.4% (n = 4) of all channels in the patch. In contrast, when the channels were pretreated with ATP, only 19.5 ± 1.7% (n = 5) of the channels were locked open.
Figure 7.

The effect of MgPPi can be modulated by altering the ligand–NBD1 interaction. (A) WT channels were exposed to 2 mM MgPPi solution immediately after washout of 2 mM ATP or 50 µM P-ATP. (B) The lock-open efficiency of MgPPi was reduced by the W401G mutation, and P-ATP can partially restore the effectiveness of MgPPi on W401g-CFTR. (C) Summary of data presented in A and B. *, P < 0.01.
We then used the same protocol to test the effect of MgPPi on the W401G mutation, which likely decreases the nucleotide-binding affinity in NBD1 (Zhou et al., 2006) because the imidazole ring of this tryptophan residue forms a ring–ring stacking interaction with the adenine ring of ATP in the crystal structure of human NBD1 (Lewis et al., 2005). As shown in Fig. 7 B, changing the solution containing 2 mM ATP immediately to one with 2 mM PPi only locked open a small fraction of the channels (6.3 ± 1.9%; n = 5). Previously, Zhou et al. (2006) showed that P-ATP may assume a tighter binding than ATP at NBD1 of W401G-CFTR. Indeed, opening of W401G-CFTR channels with P-ATP results in a higher fraction (24 ± 1.5%; n = 5) of lock-open channels (Fig. 7, B and C). Collectively, these data suggest that the binding affinity of nucleotide in NBD1 can affect the stability of the C2 state. Interestingly, we also found that S1347G, a mutation at NBD2 signature sequence, which presumably forms the ATP-binding pocket with NBD1's Walker A domain upon NBD dimer formation, greatly attenuates the stability of the C2 state, and this reduced stability can also be partly reversed by P-ATP (Fig. S1). More detailed interpretations of this result will be described in Discussion.
C2 state exists after the channel is opened by ATP
As described above, our findings suggest that upon removal of ATP, the open channel closes to the C2 state, which likely has one ATP trapped in NBD1. Then, the C2 to C1 transition is coupled to the slow dissociation of this trapped ATP (illustrated in Fig. 8 A). However, as shown in Fig. 8 B, once ATP is available to the closed channel in the C1 state, theoretically, there could be a closed state (C2*) with one ATP bound at NBD1 before the channel enters the open state. An immediate question arises of whether the two closed states (i.e., C2 and C2*) share the same property.
Figure 8.

Multiple closed states of WT-CFTR. (A) Upon washout of ATP, the first closing step (O→C2) involves ATP hydrolysis and dissociation of hydrolytic products from the NBD2 site. The second step (C2→C1) is regulated by a slow dissociation of ATP from the NBD1 site. The two closed states respond to MgPPi differently. (B) Reapplication of ATP brings the channel in the C1 state to a hypothetical C2* state with one ATP bound in NBD1. Double arrows between each state reflect the likely existence of other unidentified closed states.
To address this question, we used Y1219G-CFTR to test whether MgPPi has the same effect on these two closed states. We reasoned that because this mutant has a >50-fold lower nucleotide-binding affinity at NBD2 (Zhou et al., 2006), we can modify the distribution of channels in C2 or C2* states by using different concentrations of ATP. Treating the channels that have been closed for a long time (thus in the C1 state) with a low concentration of ATP should favor an accumulation of the C2* state because the transition rate from C2* to O is significantly decreased by the Y1219G mutation, whereas a high concentration of ATP opens the channel more frequently and thus brings more channels to the C2 state. If the two closed states respond to MgPPi in a similar manner, the subsequent addition of MgPPi to ATP solutions should result in a very similar response regardless of the proportion of channels in the C2 and C2* state.
After PKA and ATP activation and a 1-min washout, we first treated Y1219G channels with 500 µM ATP, which only elicited minimal current. Adding 2 mM MgPPi, as expected, greatly increased the channel activity. However, when the channels in the same patch were pretreated with 20 mM ATP, 2 mM MgPPi plus 20 mM ATP locked open channels much faster (Fig. 9 A). We compared the current increase 10 s and 2 min after the channels were exposed to 500 µM or 20 mM ATP plus MgPPi. In six patches, without exception, we found that more current was induced by MgPPi in the presence of 20 mM ATP (Fig. 9, B and C). These results indicate that the C2 and C2* states are two distinct closed states. Furthermore, as 20 mM ATP shifts the channel distribution to the C2 state, it is likely that the C2 state has a stronger response to MgPPi than the C2* state. The possible structural difference between the C2 and C2* states will be addressed in Discussion.
Figure 9.

Effects of PPi are facilitated by channel opening. (A) A continuous current trace of Y1219G-CFTR. After the channels were activated by PKA and ATP, phosphorylated channels were first exposed to either 500 µM or 20 mM ATP. Subsequent application of 2 mM MgPPi locked open the channels more efficiently when the channels were first opened by 20 mM ATP. (B) Comparison of the current increase 10 s after the addition of MgPPi (n = 6; P < 0.05). Paired samples statistics: 21.7 ± 7.9 pA (500 µM ATP) and 73.8 ± 30.1 pA (20 mM ATP). (C) Comparison of the current increase 2 min after the addition of MgPPi (n = 6; P < 0.05). Paired samples statistics: 84.5 ± 37.2 pA (500 µM ATP) and 190.1 ± 72.8 pA (20 mM ATP).
MgAMP-PNP locks open WT-CFTR with a similar mechanism as MgPPi
Here, we demonstrated that MgPPi binds to the ATP-primed C2 state to induce lock-open events for CFTR channels. We wondered whether this mechanism could also explain the action of nonhydrolyzable ATP analogues that were reported to lock open the channel (Hwang et al., 1994). In Fig. 10 A, we show that the closed channels were relocked open after the ATP-containing solution was switched directly to one with 2 mM MgAMP-PNP (compare Fig. 3 A). The slow current decay after MgAMP-PNP removal yields a time constant of 47.5 ± 7.3 s (n = 5). On the other hand, when the same concentration of MgAMP-PNP was applied 2 min after ATP washout, a very small current was elicited (compare Fig. 1 A). The ensemble macroscopic current relaxation could be fitted with a single-exponential function with a time constant of 1.61 s (n = 5). These results with MgAMP-PNP are qualitatively very similar to the effects of MgPPi.
Figure 10.

Modulation of CFTR gating by MgAMP-PNP. (A) A continuous recording shows that MgAMP-PNP locks open CFTR shortly after ATP removal (relaxation time constant, τ = 47.5 ± 7.3 s; n = 5). This effect was nearly completely abolished by mixing 2 mM MgAMP-PNP with 5 mM ADP. After a long nucleotide washout, 2 or 10 mM MgAMP-PNP induced short openings (ensemble current relaxation, τ = 1.61 s; n = 5). (B) WT channels were exposed to 2 mM MgAMP-PNP plus 10 mM (left) or 2 mM ATP (right) after PKA activation. Parameters yielded by double-exponential fit are Fslow = 51 ± 4.3%, τfast = 0.57 ± 0.17, and τslow = 40.2 ± 4.9 s (n = 5; 2 mM MgAMP-PNP plus 10 mM ATP) and Fslow = 73 ± 3%, τfast = 0.45 ± 0.06, and τslow = 45.7 ± 7.1 s (n = 6; 2 mM MgAMP-PNP plus 2 mM ATP). (C) Y1219G channels opened by ATP plus PKA were locked open by 2 mM MgAMP-PNP plus 2 mM ATP. The bi-exponential current decay gives Fslow = 71 ± 4%, τfast = 0.45 ± 0.03, and τslow = 36.7 ± 4.1 s (n = 6). Arrows in B and C indicate the beginning of the slow component.
Additionally, we showed that MgAMP-PNP and ATP compete for a common binding site (Fig. 10 B and not depicted), and that ADP inhibits long open bursts induced by MgAMP-PNP once ATP was withdrawn (Fig. 10 A). These effects have been demonstrated for MgPPi (compare Figs. 5 and 6). Similar experiments as shown in Fig. 7 were performed for MgAMP-PNP with W401G-CFTR and P-ATP, and virtually identical results were obtained (not depicted). Interestingly, however, we found that MgAMP-PNP, when applied minutes after the removal of ATP (thus acting on the C1 state), activates WT-CFTR with a maximally effective concentration of ∼2 mM, which is significantly lower than that of MgPPi (see Fig. 2 B). When MgAMP-PNP acts on the C2 state, there is also very little difference between 2 and 10 mM MgAMP-PNP, as the currents induced by these two concentrations of MgAMP-PNP immediately after ATP washout are 66.2 ± 3.9% (n = 5) and 77.9 ± 4.1% (n = 4) of the original ATP-induced current, respectively. These results are consistent with those reported by Vergani et al. (2003), who showed that the effect of MgAMP-PNP, similar to ATP, is saturated at millimolar concentration. Therefore, the low efficacy of MgAMP-PNP in opening the channel by itself seems to reflect its intrinsic property as a poor ligand rather than its having a lower binding affinity than ATP.
If the ring–ring stacking interaction between the side chain of Y1219 and the adenine ring of ATP (or AMP-PNP) is indeed critical in determining the affinity of nucleotide binding at the NBD2 site, removing the side chain of Y1219 is expected to lower the binding affinity for ATP as well as MgAMP-PNP. As seen in Fig. 10 (B and C), when [ATP] = [MgAMP-PNP] = 2 mM, the fractional amplitudes of the slow component upon current relaxation are 73 ± 3% (n = 6) and 71 ± 4% (n = 6) for WT-CFTR and Y1219G-CFTR, respectively. Thus, although the Y1219G mutation alters the competition between ATP and MgPPi for the NBD2 site (Fig. 5), the same mutation does not significantly affect the competition between ATP and MgAMP-PNP, suggesting that the Y1219G mutation decreases the binding affinity of ATP to a similar extent as it lowers the affinity for MgAMP-PNP.
DISCUSSION
Here, using MgPPi as a probe, we identify three distinct closed states (C1, C2, and C2*) of the CFTR chloride channels and propose a multistep channel-closing scheme after ATP hydrolysis. Three novel findings are reported. First, MgPPi alone can open CFTR despite having a much lower apparent affinity than ATP. Second, MgPPi locks open ATP-primed channels that are already closed. Third, MgPPi affects CFTR gating in a state-dependent manner. The functional significance and potential structural implications of our results will be discussed.
MgPPi as a ligand to support CFTR gating
Although it is generally agreed that ATP-dependent gating of WT-CFTR is predominantly controlled by ATP binding/hydrolysis events in NBD2 (Gadsby et al., 2006; Chen and Hwang, 2008), how ATP interacts with NBD2 to open the channel remains unclear. Nevertheless, a few clues for this question have already been provided by previous studies testing the effects of different nucleotides on CFTR. First, it was reported that CFTR channels can be opened efficiently by a wide range of nucleoside triphosphates, including ATP, GTP, ITP, CTP, and UTP (Anderson et al. 1991). These results indicate that the base of the nucleoside triphosphate may not be critical in opening CFTR. In sharp contrast, the observation that ADP acts as a competitive inhibitor of ATP (Anderson et al., 1991; Anderson and Welsh, 1992; Gunderson and Kopito, 1994; Winter et al., 1994; Schultz et al., 1995; Bompadre et al., 2005a) points to the crucial role of the phosphate group in triggering channel openings. That nucleotides with an altered bridging structure between β-γ phosphates, such as AMP-PNP, AMP-PCP, and ATPγS, only induce minimal channel activity (Anderson et al., 1991; Nagel et al., 1992; Hwang et al., 1994; Aleksandrov et al., 2000; Vergani et al., 2003) also corroborates this idea.
The importance of the phosphate group of ATP in triggering CFTR channel opening is further supported by this study where we have demonstrated that PPi, which preserves the β-γ phosphate conformation (for review see Clark and Morley, 1976), elicits opening events in the complete absence of ATP (Fig. 1). To explain early failure in observing PPi's effect on CFTR, we found that PPi opens CFTR in a Mg2+-dependent manner (Fig. 2). This result is probably not surprising, as the crystal structures of CFTR NBD1 (Lewis et al., 2004) have revealed that Mg2+ is coordinated by β-γ phosphates of ATP. The functional importance of this interaction between Mg2+ and β-γ phosphates is established by the observation that omission of Mg2+ can severely impair channel opening by ATP (Schultz et al., 1996; Aleksandrov et al., 2000; Ikuma and Welsh, 2000; Dousmanis et al., 2002).
More recent studies suggest a critical role of the evolutionarily conserved signature sequence (LSGGQ) in the signal transduction from ATP binding in NBDs to the gate of CFTR. First, several high resolution crystal structures of ABC transporter proteins show that the oxygen atom in the γ phosphate of ATP forms hydrogen bonds with residues of the signature sequence once two NBDs establish a head-to-tail dimer (Hopfner et al., 2000; Smith et al., 2002; Chen et al., 2003; Zaitseva et al., 2005). Second, the observation that the G551D mutation at the signature sequence completely abolishes ATP-dependent gating of CFTR (Bompadre et al., 2007, 2008) points to the functional significance of this interaction. Third, although G551D-CFTR cannot be opened by ATP, this mutant channel can be gated by Cd2+. Interestingly, when a cysteine residue, which is known to bind avidly to soft metals like Cd2+, is engineered into the signature sequence of CFTR's NBD1, the mutant channels can also be gated by Cd2+ (Wang et al., 2009). This latest result suggests that the interaction between the ligand (ATP or Cd2+) and the signature sequence is essential in transducing the molecular events in NBDs to the channel gate. If MgPPi is capable of binding to the same site occupied by the β-γ phosphates of ATP to interact with the signature sequence, it is not surprising that MgPPi by itself can gate CFTR.
The new result that MgPPi by itself can gate CFTR also offers clues for the role of the adenine ring of ATP in the gating process. Zhou et al. (2006) reported that Y1219G-CFTR, which presumably loses the π-electron–stacking interaction between the aromatic side chain of the tyrosine residue and the adenine ring of ATP, has a far lower ATP binding affinity compared with WT-CFTR. In addition, P-ATP, an ATP analogue with an extra benzene ring attached to adenine (Zhou et al., 2005, 2006), activates WT-CFTR at micromolar concentration. Collectively, these results suggest that the adenine ring, although not essential for ATP-dependent gating of CFTR, may help stabilize the binding of ATP molecules onto the NBD site. This idea predicts that a ligand without the base will have a lower binding affinity. Indeed, 15 mM MgPPi has yet to saturate the current response (Fig. 2 B), whereas the maximal current of WT-CFTR can be attained by 2.75 mM ATP (Zeltwanger et al., 1999). Although we cannot rule out the possibility that MgPPi has a low binding affinity simply because it does not bind to the Walker A domain as well as ATP, the observation that MgAMP-PNP elicits a maximal effect on CFTR at low millimolar concentration (Vergani et al., 2003) (Fig. 10 A), and that the binding affinity of MgAMP-PNP is weakened by Y1219G mutation (Fig. 10 C), suggests that a lack of the ring–ring interaction may have a greater impact on the ligand binding affinity than a slight structural alteration of the phosphate group.
In addition to the difference in apparent affinity, another difference between ATP and MgPPi in channel gating is the burst duration. The current relaxation after MgPPi removal yields a time constant of ∼1.5 s, whereas it yields only ∼400 ms after MgATP washout for WT-CFTR (Fig. 1 B). As PPi was reported to be hydrolyzed by many enzymes, including glucose-6-phosphatase in rat liver (Nordlie et al., 1999), alkaline phosphates of Escherichia coli (Anderson and Nordlie, 1967), and PPi-dependent phosphofructokinase in plants (Carnal and Black, 1979), we first considered the possibility that MgPPi has a lower hydrolytic rate than ATP in the NBD2 composite site, and thus the opening events are terminated more slowly. This hypothesis can be tested by comparing the gating kinetics of MgPPi in WT-CFTR and E1371S-CFTR, a mutant whose ATPase activity is abolished (Moody et al., 2002; Tombline et al., 2004; Vergani et al., 2005; Zhou et al., 2006; Stratford et al., 2007). We reasoned that if MgPPi elicits longer open bursts due to a slower hydrolysis rate, E1371S mutation should further prolong the burst duration induced by MgPPi. However, as can be seen in Fig. S2, although the E1371S mutation dramatically increases the relaxation time constant of the ATP-gated channels (τ = 126.1 ± 24.2 s; n = 5), the lifetime of MgPPi-induced openings for E1371S channels (1.65 s, ensemble current relaxation from five data) is very close to that of WT-CFTR (∼1.5 s in Fig. 1 B). This result suggests that both MgPPi-opened WT-CFTR and E1371S-CFTR channels close through a nonhydrolytic pathway. MgPPi elicits longer opening events in WT channels than ATP because ATP-induced openings are terminated by rapid ATP hydrolysis. Finally, the data also indicate that MgPPi is a poor ligand for CFTR channels because, unlike ATP, it fails to induce a stable open state with E1371S-CFTR.
MgPPi locks open CFTR channels when an ATP is bound in NBD1
The second novel finding presented here is that when the WT-CFTR channels are first opened by ATP, MgPPi applied within seconds after ATP withdrawal locks open the channels (Fig. 3). The lifetime of this lock-open state (∼30 s) is much longer than that opened by MgPPi alone after a 3-min ATP washout (∼1.5 s) as described above. These results indicate the presence of two different closed states, C1 and C2, which respond to MgPPi differently. It should be noted that this conclusion is model independent. It is solely based on the experimental observation that MgPPi exerts different effects when applied at different times during which the channels are closed.
Because the longer the washout the fewer the channels capable of entering the lock-open state and the more the channels poorly responsive to MgPPi, we postulate a dissipation of the C2 state and an accumulation of the C1 state over time during the ATP washout phase. Because the C2 to C1 transition can be slowed down when the channel is first opened by a high affinity ATP analogue, P-ATP (Fig. 7), we propose that there is at least one ATP molecule remained bound in the C2 state. We envisage that the dissociation of this bound ATP in the C2 state is associated with the transition from the C2 state to the C1 state. Thus, a more tightly bound P-ATP can stabilize the C2 state more strongly than ATP.
Two lines of evidence suggest that NBD1 is occupied by ATP during the C2 state. First, biochemical studies showed that NBD2 is a site with a high nucleotide turnover rate due to rapid ATP hydrolysis, whereas NBD1 is a site of more stable nucleotide binding (Szabo et al., 1999; Aleksandrov et al., 2001, 2002, 2008; Basso et al., 2003). Second, W401G mutation, which likely reduces ATP binding affinity in NBD1 (Zhou et al., 2006), decreases the stability of the C2 state. The fact that this mutational effect on the stability of the C2 state can be at least partially overcome by P-ATP suggests that this mutational effect can be attributed to a lower binding affinity rather than some nonspecific allosteric effects.
If the NBD1 site of the C2 state is occupied by ATP, the likely site for MgPPi's action is the presumably vacated NBD2 site after ATP hydrolysis and dissociation of the hydrolytic products. This idea is supported by the results that both ATP and ADP can compete with MgPPi for a common binding site (Figs. 5 and 6). Furthermore, the fact that MgPPi alone can open and lock open CFTR also corroborates this proposition because the NBD2 site plays a critical role in ligand-dependent opening of CFTR. Thus, the MgPPi-induced lock-open configuration of CFTR's two NBDs is composed of one MgPPi molecule in NBD2 and one ATP molecule in NBD1. Because essentially the same data were obtained for MgAMP-PNP–induced lock-open state, we propose a similar configuration of NBDs for the action of MgAMP-PNP (i.e., MgAMP-PNP in NBD2 and ATP in NBD1; see Results; Fig. 10).
So far, we have discussed how MgPPi locks open CFTR in the absence of ATP, whereas in the literature, PPi is reported to exert this effect on CFTR when applied together with ATP. It is important to note that channels opened by MgPPi or ATP plus MgPPi have a similar relaxation time constant upon washout (Fig. 3, A and B), suggesting a common lock-open state under these two conditions. However, when ATP and MgPPi are applied together, it becomes difficult to tell if MgPPi acts on the open state or the closed state. To differentiate these two scenarios, we measured the fractional amplitude of the slow component in the macroscopic current decay phase after washout of ATP plus MgPPi at different concentrations of ATP (see Fig. 5). A larger slow component indicates that the lock-open bursts occur more frequently. If MgPPi binds to the closed state to exert its effect, an increase of [ATP] should compete for binding and thus decreases the fraction of the slow component. In contrast, if MgPPi acts on the open state, the frequency of long-opening events should not be altered by varying [ATP] at the millimolar range. Our results showing that the frequency of lock-open bursts is inversely dependent on [ATP] suggest that MgPPi indeed binds to the closed state to exert its effects, even when it is applied with ATP.
For channel closure from the MgPPi-induced locked-open state, in theory, dissociation of either ATP in NBD1 or MgPPi in NBD2 may be associated with the closing step. In Fig. S3, when the channels opened by ATP are constantly exposed to MgPPi after ATP removal, the macroscopic current can remain at a constant level for >2 min. The current decay upon subsequent MgPPi withdrawal yields a single time constant of τ = 35.7 ± 6.4 s (n = 3), indicating that once the locked-open channels close, they are readily locked open again by MgPPi. This result thus suggests that it is MgPPi dissociation from NBD2, but not ATP from NBD1, that is associated with channel closing from the locked-open state. This idea again is consistent with the critical role of ligand binding/unbinding at NBD2 in CFTR gating.
Structural/biochemical implications of these results
A puzzling question is why the C1 and C2 states respond to MgPPi so differently. As the fundamental difference between the two closed states is whether ATP remains bound at NBD1 or not, it appears that ATP binding in NBD1 can help stabilize the open state induced by MgPPi. Echoing this observation, the lock-open duration of MgPPi is reduced by mutations that decrease the ATP binding affinity to NBD1, K464A, and W401G, but can be partially restored by a high affinity ATP analogue, P-ATP (unpublished data; compare Powe et al., 2002; Zhou et al., 2006).
Because the open state of CFTR represents a dimerization of its two NBDs, we ask whether the dimer completely separates when ATP-opened channels close to the C2 state. If there is a strict coupling between NBD dimerization and opening of the CFTR channel gate, the NBD dimer has to undergo some kind of dissociation upon this O→C2 transition. A complete separation of the NBD dimer for the closing step of WT-CFTR will leave one bound ATP at NBD1 well exposed to the bulk water, as seen in the monomeric ATP-NBD1 crystal structure of CFTR (Lewis et al., 2004, 2005). It seems difficult to envisage that this ATP can remain tightly bound for tens of seconds. Therefore, we speculate that the NBD dimer may only partially separate once the ligand (ADP and Pi after ATP hydrolysis, or MgPPi) dissociates from NBD2. Another ligand in NBD1 will remain trapped by the partial dimer until a complete separation of the two NBDs. Supporting this hypothesis, we demonstrated that the C2 state dissipates much faster when residues at either NBD1 (W401G; Fig. 7 B) or the signature sequence of NBD2 were mutated (S1347G; Fig. S1).
One assumption behind the partial dimer hypothesis is that the ligand in NBD1 will undergo fast binding/unbinding if not trapped in the dimer interface. Here, using the Y1219G mutation to slow down the channel opening rate, we identify another closed state (C2*) that exists before the channel is opened by ATP from the C1 state (Figs. 8 B and 9). This closed state either fails to respond to MgPPi or responds to MgPPi much more inefficiently than the C2 state. We speculate that because the NBD dimer structure is not yet formed in the C2* state as the channel is yet to open, the rather unstable binding of ATP in NBD1 results in a slower lock-open rate of MgPPi.
As described in the Results, the configuration of the C2 state proposed here seems consistent with biochemical results demonstrating that NBD1 is a site of stable nucleotide binding (Szabo et al., 1999; Aleksandrov et al., 2001, 2002; Basso et al., 2003). A careful reexamination, however, reveals two significant disagreements. First, it was shown that 8-azido-ATP can be occluded in NBD1 for tens of minutes (Basso et al., 2003; Aleksandrov et al., 2008). However, our results suggest that the ATP molecule retained in NBD1 after channel closure can almost completely dissociate within 1 min (Fig. 4). More importantly, Aleksandrov et al. (2008) recently reported that the occlusion of 8-azido-ATP occurs even when NBD2 is deleted. This latest result indicates that the intrinsic high binding affinity of NBD1 per se is sufficient to trap the nucleotide without a need of a partial dimer as we postulated above. Although one may attribute these discrepancies to very different materials and experimental procedures used in different laboratories, it is of great interest for us to understand how to reconcile functional and biochemical results in the near future.
Acknowledgments
We thank Cindy Chu and Shenghui Hu for technical assistance.
This work was supported by National Institutes of Health (NIH) grants NIHR01DK55835, NIHR01HL53455, and CFFT CLARKE06XX0 (to T.-C. Hwang). This investigation was conducted in a facility constructed with support from Research Facilities Improvement Program (grant no. C06 RR-016489-01) from the National Center for Research Resources, NIH.
Angus C. Nairn served as editor.
Footnotes
Abbreviations used in this paper: ABC, ATP-binding cassette; CHO, Chinese hamster ovary; NBD, nucleotide-binding domain; P-ATP, N6-(2-phenylethyl)-ATP; PPi, pyrophosphate; WT, wild-type.
References
- Aleksandrov A.A., Chang X., Aleksandrov L., Riordan J.R. 2000. The non-hydrolytic pathway of cystic fibrosis transmembrane conductance regulator ion channel gating.J. Physiol. 528:259–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleksandrov L., Mengos A., Chang X., Aleksandrov A., Riordan J.R. 2001. Differential interactions of nucleotides at the two nucleotide binding domains of the cystic fibrosis transmembrane conductance regulator.J. Biol. Chem. 276:12918–12923 [DOI] [PubMed] [Google Scholar]
- Aleksandrov L., Aleksandrov A.A., Chang X.B., Riordan J.R. 2002. The first nucleotide binding domain of cystic fibrosis transmembrane conductance regulator is a site of stable nucleotide interaction, whereas the second is a site of rapid turnover.J. Biol. Chem. 277:15419–15425 [DOI] [PubMed] [Google Scholar]
- Aleksandrov L., Aleksandrov A., Riordan J.R. 2008. Mg2+-dependent ATP occlusion at the first nucleotide-binding domain (NBD1) of CFTR does not require the second (NBD2).Biochem. J. 416:129–136 [DOI] [PubMed] [Google Scholar]
- Anderson M.P., Welsh M.J. 1992. Regulation by ATP and ADP of CFTR chloride channels that contain mutant nucleotide-binding domains.Science. 257:1701–1704 [DOI] [PubMed] [Google Scholar]
- Anderson M.P., Berger H.A., Rich D.P., Gregory R.J., Smith A.E., Welsh M.J. 1991. Nucleoside triphosphates are required to open the CFTR chloride channel.Cell. 67:775–784 [DOI] [PubMed] [Google Scholar]
- Anderson W.B., Nordlie R.C. 1967. Inorganic pyrophosphate-glucose phosphotransferase activity associated with alkaline phosphatase of Escherichia coli.J. Biol. Chem. 242:114–119 [PubMed] [Google Scholar]
- Basso C., Vergani P., Nairn A.C., Gadsby D.C. 2003. Prolonged nonhydrolytic interaction of nucleotide with CFTR's NH2-terminal nucleotide binding domain and its role in channel gating.J. Gen. Physiol. 122:333–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baukrowitz T., Hwang T.C., Nairn A.C., Gadsby D.C. 1994. Coupling of CFTR Cl- channel gating to an ATP hydrolysis cycle.Neuron. 12:473–482 [DOI] [PubMed] [Google Scholar]
- Berger A.L., Ikuma M., Hunt J.F., Thomas P.J., Welsh M.J. 2002. Mutations that change the position of the putative gamma-phosphate linker in the nucleotide binding domains of CFTR alter channel gating.J. Biol. Chem. 277:2125–2131 [DOI] [PubMed] [Google Scholar]
- Bompadre S.G., Ai T., Cho J.H., Wang X., Sohma Y., Li M., Hwang T.C. 2005a. CFTR gating I: characterization of the ATP-dependent gating of a phosphorylation-independent CFTR channel (ΔR-CFTR).J. Gen. Physiol. 125:361–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bompadre S.G., Cho J.H., Wang X., Zou X., Sohma Y., Li M., Hwang T.C. 2005b. CFTR gating II: effects of nucleotide binding on the stability of open states.J. Gen. Physiol. 125:377–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bompadre S.G., Sohma Y., Li M., Hwang T.C. 2007. G551D and G1349D, two CF-associated mutations in the signature sequences of CFTR, exhibit distinct gating defects.J. Gen. Physiol. 129:285–298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bompadre S.G., Li M., Hwang T.C. 2008. Mechanism of G551D-CFTR (cystic fibrosis transmembrane conductance regulator) potentiation by a high affinity ATP analog.J. Biol. Chem. 283:5364–5369 [DOI] [PubMed] [Google Scholar]
- Cai Z., Taddei A., Sheppard D.N. 2006. Differential sensitivity of the cystic fibrosis (CF)-associated mutants G551D and G1349D to potentiators of the cystic fibrosis transmembrane conductance regulator (CFTR) Cl- channel.J. Biol. Chem. 281:1970–1977 [DOI] [PubMed] [Google Scholar]
- Carnal N.W., Black C.C. 1979. Pyrophosphate-dependent 6-phosphofructokinase, a new glycolytic enzyme in pineapple leaves.Biochem. Biophys. Res. Commun. 86:20–26 [DOI] [PubMed] [Google Scholar]
- Carson M.R., Winter M.C., Travis S.M., Welsh M.J. 1995. Pyrophosphate stimulates wild-type and mutant cystic fibrosis transmembrane conductance regulator Cl- channels.J. Biol. Chem. 270:20466–20472 [DOI] [PubMed] [Google Scholar]
- Chen J., Lu G., Lin J., Davidson A.L., Quiocho F.A. 2003. A tweezers-like motion of the ATP-binding cassette dimer in an ABC transport cycle.Mol. Cell. 12:651–661 [DOI] [PubMed] [Google Scholar]
- Chen T.Y., Hwang T.C. 2008. CLC-0 and CFTR: chloride channels evolved from transporters.Physiol. Rev. 88:351–387 [DOI] [PubMed] [Google Scholar]
- Clark G.M., Morley R. 1976. Inorganic pyro-compounds Ma((X2O7)b).Chem. Soc. Rev. 5:269–295 [Google Scholar]
- Coric T., Zhang P., Todorovic N., Canessa C.M. 2003. The extracellular domain determines the kinetics of desensitization in acid-sensitive ion channel 1.J. Biol. Chem. 278:45240–45247 [DOI] [PubMed] [Google Scholar]
- Cotten J.F., Ostedgaard L.S., Carson M.R., Welsh M.J. 1996. Effect of cystic fibrosis-associated mutations in the fourth intracellular loop of cystic fibrosis transmembrane conductance regulator.J. Biol. Chem. 271:21279–21284 [DOI] [PubMed] [Google Scholar]
- Csanady L., Chan K.W., Nairn A.C., Gadsby D.C. 2005. Functional roles of nonconserved structural segments in CFTR's NH2-terminal nucleotide binding domain.J. Gen. Physiol. 125:43–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dousmanis A.G., Nairn A.C., Gadsby D.C. 2002. Distinct Mg2+-dependent steps rate limit opening and closing of a single CFTR Cl− channel.J. Gen. Physiol. 119:545–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadsby D.C., Vergani P., Csanady L. 2006. The ABC protein turned chloride channel whose failure causes cystic fibrosis.Nature. 440:477–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunderson K.L., Kopito R.R. 1994. Effects of pyrophosphate and nucleotide analogs suggest a role for ATP hydrolysis in cystic fibrosis transmembrane regulator channel gating.J. Biol. Chem. 269:19349–19353 [PubMed] [Google Scholar]
- Gunderson K.L., Kopito R.R. 1995. Conformational states of CFTR associated with channel gating: the role ATP binding and hydrolysis.Cell. 82:231–239 [DOI] [PubMed] [Google Scholar]
- Higgins C.F., Linton K.J. 2004. The ATP switch model for ABC transporters.Nat. Struct. Mol. Biol. 11:918–926 [DOI] [PubMed] [Google Scholar]
- Hopfner K.P., Karcher A., Shin D.S., Craig L., Arthur L.M., Carney J.P., Tainer J.A. 2000. Structural biology of Rad50 ATPase: ATP-driven conformational control in DNA double-strand break repair and the ABC-ATPase superfamily.Cell. 101:789–800 [DOI] [PubMed] [Google Scholar]
- Hwang T.C., Nagel G., Nairn A.C., Gadsby D.C. 1994. Regulation of the gating of cystic fibrosis transmembrane conductance regulator C1 channels by phosphorylation and ATP hydrolysis.Proc. Natl. Acad. Sci. USA. 91:4698–4702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikuma M., Welsh M.J. 2000. Regulation of CFTR Cl- channel gating by ATP binding and hydrolysis.Proc. Natl. Acad. Sci. USA. 97:8675–8680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansdell K.A., Kidd J.F., Delaney S.J., Wainwright B.J., Sheppard D.N. 1998. Regulation of murine cystic fibrosis transmembrane conductance regulator Cl- channels expressed in Chinese hamster ovary cells.J. Physiol. 512:751–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis H.A., Buchanan S.G., Burley S.K., Conners K., Dickey M., Dorwart M., Fowler R., Gao X., Guggino W.B., Hendrickson W.A., et al. 2004. Structure of nucleotide-binding domain 1 of the cystic fibrosis transmembrane conductance regulator.EMBO J. 23:282–293 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis H.A., Zhao X., Wang C., Sauder J.M., Rooney I., Noland B.W., Lorimer D., Kearins M.C., Conners K., Condon B., et al. 2005. Impact of the deltaF508 mutation in first nucleotide-binding domain of human cystic fibrosis transmembrane conductance regulator on domain folding and structure.J. Biol. Chem. 280:1346–1353 [DOI] [PubMed] [Google Scholar]
- Mense M., Vergani P., White D.M., Altberg G., Nairn A.C., Gadsby D.C. 2006. In vivo phosphorylation of CFTR promotes formation of a nucleotide-binding domain heterodimer.EMBO J. 25:4728–4739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moody J.E., Millen L., Binns D., Hunt J.F., Thomas P.J. 2002. Cooperative, ATP-dependent association of the nucleotide binding cassettes during the catalytic cycle of ATP-binding cassette transporters.J. Biol. Chem. 277:21111–21114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagel G., Hwang T.C., Nastiuk K.L., Nairn A.C., Gadsby D.C. 1992. The protein kinase A-regulated cardiac Cl- channel resembles the cystic fibrosis transmembrane conductance regulator.Nature. 360:81–84 [DOI] [PubMed] [Google Scholar]
- Nordlie R.C., Foster J.D., Lange A.J. 1999. Regulation of glucose production by the liver.Annu. Rev. Nutr. 19:379–406 [DOI] [PubMed] [Google Scholar]
- Powe A.C., Jr., Al-Nakkash L., Li M., Hwang T.C. 2002. Mutation of Walker-A lysine 464 in cystic fibrosis transmembrane conductance regulator reveals functional interaction between its nucleotide-binding domains.J. Physiol. 539:333–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramjeesingh M., Li C., Garami E., Huan L.J., Galley K., Wang Y., Bear C.E. 1999. Walker mutations reveal loose relationship between catalytic and channel-gating activities of purified CFTR (cystic fibrosis transmembrane conductance regulator).Biochemistry. 38:1463–1468 [DOI] [PubMed] [Google Scholar]
- Riordan J.R., Rommens J.M., Kerem B., Alon N., Rozmahel R., Grzelczak Z., Zielenski J., Lok S., Plavsic N., Chou J.L. 1989. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA.Science. 245:1066–1073 [DOI] [PubMed] [Google Scholar]
- Schultz B.D., Venglarik C.J., Bridges R.J., Frizzell R.A. 1995. Regulation of CFTR Cl− channel gating by ADP and ATP analogues.J. Gen. Physiol. 105:329–361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz B.D., Bridges R.J., Frizzell R.A. 1996. Lack of conventional ATPase properties in CFTR chloride channel gating.J. Membr. Biol. 151:63–75 [DOI] [PubMed] [Google Scholar]
- Scott-Ward T.S., Cai Z., Dawson E.S., Doherty A., Da Paula A.C., Davidson H., Porteous D.J., Wainwright B.J., Amaral M.D., Sheppard D.N., Boyd A.C. 2007. Chimeric constructs endow the human CFTR Cl- channel with the gating behavior of murine CFTR.Proc. Natl. Acad. Sci. USA. 104:16365–16370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith P.C., Karpowich N., Millen L., Moody J.E., Rosen J., Thomas P.J., Hunt J.F. 2002. ATP binding to the motor domain from an ABC transporter drives formation of a nucleotide sandwich dimer.Mol. Cell. 10:139–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stratford F.L., Ramjeesingh M., Cheung J.C., Huan L.J., Bear C.E. 2007. The Walker B motif of the second nucleotide-binding domain (NBD2) of CFTR plays a key role in ATPase activity by the NBD1-NBD2 heterodimer.Biochem. J. 401:581–586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo K., Szakacs G., Hegeds T., Sarkadi B. 1999. Nucleotide occlusion in the human cystic fibrosis transmembrane conductance regulator. Different patterns in the two nucleotide binding domains.J. Biol. Chem. 274:12209–12212 [DOI] [PubMed] [Google Scholar]
- Tombline G., Bartholomew L., Gimi K., Tyndall G.A., Senior A.E. 2004. Synergy between conserved ABC signature Ser residues in P-glycoprotein catalysis.J. Biol. Chem. 279:5363–5373 [DOI] [PubMed] [Google Scholar]
- Thibodeau P.H., Brautiqam C.A., Machius M., Thomas P.J. 2005. Side chain and backbone contributions of Phe508 to CFTR folding.Nat. Struct. Mol. Biol. 12:10–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergani P., Nairn A.C., Gadsby D.C. 2003. On the mechanism of MgATP-dependent gating of CFTR Cl− channels.J. Gen. Physiol. 121:17–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vergani P., Lockless S.W., Nairn A.C., Gadsby D.C. 2005. CFTR channel opening by ATP-driven tight dimerization of its nucleotide-binding domains.Nature. 433:876–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker J.E., Saraste M., Runswick M.J., Gay N.J. 1982. Distantly related sequences in the alpha- and beta-subunits of ATP synthase, myosin, kinases and other ATP-requiring enzymes and a common nucleotide binding fold.EMBO J. 1:945–951 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Bompadre S.G., Li M., Hwang T.C. 2009. Mutations at the signature sequence of CFTR create a Cd2+-gated chloride channel.J. Gen. Physiol. 133:69–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter M.C., Sheppard D.N., Carson M.R., Welsh M.J. 1994. Effect of ATP concentration on CFTR Cl- channels: a kinetic analysis of channel regulation.Biophys. J. 66:1398–1403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaitseva J., Jenewein S., Jumpertz T., Holland I.B., Schmitt L. 2005. H662 is the linchpin of ATP hydrolysis in the nucleotide-binding domain of the ABC transporter HlyB.EMBO J. 24:1901–1910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeltwanger S., Wang F., Wang G.T., Gillis K.D., Hwang T.C. 1999. Gating of cystic fibrosis transmembrane conductance regulator chloride channels by adenosine triphosphate hydrolysis. Quantitative analysis of a cyclic gating scheme.J. Gen. Physiol. 113:541–554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z., Wang X., Li M., Sohma Y., Zou X., Hwang T.C. 2005. High affinity ATP/ADP analogues as new tools for studying CFTR gating.J. Physiol. 569:447–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z., Wang X., Liu H.Y., Zou X., Li M., Hwang T.C. 2006. The two ATP binding sites of cystic fibrosis transmembrane conductance regulator (CFTR) play distinct roles in gating kinetics and energetics.J. Gen. Physiol. 128:413–422 [DOI] [PMC free article] [PubMed] [Google Scholar]