

Abstract
This study was undertaken to investigate the effects of prenatal and postnatal exposure to high fat diet on the brain. Female rats were divided into high fat diet (HFD) and control diet (CD) groups 4 weeks prior to breeding and throughout gestation and lactation. After weaning, male progeny were placed on a chow diet until 8 weeks old, and then segregated into HFD or CD groups. At 20 weeks old, rats were evaluated in the Morris water maze, and markers of oxidative stress and inflammation were documented in brain. In comparison to rats fed CD, cognitive decline in HFD progeny from HFD dams manifested as a decline in retention, but not acquisition, in the water maze. HFD was also associated with significant increases in 3-nitrotyrosine, inducible nitric oxide synthase, IL-6, and glial markers Iba-1 and GFAP, with the largest increases frequently observed in HFD animals born to HFD dams. Thus, these data collectively suggest that HFD increases oxidative and inflammatory signaling in brain, and further indicate that maternal HFD consumption might sensitize offspring to the detrimental effects of HFD.
Keywords: cognition, dementia, fetal programming, neurodegeneration, obesity, reactive gliosis
INTRODUCTION
Obesity is clinically identified based on measurements of body mass index (Mei et al., 2002), but can be generally defined as the physiological condition in which excess body fat has accumulated to an extent that detrimentally affects overall health. This working definition is based on the dramatically enhanced risk for a myriad of diseases associated with obesity, including type 2 diabetes, cardiovascular disease, gastrointestinal and respiratory difficulties, and many types of cancer (reviewed in Haslam et al., 2005). Emerging data also show that obesity in human populations is associated with cognitive decline and enhanced vulnerability to brain injury, while experimental studies in animal models confirm a profile of heightened brain vulnerability and decreased cognitive function. For example, studies have reported deficits in learning, memory, and executive function in obese as compared to nonobese patients (Elias et al., 2003), (Elias et al., 2005), (Waldstein et al., 2006). Other studies of young and middle-aged adults have confirmed an association of obesity with declines in executive function (Gunstad et al., 2007). Likewise, animal studies indicate that experimental obesity is associated with cognitive decline and specific decreases in dendritic spine density (Stranahan et al., 2008) and in the expression of synaptic marker proteins such as synapsin and GAP-43 (Molteni et al., 2002), (Wu et al., 2004). Although the physiologic mechanisms whereby obesity adversely affects the brain are poorly understood, both experimental and human studies have shown that obesity is associated with increased oxidative stress (Mattson et al., 2003), (Zhang et al., 2005), (Souza et al., 2007), which is strongly implicated in cognitive decline caused by brain injury (Ansari et al., 2008; Ansari et al., 2008) or neurodegenerative disease (Joseph et al., 1998), (Kamat et al., 2008), (Sayre et al., 2008).
While it is generally accepted that obesity is associated with adverse health outcomes, studies have also shown that maternal obesity is specifically associated with a variety of pregnancy complications, including labor and delivery difficulties, fetal and neonatal death, maternal hypertension, preeclampsia, and gestational diabetes (Lu et al., 2001), (Bhattacharya et al., 2007), (Thompson et al., 2008). Furthermore, it is possible that maternal obesity might precipitate adverse outcomes that persist through adulthood in offspring. Indeed, evidence suggests that epigenetic events initiated during the prenatal period can result in persistent adaptations in structure, physiology and metabolism that predispose offspring towards disease and impaired physiology (Barker, 1995), (Lucas, 1998). For examples, animal studies have shown that maternal overnutrition and/or neonatal overfeeding predispose offspring to obesity (Levin et al., 1998), (Plagemann et al., 1999), (Shankar et al., 2008). Specifically related to neurological function, prenatal exposure to cigarette smoke, cocaine, or alcohol all impair cognitive function (Bredy et al., 2004), (Singer et al., 2004), (Willford et al., 2006), (Toro et al., 2008). Thus, the possibility exists that maternal obesitycould enhance both the propensity towards, and the neurological consequences of, obesity in adult offspring.
This study was undertaken to investigate the effects of diet-induced obesity on the brain, with additional emphasis on the contributions of maternal diet to the neurological consequences of obesity. Experimental high fat diet (HFD) consumption can induce obesity, hyperglycemia, whole-body insulin resistance, and generalized metabolic syndrome in rodents (Tschöp et al., 2001), (Oakes et al., 1997), (Buettner et al., 2007) suggesting that HFD per se is sufficient to induce obesity and metabolic disease. To determine the effects of both maternal and progeny diet-induced obesity on brain function, breeding age female Long Evans rats were divided into 2 groups and fed either HFD or control diet (CD) from 4 weeks prior to breeding and throughout gestation and lactation. After weaning, male progeny were placed on a chow diet until 8 weeks old, at which time they were segregated into HFD or CD diet groups, and maintained on their respective diets until 20 weeks of age. Body weight and retroperitoneal fat pad weight were measured at the end of study, as was cognitive performance and levels of specific markers of oxidative stress and inflammation in the cortex.
EXPERIMENTAL PROCEDURES
Animal treatments and tissue preparation
The Institutional Animal Care and Use Committee approved all experimental protocols which were consistent with the NIH guidelines on the use of experimental animals. Rats used in this study were a subset of a larger study group designed to determine the role of maternal adiposity on fetal programming related to body weight regulation (White et al., 2009). Twenty 8-week old virgin female Long Evans rats (Harlan Laboratories, Indianapolis, IN) were singly housed with a 12-hour light/dark cycle, and access to food (either HFD or CD) and water ad libitum. The HFD (Research Diets D12492) consisted of 60% fat energy, 20% carbohydrate energy and 20% protein energy and had an energy density of 5.24 kcals/gm. The CD (Research Diets D12450B) consisted of 10% fat energy, 70% carbohydrate energy and 20% protein energy with an energy density of 3.85 kcals/gm. After 4 weeks of diet exposure, dams were bred to 10-week old virgin male chow-fed Long Evans rats. Dams continued on their respective diets throughout gestation and lactation. Within 24 hours of birth, litter size was adjusted to 10 pups per litter. After 3 weeks, male pups from both HFD and CD dams were weaned onto chow (LabDiet 5001) and multi-housed in shoebox cages until 8 weeks, at which time the rats were further divided into HFD and CD groups, and maintained until 20 weeks of age. Thus, there were 4 groups of animals used in this study: rats born to obese HFD dams and consuming a HFD diet (HFD/HFD), rats born to obese HFD dams but given CD (HFD/CD), rats born to CD dams and consuming CD (CD/CD), and rats born to CD dams but given HFD in adulthood (CD/HFD). At the end of the study, rats were tested in the Morris water maze, then euthanized, and brains were isolated. The left half of each brain was used for histological analyses, while tissues from the cortex (predominately frontal cortical area) of the remaining right side were collected and used for biochemical assays.
Body weight and body composition analysis
Body weight was measured weekly on the dams pre-conception and during gestation. During lactation, both the dams and pups were weighed on days 2, 7, 14 and 20 post-partum. Pups were then weighed weekly throughout the remainder of the study. At 20 weeks of age, rats were euthanized and retroperitoneal adipose tissue was collected and weighed.
Morris water maze
Cognitive spatial ability was evaluated in all rats using the Morris water maze as previously described (Janas et al., 2005). Rats were tested during the day (light phase), and the water maze tank was 165 cm in diameter, and extra-maze cues were placed on the walls of the room. To evaluate learning and memory function, rats trained and tested at 19.5 weeks old, and the training paradigm extended over 4 days. For the first 3 days the platform was hidden below the surface of the water in one of the 4 quadrants, and each rat received four acquisition trials/day. The maximum trial length was 90 sec with an approximate 15 min intertrial interval, and between trials rats were placed in a holding cage with a dry towel. For each trial, the rat was placed into the water facing the wall of the pool, with each trial starting from a different start location. If a rat failed to find the platform within the 90 sec, it was placed on the platform, and all rats remained on the platform for 30 sec at the end of each trial. The actual location of the platform was counterbalanced across groups, but its location remained constant across all days and trials for an individual animal. During the 3-day acquisition period, latency and distance traveled to reach the platform were measured as indicators of learning. Task retention over training days was also assessed, and was calculated based on the difference between performance (latency or distance traveled) on the last trail of day 1 or day 2 from the first trail of day 2 or 3, respectively. As a specific example, a numerical retention index would be calculated by subtracting the distance traveled by a specific rat on the last trial of day 1 from distance traveled by that rat on the first trail of day 2, and was used a measure of retention. For the probe trial on day 4, the platform was removed and each rat was allowed to swim for 60 sec. Percent time spent in the target quadrant was utilized as the measure of learning in the probe trial. The rats’ behavior was tracked and recorded using video tracking software (Viewpoint Lifesciences, Montreal, Quebec, Canada).
Analyses of protein nitration and carbonylation
Overall oxidative modifications to proteins were quantitatively evaluated by measuring levels of 3-nitrotyrosine (3-NT) and protein carbonylation in brain tissue homogenates by dot blot analyses. Briefly, samples were homogenized in Tris-buffered saline (pH 7.4) lysis buffer containing 0.1% Triton X-100, 5 mM EDTA, and protease inhibitor cocktail (Sigma-Aldrich, St.Louis, MO). Exactly 30 μg of total protein was blotted directly to PVDF membranes using a vacuum-powered dot blot apparatus (Bio-Rad Laboratories, Hercules, CA). Protein carbonylation was evaluated using a commercially available OxyBlot protein carbonyl detection system (Millipore, Billerica, MA), following the manufacturers’ instruction as described previously (Keller et al., 2005). This assay is based on the immunochemical detection of protein carbonyl groups derivatized with 2,4-dinitrophenyl hydrazine. For protein nitration, blots were developed using a primary antibody to 3-NT (Upstate, Charlottesville, VA) followed by a horseradish peroxidase-conjugated secondary antibody, and visualized using a chemiluminescence system (Amersham Biosciences, Piscataway, NJ). All samples were analyzed in triplicate, and blot images were scanned and densitometrically analyzed for quantification.
Western blot
Brain tissues were homogenized in a Tris-buffered saline (pH 7.4) lysis buffer containing 0.1% Triton X-100, 5 mM EDTA, and protease inhibitor cocktail (Sigma-Aldrich). Samples were then denatured in SDS, and equivalent amounts of protein were electrophoretically separated in polyacrylamide gels and blotted onto nitrocellulose. Blots were processed using the following primary antisera: anti-GFAP (1:500, Dako, Carpinteria, CA); anti-Iba-1 (1:100, Wako Chemicals USA, Inc. Richmond, VA); and anti-iNOS (1:100, Abcam Inc. Cambridge, MA) After incubation with primary antibodies, blots were washed and exposed to horseradish peroxidase-conjugated secondary antibodies, and visualized using a chemiluminescence system (Amersham Biosciences). Blot images were scanned and densitometrically analyzed for quantification, and all data are presented as a ratio of expression over that of tubulin, which was included as a loading control for all Western blot analyses.
ELISA assays
Brain tissues were homogenized in lysis buffer (Tris-buffered saline supplemented with 1% triton X-100, 5 mM EDTA, and protease inhibitor cocktail (Sigma-Aldrich) centrifuged at 12,000 × g for 10 min, and used for ELISA as described previously (Bruce-Keller et al., 2001). IL-6 and TNF-α were measured using a Duoset kit (R&D Systems, Minneapolis, MN), while MCP-1 was also measured using commercially available kits (BD Biosciences, San Jose, CA) in accordance with the manufacturer’s assay protocol. Briefly, 96-well Immulon 4 plates (Dynex Technologies, Chantilly, VA) were coated overnight with 1 μg/ml capture antibody and blocked with PBS + 1% BSA. Cytokines in cell supernatants were detected with 0.5 μg/ml biotinylated antibody. Antibody staining was developed using streptavidin-HRP (1:200) and reacted with SureBlue Reserve TMB Microwell Peroxidase Substrate (KPL, Gaithersburg, Maryland) to visualize staining intensity. Reactions were terminated with TMB Stop solution (KPL), and plates were read on a Victor plate reader (Perkin Elmer, Waltham, MA) set at 450nm with wavelength correction at 540nm.
Histological analyses
For histology, hemi-brains were emersion-fixed in 4% paraformaldehyde for 24 hrs, after which the middle 2/3 of the brain was blocked, and processed for paraffin embedding. 6 μm coronal sections were cut at the level of the striatum/lateral ventricle and were collected for analysis. The slides were then processed for immunohistochemistry using the following primary antisera: anti-GFAP (1:500, Dako); anti-Iba-1 (1:100, Wako Chemicals); anti-iNOS (1:100, Abcam Inc.), and anti 3-NT) (1:100, Upstate). For overall qualitative evaluations of the patterns of immunoreactivity, sections were incubated with biotinylated secondary antibodies, and then visualized using DAB as a chromagen following manufacturers’ instructions (Vector Laboratories, Burlingame, CA). To document non-specific staining, the primary antibody was either pre-incubated with excess epitope or omitted from the staining protocol.
Statistical analyses
All data are shown as mean ± standard error of measurement. Maternal body weight was analyzed with 1-way analyses of variance (ANOVA) to determine effects of HFD. All other data were analyzed with 2-way ANOVA to evaluate potential main effects or interaction of maternal and progeny HFD. In addition, for the Morris water maze data, planned comparisons of the CD/HFD, HFD/CD and HFD/HFD group to the CD/CD group were made using the LSD procedure. Statistical significance was accepted at p<0.05.
RESULTS
Effects of HFD on body weight and composition
Female rats were fed HFD or CD beginning 4 weeks before breeding and continuing throughout gestation and lactation. By 2 weeks on the diet, the HFD dams weighed significantly more than the CD rats, and this difference continued through the end of lactation (Table 1).
Table 1. Body weight in dams before breeding, during gestation and during lactation.
Female Long Evans rats were given HFD or CD as described in Methods for 4 weeks and then bred, continuing on their respective diets throughout lactation. Data are expressed as mean body weights ± SEM with 6-7 rats per group.
| Time of weighing | CD dams | HFD dams | |
|---|---|---|---|
| Before breeding | wk 0 | 185.3 ± 2.6 | 188 ± 2.8 |
| wk 2 | 221.4 ± 3.8 | 239.6 ± 8.1*** | |
| wk 4 | 233.1 ± 4.9 | 263.6 ± 10.5*** | |
| Gestation | gd 15 | 302.8 ± 5.9 | 341.0 ± 15.0*** |
| Lactation | pnd 2 | 293.1 ± 7.5 | 329.6 ± 15.4*** |
| pnd 7 | 295.5 ± 5.4 | 327.9 ± 13.8*** | |
| pnd 14 | 298.0 ± 6.1 | 330.2 ± 12.4*** | |
| pnd 20 | 295.7 ± 4.8 | 318.5 ± 10.8*** | |
p<0.001 when compared to CD group.
Male pups from HFD and CD dams were all weaned to standard labchow at 3 weeks old, maintained on this diet until 8 weeks old, at which time they were segregated into HFD and CD groups. During this period, body weights progressively diverged such that pups born to obese HFD dams and consuming a HFD diet (HFD/HFD) weighed more than any other group, whereas pups born to CD dams and consuming CD (CD/CD) weighed the least (Table 2). Intermediate between these groups were rats born to obese HFD dams but given CD (HFD/CD), or rats born to CD dams but given HFD in adulthood (CD/HFD). Statistically, ANOVA for Maternal Diet × Offspring Diet indicated there was a main effect of Offspring Diet on body weight at 20 weeks of age (F(1,34) = 5.23, p = 0.028). The main effect of Maternal Diet approached significance (F(1,34) = 3.47, p = 0.071), but the interaction was not significant. In addition to body weight, there were significant differences in body adiposity. As an index of body fat, the retroperitoneal adipose tissue was collected and weighed at 20 weeks. Data show that, as was observed for body weight, HFD/HFD rats had the heaviest retroperitoneal fat pads (Table 2). Results of a 2-way ANOVA confirmed a significant main effect of Maternal Diet (F(1,34) = 6.19, p = 0.0179) and Offspring Diet (F(1,34) = 29.62, p = 0.0001) on body adiposity, but no interaction.
Table 2. Body weight and retroperitoneal fat pad weight experimental animals over adult development.
Male pups born to dams given HFD or CD were separated and given either CD or HFD as described in Methods, and were maintained on their respective diets throughout the experiment. Body Weight data are expressed as mean body weight in grams ± SEM with 9-10 rats per group, while retroperitoneal fat weight is also expressed as mean weight in grams ± SEM.
| Age of weighing | CD/CD | CD/HDF | HFD/CD | HFD/HFD | |
|---|---|---|---|---|---|
| Body Weight | wk 8 | 237.6 ± 4.6 | 241.1 ± 6.6 | 262.5 ± 8.3 | 263.1 ± 8.4 |
| wk 13 | 384.1 ± 9.0 | 410.1 ± 12.1 | 413.8 ± 13.8 | 446.3 ± 18.0 | |
| wk 20 | 457.9 ± 9.8 | 498.1 ± 11.6 | 491.6 ± 16.6 | 521.2 ± 19.3 | |
| Retro Fat | wk 20 | 4.6 ± 0.2 | 8.1 ± 0.6 | 6.1 ± 0.5 | 9.9 ± 1.0 |
Effects of HFD on spatial learning
While epidemiological studies in humans suggest that obesity is associated with disruptions in cognitive ability (Elias et al., 2003), (Elias et al., 2005), (Waldstein et al., 2006), results from animal studies have been mixed, with some reports describing significant effects of HFD-induced obesity on performance in the Morris maze (Farr et al., 2008) and others failing to detect effects of HFD on spatial learning in this task (Mielke et al., 2006), (Jurdak et al., 2008). Thus, to determine if HFD administration to either dams and/or offspring could affect spatial learning, the four groups of rats were tested behaviorally in this task at 20 weeks of age using a 3-day acquisition protocol followed by a probe trial as described in Methods. Data show that rats in all four groups were able to learn the location of the hidden platform as evidenced by decreased latency and distance traveled to reach the platform (Fig. 1A and B). In addition, data from the the probe trial indicated that all four groups exhibited a preference for the target quadrant (Fig. 1C). An impact of diet was detected on task retention over training days, as indicated by evaluation of retention indices. Retention indices were calculated based on differences in performance on the last trail of a training day as compared to the first trail of the following training day, as described in Methods. Multiple comparisons for Group for the trial 4-5 difference score indicated no differences (p > 0.05) compared to the CD/CD group for both latency and distance traveled (see inset for Fig. 1 and B), with all groups exhibiting some degree of retention deficit. However, the comparison of Group for the Day 8-9 difference scores indicated the HFD/HFD group was unable to retain the previous days training as compared to CD/CD rats (p < 0.05), while the HFD/CD and CD/HFD diet groups were not different from CD/CD rats (p > 0.05).
Figure 1. Effects of maternal and offspring diet on cognitive performance in Morris water maze.

Male rats were segregated into 4 groups: CD/CD (rats from CD dams given CD), CD/HFD (rats from CD dams given HFD), HFD/CD (rats from HFD dams given CD), and HFD/HFD (rats from HFD dams given HFD) groups, and the effects of maternal and offspring diet on visuo-spatial learning in 20-week old animals (n = 9-10 per group) was evaluated using the Morris water maze as described in Methods. (A) Mean latency to reach the hidden platform across was measured, and data were analyzed by ANOVA as described in Methods. There was no effect of Maternal Diet or Offspring Diet during acquisition. However, planned comparisons of retention indices (Mean Difference) based on differences in performance between training days as described in Methods indicated that the HFD/HFD group exhibited a significant (p < 0.05) retention deficit compared to the CD/CD group (see graph insert). (B) Mean distance traveled to reach the hidden platform was measured, and data were analyzed by ANOVA. There was no effect of Maternal Diet or Offspring Diet during acquisition. However, planned comparisons of retention indices (Mean Difference) indicated that the HFD/HFD group exhibited a significant (p < 0.05) retention deficit compared to the CD/CD group (see graph insert). (C) Percentage of time spent searching in each quadrant during a 60 sec probe trial was measured, and statistical analyses did not reveal any difference in quadrant preference between the 4 groups.
Effects of HFD on brain oxidative stress
Previous reports have suggested that both diet-induced and genetic models of obesity can increase free radical generation and oxidative stress in brains of rodents (Zhang et al., 2005), (Chinen et al., 2007), (Souza et al., 2007). To determine if maternal and/or offspring diet could induce oxidative stress in brain, initial experiments were designed to evaluate the degree of protein oxidation in brain tissues from HFD and CD rats born from HFD and CD dams. Specifically, the levels of both protein carbonylation and protein nitration from the four groups of rats were evaluated using dot blot analyses as described in Methods. Data show that there was no significant effect of either maternal or offspring diet on levels of protein carbonyls in brains as indicated by levels of DNP (Fig. 2A). However, 3-NT levels were affected by HFD (Fig 2B). Specifically, an ANOVA for Maternal Diet × Offspring Diet indicated there was an main effect of Offspring Diet on 3-NT levels in the cortex (F(1,37) = 13.99, p = 0.0006). The overall pattern of 3-NT expression in the cortex was then examined histologically in DAB-stained sections taken at the level of the lateral ventricle. These qualitative analyses also demonstrated increases in 3-NT levels in HFD/HFD rats as compared to CD/CD, and indicated that 3-NT immunoreactivity was associated both with brain vasculature and individual cells (Fig. 2C).
Figure 2. Effects of maternal and offspring diet on oxidative modifications to proteins in brain.
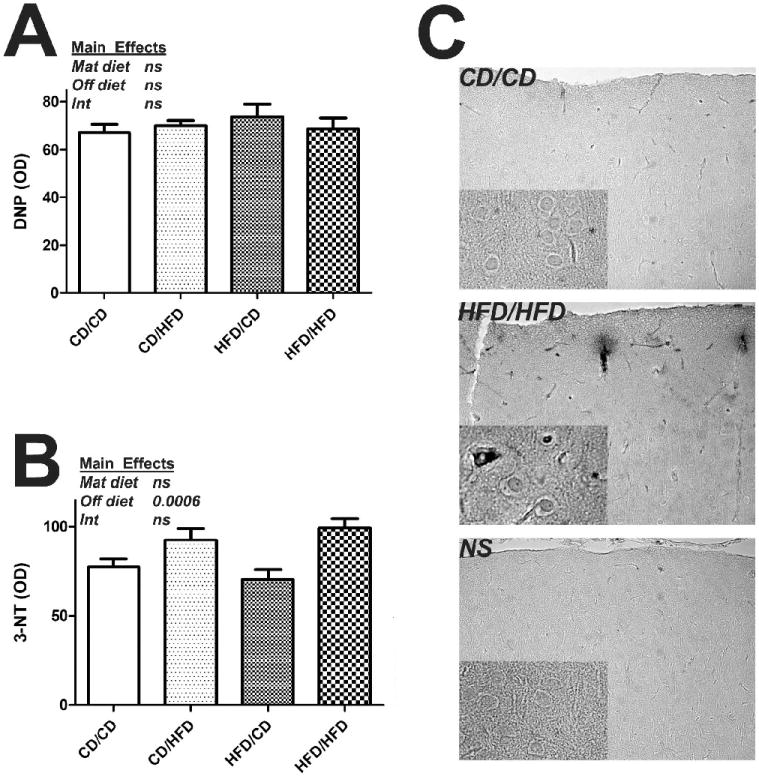
Male rats were segregated into CD/CD, CD/HFD, HFD/CD and HFD/HFD groups as described in Methods, and the effects of maternal and offspring diet on protein carbonylation and nitration in the brains of 20 week old rats was evaluated by dot blot analyses as described in Methods. (A) The accumulation of protein carbonyls in cortical tissue homogenates was measured by evaluating dinitrophenyl (DNP) immunoreactivity levels. Data are means and SEM, with 9-10 individual rats per group, and all samples measured in triplicate. 2-way ANOVA indicated that there was no effect of either Maternal diet or Offspring diet on DNP levels (text insert). (B) The accumulation of 3-nitrotyrosine (3-NT) in cortical tissue homogenates was measured. Data are means and SEM, with 9-10 individual rats per group, with all samples measured in triplicate. 2-way ANOVA indicated an overall significant effect of Offspring Diet on 3-NT levels in the cortex (text insert). (C) Representative low-magnification and high-magnification (insert) images depict the pattern of 3-NT staining in cortex as observed in sections taken at the level of the lateral ventricle.
The observed specific increases in 3-NT suggest that HFD administration could be associated with nitrosative stress, a pathological condition in which the production of highly reactive nitrogen species exceeds the ability of cells to eliminate them, leading to alterations (i.e., nitration) in protein structure (reviewed in Martínez et al., 2009). Thus, to determine if HFD administration might increase the production of reactive nitrogen species, the expression of inducible nitric oxide synthase (iNOS), a pro-oxidant/ pro-inflammatory enzyme that generates nitric oxide, was measured in the four groups of rats using Western blot analyses as described in Methods. Data show that iNOS expression was increased by both dam and offspring diet (Fig 3A). Specifically, an ANOVA for Maternal Diet × Offspring Diet indicated there was an main effect of both Maternal Diet (F(1,37) = 45.38, p = 0.026, Fig. 3A) and also Offspring Diet on iNOS levels in the cortex (F(1,37) = 6.98, p = 0.012) but no interaction. While ANOVA did not reveal a significant statistical interaction between maternal and progeny diet, mean iNOS expression was much higher in brains from HFD/HFD rats as compared to the other 3 groups of rats (Fig. 3A). This observation suggests that HFD administration preferentially increases iNOS expression in rats born to HFD dams. The overall pattern of iNOS expression was then examined histologically in DAB-stained sections taken at the level of the lateral ventricle. These qualitative analyses revealed a punctuate, cell-associated pattern of iNOS expression, and also demonstrated an increased number of iNOS-positive cells in the cortex of HFD/HFD rats (Fig. 3B).
Figure 3. Effects of maternal and offspring diet on iNOS expression in rat brain.

Male rats were segregated into CD/CD, CD/HFD, HFD/CD and HFD/HFD groups as described in Methods, and the effects of maternal and offspring diet on iNOS expression was evaluated by Western blot and immunocytochemistry as described in Methods. (A) The levels of iNOS protein measured by Western blot. Data are means and SEM, with 9-10 individual rats per group, and 2-way ANOVA indicated an overall significant effect of both Maternal diet and Offspring diet on iNOS expression in the cortex (text insert). (B) Representative low-magnification and high-magnification (insert) images depict the pattern of iNOS staining in cortex as observed in sections taken at the level of the lateral ventricle.
Effects of HFD on markers of brain inflammation
While the precise mechanisms underlying the ability of obesity to increase brain oxidative stress are unknown, several studies suggest that experimental obesity in rodents is associated with glial inflammation and hypertrophy (Choi et al., 2005), (Dunbar et al., 2005), (Sriram et al., 2002). Furthermore, inflammatory reactions in glia, particularly microglia, are well known to use reactive oxygen and nitrogen species as intermediates (Mhatre et al., 2004), (Reynolds et al., 2007). Thus, to clarify the role of glial reactivity in models of obesity and to specifically determine if maternal and/or offspring diet could modulate gliosis in rats, markers of glial reactivity in the four groups of rats were evaluated using Western blot and immunohistochemical analyses as described in Methods. The intermediate filament protein glial fibrillary acidic protein (GFAP) was used to evaluate astrocyte hypertrophy (O’Callaghan et al., 2005). To quantify GFAP expression in the cortex of mice in an unbiased manner, Western blot analyses were undertaken. Quantification and statistical analysis of GFAP band intensities (expressed as a ratio of GFAP over the expression of the protein tubulin) confirmed a significant main effect of Offspring Diet on GFAP expression in the cortex (F(1,37) = 4.60, p = 0.0387; Fig. 4A), but no effect of Maternal Diet nor any interaction. The overall pattern of GFAP immunoreactivity was evaluated in DAB-stained sections taken at the level of the lateral ventricle. Qualitative analyses of GFAP-stained sections also suggested that administration of HFD to offspring modestly increased overall glial hypertrophy regardless of maternal diet (Fig. 4B).
Figure 4. Effects of maternal and offspring diet on GFAP expression in rat brain.

Male rats were segregated into CD/CD, CD/HFD, HFD/CD and HFD/HFD groups as described in Methods, and the effects of maternal and offspring diet on expression of the astrocyte hypertrophy marker GFAP was evaluated by immunocytochemistry and Western blot as described in Methods. (A) The levels of GFAP protein measured by Western blot. Data are means and SEM, with 9-10 individual rats per group, and 2-way ANOVA indicated an overall significant effect of Offspring diet on GFAP expression in the cortex (text insert). (B) Representative low-magnification and high-magnification (insert) images depict the pattern of GFAP staining in cortex as observed in sections taken at the level of the lateral ventricle.
Microglial reactivity was evaluated by measuring expression of Iba-1, which is a 17-kDa calcium binding protein that is specifically expressed in macrophages/ microglia and is upregulated during the in vivo activation of these cells (Hilton et al., 2008), (Lee et al., 2008), (Zecca et al., 2008). This particular marker is widely used to evaluate microglial reactivity as it can be used both in histological, paraffin-processed tissue sections and also in denatured cell lysates (Ahmed et al., 2007) (Vega-Avelaira et al., 2007). Western blot analyses were thus used to quantify Iba-1 expression in the cortex of mice in an unbiased manner. Quantification and statistical analysis of Iba-1 band intensities (expressed as a ratio over tubulin levels) confirmed a significant main effect of Offspring Diet on Iba-1 expression in the cortex (F(1,37) = 9.30, p = 0.0042; Fig. 5A). The overall pattern of Iba-1 immunoreactivity was then evaluated in DAB-stained sections. Qualitative analyses suggested that, like GFAP immunoreactivity, increased Iba-1immunoreactivity was associated HFD administration (Fig. 5B).
Figure 5. Effects of maternal and offspring diet on Iba-1 expression in rat brain.

Male rats were segregated into CD/CD, CD/HFD, HFD/CD and HFD/HFD groups as described in Methods, and the effects of maternal and offspring diet on expression of the microglia marker Iba-1 was evaluated by immunocytochemistry and Western blot as described in Methods. (A) The levels of Iba-1 protein measured by Western blot. Data are means and SEM, with 9-10 individual rats per group, and ANOVA indicated an overall significant effect of Offspring diet on Iba-1 expression in the cortex (text insert). (B) Representative low-magnification and high-magnification (insert) images depict the pattern of Iba-1 staining in cortex as observed in sections taken at the level of the lateral ventricle.
Experimental obesity is generally associated with enhanced expression of proinflammatory cytokines (Ahima et al., 2008), (Rasouli et al., 2008), and excessive cytokine expression in the brain has been proposed to interfere with cognitive ability (Wang et al., 2002), (Wilson et al., 2002). Thus, the protein levels of TNFα and IL-6 in brain tissue homogenates were evaluated by ELISA as described in Methods. Results of ANOVA indicated that there were no significant effects of either Maternal Diet or Offspring Diet on TNFα expression in the cortex (Fig. 6A). However, 2-way ANOVA revealed a significant main effect of Maternal Diet on IL-6 levels in the cortex (F(1,37) = 7.317, p = 0.0103), but no overall effect of Offspring Diet (Fig. 6B). While ANOVA did not reveal a significant statistical interaction between maternal and progeny diet, mean IL-6 expression was much higher in brains from HFD/HFD rats as compared to the other groups of rats (Fig. 6B). This observation suggests that maternal HFD administration can sensitive offspring to the proinflammatory effects of HFD administration. Finally, protein levels of the chemokine MCP-1 and the growth factor BDNF were also measured by ELISA, but statistical analyses indicated that these signals were not affected by either Maternal Diet or Offspring Diet (data not shown)
Figure 6. Effects of maternal and offspring diet on cytokine levels in rat brain.

Male rats were segregated into CD/CD, CD/HFD, HFD/CD and HFD/HFD groups as described in Methods, and the effects of maternal and offspring diet on cytokine expression in the brains of 20 week old rats was evaluated by ELISA as described in Methods. (A) TNFα levels in cortical homogenates were measured. Data are means and SEM, with 9-10 individual rats per group, and 2-way ANOVA indicated that there was no effect of either Maternal diet or Offspring diet on TNF expression (text insert). (B) Expression of IL-6 in the cortex of rats was measured by ELISA. Data are means and SEM, with 9-10 individual rats per group, and 2-way ANOVA indicated an overall significant effect of Maternal diet on IL-6 expression in the cortex (text insert).
DISCUSSION
In this manuscript, data obtained from rats born to obese HFD dams and consuming either a HFD (HFD/HFD) or CD (HFD/CD), and rats born to CD dams and consuming either CD (CD/CD) or HFD in adulthood (CD/HFD) are described. Experiments were designed to evaluate if the interaction of maternal and offspring HFD could alter cognitive function and perturb brain homeostasis in vivo. Using the Morris water maze to evaluate cognitive ability, data show that retention across days of training was impaired in HFD/HFD rats when compared to CD/CD rats, although diet had no impact on other measures of performance in this task. These findings suggest that the combination of HFD administration with a maternal history of HFD administration, but not administration of this particular HFD to either group alone for the designated periods of time, could encumber memory retention. Data also show that this paradigm of HFD administration was able to increase body weight and retroperitoneal fat pad weight, as well as protein nitration, iNOS expression, glial activation, and IL-6 expression. Furthermore, results suggest that some indices, particularly retroperitoneal fat pad weight, iNOS expression, and IL-6 levels, were induced by HFD to a greater extent in animals born to HFD dams. Thus, our findings raise the possibility that prenatal exposure to HFD increases the sensitivity of offspring to the deleterious proinflammatory effects of postnatal diet-induced obesity. Overall, these data are in general agreement with published studies showing that maternal consumption of a high fat diet prior to and during pregnancy increases obesity in adult offspring, and further extend these studies by demonstrating that both maternal and offspring obesity can lead to behavioral and biochemical alterations within the central nervous system.
Data reported here are in general agreement with previous studies on maternal obesity and/or fat intake on obesity in offspring (Guo et al., 1995), (Khan et al., 2004), (Srinivasan et al., 2006), (Bayol et al., 2007), (Chen et al., 2008), (Shankar et al., 2008). It should be pointed out, however, that other studies have failed to detect increased body weight in pups born to obese dams (Buckley et al., 2005) (Beck et al., 2006), (Rolls et al., 1982). Several experimental issues could explain this incongruence in results, including the use of different fat sources (lard, vegetable oil, cafeteria diet, margarine, etc) and/or the different durations of diet exposure. Nonetheless, our findings indicate that maternal HFD significantly increases body weight in offspring, particularly when progeny are fed HFD. This observation is important as it is becoming increasingly evident that diet-induced, developmental alterations can impinge on neurological systems in adulthood. For example, reports have shown that maternal HFD can alter the developmental profile of precursor cells in the embryonic hypothalamus such that a greater proportion of the new neurons express orexigenic peptides (Chang et al., 2008). This particular pattern of peptide expression is in turn postulated to lead to increased risk for overeating and obesity in adulthood (Chang et al., 2008). Furthermore, other studies have found that HFD administration to breeding female rats leads to increased obesity and hyperlipidemia in newborn pups, which is also associated with increased lipid peroxidation in serum as well as in the hippocampus (Tozuka et al., 2009). Furthermore, neurogenesis in the offspring of HFD-fed animals is reduced during postnatal development (Lindqvist et al., 2006), (Tozuka et al., 2009), which could be related to increased lipid peroxidation, as malondialdehyde, a product of peroxidized lipids, has been shown to reduce proliferation of hippocampal progenitor cells in vitro (Tozuka et al., 2009).
While not directly addressed in this study, many potential mechanisms could be proposed to explain how maternal obesity predisposes offspring to CNS abnormalities. One possibility could be gestational diabetes, although this is unlikely as there was no evidence that the dams in this study had gestational diabetes (White et al., 2009). Indeed, increased birth weight is nearly always associated with gestational diabetes (Pezzarossa et al., 1996), (Ben-Haroush et al., 2008), but birth weights in these rats did not differ significantly (White et al., 2009). Alternatively, alterations in adipokines and adipokine signaling could affect brain development during the perinatal and postnatal period in rodents. For example, IL-6 and placental inflammatory mediators are elevated with maternal obesity (Challier et al., 2008), (Ramsay et al., 2002), and maternal IL-6 levels are predictive of obesity in the neonatal offspring (Dahlgren et al., 2001). In our study, brain IL-6 expression was one of the few indices measured that showed a significant main effect of maternal diet, and increased IL-6 levels in the immature brain have been proposed to lead to white matter damage (Saliba et al., 2001). Conversely, maternal leptin could also affect fetal development during the perinatal period. Leptin is best known for its action as an afferent adiposity signal to the brain that suppresses appetite and increases energy expenditure (Friedman et al., 1998), and it is well-known that leptin acts on hypothalamic centers to regulate feeding behavior (reviewed in Figlewicz et al., 2009). Interestingly, however, even though leptin levels are elevated in models of obesity, hypothalamic cells lose the ability to respond to leptin, a phenomena known as leptin resistance (reviewed in Morrison, 2008). Central resistance to peripheral leptin is the reason behind the seemingly paradoxical situation in models of obesity, in which obese or overweight individuals have high plasma leptin concentrations related to the size of adipose tissue, but this elevated leptin signal does not induce the expected responses (i.e., a reduction in food intake and/ or an increase in energy expenditure). Interestingly, leptin receptors (OBR) are widely expressed in numerous extra-hypothalamic regions of the brain, including the hippocampus, cerebellum, amygdala, and brain stem (Tartaglia et al., 1995), (Fei et al., 1997), (Elmquist et al., 1998). While the exact role that leptin plays in these non-hypothalamic nuclei is not clear, data suggest that leptin may act on the fetal cerebral cortex and may affect maintenance and differentiation of neural stem cells, glial-restricted progenitor cells, and/or neuronal lineage cells (Udagawa et al., 2007). These data are in keeping with a number of reports that have suggested a role for leptin in cognitive processes (Harvey et al., 2005) (Morrison, 2009). For example, there is evidence that leptin influences neuronal excitability via the activation and trafficking of potassium channels in several brain regions, (Diano et al., 2008), while direct administration of leptin enhances long-term potentiation (Wayner et al., 2004), and facilitates behavioral performance in passive avoidance and Morris water maze tasks (Oomura et al., 2006). Collectively, these experimental studies suggest that leptin may regulate neuronal survival and function (Signore et al., 2008). This potential scenario is supported by recent findings in humans that suggest that decreased or perturbed aspects of metabolism, including impaired leptin signaling, could relate to the prevalence and severity of dementia (Erol, 2008), (Greco et al., 2009). Indeed, neuropathology reminiscent of Alzheimer’s -type neurodegeneration can be produced experimentally by selectively impairing insulin/IGF functions in the brain (Lester-Coll et al., 2006), and it has even been suggested that Alzheimer’s disease could represent a type 3 form of diabetes (de la Monte et al., 2006).
While the developing brain can be affected by maternal nutrition status (over nutrition or under nutrition) during pregnancy, it is not clear whether the observed abnormalities noted in HFD/HFD rats reflect developmental changes in utero that persist into adulthood, or rather are a consequence of the increased body weight and adiposity in these rats. Offspring of maternally obese dams weighed significantly more than the offspring of non-obese dams at weaning and during the entire time they were on chow (White et al., 2009), and thus increased obesity per se could potentially mediate neurological perturbations noted in this study. Indeed, epidemiological and experimental data have established that the brain is sensitive to obesity and obesity-induced metabolic dysfunction (reviewed in Bruce-Keller et al., 2008). For example, one of the earliest reports of adverse effects of experimental obesity on brain pathology reported that genetically obese (ob/ob) mice had decreased overall levels of myelin and marked alterations to the fatty acid composition of myelin as compared to wild type mice (Sena et al., 1985). The use of a genetic model for these studies is significant in that it suggests that increased adiposity, rather than increase in dietary lipids, is sufficient to alter the composition of brain myelin. This study is supported by more recent imaging studies in humans that also have revealed changes in white matter and myelin abnormalities in association with obesity (Jagust, 2007), (Gazdzinski et al., 2008). While the human studies have not been confirmed by pathological data to identify the actual molecular and biochemical changes to human myelin in obese patients, an obvious consequence of altered myelination would be altered axonal transmission. Indeed, animal studies have confirmed that experimental obesity is associated with a wide array of cognitive abnormalities (Greenwood et al., 2005), (Winocur et al., 2005). Furthermore, animal studies have documented that experimental obesity can be associated with different types of cognitive abnormalities (Greenwood et al., 2005), (Winocur et al., 2005), including alterations in spatial learning skills in rats (Molteni et al., 2002), (Stranahan et al., 2008), (Jurdak et al., 2008). Although biochemical and histological indices of synaptic dysfunction were not addressed in the current study, previous reports have suggested that obesity in rodents is associated with decreases in dendritic spine density (Stranahan et al., 2008) and in the expression of synaptic marker proteins such as synapsin and GAP-43 (Molteni et al., 2002), (Wu et al., 2004). Thus, the behavioral decline noted in this study associated with HFD/HFD rats might be secondary to structural alterations induced by maternal and/or progeny diet, although further studies with longer exposures to the diets would be necessary to fully clarify this point.
Experimental and human studies have shown that obesity is associated with increased oxidative stress, and observations in this report document increased protein nitration in the brains of HFD rats. Protein nitration is a common characteristic of oxidative injury, and the formation of 3-nitrotyrosine in specific tyrosine residues, via the action of peroxynitrite (ONOO–) on proteins, can alter protein structure and function (reviewed in Martínez et al., 2009). Oxidative stress has been implicated in neurodegenerative disease (Joseph et al., 1998), (Kamat et al., 2008), (Sayre et al., 2008), and oxidative modifications to proteins have been documented in both aging (Stadtman et al., 2003) and Alzheimer’s disease (Smith et al., 1996). Furthermore, age-related oxidative stress in the brain has indeed been suggested to be altered by diet (Mattson et al., 2003), (Perry et al., 2003). For example, several studies have shown that administration of high fat or high calorie diets to rodents increases free radical generation (Zhang et al., 2005) and protein oxidation (Souza et al., 2007) in the brain. Furthermore, it has been reported that endothelial cell dysfunction in the Zucker genetic model of rat obesity is caused by oxidative stress secondary to overactivation of NADPH oxidase, which in turn is caused by circulating free fatty acids (Chinen et al., 2007). Observations of increased protein oxidation in brain aging and in Alzheimer’s disease (Smith et al., 1996), (Stadtman et al., 2003) as well as in models of HFD raise the possibility that HFD might accelerate the aging phenotype of the brain. Finally, it has been demonstrated that administration of the lipid-soluble antioxidant Vitamin E not only reverses oxidative modification to proteins in the brains of rats given a high-fat diet, but also normalizes levels of synapsin I and cyclic AMP-response element-binding protein (CREB), and prevents obesity-induced alterations to cognitive function (Wu et al., 2004). However, it remain uncertain if oxidative stress in genetic or diet-induced models of obesity is a result of obesity per se, or is more related to obesity-related metabolic syndrome. Available data from animal models indicate that obesity and/or obesity-induced metabolic syndrome can induce neuropathology and cognitive dysfunction and can also synergize with pathological conditions and/or environmental toxins to accelerate CNS dysfunction. Thus, while investigation into the dialog of peripheral signals of adiposity with the CNS is necessary to identify the mechanisms of obesity-induced brain pathology, our results provide clear support for the hypothesis that excess dietary fat, particularly when present both in maternal and progeny diet, can have widespread significant detrimental effects on brain homeostasis.
Acknowledgments
The authors are grateful to Kenneth Ballard for expert technical assistance and animal handling. This work was supported by grants from the NIH (NS46267, DA19398, and AG05119 to ABK; and DK07246, RR021945, and NS051570 to CDM).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahima RS, Osei SY. Adipokines in obesity. Front Horm Res. 2008;36:182–97. doi: 10.1159/000115365. [DOI] [PubMed] [Google Scholar]
- Ahmed Z, Shaw G, Sharma VP, Yang C, McGowan E, Dickson DW. Actin-binding proteins coronin-1a and IBA-1 are effective microglial markers for immunohistochemistry. J Histochem Cytochem. 2007;55:687–700. doi: 10.1369/jhc.6A7156.2007. [DOI] [PubMed] [Google Scholar]
- Ansari MA, Roberts KN, Scheff SW. Oxidative stress and modification of synaptic proteins in hippocampus after traumatic brain injury. Free Radic Biol Med. 2008;45:443–52. doi: 10.1016/j.freeradbiomed.2008.04.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ansari MA, Roberts KN, Scheff SW. A time course of contusion-induced oxidative stress and synaptic proteins in cortex in a rat model of TBI. J Neurotrauma. 2008;25:513–26. doi: 10.1089/neu.2007.0451. [DOI] [PubMed] [Google Scholar]
- Barker DJ. Intrauterine programming of adult disease. Mol Med Today. 1995;1:418–23. doi: 10.1016/s1357-4310(95)90793-9. [DOI] [PubMed] [Google Scholar]
- Bayol SA, Farrington SJ, Stickland NC. A maternal ‘junk food’ diet in pregnancy and lactation promotes an exacerbated taste for ‘junk food’ and a greater propensity for obesity in rat offspring. Br J Nutr. 2007;98:843–851. doi: 10.1017/S0007114507812037. [DOI] [PubMed] [Google Scholar]
- Beck B, Kozak R, Moar KM, Mercer JG. Hypothalamic orexigenic peptides are overexpressed in young Long-Evans rats after early life exposure to fat-rich diets. Biochem Biophys Res Commun. 2006;342:452–458. doi: 10.1016/j.bbrc.2006.01.158. [DOI] [PubMed] [Google Scholar]
- Ben-Haroush A, Hadar E, Chen R, Hod M, Yogev Y. Maternal obesity is a major risk factor for large-for-gestational-infants in pregnancies complicated by gestational diabetes. Arch Gynecol Obstet. 2008 doi: 10.1007/s00404-008-0767-4. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Bhattacharya S, Campbell DM, Liston WA. Effect of Body Mass Index on pregnancy outcomes in nulliparous women delivering singleton babies. BMC Public Health. 2007;7:168. doi: 10.1186/1471-2458-7-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bredy TW, Lee AW, Meaney MJ, Brown RE. Effect of neonatal handling and paternal care on offspring cognitive development in the monogamous California mouse (Peromyscus californicus) Horm Behav. 2004;46:30–38. doi: 10.1016/j.yhbeh.2003.09.017. [DOI] [PubMed] [Google Scholar]
- Bruce-Keller AJ, Barger SW, Moss NI, Pham JT, Keller JN, Nath A. Pro-inflammatory and pro-oxidant properties of the HIV protein Tat in a microglial cell line: attenuation by 17beta-estradiol. J Neurochem. 2001;78:1315–1324. doi: 10.1046/j.1471-4159.2001.00511.x. [DOI] [PubMed] [Google Scholar]
- Bruce-Keller AJ, Keller JN, Morrison CD. Obesity and vulnerability of the CNS. Biochim Biophys Acta. 2008 doi: 10.1016/j.bbadis.2008.10.004. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley AJ, Keseru B, Briody J, Thompson M, Ozanne SE, Thompson CH. Altered body composition and metabolism in the male offspring of high fat-fed rats. Metabolism. 2005;54:500–507. doi: 10.1016/j.metabol.2004.11.003. [DOI] [PubMed] [Google Scholar]
- Buettner R, Schölmerich J, Bollheimer LC. High-fat diets: modeling the metabolic disorders of human obesity in rodents. Obesity (Silver Spring) 2007;15:798–808. doi: 10.1038/oby.2007.608. [DOI] [PubMed] [Google Scholar]
- Challier JC, Basu S, Bintein T, Minium J, Hotmire K, Catalano PM, Hauguel-de Mouzon S. Obesity in pregnancy stimulates macrophage accumulation and inflammation in the placenta. Placenta. 2008;29:274–81. doi: 10.1016/j.placenta.2007.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang GQ, Gaysinskaya V, Karatayev O, Leibowitz SF. Maternal high-fat diet and fetal programming: increased proliferation of hypothalamic peptide-producing neurons that increase risk for overeating and obesity. J Neurosci. 2008;28:12107–19. doi: 10.1523/JNEUROSCI.2642-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Simar D, Lambert K, Mercier J, Morris MJ. Maternal and postnatal overnutrition differentially impact appetite regulators and fuel metabolism. Endocrinology. 2008;149:5348–56. doi: 10.1210/en.2008-0582. [DOI] [PubMed] [Google Scholar]
- Chinen I, Shimabukuro M, Yamakawa K, Higa N, Matsuzaki T, Noguchi K, Ueda S, Sakanashi M, Takasu N. Vascular lipotoxicity: endothelial dysfunction via fatty-acid-induced ROS overproduction in obese Zucker diabetic fatty rats. Endocrinology. 2007;148:160–5. doi: 10.1210/en.2006-1132. [DOI] [PubMed] [Google Scholar]
- Choi JY, Jang EH, Park CS, Kang JH. Enhanced susceptibility to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine neurotoxicity in high-fat diet-induced obesity. Free Radic Biol Med. 2005;38:806–16. doi: 10.1016/j.freeradbiomed.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Dahlgren J, Nilsson C, Jennische E, Ho HP, Eriksson E, Niklasson A, Björntorp P, Albertsson Wikland K, Holmäng A. Prenatal cytokine exposure results in obesity and gender-specific programming. Am J Physiol Endocrinol Metab. 2001;281:E326–34. doi: 10.1152/ajpendo.2001.281.2.E326. [DOI] [PubMed] [Google Scholar]
- de la Monte SM, Tong M, Lester-Coll N, Plater MJ, Wands JR. Therapeutic rescue of neurodegeneration in experimental type 3 diabetes: relevance to Alzheimer’s disease. J Alzheimers Dis. 2006;10:89–109. doi: 10.3233/jad-2006-10113. [DOI] [PubMed] [Google Scholar]
- Diano S, Horvath TL. Anticonvulsant effects of leptin in epilepsy. J Clin Invest. 2008;118:26–8. doi: 10.1172/JCI34511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunbar J, Lapanowski K, Barnes M, Rafols J. Hypothalamic agouti-related protein immunoreactivity in food-restricted, obese, and insulin-treated animals: evidence for glia cell localization. Exp Neurol. 2005;191:184–92. doi: 10.1016/j.expneurol.2004.09.002. [DOI] [PubMed] [Google Scholar]
- Elias MF, Elias PK, Sullivan LM, Wolf PA, D’Agostino RB. Lower cognitive function in the presence of obesity and hypertension: the Framingham Heart Study. Int J Obes Relat Metab Disord. 2003;27:260–268. doi: 10.1038/sj.ijo.802225. [DOI] [PubMed] [Google Scholar]
- Elias MF, Elias PK, Sullivan LM, Wolf PA, D’Agostino RB. Obesity, diabetes and cognitive deficit: the Framingham Heart Study. Neurobiol Aging. 2005;26:11–16. doi: 10.1016/j.neurobiolaging.2005.08.019. [DOI] [PubMed] [Google Scholar]
- Elmquist JK, Bjorbaek C, Ahima RS, Flier JS, Saper CB. Distributions of leptin receptor mRNA isoforms in the rat brain. J Comp Neurol. 1998;395:535–547. [PubMed] [Google Scholar]
- Erol A. An integrated and unifying hypothesis for the metabolic basis of sporadic Alzheimer’s disease. J Alzheimers Dis. 2008;13:241–53. doi: 10.3233/jad-2008-13302. [DOI] [PubMed] [Google Scholar]
- Farr SA, Yamada KA, Butterfield DA, Abdul HM, Xu L, Miller NE, Banks WA, Morley JE. Obesity and hypertriglyceridemia produce cognitive impairment. Endocrinology. 2008;149:2628–36. doi: 10.1210/en.2007-1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei H, Okano HJ, Li C, Lee GH, Zhao C, Darnell R, Friedman JM. Anatomic localization of alternatively spliced leptin receptors (Ob-R) in mouse brain and other tissues. Proc Natl Acad Sci U S A. 1997;94:7001–5. doi: 10.1073/pnas.94.13.7001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figlewicz DP, Benoit SC. Insulin, leptin, and food reward: update 2008. Am J Physiol Regul Integr Comp Physiol. 2009;296:R9–R19. doi: 10.1152/ajpregu.90725.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JM, Halaas JL. Leptin and the regulation of body weight in mammals. Nature. 1998;395:763–770. doi: 10.1038/27376. [DOI] [PubMed] [Google Scholar]
- Gazdzinski S, Kornak J, Weiner MW, Meyerhoff DJ. Body mass index and magnetic resonance markers of brain integrity in adults. Ann Neurol. 2008;63:652–7. doi: 10.1002/ana.21377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greco SJ, Sarkar S, Johnston JM, Tezapsidis N. Leptin regulates tau phosphorylation and amyloid through AMPK in neuronal cells. Biochem Biophys Res Commun. 2009;380:98–104. doi: 10.1016/j.bbrc.2009.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenwood CE, Winocur G. High-fat diets, insulin resistance and declining cognitive function. Neurobiol Aging. 2005;26:42–5. doi: 10.1016/j.neurobiolaging.2005.08.017. [DOI] [PubMed] [Google Scholar]
- Gunstad J, Paul RH, Cohen RA, Tate DF, Spitznagel MB, Gordon E. Elevated body mass index is associated with executive dysfunction in otherwise healthy adults. Compr Psychiatry. 2007;48:57–61. doi: 10.1016/j.comppsych.2006.05.001. [DOI] [PubMed] [Google Scholar]
- Guo F, Jen KL. High-fat feeding during pregnancy and lactation affects offspring metabolism in rats. Physiol Behav. 1995;57:681–686. doi: 10.1016/0031-9384(94)00342-4. [DOI] [PubMed] [Google Scholar]
- Harvey J, Shanley LJ, O’Malley D, Irving AJ. Leptin: a potential cognitive enhancer? Biochem Soc Trans. 2005;33:1029–32. doi: 10.1042/BST20051029. [DOI] [PubMed] [Google Scholar]
- Haslam DW, James WP. Obesity. Lancet Neurol. 2005;366:1197–209. doi: 10.1016/S0140-6736(05)67483-1. [DOI] [PubMed] [Google Scholar]
- Hilton GD, Stoica BA, Byrnes KR, Faden AI. Roscovitine reduces neuronal loss, glial activation, and neurologic deficits after brain trauma. J Cereb Blood Flow Metab. 2008;28:1845–59. doi: 10.1038/jcbfm.2008.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagust W. What can imaging reveal about obesity and the brain? Curr Alzheimer Res. 2007;4:135–9. doi: 10.2174/156720507780362146. [DOI] [PubMed] [Google Scholar]
- Janas A, Duffy K, Devan B, Greig N, Holloway H, Yu QS, Markowska A, Cunningham S, Ingram DK, Spangler E. The cholinesterase inhibitor, phenserine, improves Morris water maze performance of scopolamine-treated rats. Life Sciences. 2005;76:1073–81. doi: 10.1016/j.lfs.2004.06.028. [DOI] [PubMed] [Google Scholar]
- Joseph JA, Denisova N, Fisher D, Bickford P, Prior R, Cao G. Age-related neurodegeneration and oxidative stress: putative nutritional intervention. Neurol Clin. 1998;16:747–55. doi: 10.1016/s0733-8619(05)70092-x. [DOI] [PubMed] [Google Scholar]
- Jurdak N, Lichtenstein AH, Kanarek RB. Diet-induced obesity and spatial cognition in young male rats. Nutr Neurosci. 2008;11:48–54. doi: 10.1179/147683008X301333. [DOI] [PubMed] [Google Scholar]
- Kamat CD, Gadal S, Mhatre M, Williamson KS, Pye QN, Hensley K. Antioxidants in central nervous system diseases: preclinical promise and translational challenges. J Alzheimers Dis. 2008;15:473–93. doi: 10.3233/jad-2008-15314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller JN, Schmitt FA, Scheff SW, Ding Q, Chen Q, Butterfield DA, Markesbery WR. Evidence of increased oxidative damage in subjects with mild cognitive impairment. Neurology. 2005;64:1152–6. doi: 10.1212/01.WNL.0000156156.13641.BA. [DOI] [PubMed] [Google Scholar]
- Khan I, Dekou V, Hanson M, Poston L, Taylor P. Predictive adaptive responses to maternal high-fat diet prevent endothelial dysfunction but not hypertension in adult rat offspring. Circulation. 2004;110:1097–1102. doi: 10.1161/01.CIR.0000139843.05436.A0. [DOI] [PubMed] [Google Scholar]
- Lee CH, Hwang IK, Lee IS, Yoo KY, Choi JH, Lee BH, Won MH. Differential immunoreactivity of microglial and astrocytic marker protein in the hippocampus of the seizure resistant and sensitive gerbils. J Vet Med Sci. 2008;70:1405–9. doi: 10.1292/jvms.70.1405. [DOI] [PubMed] [Google Scholar]
- Lester-Coll N, Rivera EJ, Soscia SJ, Doiron K, Wands JR, de la Monte SM. Intracerebral streptozotocin model of type 3 diabetes: relevance to sporadic Alzheimer’s disease. J Alzheimers Dis. 2006;9:13–33. doi: 10.3233/jad-2006-9102. [DOI] [PubMed] [Google Scholar]
- Levin BE, Govek E. Gestational obesity accentuates obesity in obesity-prone progeny. Am J Physiol. 1998;275:R1374–1379. doi: 10.1152/ajpregu.1998.275.4.R1374. [DOI] [PubMed] [Google Scholar]
- Lindqvist A, Mohapel P, Bouter B, Frielingsdorf H, Pizzo D, Brundin P, Erlanson-Albertsson C. High-fat diet impairs hippocampal neurogenesis in male rats. Eur J Neurol. 2006;13:1385–1388. doi: 10.1111/j.1468-1331.2006.01500.x. [DOI] [PubMed] [Google Scholar]
- Lu GC, Rouse DJ, DuBard M, Cliver S, Kimberlin D, Hauth JC. The effect of the increasing prevalence of maternal obesity on perinatal morbidity. Am J Obstet Gynecol. 2001;185:845–9. doi: 10.1067/mob.2001.117351. [DOI] [PubMed] [Google Scholar]
- Lucas A. Programming by early nutrition: an experimental approach. J Nutr. 1998;128:401S–406S. doi: 10.1093/jn/128.2.401S. [DOI] [PubMed] [Google Scholar]
- Martínez MC, Andriantsitohaina R. Reactive Nitrogen Species: Molecular Mechanisms and Potential Significance in Health and Disease. Antioxid Redox Signal. 2009 doi: 10.1089/ars.2007.1993. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Mattson MP, Duan W, Guo Z. Meal size and frequency affect neuronal plasticity and vulnerability to disease: cellular and molecular mechanisms. J Neurochem. 2003;84:417–431. doi: 10.1046/j.1471-4159.2003.01586.x. [DOI] [PubMed] [Google Scholar]
- Mei Z, Grummer-Strawn LM, Pietrobelli A, Goulding A, Goran MI, Dietz WH. Validity of body mass index compared with other body-composition screening indexes for the assessment of body fatness in children and adolescents. Am J Clin Nutr. 2002;75:978–85. doi: 10.1093/ajcn/75.6.978. [DOI] [PubMed] [Google Scholar]
- Mhatre M, Floyd RA, Hensley K. Oxidative stress and neuroinflammation in Alzheimer’s disease and amyotrophic lateral sclerosis: common links and potential therapeutic targets. J Alzheimers Dis. 2004;6:147–57. doi: 10.3233/jad-2004-6206. [DOI] [PubMed] [Google Scholar]
- Mielke JG, Nicolitch K, Avellaneda V, Earlam K, Ahuja T, Mealing G, Messier C. Longitudinal study of the effects of a high-fat diet on glucose regulation, hippocampal function, and cerebral insulin sensitivity in C57BL/6 mice. Behav Brain Res. 2006;175:374–82. doi: 10.1016/j.bbr.2006.09.010. [DOI] [PubMed] [Google Scholar]
- Molteni R, Barnard R, Ying Z, Roberts C, Gómez-Pinilla F. A high-fat, refined sugar diet reduces hippocampal brain-derived neurotrophic factor, neuronal plasticity, and learning. Neuroscience. 2002;112:803–14. doi: 10.1016/s0306-4522(02)00123-9. [DOI] [PubMed] [Google Scholar]
- Morrison C. Leptin signaling in brain: A link between nutrition and cognition? Biochim Biophys Acta. 2009 doi: 10.1016/j.bbadis.2008.12.004. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison CD. Leptin resistance and the response to positive energy balance. Physiol Behav. 2008;94:660–3. doi: 10.1016/j.physbeh.2008.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Callaghan JP, Sriram K. Glial fibrillary acidic protein and related glial proteins as biomarkers of neurotoxicity. Expert Opin Drug Saf. 2005;4:433–42. doi: 10.1517/14740338.4.3.433. [DOI] [PubMed] [Google Scholar]
- Oakes ND, Cooney GJ, Camilleri S, Chisholm DJ, Kraegen E. Mechanisms of liver and muscle insulin resistance induced by chronic high-fat feeding. Diabetes. 1997;46:1768–74. doi: 10.2337/diab.46.11.1768. [DOI] [PubMed] [Google Scholar]
- Oomura Y, Hori N, Shiraishi T, Fukunaga K, Takeda H, Tsuji M, Matsumiya T, Ishibashi M, Aou S, Li X, Kohno D, Uramura K, Sougawa H, Yada T, Wayner M, Sasaki K. Leptin facilitates learning and memory performance and enhances hippocampal CA1 long-term potentiation and CaMK II phosphorylation in rats. Peptides. 2006;11:2738–49. doi: 10.1016/j.peptides.2006.07.001. [DOI] [PubMed] [Google Scholar]
- Perry G, Nunomura A, Raina AK, Aliev G, Siedlak SL, Harris PL, Casadesus G, Petersen RB, Bligh-Glover W, Balraj E, Petot GJ, Smith MA. A metabolic basis for Alzheimer disease. Neurochem Res. 2003;28:1549–52. doi: 10.1023/a:1025678510480. [DOI] [PubMed] [Google Scholar]
- Pezzarossa A, Orlandi N, Baggi V, Dazzi D, Ricciarelli E, Coppola F. Effects of maternal weight variations and gestational diabetes mellitus on neonatal birth weight. J Diabetes Complications. 1996;10:78–83. doi: 10.1016/1056-8727(94)00065-4. [DOI] [PubMed] [Google Scholar]
- Plagemann A, Harder T, Rake A, Voits M, Fink H, Rohde W, Dorner G. Perinatal elevation of hypothalamic insulin, acquired malformation of hypothalamic galaninergic neurons, and syndrome x-like alterations in adulthood of neonatally overfed rats. Brain Res. 1999;836:146–155. doi: 10.1016/s0006-8993(99)01662-5. [DOI] [PubMed] [Google Scholar]
- Ramsay JE, Ferrell WR, Crawford L, Wallace AM, Greer IA, Sattar N. Maternal obesity is associated with dysregulation of metabolic, vascular, and inflammatory pathways. J Clin Endocrinol Metab. 2002;87:4231–7. doi: 10.1210/jc.2002-020311. [DOI] [PubMed] [Google Scholar]
- Rasouli N, Kern PA. Adipocytokines and the metabolic complications of obesity. J Clin Endocrinol Metab. 2008;93:S64–73. doi: 10.1210/jc.2008-1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds A, Laurie C, Mosley RL, Gendelman HE. Oxidative stress and the pathogenesis of neurodegenerative disorders. Int Rev Neurobiol. 2007;82:297–325. doi: 10.1016/S0074-7742(07)82016-2. [DOI] [PubMed] [Google Scholar]
- Rolls BJ, Rowe EA. Pregnancy and lactation in the obese rat: effects on maternal and pup weights. Physiol Behav. 1982;28:393–400. doi: 10.1016/0031-9384(82)90130-5. [DOI] [PubMed] [Google Scholar]
- Saliba E, Henrot A. Inflammatory mediators and neonatal brain damage. Biol Neonate. 2001;79:224–7. doi: 10.1159/000047096. [DOI] [PubMed] [Google Scholar]
- Sayre LM, Perry G, Smith MA. Oxidative stress and neurotoxicity. Chem Res Toxicol. 2008;21:172–88. doi: 10.1021/tx700210j. [DOI] [PubMed] [Google Scholar]
- Sena A, Sarliève LL, Rebel G. Brain myelin of genetically obese mice. J Neurol Sci. 1985;68:233–43. doi: 10.1016/0022-510x(85)90104-2. [DOI] [PubMed] [Google Scholar]
- Shankar K, Harrell A, Liu X, Gilchrist JM, Ronis MJ, Badger TM. Maternal obesity at conception programs obesity in the offspring. Am J Physiol Regul Integr Comp Physiol. 2008;294:R528–538. doi: 10.1152/ajpregu.00316.2007. [DOI] [PubMed] [Google Scholar]
- Signore AP, Zhang F, Weng Z, Gao Y, Chen J. Leptin neuroprotection in the CNS: mechanisms and therapeutic potentials. J Neurochem. 2008;106:1977–90. doi: 10.1111/j.1471-4159.2008.05457.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singer LT, Minnes S, Short E, Arendt R, Farkas K, Lewis B, Klein N, Russ S, Min MO, Kirchner HL. Cognitive outcomes of preschool children with prenatal cocaine exposure. J A M A. 2004;291:2448–56. doi: 10.1001/jama.291.20.2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MA, Perry G, Richey PL, Sayre LM, Anderson VE, Beal MF, Kowall N. Oxidative damage in Alzheimer’s. Nature. 1996;382:120–121. doi: 10.1038/382120b0. [DOI] [PubMed] [Google Scholar]
- Souza CG, Moreira JD, Siqueira IR, Pereira AG, Rieger DK, Souza DO, Souza TM, Portela LV, Perry ML. Highly palatable diet consumption increases protein oxidation in rat frontal cortex and anxiety-like behavior. Life Sci. 2007;81:198–203. doi: 10.1016/j.lfs.2007.05.001. [DOI] [PubMed] [Google Scholar]
- Srinivasan M, Katewa SD, Palaniyappan A, Pandya JD, Patel MS. Maternal high-fat diet consumption results in fetal malprogramming predisposing to the onset of metabolic syndrome-like phenotype in adulthood. Am J Physiol Endocrinol Metab. 2006;291:E792–799. doi: 10.1152/ajpendo.00078.2006. [DOI] [PubMed] [Google Scholar]
- Sriram K, Benkovic SA, Miller DB, O’Callaghan JP. Obesity exacerbates chemically induced neurodegeneration. Neuroscience. 2002;115:1335–46. doi: 10.1016/s0306-4522(02)00306-8. [DOI] [PubMed] [Google Scholar]
- Stadtman ER, Levine RL. Free radical-mediated oxidation of free amino acids and amino acid residues in proteins. Amino Acids. 2003;25:207–18. doi: 10.1007/s00726-003-0011-2. [DOI] [PubMed] [Google Scholar]
- Stranahan AM, Norman ED, Lee K, Cutler RG, Telljohann RS, Egan JM, Mattson MP. Diet-induced insulin resistance impairs hippocampal synaptic plasticity and cognition in middle-aged rats. Hippocampus. 2008 doi: 10.1002/hipo.20470. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tartaglia LA, Dembski M, Weng X, Deng N, Culpepper J, Devos R, Richards GJ, Campfield LA, Clark FT, Deeds J, Muir C, Sanker S, Moriarty A, Moore KJ, Smutko JS, Mays GG, Wool EA, Monroe CA, Tepper RI. Identification and expression cloning of a leptin receptor. OB-R Cell. 1995;83:1263–1271. doi: 10.1016/0092-8674(95)90151-5. [DOI] [PubMed] [Google Scholar]
- Thompson DR, Clark CL, Wood B, Zeni MB. Maternal obesity and risk of infant death based on Florida birth records for 2004. Public Health Rep. 2008;123:487–93. doi: 10.1177/003335490812300410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toro R, Leonard G, Lerner JV, Lerner RM, Perron M, Pike GB, Richer L, Veillette S, Pausova Z, Paus T. Prenatal exposure to maternal cigarette smoking and the adolescent cerebral cortex. Neuropsychopharmacology. 2008;33:1019–27. doi: 10.1038/sj.npp.1301484. [DOI] [PubMed] [Google Scholar]
- Tozuka Y, Wada E, Wada K. Diet-induced obesity in female mice leads to peroxidized lipid accumulations and impairment of hippocampal neurogenesis during the early life of their offspring. FASEB J. 2009 doi: 10.1096/fj.08-124784. Epub ahead of print. [DOI] [PubMed] [Google Scholar]
- Tschöp M, Heiman ML. Rodent obesity models: an overview. Exp Clin Endocrinol Diabetes. 2001;109:307–19. doi: 10.1055/s-2001-17297. [DOI] [PubMed] [Google Scholar]
- Udagawa J, Hatta T, Hashimoto R, Otani H. Roles of leptin in prenatal and perinatal brain development. Congenit Anom (Kyoto) 2007;47:77–83. doi: 10.1111/j.1741-4520.2007.00150.x. [DOI] [PubMed] [Google Scholar]
- Vega-Avelaira D, Moss A, Fitzgerald M. Age-related changes in the spinal cord microglial and astrocytic response profile to nerve injury. Brain Behav Immun. 2007;21:617–23. doi: 10.1016/j.bbi.2006.10.007. [DOI] [PubMed] [Google Scholar]
- Waldstein SR, Katzel LI. Interactive relations of central versus total obesity and blood pressure to cognitive function. Int J Obes (Lond) 2006;30:201–207. doi: 10.1038/sj.ijo.0803114. [DOI] [PubMed] [Google Scholar]
- Wang CX, Shuaib A. Involvement of inflammatory cytokines in central nervous system injury. Prog Neurobiol. 2002;67:161–172. doi: 10.1016/s0301-0082(02)00010-2. [DOI] [PubMed] [Google Scholar]
- Wayner MJ, Armstrong DL, Phelix CF, Oomura Y. Orexin-A (Hypocretin-1) and leptin enhance LTP in the dentate gyrus of rats in vivo. Peptides. 2004;25:991–996. doi: 10.1016/j.peptides.2004.03.018. [DOI] [PubMed] [Google Scholar]
- White CL, Purpera MN, Morrison CD. Maternal obesity is necessary for the programming effect of a high-fat diet on offspring. Am J Physiol Regul Integr Comp Physiol. 2009 doi: 10.1152/ajpregu.91015.2008. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willford J, Leech S, Day N. Moderate prenatal alcohol exposure and cognitive status of children at age 10. Alcohol Clin Exp Res. 2006;30:1051–1059. doi: 10.1111/j.1530-0277.2006.00119.x. [DOI] [PubMed] [Google Scholar]
- Wilson CJ, Finch CE, Cohen HJ. Cytokines and cognition--the case for a head-to-toe inflammatory paradigm. J Am Geriatr Soc. 2002;50:2041–2056. doi: 10.1046/j.1532-5415.2002.50619.x. [DOI] [PubMed] [Google Scholar]
- Winocur G, Greenwood CE. Studies of the effects of high fat diets on cognitive function in a rat model. Neurobiol Aging. 2005;26:46–9. doi: 10.1016/j.neurobiolaging.2005.09.003. [DOI] [PubMed] [Google Scholar]
- Wu A, Ying Z, Gomez-Pinilla F. The interplay between oxidative stress and brain-derived neurotrophic factor modulates the outcome of a saturated fat diet on synaptic plasticity and cognition. Eur J Neurosci. 2004;19:1699–707. doi: 10.1111/j.1460-9568.2004.03246.x. [DOI] [PubMed] [Google Scholar]
- Zecca L, Wilms H, Geick S, Claasen JH, Brandenburg LO, Holzknecht C, Panizza ML, Zucca FA, Deuschl G, Sievers J, Lucius R. Human neuromelanin induces neuroinflammation and neurodegeneration in the rat substantia nigra: implications for Parkinson’s disease. Acta Neuropathol. 2008;116:47–55. doi: 10.1007/s00401-008-0361-7. [DOI] [PubMed] [Google Scholar]
- Zhang X, Dong F, Ren J, Driscoll MJ, Culver B. High dietary fat induces NADPH oxidase-associated oxidative stress and inflammation in rat cerebral cortex. Exp Neurol. 2005;191:318–25. doi: 10.1016/j.expneurol.2004.10.011. [DOI] [PubMed] [Google Scholar]