

Abstract
The endogenous catecholamine norepinephrine (NE) exhibits anti-epileptic properties, however it is not well understood which adrenergic receptor (AR) mediates this effect. The aim of this study was to investigate α1-adrenergic receptor (AR) activation in region CA1 of the hippocampus, a subcortical structure often implicated in temporal lobe epilepsies. Using cell-attached and whole-cell recordings in rat hippocampal slices, we confirmed that selective α1-AR activation increases action potential firing in a subpopulation of CA1 interneurons. We found that this response is mediated via the α1A-AR subtype, initiated by sodium influx, and appears independent of second messenger signaling. In CA1 pyramidal cells, α1A-AR activation decreases activity due to increased pre-synaptic GABA and somatostatin release. Examination of post-synaptic receptor involvement revealed that while GABAA receptors mediate the majority of α1A-adrenergic effects on CA1 pyramidal cells, significant contributions are also made by GABAB and somatostatin receptors. Finally, to test whether α1A-AR activation could have potential therapeutic implications, we performed AR agonist challenges using two in vitro epileptiform models. When GABAA receptors were available, α1A-AR activation significantly decreased epileptiform bursting in CA1. Together, our findings directly link stimulation of the α1A-AR subtype to release of GABA and somatostatin at the single cell level and suggest that α1A-AR activation may represent one mechanism by which NE exerts anti-epileptic effects within the hippocampus.
1. Introduction
A beneficial role for catecholamines in seizure susceptibility was first suggested in 1954, when a study utilizing reserpine revealed a decreased convulsive threshold for caffeine-and pentylenetetrazole-induced seizures in experimental animals (Chen et al., 1954). Building from this initial experiment, numerous investigations have since established norepinephrine (NE) as the specific catecholamine capable of decreasing hyperexcitability in many in vivo and in vitro epilepsy models, including kindling (McIntyre et al., 1982; Jimenez-Rivera et al., 1986), electroshock (Mason and Corcoran, 1979), audiogenic (Jerlicz et al., 1978), and focal penicillin (Ferraro et al., 1994) induced seizures (for review see Giorgi et al., 2004). Moreover, the noradrenergic system has been found to contribute to the effectiveness of certain non-pharmacological anti-epileptic treatments, including vagal nerve stimulation (Krahl et al., 1998) and the ketogenic diet (Szot et al., 2001; Szot, 2004). Manipulation of the noradrenergic system, particularly through activation of specific adrenergic receptors (ARs), clearly holds therapeutic potential, however the precise mechanisms underlying NE’s anticonvulsant properties remain poorly understood and thus clinical application is currently limited.
One area of the brain where NE exhibits anticonvulsant properties in vitro and in vivo is the hippocampus, a C-shaped subcortical structure adjacent to the lateral ventricles (Nishi et al., 1981; Wu et al., 1987; Kokaia et al., 1989). The hippocampus is structurally well-defined, consisting of a distinct principal pyramidal cell layer surrounded above and below by small numbers of widely dispersed interneurons. The principal pyramidal neurons, particularly in CA3, exhibit intrinsic bursting properties and form excitatory recurrent circuits (Brown et al., 1979; Kandel and Spencer, 1961), making the hippocampus susceptible to epileptogenesis. Indeed, the structure is implicated in a majority of temporal lobe epilepsies (McNamara, 1994). CA1 hippocampus is particularly vulnerable to neuronal damage following seizure activity (Freund et al., 1991; Cavazos et al., 1994; Kaur et al., 2007), which can have dire consequences as this region is heavily implicated in synaptic plasticity associated with learning and episodic memory (for review see Neves et al., 2008). A strategy to decrease hyperexcitability in the hippocampus, with attention to CA1, might therefore prove important in the treatment of temporal lobe epilepsies.
NE’s ability to decrease heightened activity in the hippocampus has been attributed to activation of the α1- and/or α2-AR (Pang and Rose, 1987; Curet and de Montigny, 1988; Mynlieff and Dunwiddie, 1988; Bergles et al., 1996; Boehm, 1999), however further characterization of the specific α-AR subtype(s) involved is lacking. The ARs comprise a family of 7-transmembrane spanning G protein-coupled receptors broadly defined into α1-, α2-, and β-AR subfamilies. Three subtypes for each the α1-, α2-, and β-AR have been cloned and characterized: α1A, α1B, α1D, α2A, α2B, α2C, β1, β2, and β3 (Bylund et al., 1992; Hieble et al., 1995). The α1-, α2-, and β-AR are traditionally coupled to the G protein isotypes Gq, Gi and Gs, respectively, though novel signaling paradigms are increasingly being described (for review see Perez, 2006).
Previous studies, including our own, have identified a subpopulation of CA1 interneurons that are depolarized by NE via activation of an α1-AR, particularly the α1A-AR subtype (Bergles et al., 1996; Hillman et al., 2007). These interneurons are GABAergic and express the neuropeptide somatostatin (Hillman et al., 2005a), raising the possibility that an α1A-AR-mediated depolarization results in GABA and somatostatin release onto nearby pyramidal neurons. This perhaps represents one mechanism by which NE decreases excitability in the hippocampus. In this study we aimed to examine the functional consequences of α1A-AR activation on both CA1 interneuron and pyramidal neuron activity, and test potential therapeutic implications to epilepsy by performing an α1-AR agonist challenge in two in vitro epilepsy models.
2. Methods
2.1 Reagents
The following were obtained from Sigma-Aldrich (St. Louis, MO): D-(−)-2-amino-5-phosphonopentanoic acid (APV), atropine, 1,2-bis(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid tetrapotassium salt (BAPTA), barium, BMY7378, cesium, CGP35348, cyclosomatostatin, 6,7-dinitroquinoxaline-2,3-dione (DNQX), L765,314, lanthanum, 5-methylurapidil, MgATP, NaGTP, PD98059, pertussis toxin, (R)-(−)-phenylephrine (PE), picrotoxin (PTX), QX-314, Ro31-8220, SKF96365, SQ22,536, tetraethylammonium (TEA), tetrodotxoin (TTX), and U73122. Isoflurane was ordered from Abbott Diagnostics (Chicago, IL). All other chemical reagents were of biological grade and ordered through J.T. Baker, Inc. (Phillipsburg, NJ) or Fisher Scientific (Fairlawn, NJ).
2.2 Research Animals
Male Sprague-Dawley rat pups were obtained from Harlan (Indianapolis, IN) and housed with their mothers before weaning. After arrival, rats were allowed to acclimate for at least two days before experimentation. Animals were maintained on a 12-hr light/dark cycle and provided standard rat chow and water ad libitum. All protocols described have been approved by the Institutional Animal Care and Use Committee at the University of North Dakota, which is in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
2.3 Hippocampal Slice Preparation
Sprague-Dawley rat pups (P14–P22, 25–65 g) were deeply anesthetized with isoflurane and decapitated. Brains were rapidly removed and placed in chilled solution containing (in mM): 110 choline chloride, 25 NaHCO3, 25 D-glucose, 11.6 sodium ascorbate, 7 MgSO4, 3.1 sodium pyruvate, 2.5 KCl, 1.25 NaPO4, and 0.5 CaCl2. Using an HM650V vibratome (Microm, Walldorf, Germany), 400 μm coronal sections were cut and transferred to a holding solution of (in mM): 130 NaCl, 3.5 KCl, 5 MgCl2, 0.5 CaCl2, 1.25 NaH2PO4, 24 NaHCO3, and 10 D-glucose. Slices were incubated at 37°C for 30 min and then moved to room temperature. For experimentation, the hippocampus was visualized under infrared-differential interference contrast (IR-DIC) optics using a BX51WI upright microscope (Olympus, Melville, NY). Pyramidal cells (n=64) were identified as such based on their location within stratum pyramidale, characteristic pyramidal morphology, and low input resistance. Interneurons were identified as such based on their location above and below stratum pyramidale (stratum oriens n=104, stratum radiatum n=37), elongated shape, and high input resistance. Slices were continually perfused with artificial cerebral spinal fluid (aCSF) consisting of (in mM): 119 NaCl, 5 KCl, 1.3 MgSO4, 2.5 CaCl2, 1 NaH2PO4, 26.2 NaHCO3, and 11 D-glucose. High KCl (5 mM) was chosen over a more physiological concentration so that a suitable level of baseline activity was consistently present, particularly for cell-attached studies. For cell-attached and whole-cell experiments, 1 μM atropine, 10 μM DNQX, and 50 μM APV were included in the aCSF to pharmacologically isolate the cell of interest from primary excitatory inputs. All solutions were continually aerated with a mixture of 95% O2, 5% CO2.
2.4 Cell-Attached Recording
Micropipettes (4–6 MΩ) were prepared from borosilicate glass using a vertical PP-830 puller (Narishige, Tokyo, Japan). Pipettes were filled with an internal solution of (in mM): 135 KCH3SO4, 8 NaCl, 10 HEPES, 2 MgATP, 0.3 NaGTP, and 0.1 BAPTA. Gigaohm seals were formed on the soma of candidate CA1 neurons, enabling isolated recordings without disrupting the integrity of the cell membrane. Baseline action potential frequency was recorded for 25 min, allowing time for equilibration of a receptor antagonist if warranted by the experiment. Select concentrations of PE were then added to the perfusion line in 8 min increments. In preliminary experiments (data not shown), we trialed 8, 12, 15 and 20+ min PE applications and found that cellular responses to PE at 8 min were no different than at 12 or 15 min; infusions lasting 20+ min generally resulted in a decrement of activity. Therefore, an 8 min PE application was used for all subsequent experiments. Changes in action potential frequency were visualized in real time and recorded for subsequent analysis. To avoid issues of receptor desensitization and depolarization block, only one hippocampal slice was used per experimental paradigm. After data was obtained from one CA1 neuron, generating an n=1, the hippocampal slice was discarded and a new slice equilibrated in a CSF.
Action potentials were detected using an Axoclamp 700B (Molecular Devices Corporation, Sunnyvale, CA), digitized with a Digidata 1322A analog-to-digital converter (Molecular Devices), and recorded using Axoscope 9.2 software (Molecular Devices). Postexperimental analysis was completed using Mini Analysis 5.0 (Synaptosoft, Decatur, GA) and Prism 4.03 (GraphPad Software Inc., San Diego, CA). Action potentials recorded during the course of each functional experiment were counted and binned at 60 sec intervals. Given the variation in baseline activity between cells, action potential activity was normalized for each individual cell, considering baseline activity as 100%. Significance between treatment groups was tested using a one-way ANOVA with post-hoc Tukey test. All values are reported as the mean±S.E., n≥3 as indicated. In figures, significance is denoted as *p<0.05, **p<0.01, ***p<0.001.
2.5 Whole-Cell Recording
For recording of holding current from CA1 interneurons, micropipettes (4–6 MΩ) were filled with an internal solution of (in mM): 100 potassium gluconate, 0.6 EGTA, 5 MgCl2, 8 NaCl, 2 MgATP, 0.3 NaGTP, and 40 HEPES. In experiments utilizing inhibitory compounds (e.g. cesium, SKF96365), slices were equilibrated with the compounds for at least 1 hour prior to whole-cell recording, with the following exceptions: 1) for pertussis toxin experimentation, slices were equilibrated with the compound for ≥4 hours; and 2) for Ro31-8220 and BAPTA experimentation, the compounds were delivered via diffusion from the micropipette. After achieving whole-cell configuration, cells were voltage clamped at −65 mV and current was continuously recorded online. Access resistance (range 4–31 MΩ) was checked regularly; recordings were discarded if resistance changed more than 20% during the course of the experiment. Following a 4 min baseline, a single 8 min PE challenge was delivered via the aCSF perfusion. Current was detected, digitized and recorded using the equipment and software described above. Current values were compiled in 30 sec bins, with the last 30 sec bin from baseline and PE challenge periods being used to calculate change in holding current. Significance between treatment groups was tested using a one-way ANOVA with post-hoc Tukey test. All values are reported as the mean±S.E., n ≥ 5 as indicated.
For recording of inhibitory post-synaptic currents (IPSCs) from CA1 pyramidal cells, micropipettes (4–6 MΩ) were filled with an internal solution of (in mM): 100 cesium gluconate, 0.6 EGTA, 5 MgCl2, 8 NaCl, 2 MgATP, 0.3 NaGTP, 40 HEPES and 1 QX-314. For recording of miniature IPSCs, 0.5 μM TTX was added to the aCSF. After achieving whole-cell configuration at −60 mV, the holding potential was moved to +20 mV for monitoring IPSCs. PE challenges (10, 50, and 100 μM) were delivered in 8 min increments via the aCSF perfusion. IPSCs recorded during the course of each functional experiment are expressed as percentage of baseline frequency. Significance between experimental groups was tested using a one-way ANOVA with post-hoc Tukey test. All values are reported as the mean±S.E., n ≥ 3 as indicated.
For recording of evoked IPSCs, stimulating micropipettes (2–3 MΩ) were filled with aCSF solution, and recording micropipettes (4–6 MΩ) were filled with (in mM): 100 cesium gluconate, 0.6 EGTA, 5 MgCl2, 8 NaCl, 2 MgATP, 0.3 NaGTP, and 40 HEPES. Evoked IPSCs were stimulated from CA1 stratum oriens and recorded from CA1 stratum pyramidale. Following 5 min of baseline recording, a single 10 min PE challenge (50 or 100 μM) was delivered via the aCSF perfusion. Evoked IPSC amplitudes were averaged per minute and normalized to baseline. Significance between the two experimental conditions was tested using an unpaired, two-tailed Student’s t test. All values are reported as the mean±S.E., n=8.
2.6 Field Potential Recording
For recording of spontaneous epileptiform burst activity, micropipettes (4–6 MΩ) were filled with 2 M NaCl and lowered into CA1 stratum pyramidale. The aCSF solution contained either 100 μM PTX or lacked MgSO4, as outlined by the experiment. Following 25 min of baseline recording, a single 8 min PE challenge was delivered via the aCSF perfusion. Epileptiform burst frequency was averaged per minute and normalized to baseline. Significance between experimental groups was tested using a one-way ANOVA with post-hoc Tukey test. All values are reported as the mean±S.E., n=8.
3. Results
We have previously reported cell-specific, AR subtype-specific expression in CA1 hippocampus; namely, CA1 pyramidal cells predominately express β2-ARs, while a subpopulation of somatostatin-positive CA1 interneurons predominately express α1A-ARs (Hillman et al., 2005b; Hillman et al., 2007). To examine the functional consequences of activation for the latter receptor subtype, we first used cell-attached configuration to record action potentials from individual CA1 interneurons as well as pyramidal neurons. Cells that provided a stable baseline recording for 25 minutes were challenged with increasing concentrations of the selective α1-AR agonist PE, and the effect on action potential frequency noted. Bergles et al. (1996) previously demonstrated that α1-AR activation results in increased action potential firing in CA1 interneurons; here we not only substantiate those findings, but demonstrate that α1-AR activation modulates both interneuron and pyramidal neuron firing in a concentration-dependent, α1A-AR subtype-dependent manner.
As shown in Figure 1, PE (10, 50 and 100 μM) caused a concentration-dependent increase in interneuron action potential firing (142±5, 203±4, 211±11% of baseline, respectively, mean baseline frequency 0.4 Hz, range 0.1–0.8 Hz) and a concomitant decrease in pyramidal neuron activity (76±5, 36±2, 11±3% of baseline, respectively, mean baseline frequency 0.9 Hz, range 0.5–2.1 Hz). When slices were pre-treated with 100 nM of the selective α1A-AR antagonist 5-methylurapidil (5-MU), the ability of PE to modulate action potential frequency was significantly altered in both cell types (104±2, 141±8, 165±5% of baseline for interneurons; 99±1, 71±5, 38±4% of baseline for pyramidal cells). Conversely, when slices were pre-treated with 100 nM of the selective α1B-AR antagonist L765,314 – a concentration 18 times greater than its affinity for the α1B-AR subtype – PE’s ability to modulate action potential frequency in both cell types was no different from control (for 50 μMPE: 191±13% of baseline for interneurons, 50±5% of baseline for pyramidal cells; data not shown). Similarly, pre-treatment with 100 nM of the selective α1D-AR antagonist BMY7378 – a concentration 23 times greater than its affinity for the α1D-AR subtype – provided no significant effect (for 50 μM PE: 195±7% of baseline for interneurons, 31±9% of baseline for pyramidal cells; data not shown). Given that we have extensively characterized these three subtype-selective α1-AR antagonists as part of a rigorous pharmacological study in CA1 hippocampus (Hillman et al., 2007), we were assured that a concentration of 100 nM for each receptor antagonist would effectively and moreover selectively block the α1-AR subtype of interest (i.e., 5-MU=α1A-AR, L765,314=α1B-AR and BMY7378=α1D-AR subtype). Thus, we were confident that our negative results obtained with L765,314 and BMY7378 experimentation accurately reflected no involvement of the α1B- or α1D-AR in the PE-mediated increase in interneuron activity. Taken together, these data suggest that the α1A-AR is the AR subtype primarily responsible for PE’s effect.
Figure 1.

Differing effects of α1-AR activation on CA1 neuron activity. In cell-attached configuration, individual neurons were challenged with increasing concentrations of the selective α1-AR agonist PE in a pharmacologically isolated system. (A) In CA1 interneurons, PE at 10, 50 and 100 μM produced a concentration-dependent increase in action potential firing (142±5, 203±4, 211±11% of baseline, respectively) which was attenuated by pre-treatment with 100 nM of the selective α1A-AR antagonist 5-MU (104±2, 141±8, 165±5%). (B) Time course from one stratum oriens interneuron illustrating the concentration-dependent increase in action potential frequency produced by PE. (C) In CA1 pyramidal cells, PE caused a concentration dependent decrease in activity (76±5, 36±2, 11±3% of baseline) which was also significantly affected by pre-treatment with 100 nM of 5-MU (99±1, 71±5, 38±4% of baseline). (D) Time course from one pyramidal cell illustrating the concentration-dependent decrease in action potential frequency produced by PE. For (B) and (D), bins represent average frequency per 1 min interval. For each experiment n=5.
Though the data presented in Figure 1 clearly illustrates the ability of PE to decrease CA1 pyramidal neuron activity, it is unlikely that this is due to direct activation of an α1-AR on the pyramidal cell itself. Our previous studies found no evidence of α-ARs on CA1 pyramidal cells either transcriptionally (Hillman et al., 2005a) or functionally (Hillman et al., 2005b). Furthermore, activation of the α1-AR is generally associated with excitatory effects via Gq/Phospholipase C (PLC) signaling. A more plausible explanation behind PE’s ability to modulate pyramidal cell activity would be via a pre-synaptic mechanism, such as α1-AR activation of pre-synaptic inhibitory interneurons. Indeed, Bergles et al. (1996) have previously reported that α-AR agonists increase inhibitory post-synaptic current (IPSC) frequency in pyramidal neurons. To test this possibility in our set-up, we recorded spontaneous IPSCs from CA1 pyramidal neurons in whole-cell, voltage-clamp configuration. As shown in Figure 2, increasing concentrations of PE (10, 50 and 100 μM) produced a concentration-dependent increase in spontaneous IPSCs (131±6, 153±10, 182±2% of baseline, respectively, mean baseline frequency 1.2 Hz, range 0.6–4.5 Hz). Observed IPSCs were GABAA-mediated, as currents were abolished by application of the selective GABAA receptor antagonist PTX. The PE-dependent increase in spontaneous IPSCs was significantly altered when slices were pre-treated with 100 nM 5-MU (106±3, 115±8, 157±4% of baseline; Figure 2D). Together these results confirm Bergles et al. original finding (1996) in our set-up, and moreover demonstrate that it is specific activation of the α1A-AR subtype which leads to an increase in GABAA receptor-mediated synaptic transmission onto CA1 pyramidal neurons.
Figure 2.

Spontaneous IPSC frequency is increased by PE in CA1 pyramidal neurons. (A) Representative traces recorded from CA1 pyramidal cells demonstrating a concentration-dependent increase in spontaneous IPSC frequency following PE application. (B) Pre-treatment of slices with 10 μM PTX effectively abolished IPSCs, indicating currents were primarily GABAA receptor mediated. (C) Time course from one pyramidal cell illustrating increased spontaneous IPSC frequency following consecutive PE challenges. (D) PE at 10, 50 and 100 μM produced a concentration-dependent increase in pyramidal cell IPSC frequency (131±6, 153±10, 182±2% of baseline respectively, n=5), an effect that was significantly affected by pre-treatment of slices with 100 nM of 5-MU (106±3, 115±8, 157±4% of baseline, n=5). Miniature IPSC frequency, recorded in the presence of 500 nM TTX, did not significantly differ from baseline following application of 10, 50 and 100 μM PE (101±3, 112±5, 107±5% of baseline respectively, n=3). (E) Cumulative probability plotting revealed no measurable change in miniature IPSC amplitude following 100 μM PE application. (F) Time course from evoked IPSC experiments, illustrating no significant change in eIPSC amplitude following 100 μM PE application (0.97±0.06 versus 1.06±0.05 control, p=0.19, n=8). Arrows indicate time points used for amplitude comparison.
As mentioned above, PE could be increasing spontaneous IPSCs in pyramidal cells by prompting pre-synaptic GABA release from CA1 interneurons. Alternatively, PE could be increasing spontaneous IPSCs by modulating GABAA receptors present on the pyramidal neurons themselves. To determine whether the effect of PE on CA1 pyramidal cells was pre-or post-synaptic, we recorded miniature and evoked IPSCs. Miniature IPSCs – recorded in the presence of 500 nM TTX – reflect GABAA receptor-mediated chloride currents due to quantal, pre-synaptic GABA release that is not action potential dependent. In our system, miniature IPSC frequency was not significantly increased in response to PE application (101±3, 112±5, 107±5% of baseline, mean baseline frequency 0.3 Hz, range 0–2.3 Hz; Figure 2D), and cumulative probability plotting revealed no PE-mediated change in miniature IPSC amplitude (Figure 2E). Evoked IPSCs – stimulated from stratum oriens – also failed to exhibit a significant change in amplitude following application of 50 μM (data not shown) or 100 μM PE (0.97±0.06 versus 1.06±0.05 control; Figure 2F). Taken together, these whole cell experiments demonstrate that PE modulates pyramidal cell activity by increasing pre-synaptic, action potential mediated release of GABA.
As evidenced in Figure 2, PE’s effect on pyramidal neuron activity involves pre-synaptic GABA release and consequent post-synaptic GABAA receptor activation. We were curious, however, if other post-synaptic receptors were involved. In previous RT-PCR studies we localized α1A-AR mRNA to a subpopulation of somatostatin-containing, GABAergic interneurons (Hillman et al., 2005a), raising the possibility that GABA and somatostatin may both be involved in the PE-mediated decrease in pyramidal neuron activity. Moreover, pre-synaptic release of GABA likely affects both GABAA and GABAB receptors present on the post-synaptic cell. To define involvement of GABAA, GABAB and somatostatin receptors we utilized selective receptor antagonists – PTX, CGP35348, and cyclosomatostatin, respectively – in combination with a PE challenge. As shown in Figure 3, in control aCSF conditions, 50 μM PE produces an ~75% decrease in pyramidal neuron action potential firing (mean baseline frequency 1.2 Hz, range 0.3–3.6 Hz). This effect was significantly attenuated by pre-treatment with 10 μM PTX, demonstrating functional GABAA receptors are indeed needed for PE to modulate pyramidal neuron activity (73±3 versus 25±3% control). Interestingly, including 30 μM of the selective GABAB receptor antagonist CGP35348 or 1 μM of the somatostatin receptor antagonist cyclosomatostatin in the aCSF also significantly altered the ability of PE to decrease pyramidal neuron activity as compared to control (45±7 and 46±6%, respectively), suggesting that GABAB and somatostatin receptors also individually contribute to PE’s effect.
Figure 3.

GABA and somatostatin are required for the PE-mediated decrease in pyramidal activity. Using cell-attached configuration, action potentials were recorded from individual CA1 pyramidal cells during a 50 μM PE challenge. In control aCSF, this challenge decreased action potential frequency to 25±3% of baseline. This PE-mediated decrease in activity was significantly attenuated by pre-treatment of slices with either 10 μM PTX (73±3% of baseline), 30 μM CGP35348 (45±7%), 1 μM cyclosomatostatin (46±6%), or dual application of 10 μM PTX/30 μM CGP35348 (81±2%). Combined use of PTX, CGP35348 and cyclosomatostatin effectively abolished the ability of PE to modulate CA1 pyramidal neuron activity, restoring action potential frequency to 99±1% of baseline. For each experiment n=5.
When both GABA receptor subtypes are blocked by dual inclusion of PTX and CGP35348 in the aCSF, the ability of PE to decrease pyramidal neuron activity was not significantly different than when PTX was used alone (81±2 versus 73±3%, p=0.07), indicating that when both receptors are available the GABAA subtype predominates functionally. Of note, when both GABA receptor subtypes were blocked PE was still able to produce an ~20% decrease in pyramidal neuron activity. Only when GABAA, GABAB, and somatostatin receptors were pharmacologically blocked in unison was the ability of PE to modulate pyramidal neuron activity essentially abolished (99±1% of baseline).
PE’s ability to decrease pyramidal neuron activity via modulation of pre-synaptic GABA and somatostatin release could have potential therapeutic implications as the hippocampus is often implicated in temporal lobe epilepsies. To determine if the PE-mediated release of GABA and somatostatin onto pyramidal neurons is sufficient to decrease epileptic-type events in CA1, we performed PE challenges using two in vitro epileptic models. In the first model, spontaneous seizure activity was generated in hippocampal slices by omitting Mg2+ from the aCSF, enabling heightened glutamatergic activity. In the second model, spontaneous seizure activity was generated by including 100 μM PTX in the aCSF to block basal inhibitory tone. As illustrated in Figure 4, in zero Mg2+ aCSF, PE provided an ~30% decrease in epileptic-type events, an effect attenuated by pre-treatment of slices with 100 nM 5-MU. In 100 μM PTX aCSF, PE produced a minimal but not significant decrease in event frequency (93±2% of baseline). This finding was not too surprising given, as shown earlier in Figure 3, GABAA receptors are the largest contributor to the PE-mediated decrease in pyramidal action potential frequency. Taken together these experiments suggest that if GABAA receptors are available, PE can significantly decrease hyperexcitability in CA1 via activation of the α1A-AR.
Figure 4.

PE decreases CA1 epileptiform bursts in one in vitro epilepsy model. Field recordings were made from CA1 stratum pyramidale in either zero Mg2+ aCSF or aCSF containing 100 μM PTX. (A) Representative traces of population spikes recorded from CA1 stratum pyramidale in zero Mg2+ aCSF before, during, and after a 50 μM PE challenge. (B) In zero Mg2+ aCSF, 50 μM PE produced a significant decrease in population spike frequency (70±5% of baseline), an effect that was blocked by pre-treatment of slices with 100 nM of 5-MU (96±0.3%). (C) In aCSF containing 100 μM PTX, 50 μM PE provided a minimal but not significant decrease in population spike frequency (93±2% of baseline). For each experiment n=8.
Thus far we have focused on the post-synaptic consequences of α1A-AR activation, namely a decrease in pyramidal neuron activity via GABA and somatostatin receptor activation. In the last portion of our study we wanted to examine the pre-synaptic consequences of α1A-AR activation, that is the mechanism underlying the PE-mediated increase in interneuron activity shown earlier in Figure 1. α1-ARs traditionally signal through the Gq G protein, prompting increased PLC activity leading to downstream activation of protein kinase C (PKC) and inositol 1,4,5-trisphosphate-mediated calcium release. To test if this was indeed the signaling cascade responsible for the α1A-AR-mediated increase in action potential firing seen in CA1 interneurons, we utilized pharmacological inhibitors of key signaling molecules in combination with whole-cell voltage clamp recordings. As shown in Figure 5, in responsive CA1 interneurons PE (50 μM) initiated a negative change in holding current (−21±2.5 pA), indicative of inward current flux. Surprisingly, this observed PE-mediated change in holding potential was not altered by inhibition of PLC (−18±1.6 pA, 50 μM U73122), inhibition of PKC (−20±6.9 pA, 100 nM Ro31-8220), or chelation of intracellular calcium (−19±3.1 pA, 10 mM BAPTA), suggesting that PE’s effect is independent of canonical PLC signaling. Similar concentrations of U73122, Ro31-8220 and BAPTA used for our study have been applied by others in neuronal cells to inhibit PLC, PKC and chelate calcium, respectively (Chung et al., 2006; Singaravelu and Deitmer, 2006; Huang and van den Pol 2007). The PE-mediated change in holding current was also unaltered by inhibition of MAPK (−18±3.3 pA, 10 μM PD98059) and adenylate cyclase (−18±3.9 pA, 20 μM SQ22,536), second messengers which can also be utilized by the α1-AR and whose inhibition through use of these compounds has been shown in neurons previously (Horie et al., 1995; Gutkind, 1998; Chung et al., 2006; Bonsi et al., 2007). Interestingly, pre-treatment with 100 μM pertussis toxin, an inhibitor of the Gi/o G-protein, significantly decreased the inward current seen after PE application (−6±1.3 pA). Together these data suggest that in certain CA1 interneurons, α1-AR activation initiates an inward current flux that is pertussis toxin-sensitive and seemingly independent of characteristic α1-AR second messenger systems.
Figure 5.
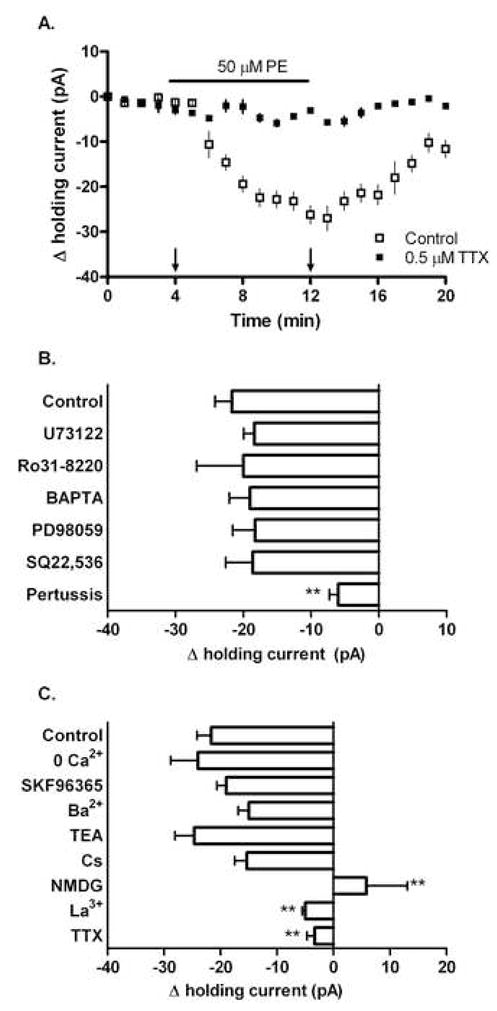
PE initiates a pertussis-sensitive inward sodium current in responsive CA1 interneurons. Whole-cell recordings of holding current in CA1 interneurons voltage clamped to −65 mV. In control aCSF, 50 μM PE produced a measurable inward current (−21±2.5 pA) in a subpopulation of CA1 interneurons. (A) Time course illustrating the PE-mediated inward current in control and TTX conditions. Arrows indicate time points used for Δ holding current calculations shown in (B) and (C). (B) The PE-mediated inward current was not significantly altered by pre-treatment with 50 μM U73122 (−18±1.6 pA), 100 nM Ro31-8220 (−20±6.9 pA), 10 mM BAPTA (−19±3.1 pA), 10 μM PD98059 (−18±3.3 pA), or 20 μM SQ22,536 (−18±3.9 pA). Pre-treatment with 100 μM pertussis toxin however significantly reduced the magnitude of the PE-mediated current (−6.0±1.3 pA). (C) Manipulations of Ca2+, Na+ and K+ availability differentially affected the observed PE-mediated inward current. In control aCSF, 50 μM PE produced a measurable inward current in a subpopulation of CA1 interneurons (−21±2.5 pA). This effect was not altered by removal of extracellular calcium from the aCSF (−24±4.8), or by pre-treatment with 100 μM SKF96365 (−19±1.7 pA), 2 mM barium (−15±1.8 pA), 10 μM TEA (−24±3.4 pA), or 3 mM cesium (−15±2.1 pA). However, significant reductions in the magnitude of the PE-mediated current were seen when NMDG was substituted for extracellular sodium (5.8±7.2 pA), or when slices were pre-treated with 10 μM lanthanum (−5.0±0.52 pA) or 0.5 μM TTX (−3.0±1.3 pA). For each experiment n=8.
To identify the cationic contributors of the PE-mediated inward current, we manipulated the availability of Ca2+, Na+, and K+ by either altering aCSF composition or utilizing channel inhibitors. As shown in Figure 5C, in aCSF lacking calcium, as well as in aCSF containing 100 μM of the calcium channel inhibitor SKF96365, a concentration previously shown to constrain hippocampal calcium mobilization (Wang et al., 2007), the inward current generated by a 50 μM PE challenge was not significantly different (−24±4.8 and −19±1.7 pA, respectively) from control (−21±2.5 pA). PE’s ability to generate inward current was also unaffected in aCSF containing non-selective potassium channel blockers; neither 2 mM barium, 10 μM TEA, or 3 mM cesium significantly altered the observed inward current (−15±1.8, −24±3.4 and −15±2.1 pA, respectively). However, in aCSF lacking sodium (replacement of NaCl with N-methyl-D-glucamine (NMDG)), as well as in aCSF containing 10 μM of the non-selective sodium channel blocker lanthanum, PE was unable to elicit the negative change in holding current seen in control conditions (5.8±7.2 and −5.0±0.52 pA respectively). Moreover, PE’s ability to induce inward current was significantly attenuated in aCSF containing 500 nM of the sodium channel blocker TTX (−3.0±1.3 pA). These results suggest that the PE-mediated inward current observed in responsive CA1 interneurons is likely sodium dependent.
4. Discussion
Previous studies, including our own, have identified a subpopulation of CA1 interneurons which are depolarized by NE via activation of an α1-AR, specifically the α1A-AR subtype (Bergles et al., 1996; Hillman et al., 2007). These interneurons predominate in stratum oriens, contain the GABAergic enzyme GAD65, and consistently express the neuropeptide somatostatin (Hillman et al., 2005); ~75% of stratum oriens interneurons and ~25% of stratum radiatum interneurons respond to α1-adrenergic agonists (Hillman et al., 2007). Given these interneurons project axon collaterals to neighboring pyramidal cells (Knowles, 1992), it is likely that activation of the α1A-AR not only modulates firing of CA1 interneurons, but also has downstream effects on pyramidal neuron activity. The aim of this study was therefore to investigate the functional consequences of α1A-AR activation in CA1 hippocampus, in terms of the pre- and post-synaptic intracellular effects. Based on our previous molecular and functional characterization studies, we set forth the hypothesis that α1A-AR activation initiates classical PLC signaling in CA1 interneurons, causing depolarization and release of GABA and somatostatin onto neighboring CA1 pyramidal neurons.
α1A-AR activation decreases CA1 pyramidal activity via pre-synaptic release of GABA and somatostatin
In our initial cell-attached recording of CA1 neurons, shown in Figure 1, the selective α1-AR agonist PE produced a concentration-dependent increase in interneuron activity and a converse decrease in pyramidal neuron activity. Here we demonstrate that the PE-mediated decrease in pyramidal firing is due to increased pre-synaptic release of GABA and somatostatin from CA1 interneurons, an effect mediated via the α1A-AR subtype. As shown in Figure 2, PE increases spontaneous GABAA-mediated IPSCs in pyramidal cells but does not alter the frequency or amplitude of miniature or evoked IPSCs. As shown in Figure 3, the ability of PE to decrease pyramidal neuron firing is dependent on functional post-synaptic GABAA, GABAB and somatostatin receptors. Of particular note, the ability of PE to decrease pyramidal cell activity is essentially abolished only when all three receptor types –GABAA, GABAB, and somatostatin – are pharmacologically blocked simultaneously. Taken together, these findings suggest that both GABA and somatostatin are significant in mediating PE’s effect on CA1 pyramidal cells.
The ability of PE to mediate GABA and neuropeptide release provides new insight into understanding noradrenergic modulation of neuronal excitability in the hippocampus. While NE has previously been shown to promote GABA release in a number of brain regions, including hippocampus (Bergles et al., 1996), piriform cortex (Gellman and Aghajanian, 1993), entorhinal cortex (Lei et al., 2007), frontal cortex (Kawaguchi and Shindou, 1998), and basolateral amygdala (Braga et al., 2004), few studies have ventured further to examine if NE also promotes co-release of neuropeptide in these regions. This is a reasonable line of investigation, given immunocytochemical studies in the brain have frequently co-localized GABA with neuropeptides like somatostatin, neuropeptide Y, cholecystokinin or vasoactive intestinal polypeptide (Hendry et al., 1984; Somogyi et al., 1984; McDonald and Pearson, 1989) and instances of co-release have been reported (Sun et al., 2003). The results presented in this study are unique in that they directly link α1A-AR activation to dual release of GABA and somatostatin on the single cell level.
NE’s ability to initiate neurotransmitter as well as neuropeptide release from CA1 interneurons gives the noradrenergic system a heightened degree of both temporal and mechanistic control on CA1 pyramidal activity. NE’s activation of downstream GABAA, GABAB, and somatostatin receptors theoretically enables rapid- as well as delayed-onset inhibition given the ionotropic/metabotropic differences in the receptors. In addition to such temporal staging of inhibition, NE’s activation of downstream GABAA, GABAB, and somatostatin receptors also provides pleiotropic signaling effectors from which to increase inhibitory tone. Ionotropic GABAA receptors mediate chloride conductance while metabotropic GABAB receptors can modulate K+ or Ca2+ conductance via G protein activation (for reviews see Emson, 2007; Goetz et al., 2007). Furthermore, metabotropic somatostatin receptors can alter K+ conductance, adenylate cyclase production, or pre-synaptic glutamatergic transmission (for review see Baraban and Tallent, 2004). Thus the α1A-AR, acting via downstream GABAA, GABAB and somatostatin receptors, can increase inhibitory tone in CA1 hippocampus through a multitude of diverse signaling effectors.
α1A-AR activation significantly decreases epileptic burst activity in one in vitro epilepsy model
The ability of the α1A-AR to activate diverse downstream, inhibitory effectors in the hippocampus – both rapid- and delayed-onset – may be particularly advantageous in reducing the hyperexcitability characteristic of certain temporal lobe epilepsies. As illustrated in Figure 4, in the two in vitro epilepsy model conditions we examined – zero Mg2+ aCSF and 100 μM PTX aCSF – PE was able to significantly decrease epileptic bursts only in the zero Mg2+ condition. While this was not surprising, given the large contribution of GABAA receptor activation observed in Figure 3, we were somewhat disappointed that PE did not provide a significant reduction in epileptiform bursts in both models tested. Recall that at the single cell level, GABAA, GABAB and somatostatin receptors all demonstrated identifiable individual contributions to the PE-mediated decrease in pyramidal activity, and blockade of GABA receptors alone (combined PTX and bicuculline treatment) was not sufficient to abolish PE’s effect. We had hoped therefore that even with GABAA receptors functionally blocked, PE would still be able to decrease epileptiform bursting via downstream activation of GABAB and/or somatostatin receptors. While this was not the case in the experiment reported here, it is possible that increasing the duration of PE application – theoretically allowing more time for neuropeptide release and metabotropic receptor activation – might provide a significant effect for PE in the hippocampal PTX model. Interestingly, a recent study examining AR activation in a different region of the brain found that a 20 min NE application, but not a 10 min NE application, induced an α1-AR-mediated long-term depression of excitatory transmission (McElligot and Winder, 2008). Thus a lengthened PE challenge in the PTX model is certainly an avenue for future investigation, and indeed α1-AR-mediated long term depression likely represents an alternative if not synergistic mechanism contributing to the therapeutic potential of the α1-AR in temporal lobe epilepsy (Scheiderer et al., 2004). For the purpose of consistency however we did not alter the agonist application duration in the current study. Based on the results presented here, we can conclude that in periods of excessive glutamatergic activity, mimicking epileptiform activity, PE can decrease bursting if GABAA receptors are available.
While we were disappointed that PE-mediated somatostatin release was not sufficient to decrease epileptiform bursting in our PTX model, other groups have reported potent antiepileptic properties of somatostatin, albeit not in relation to the noradrenergic system. Direct application of somatostatin to hippocampal slices dramatically reduces CA1/CA3 epileptiform bursting in a number of in vitro epileptic models, including 0 Mg2+, 15 μM bicuculline, and 0 Mg2+/4-aminopyridine (Tallent and Siggins, 1999; Cammalleri et al., 2004). Moreover, in vivo studies demonstrate that somatostatin decreases seizure incidence, particularly in the hippocampus (Vezzani et al., 1991; Mazarati and Telegdy, 1992). From these studies and others it is clear that somatostatin possesses anticonvulsant properties, however the amount released in CA1 following α1A-AR activation appears to be, at least in our studies, insufficient to decrease epileptiform activity in and of itself. A potentially interesting inquiry might be to examine if combinatorial application of PE and somatostatin provides a synergistic antiepileptic effect in hippocampal models of epilepsy.
α1A-AR activation produces a pertussis toxin-sensitive, sodium dependent current in CA1 interneurons
To delineate the intracellular mechanism underlying the PE-mediated increase in interneuron action potential frequency, we utilized whole-cell recording in combination with a 50 μM PE challenge. In preliminary whole-cell current clamp experiments (data not shown), PE caused a significant depolarization in responsive CA1 interneurons (14.6±2.2 mV), often sufficient to generate action potentials. Here we report that in whole-cell voltage clamp configuration (Vm = −65 mV), PE initiates a transient negative change in holding current (−21.7±2.5 pA), indicative of inward current flux. To determine if the change in holding current was due to activation of a cationic channel or inhibition of a resting K+ conductance, we manipulated availability of Na+, Ca2+, and K+ using channel inhibitors and aCSF modifications. As shown in Figure 5, manipulations of sodium availability greatly attenuated this PE-mediated change in holding current. Lanthanum pre-treatment, TTX pre-treatment, or substitution of NMDG for NaCl were all conditions which hampered the ability of PE to generate inward current. On the contrary, manipulations of Ca2+ and K+ availability were of little to no effect. Together these findings suggest that in a subpopulation of CA1 interneurons, α1-AR activation elicits an inward sodium current sufficient to raise the Vm ~15 mV.
Our finding that Na+ manipulation, but not K+ or Ca2+ manipulation, altered PE’s ability to generate inward current in CA1 interneurons is at odds with a previous study examining adrenergic-mediated depolarization in the same cells. Bergles et al. (1996) attributed the NE-mediated depolarization they observed in CA1 interneurons primarily to α-adrenergic modulation of resting K+ conductance. Their conclusion, based on more sensitive electrophysiological experimentation, is well founded and indeed similar mechanisms are evidenced in other areas of the brain (Aghajanian, 1985; Stevens et al., 1994). Likely it is disparities in experimental design between our own study and Bergles et al. that explain our differing results. Foremost, their voltage-clamp experiments were all conducted in TTX-containing aCSF, which would mask any potential sodium channel involvement. Since we did not routinely use TTX in our recording aCSF preparation, we were able to observe a previously unreported α1-AR-dependent sodium current in this subpopulation of CA1 interneurons. On the contrary, our failure to routinely use TTX-containing aCSF may have enabled a heightened level of baseline noise that masked the smaller but significant K+ contributions previously reported. As shown in Figure 5C, cesium and barium pre-treatments both produced a small reduction in the PE-mediated current, albeit not a statistically significant effect. It is possible therefore that in our study K+ channel-mediated effects were indeed occurring in response to PE, however our use of zero TTX aCSF shifted focus – and statistical significance – towards Na+ involvement.
Additional discrepancies that should be noted between our study and that of Bergles et al. include the age of the experimental animals and the AR agonists tested. Their studies utilized adult rats (150–250 g), and primarily EPI and NE. Our studies utilized young rats (35–60 g), and the selective α1-AR agonist PE. As rats age, AR expression as well as adrenergic agonist efficacy can change (Burnett et al., 1990; Erdtsieck-Ernst et al., 1991; Mitchell et al., 2003; Happe et al., 2004), however it is unlikely that the ion channels associated with AR activation would also change. Interestingly though a recent study by Thomas et al. (2008) demonstrates that isoforms of the K2P2.1 leak channel are actually permeable to sodium, and such isoforms are differentially expressed in rat CNS throughout development. Although highly speculative at this point, it would be interesting to investigate if such a K2P isoform shift occurs in CA1 interneurons of the hippocampus at some point in development. At this stage however it would be most logical to first investigate – using PE and young animals – the parameters studied by Bergles et al. (1996), namely PE’s influence on input conductance, voltage ramps, and inward rectifier currents, the latter in the presence and absence of NMDG, La3+, and TTX. It is quite possible that alterations in both K+ and Na+ conductance participate in the α1-AR-mediated depolarization of CA1 interneurons; clearly deciphering the contributions of either/both ions represents an independent follow-up investigation which may potentially produce intriguing results.
To begin to delineate the signaling mechanisms underlying this α1-AR-mediated depolarization, we continued to use whole-cell, voltage-clamp recording in combination with a 50 μM PE challenge. Gq is the G protein traditionally implicated in α1-AR signaling; Gqα activation of PLC leads to generation of inositol 1,4,5-trisphosphate and diacylglycerol, leading to intracellular calcium release and downstream activation of PKC (Berridge and Irvine, 1984; Nishizuka, 1992). We assumed this was indeed the signaling pathway initiated by α1A-AR activation in CA1 interneurons, however the data presented in Figure 5 suggests otherwise. Pre-treatment with 10 μM (not shown) or 50 μM of the PLC inhibitor U73122 did not significantly affect the ability of PE to generate inward current (−22±2.4 and −18.3±1.6 pA, respectively). Furthermore, PE was still able to generate inward current when cells had been treated with the PKC inhibitor Ro31-8220 or the calcium chelator BAPTA. Since α1-AR activation has also been linked to adenylate cyclase and MAPK activation (Horie et al., 1995; Gutkind, 1998), we tested possible involvement of these pathways but found inhibition of either second messenger produced no significant effect on the PE-mediated inward current. Together our results suggest that the PE-mediated depolarization of CA1 interneurons does not critically depend on second messenger signaling through PLC, adenylate cyclase, or MAPK pathways.
We could not examine direct involvement of Gq in the PE-mediated depolarization given highly selective Gq inhibitors are proprietary, however our results with U73122 imply that canonical Gq/PLC signaling is not occurring. We did examine the effect of pertussis toxin, an inhibitor of Gi/Go, and to our surprise pre-treatment with pertussis toxin significantly attenuated the PE-mediated depolarization (−7.2±1.4 pA). While surprising, this result was not implausible given others have reported α1-AR-mediated effects, particularly α1A-AR-mediated effects, that are pertussis toxin-sensitive (reviewed by Wilson and Minneman, 1990), however these studies were examining PLC activation. While the α1A-AR-mediated depolarization of CA1 interneurons noted in our studies does not appear to involve PLC activity, given the data presented in Figure 5, it is interesting to consider that we may have identified a novel signaling mechanism of the α1A-AR in hippocampal neurons. The data presented here provide an initial suggestion that α1A-AR agonist stimulation in these cells activates a pertussis toxin-sensitive Gi/o protein, which possibly directly facilitates sodium channel opening. Others have demonstrated that α1A-AR activation can activate nonselective cation channels independent of PLC (Kawanabe et al., 2004), and furthermore in non-neuronal cell types α1-AR has been linked to sodium channel activity via a pertussis toxin-sensitive G protein (Bubien et al., 1998). Therefore it is possible that in a subpopulation of somatostatin-positive CA1 interneurons α1A-AR activation may directly couple to sodium channel activity via Gi/o, independent of a second messenger system. Future studies are clearly warranted to more thoroughly examine this possibility.
In summary, we report here one mechanism by which NE exerts antiepileptic properties in hippocampus, namely that activation of the CA1 interneuron α1A-AR prompts release of GABA and somatostatin onto CA1 pyramidal cells. α1A-AR-mediated pre-synaptic depolarization involves sodium influx, and appears independent of canonical second messenger systems; α1A-AR-mediated post-synaptic inhibition involves GABAA, GABAB, and somatostatin receptor activation, and is sufficient to decrease epileptiform activity induced by 0 Mg2+ conditions. The research outlined in this study is significant in that it identifies a mechanism by which inhibitory input (GABAergic and somatostatinergic) can be selectively increased via an alternative neurotransmitter system (noradrenergic). For decades the mainstay of antiepileptic therapy has been increasing GABAergic inhibition, but the ubiquitous nature of the GABA receptor has caused numerous complications in this approach. This project suggests a potential therapeutic strategy for temporal lobe epilepsy in that activation of the α1A-AR subtype would increase inhibition in CA1 hippocampus, an area susceptible to epileptogenic activity and subsequent neuronal damage. It is hoped the results presented in this study will not only advance understanding of NE’s role in the hippocampus, but provide a target receptor for translational anti-epileptic drug development research.
Acknowledgments
Funding to conduct and present these studies was provided by a predoctoral fellowship from the American Epilepsy Society (KLH), by the North Dakota Experimental Program to Stimulate Competitive Research through National Science Foundation Grant EPS-0447679 (KLH, VAD, JEP) and by National Science Foundation CAREER Grant 0347259 (VAD). The authors would like to thank Shanshan Lee for assistance with the evoked IPSC experiments.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
5. Literature Cited
- Aghajanian GK. Modulation of a transient outward current in serotonergic neurons by α1-adrenoceptors. Nature. 1985;315:501–503. doi: 10.1038/315501a0. [DOI] [PubMed] [Google Scholar]
- Baraban SC, Tallent MK. Interneuron diversity series: Interneuronal neuropeptides –endogenous regulators of neuronal excitability. Trends Neurosci. 2004;27:135–142. doi: 10.1016/j.tins.2004.01.008. [DOI] [PubMed] [Google Scholar]
- Bergles DE, Doze VA, Madison DV, Smith SJ. Excitatory actions of norepinephrine on multiple classes of hippocampal CA1 interneurons. J Neurosci. 1996;16:572–585. doi: 10.1523/JNEUROSCI.16-02-00572.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ, Irvine RF. Inositol trisphosphate, a novel second messenger in cellular signal transduction. Nature. 1984;312:315–321. doi: 10.1038/312315a0. [DOI] [PubMed] [Google Scholar]
- Boehm S. Presynaptic alpha2-adrenoceptors control excitatory, but not inhibitory, transmission at rat hippocampal synapses. J Physiol. 1999;519:439–449. doi: 10.1111/j.1469-7793.1999.0439m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonsi P, Cuomo D, Ding J, Sciamanna G, Ulrich S, Tscherter A, Bernardi G, Surmeier DF, Pisani A. Endogenous serotonin excites striatal cholinergic interneurons via the activation of 5-HT 2C, 5-HT6 and 5-HT7 serotonin receptors: Implications for extrapyramidal side effects of serotonin reuptake inhibitors. Neuropsychopharmacology. 2007;32:1840–1854. doi: 10.1038/sj.npp.1301294. [DOI] [PubMed] [Google Scholar]
- Braga MF, Aroniadou-Anderjaska V, Manion ST, Hough CJ, Li H. Stress impairs alpha(1A) adrenoceptor-mediated noradrenergic facilitation of GABAergic transmission in the basolateral amygdale. Neuropsychopharmacology. 2004;29:45–58. doi: 10.1038/sj.npp.1300297. [DOI] [PubMed] [Google Scholar]
- Brown TH, Wong RKS, Prince DA. Spontaneous miniature synaptic potentials in hippocampal neurons. Brain Res. 1979;177:194–199. doi: 10.1016/0006-8993(79)90931-4. [DOI] [PubMed] [Google Scholar]
- Bubien JK, Cornwell T, Bradford AL, Fuller CM, DuVall MD, Benos DJ. Alpha-adrenergic receptors regulate human lymphocyte amiloride-sensitive sodium channels. Am J Physiol. 1998;275:C701–710. doi: 10.1152/ajpcell.1998.275.3.C702. [DOI] [PubMed] [Google Scholar]
- Burnett DM, Bowyer JF, Masserano JM, Zahniser NR. Effect of aging on alpha-1 adrenergic stimulation of phosphoinositide hydrolysis in various regions of rat brain. J Pharmacol Exp Ther. 1990;255:1265–1270. [PubMed] [Google Scholar]
- Bylund DB, Blaxall HS, Iversen LJ, Caron MG, Lefkowitz RJ, Lomasney JW. Pharmacological characteristics of α2-adrenergic receptors: comparison of pharmacologically defined subtypes with subtypes identified by molecular cloning. Mol Pharmacol. 1992;42:1–5. [PubMed] [Google Scholar]
- Cammalleri M, Cervia D, Langenegger D, Liu Y, Dal Monte M, Hoyer D, Bagnoli P. Somatostatin receptors differentially affect spontaneous epileptiform activity in mouse hippocampal slices. Eur J Neurosci. 2004;20:2711–2721. doi: 10.1111/j.1460-9568.2004.03741.x. [DOI] [PubMed] [Google Scholar]
- Cavazos JE, Das I, Sutula TP. Neuronal loss induced in limbic pathways by kindling: evidence for induction of hippocampal sclerosis by repeated brief seizures. J Neurosci. 1994;14:3106–3121. doi: 10.1523/JNEUROSCI.14-05-03106.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Ensor GF, Bohner BA. A facilitation of reserpine on the central nervous system. Proc Soc Exp Biol Med. 1954;86:507–510. doi: 10.3181/00379727-86-21149. [DOI] [PubMed] [Google Scholar]
- Chung T-W, Koo B-S, Kim K-O, Jeong H-S, Kim M-G, Chung K-H, Lee I-S, Kim C-H. Salviae miltiorrhizae BGE radix increases rat striatal K+-stimulated dopamine release and activates the dopamine release with protection against hydrogen peroxide-induced injury in rat pheochromocytoma PC12 cells. Neurochem Res. 2006;31:109–120. doi: 10.1007/s11064-005-9264-3. [DOI] [PubMed] [Google Scholar]
- Curet O, de Montigny C. Electrophysiological characterization of adrenoceptors in the rat dorsal hippocampus. II. Receptors mediating the effect of synaptically released norepinephrine. Brain Res. 1988;475:47–57. doi: 10.1016/0006-8993(88)90197-7. [DOI] [PubMed] [Google Scholar]
- Emson PC. GABAB receptors: structure and function. Prog Brain Res. 2007;160:43–57. doi: 10.1016/S0079-6123(06)60004-6. [DOI] [PubMed] [Google Scholar]
- Erdtsieck-Ernst BH, Feenstra MG, Boer GJ. Pre- and postnatal developmental changes of adrenoceptor subtypes in rat brain. J Neurochem. 1991;57:897–903. doi: 10.1111/j.1471-4159.1991.tb08235.x. [DOI] [PubMed] [Google Scholar]
- Ferraro G, Sardo P, Sabatino M, La Grutta V. Locus coeruleus noradrenaline system and focal penicillin hippocampal epilepsy: neurophysiological study. Epilepsy Res. 1994;19:215–220. doi: 10.1016/0920-1211(94)90064-7. [DOI] [PubMed] [Google Scholar]
- Freund TF, Ylinen A, Miettinen R, Pitakanen A, Lahtinen H, Baimbridge KG, Riekkinen PJ. Pattern of neuronal death in the rat hippocampus after status epilepticus. Relationship to calcium binding protein content and ischemic vulnerability. Brain Res Bulletin. 1991;28:27–38. doi: 10.1016/0361-9230(92)90227-o. [DOI] [PubMed] [Google Scholar]
- Gellman RL, Aghajanian GK. Pyramidal cells in piriform cortex receive a convergence of inputs from monoamine activated GABAergic interneurons. Brain Res. 1993;600:63–73. doi: 10.1016/0006-8993(93)90402-9. [DOI] [PubMed] [Google Scholar]
- Giorgi FS, Pizzanelli C, Biagioni F, Murri L, Fornai F. The role of norepinephrine in epilepsy: from the bench to the bedside. Neurosci Bio Rev. 2004;28:507–524. doi: 10.1016/j.neubiorev.2004.06.008. [DOI] [PubMed] [Google Scholar]
- Goetz T, Arslan A, Wisden W, Wulff P. GABAA receptors: structure and function in the basal ganglia. Prog Brain Res. 2007;160:21–41. doi: 10.1016/S0079-6123(06)60003-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutkind JS. The pathways connecting G protein-coupled receptors to the nucleus through divergent mitogen-activated protein kinase cascades. J Biol Chem. 1998;273:1839–1842. doi: 10.1074/jbc.273.4.1839. [DOI] [PubMed] [Google Scholar]
- Happe HK, Coulter CL, Gerety ME, Sanders JD, O’Rourke M, Bylund DB, Murrin LC. Alpha-2 adrenergic receptor development in rat CNS: an autoradiographic study. Neuroscience. 2004;123:167–178. doi: 10.1016/j.neuroscience.2003.09.004. [DOI] [PubMed] [Google Scholar]
- Hendry SH, Jones EG, DeFelipe J, Schmechel D, Brandon C, Emson PC. Neuropeptide-containing neurons of the cerebral cortex are also GABAergic. Proc Natl Acad Sci USA. 1984;81:6526–6530. doi: 10.1073/pnas.81.20.6526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hieble JP, Bylund DB, Clarke DE, Eikenburg DC, Langer SZ, Lefkowitz RJ, Minneman KP, Ruffolo RR., Jr Recommendation for nomenclature of α1-adrenoceptors: consensus update. Pharmacol Rev. 1995;47:267–270. [PubMed] [Google Scholar]
- Hillman KL, Knudson CA, Carr PA, Doze VA, Porter JE. Adrenergic receptor characterization of CA1 hippocampal neurons using real time single cell RT-PCR. Brain Res Mol Brain Res. 2005a;139:267–276. doi: 10.1016/j.molbrainres.2005.05.033. [DOI] [PubMed] [Google Scholar]
- Hillman KL, Doze VA, Porter JE. Functional characterization of the beta-adrenergic receptor subtypes expressed by CA1 pyramidal cells in the rat hippocampus. J Pharmacol Exp Ther. 2005b;314:561–567. doi: 10.1124/jpet.105.084947. [DOI] [PubMed] [Google Scholar]
- Hillman KL, Doze VA, Porter JE. Alpha1A-adrenergic receptors are functionally expressed by a subpopulation of cornu ammonis 1 interneurons in rat hippocampus. J Pharmacol Exp Ther. 2007;321:1062–1068. doi: 10.1124/jpet.106.119297. [DOI] [PubMed] [Google Scholar]
- Horie K, Itoh H, Tsujimoto G. Hamster α1B-adrenergic receptor directly activates Gs in the transfected Chinese hamster ovary cells. Mol Pharmacol. 1995;48:392–400. [PubMed] [Google Scholar]
- Huang H, van den Pol AN. Rapid direct excitation and long-lasting enhancement of NMDA response by group I metabotropic glutamate receptor activation of hypothalamic melanin-concentrating hormone neurons. J Neurosci. 2007;27:11560–11572. doi: 10.1523/JNEUROSCI.2147-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerlicz M, Kostowski W, Bidzinski A, Hauptman M, Dymecki J. Audiogenic seizures in rats: relation to noradrenergic neurons of the locus coeruleus. Acta Physiol Pol. 1978;29:409–412. [PubMed] [Google Scholar]
- Jimenez-Rivera C, Voltura A, Weiss GK. Effect of locus coeruleus stimulation on the development of kindled seizures. Exp Neurol. 1986;95:13–20. doi: 10.1016/0014-4886(87)90002-1. [DOI] [PubMed] [Google Scholar]
- Kandel ER, Spencer WA. Electrophysiology of hippocampal neurons. I. Afterpotentials and repetitive firing. J Neurophysiol. 1961;24:243–259. doi: 10.1152/jn.1961.24.3.243. [DOI] [PubMed] [Google Scholar]
- Kaur J, Keesey R, Magrys B, Liu H, Friedman LK. NR1 knockdown reveals CA1 injury during a developmental period of high seizure susceptibility despite reduced seizure activity. Neuromolecular Med. 2007;9:298–314. doi: 10.1007/s12017-007-8009-7. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, Shindou T. Noradrenergic excitation and inhibition of GABAergic cell types in rat frontal cortex. J Neurosci. 1998;18:6963–6976. doi: 10.1523/JNEUROSCI.18-17-06963.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawanabe Y, Hashimoto N, Masaki T. Characterization of G proteins involved in activation of nonselective cation channels and arachidonic acid release by norepinephrine/alpha1A-adrenergic receptors. Am J Physiol Cell Physiol. 2004;286:C596–600. doi: 10.1152/ajpcell.00359.2003. [DOI] [PubMed] [Google Scholar]
- Knowles WD. Normal anatomy and neurophysiology of the hippocampal formation. J Clin Neurophysiol. 1992;9:252–263. [PubMed] [Google Scholar]
- Kokaia M, Bengzon J, Kalen P, Lindvall O. Noradrenergic mechanisms in hippocampal kindling with rapidly recurring seizures. Brain Res. 1989;491:398–402. doi: 10.1016/0006-8993(89)90079-6. [DOI] [PubMed] [Google Scholar]
- Krahl SE, Clark KB, Smith DC, Browning RA. Locus coeruleus lesions suppress the seizure-attenuating effects of vagus nerve stimulation. Epilepsia. 1998;29:709–714. doi: 10.1111/j.1528-1157.1998.tb01155.x. [DOI] [PubMed] [Google Scholar]
- Lei S, Deng PY, Porter JE, Shin HS. Adrenergic facilitation of GABAergic transmission in rat entorhinal cortex. J Neurophysiol. 2007;98:2868–2877. doi: 10.1152/jn.00679.2007. [DOI] [PubMed] [Google Scholar]
- Mason ST, Corcoran ME. Catecholamines and convulsions. Brain Res. 1979;170:497–507. doi: 10.1016/0006-8993(79)90967-3. [DOI] [PubMed] [Google Scholar]
- Mazarati AM, Telegdy G. Effects of somatostatin and anti-somatostatin serum on picrotoxin-kindled seizures. Neuropharmacology. 1992;31:793–797. doi: 10.1016/0028-3908(92)90043-o. [DOI] [PubMed] [Google Scholar]
- McDonald AJ, Pearson JC. Coexistence of GABA and peptide immunoreactivity in non-pyramidal neurons of the basolateral amygdale. Neurosci Lett. 1989;100:53–58. doi: 10.1016/0304-3940(89)90659-9. [DOI] [PubMed] [Google Scholar]
- McElligott ZA, Winder DG. Alpha1-adrenergic receptor-induced heterosynaptic long-term depression in the bed nucleus of the stria terminalis is disrupted in mouse models of affective disorders. Neuropsychopharmacology. 2008;33:2313–2323. doi: 10.1038/sj.npp.1301635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIntyre DC, Pusztay W, Edson N. Effects of flurazepam on kindled amygdala convulsion in catecholamine-depleted rats. Exp Neurol. 1982;77:78–85. doi: 10.1016/0014-4886(82)90144-3. [DOI] [PubMed] [Google Scholar]
- McNamara JO. Cellular and molecular basis of epilepsy. J Neurosci. 1994;14:3413–3425. doi: 10.1523/JNEUROSCI.14-06-03413.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mynlieff M, Dunwiddie TV. Noradrenergic depression of synaptic responses in hippocampus of rat: evidence for mediation by alpha 1-receptors. Neuropharmacology. 1988;27:391–398. doi: 10.1016/0028-3908(88)90148-7. [DOI] [PubMed] [Google Scholar]
- Mitchell VA, Christie MJ, Vaughan CW. Developmental changes in the alpha-adrenergic responses of rat periaqueductal grey neurons. Neuroreport. 2003;14:1637–1639. doi: 10.1097/00001756-200308260-00019. [DOI] [PubMed] [Google Scholar]
- Neves G, Cooke SF, Bliss TV. Synaptic plasticity, memory and the hippocampus: a neural network approach to causality. Nat Rev Neurosci. 2008;9:65–75. doi: 10.1038/nrn2303. [DOI] [PubMed] [Google Scholar]
- Nishi H, Watanabe S, Ueki S. Effects of monoamines injected into the hippocampus on hippocampal seizure discharge in the rabbit. J Pharmacobiodyn. 1981;4:7–14. doi: 10.1248/bpb1978.4.7. [DOI] [PubMed] [Google Scholar]
- Nishizuka Y. Intracellular signalling by hydrolysis of phospholipids and activation of protein kinase C. Science. 1992;258:607–614. doi: 10.1126/science.1411571. [DOI] [PubMed] [Google Scholar]
- Pang K, Rose GM. Differential effects of norepinephrine on hippocampal complex-spike and theta-neurons. Brain Res. 1987;425:146–158. doi: 10.1016/0006-8993(87)90493-8. [DOI] [PubMed] [Google Scholar]
- Perez DM, editor. The adrenergic receptors: in the 21st century. Humana Press; New Jersey: 2006. [Google Scholar]
- Scheiderer CL, Dobrunz LE, McMahon LL. Novel form of long-term synaptic depression in rat hippocampus induced by activation of alpha 1 adrenergic receptors. J Neurophysiol. 2004;91:1071–1077. doi: 10.1152/jn.00420.2003. [DOI] [PubMed] [Google Scholar]
- Singaravelu K, Deitmer JW. Calcium mobilization by nicotinic acid adenine dinucleotide phosphate (NAADP) in rat astrocytes. Cell Calcium. 2006;39:143–153. doi: 10.1016/j.ceca.2005.10.001. [DOI] [PubMed] [Google Scholar]
- Somogyi P, Hodgson AJ, Smith AD, Nunzi MG, Gorio A, Wu JY. Different populations of GABAergic neurons in the visual cortex and hippocampus of cat contain somatostatin- or cholecystokinin-immunoreactive material. J Neurosci. 1984;4:2590–2603. doi: 10.1523/JNEUROSCI.04-10-02590.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens DR, McCarley RW, Green RW. The mechanism of noradrenergic α1 excitatory modulation of pontine reticular formation neurons. J Neurosci. 1994;14:6481–6487. doi: 10.1523/JNEUROSCI.14-11-06481.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun QQ, Baraban SC, Prince DA, Huguenard JR. Target specific neuropeptide-Y-ergic synaptic inhibition and its network consequences within the mammalian thalamus. J Neurosci. 2003;23:9639–9649. doi: 10.1523/JNEUROSCI.23-29-09639.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szot P, Weinshenker D, Rho JM, Storey TW, Schwartzkroin PA. Norepinephrine is required for the anticonvulsant effect of the ketogenic diet. Brain Res Dev Brain Res. 2001;129:211–214. doi: 10.1016/s0165-3806(01)00213-9. [DOI] [PubMed] [Google Scholar]
- Szot P. The role of norepinephrine in the anticonvulsant mechanism of action of the ketogenic diet. In: Stafstrom CE, Rho JM, editors. Epilepsy and the Ketogenic Diet. Humana Press; New Jersey: 2004. pp. 265–278. [Google Scholar]
- Tallent MK, Siggins GR. Somatostatin acts in CA1 and CA3 to reduce hippocampal epileptiform activity. J Neurophysiol. 1999;81:1626–1635. doi: 10.1152/jn.1999.81.4.1626. [DOI] [PubMed] [Google Scholar]
- Thomas D, Plant LD, Wilkens CM, McCrossan ZA, Goldstein SA. Alternative translation initiation in rat brain yields K2P2.1 potassium channels permeable to sodium. Neuron. 2008;58:859–870. doi: 10.1016/j.neuron.2008.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzani A, Serafini R, Stasi MA, Vigano G, Rizzi M, Samanin R. A peptidase-resistant cyclic octapeptide analogue of somatostatin (SMS 201-995) modulates seizures induced by quinolinic and kainic acids differently in the rat hippocampus. Neuropharmacology. 1991;30:345–352. doi: 10.1016/0028-3908(91)90059-k. [DOI] [PubMed] [Google Scholar]
- Wang M, Bianchi R, Chuang S-C, Zhao W, Wong RKS. Group I metabotropic glutamate receptor-dependent TRPC channel trafficking in hippocampal neurons. J Neurochem. 2007;101:411–421. doi: 10.1111/j.1471-4159.2006.04377.x. [DOI] [PubMed] [Google Scholar]
- Wilson KM, Minneman KP. Pertussis toxin inhibits norepinephrine-stimulated inositol phosphate formation in primary brain cell cultures. Mol Pharmacol. 1990;38:274–281. [PubMed] [Google Scholar]
- Wu HQ, Tullii M, Samanin R, Vezzani A. Norepinephrine modulates seizures induced by quinolinic acid in rats: selective and distinct roles of alpha-adrenoceptor subtypes. Eur J Pharmacol. 1987;138:309–318. doi: 10.1016/0014-2999(87)90468-7. [DOI] [PubMed] [Google Scholar]