

Abstract
Anti-platelet integrin GPIIIa49-66 antibody (Ab) induces complement-independent platelet oxidative fragmentation and death by generation of platelet peroxide following NADPH oxidase activation. A C-terminal 385–amino acid fragment of ADAMTS-18 (a disintegrin metalloproteinase with thrombospondin motifs produced in endothelial cells) induces oxidative platelet fragmentation in an identical kinetic fashion as anti–GPIIIa49-66 Ab. Endothelial cell ADAMTS-18 secretion is enhanced by thrombin and activated by thrombin cleavage to fragment platelets. Platelet aggregates produced ex vivo with ADP or collagen and fibrinogen are destroyed by the C-terminal ADAMTS-18 fragment. Anti–ADAMTS-18 Ab shortens the tail vein bleeding time. The C-terminal fragment protects against FeCI3-induced carotid artery thrombosis as well as cerebral infarction in a postischemic stroke model. Thus, a new mechanism is proposed for platelet thrombus clearance, via platelet oxidative fragmentation induced by thrombin cleavage of ADAMTS-18.
Introduction
Patients with early-onset HIV-1 or hepatitis C virus (HCV)–related thrombocytopenia (HIV-1-ITP)1,2 develop a unique anti-platelet GPIII Ab directed against the GPIIIa49-66 epitope capable of fragmenting human and mouse platelets in the absence of complement and inducing thrombocytopenia in mice.2–4 In both HIV-1-ITP and HCV-ITP, the anti-GPIIIa Ab correlates inversely with platelet count.5 The development of thrombocytopenia in mice can be inhibited/reversed with GPIIIa 49-66 peptide conjugated to albumin.5 A similar Ab with similar properties can be raised in rabbits immunized against GPIIIa 49-66 peptide.4
Recent studies have revealed that the Ab-induced platelet fragmentation is induced by reactive oxygen species (peroxides) generated from a platelet NADPH oxidase pathway and 12-lipoxygenase (12-LO).3 The Ab-induced mechanism requires Ca2+ flux and activation of platelet 12-LO and phospholipase A2.6 In a recent report we demonstrated that host production of this Ab is secondary to molecular mimicry with polymorphic HIV-1 peptides.7 These unique findings prompted a search for a physiologic ligand capable of inducing this reaction, since we hypothesized that it could regulate platelet thrombus formation. Search for a physiologic ligand was investigated by panning the GPIIIa 49-66 peptide with a phage surface display 7-mer peptide library. Twenty positive peptide clones were isolated that reacted with GPIIIa 49-66. One of these peptides had 70% homology with the C-terminal end of ADAMTS-18 (1148-1152), a disintegrin and metalloproteinase with thrombospondin (TSR)–like motifs.
The human ADAMTSs are a family of 19 secreted Zn-metalloproteinases, which have multidomain structural components in common. These include an N-terminal signal peptide, followed by a prodomain, a metalloproteinase catalytic domain with a zinc binding motif, a disintegrin-like domain, a central thrombospondin type-1-like repeat (TSR), a cysteine-rich domain (high sequence homology), a spacer region, and a variable number of C-terminal TSR repeats. Some have further C-terminal domains. Several ADAMTSs have known functions. These include N-terminal procollagen processing (ADAMTS-2, -3, -14),8–11 spermatogenesis (ADAMTS 2),12 inhibition of angiogenesis (ADAMTS-1, -8, and -9),13,14 follicular rupture and ovulation (ADAMTS- 1),15 cleavage of matrix proteoglycans aggrecan, versican, and brevican (ADAMTS-1, -4, -5, -8, -9, -15),16–19 degradation of cartilage oligomeric matrix protein (ADAMTS-7, ADAMTS-12), and cleavage of ultralarge molecular weight von Willebrand factor (ADAMTS-13). All known ADAMTSs (except 10 and 12) contain a subtilisin-like pro–protein convertase cleavage site in their prodomains that are furin recognition sequences.20 It is suggested that autocatalytic sequential processing of the N-terminus may regulate their intracellular distribution, release, or activity.21,22 The C-terminal domains can also be enzymatically processed to affect substrate specificity, extracellular matrix binding, and bioactivity.23–26 ADAMTSs with no known function or substrate have been termed “orphan ADAMTSs.”17–20 ADAMTS-18 has recently been shown to be epigenetically silenced in multiple carcinomas and to have tumor suppressor activity.27 Mutation of ADAMTS-18 is strongly associated with colorectal cancer.28 Our studies focus on the C-terminal fragment of ADAMTS-18, encompassing the terminal TSR domain that binds to platelet GPIIIa49-66 and oxidatively fragments platelets.
In this report, we demonstrate the following: (1) Endothelial cells constitutively secrete ADAMTS-18. (2) Thrombin enhances endothelial ADAMTS-18 secretion in vitro and in vivo. (3) Thrombin cleaves ADAMTS-18 to produce a C-terminal 45-kDa platelet-active fragment. (4) C-terminal ADAMTS-18 peptide induces the same oxidative fragmentation of platelets as anti–GPIIIa49-66 Ab platelet activation. (5) C-terminal ADAMTS-18 peptide dissolves ex vivo platelet aggregates, regulates the in vivo bleeding time, inhibits carotid artery platelet thrombus formation, and protects against postischemic cerebral stroke in mice. It is hypothesized that platelet thrombi are cleared through thrombin-induced secretion and cleavage of ADAMTS-18.
Methods
Experimental procedures
Human population.
Patient sera were obtained with informed consent acquired in accordance with the Declaration of Helsinki from early-onset HIV-1–infected thrombocytopenic patients without AIDS and control subjects (healthy laboratory personnel). These studies were approved by the New York University Medical Center Institutional Review Board.
Mice.
Platelet counts were measured by phase-contrast microscopy as previously described.3 GPIIIa−/− mouse platelets with C57/BL6 background and BALB/C mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and genotype was confirmed by polymerase chain reaction (PCR). Swiss Webster mice were obtained from The Jackson Laboratory.
Reagents.
All reagents were obtained from Sigma-Aldrich (St Louis, MO) unless otherwise designated. 7-Dicholordihydrofluorescine diacetate (DCFH-DA) was obtained from Molecular Probes (Eugene, OR). Hirudin was obtained from Hoechst Marion Rousel (Kansas City, MO). ADAMTS-18 peptide conjugated to biotin, Bio-VQTRSVHCVQQGRPSSSC-OH, ADAMTS-18 scrambled peptide, Bio-VQTRSVQVHCQGRPSSSC-OH, GPIIIa49-66 conjugated to biotin (Bio-CAPESIEFPVSEARVLED), and GPIIIa49-66 (CAPESIEFPVSEARVLED) were synthesized by Biosynthesis (Lewisville, TX). scFv A11 was made from a human scFv phage surface display monoclonal library as described.29 Anti–N-terminal ADAMTS-18 is a polyclonal Ab, kindly provided by Dr Andrew J. Connolly, Stanford Medical School (Stanford, CA).
Immune complexes.
Serum complexes were isolated from sera by polyethylene glycol precipitation and dissolved in one fifth their serum volume in 0.01 M PBS, pH 7.4.30 Purified IgG was isolated from immune complexes and affinity purified on fixed platelets as described.31
Affinity purification of anti–platelet GPIIIa49-66.
Peptide GPIIIa49-66 (CAPESIEFPV-SEARVLED) was coupled to an affinity column with the heterobifunctional cross-linker sulfo-succinimidyl 4-(N-maleimidomethyl) cyclohexane-1 carboxylase as recommended by the manufacturer (Pierce Biotechnology, Rockford, IL). This cross-linked the resin with the NH2-terminal cysteine of the peptide, which was then incubated with 0.4 mL platelet affinity-purified IgG31 overnight at 4°C. The column was then washed, eluted at pH 2.5, and neutralized as described.31
Induction of platelet particles.
Gel-filtered normal platelets were prepared from platelet-rich plasma obtained from blood collected in 0.38% sodium citrate using a sepharose 2B column preincubated with Tyrode buffer (pH 7.4). Gel-filtered platelets (107/mL) were either unlabeled or labeled with an anti–GPIIIa-FITC mAb (Ancell, Bayport, MN), 10 μg/mL for 30 minutes at 4°C, centrifuged at 1000g for 6 minutes at room temperature, and resuspended in Tyrode buffer. Ten microliters of unlabeled or FITC-labeled platelets (107/mL) were then incubated with affinity-purified anti–GPIIIa49-66 or rADAMTS-18 (385 AAs; 25-50 μg/mL), in 100 μL volume Tyrode buffer for 0 to 4 hours at 37°C and then stored in an ice bucket prior to measurement of percentage of platelet particles by flow cytometry. Further particle formation is arrested at 0°C.
Assay of platelet particle formation.
Fluorescent-labeled particles were measured by flow cytometry, using a FACScan (Becton Dickinson, Mountain View, CA). Debris and dead cells were excluded using scatter gates. Only cells with low orthogonal light scattering were included in the sorting gates. Gates were adjusted for control platelets by exclusion of other blood cells. Fluorescent-labeled intact platelets were monitored in the right upper quadrant (RUQ) with the y-axis measuring forward scatter and the x-axis measuring fluorescence. A shift in fluorescent or nonfluorescent particles from RUQ to LUQ and LLQ reflected percentage of platelet particle induction of 10 000 counted platelets/particles.
Assay of LDH.
LDH was assayed using the commercial LDH Kit supplied by Biotrin Diagnostics (Hemet, CA).
Assay of platelet oxidation.
Gel-filtered platelets were loaded with 10 μM DCFH-DA for 30 minutes at 37°C as described4 and challenged with control or anti–GPIIIa49-66 IgG. Oxidation was quantitated by measuring the increase in mean fluorescence with flow cytometry.
Screening phage display peptide library.
The PhD-7 phage library was obtained from New England Biolabs (Cambridge, MA). Biotin-conjugated GPIIIa 49-66 peptide (13 ng) was incubated with 2 × 1011 pfu phage in 400 μL TBST buffer (Tris 50 mM, NaCl 150 mM, Tween-20 0.1%, pH 7.5) at 4°C overnight. Positive phage clones were pulled down and washed with TBST by adding 20 μL avidin-agarose beads (Sigma-Aldrich). Phage clones with specific binding to GPIIIa49-66 were eluted by adding 13 μg non–biotin-conjugated GPIIIa 49-66 in 200 μL TBST. The eluted phage was amplified in E coli ER2738. The amplified phages were titered on LB/IPTG/X-gal plates and the titer, derived from the number of blue phage plaques, was used to calculate an input volume corresponding to 2 × 1011 pfu phages for the next round of panning. After the third round of panning, 20 clones were randomly selected for sequencing. Phage peptide sequences were analyzed for sequence similarity to other proteins using the BLAST algorithm of the Blast program and the database of the National Center for Biotechnology Information (NCBI).32
Expression constructs.
Full-length ADAMTS-18 cDNA coding sequence was cloned into mammalian expression vector pBudCE4.1 from Invitrogen (Carlsbad, CA). Two constructs with and without the His-tag were made. Three constructs were made to express the C-terminal domains of ADAMTS-18 in pGEX-4T-2 vector from GE Healthcare (Piscataway, NJ). These constructs express the C-terminal fragments of ADAMTS-18 covering 837 to 1221 (385 AAs), 1034-1221 (188 AAs), and 1156-1221 (66 AAs). The 385-AA construct was also scrambled at 1149 to 1152 in which HCVQQ was mutated to QVHCQ (sc 385 AAs).
Reverse transcription–PCR of human umbilical vein endothelial cells (HUVECs) for ADAMTS-18.
Nestled primers were used for ADAMTS-18. The first round was as follows: forward: 5′-cgaagttgtaccatcaatgag-3′ and reverse: 5′-agagcctagttcatgatcac-3′; and second round: forward: 5′-atggcaatttggagcaaaag-3′ and reverse: 5′-gcttggggttattgcagtgt-3′. A PCR product of 295 bp was obtained.
Purification of GST-ADAMTS-18 fragments from E coli.
Escherichia coli BL 21 transformed with the pGEX-4T-2 construct plasmid were grown at 37°C in LB medium with 50 μg/mL to an OD600 of 0.5. IPTG (0.2 mM) was then added to induce expression of the GST fusion gene in culture overnight, at 25°C. Cells were harvested, resuspended in phosphate-buffered saline, and sonicated with a sonicator. The cell debris was removed by centrifugation. The cell-free extract was loaded onto glutathione-agarose beads from Sigma-Aldrich. The purified GST proteins were then examined by SDS–polyacrylamide gel electrophoresis (PAGE).
Cell lines.
HUVECs were obtained from Cambrex Biosciences (Walkersville, MD). Human brain microvascular endothelial cells (BMECs) were a gift of Dr George Ghiso, NYU Medical Center. HEK293T cells were obtained from the ATCC (Manassas, VA).
Assay of ADAMTS-18.
A standard curve was prepared with the ADAMTS-18 C-terminal 385-AA peptide applied overnight at 4°C to a microtiter plate in 0.01 M bicarbonate buffer, pH 9.5. The plate was washed and blocked twice with TBS + 1% BSA and incubated overnight at 4°C. A polyclonal rabbit anti–ADAMTS-18-mer IgG (> 93% specificity, determined by Ag absorption) was then applied overnight. The plate was then washed and treated with donkey anti–rabbit IgG. A standard curve was sensitive at 10 ng. Test material was assayed at various dilutions, at which preimmune sera or second Ab alone had no effect.
Immunochemistry.
This was performed on formalin-fixed paraffin-embedded tissue. Five-micrometer-thick tissue sections were deparaffinized (2 washes of xylenes, graded alcohols, and buffered saline) and stained with a specific polyclonal rabbit Ab against ADAMTS-18 at 50 μg/mL following a 10-minute heat-induced epitope retrieval in boiling 10 mM citrate buffer, pH6. The primary Ab was incubated overnight and detected using a conventional ABC method with 3′-3′diaminobenzidene as the precipitating chromagen. Negative controls were with preimmune serum or without the primary Ab. No positive staining was seen with negative controls.
Preparation of platelet-fibrinogen aggregates in vitro.
For ADP-induced aggregates, 107 gel-filtered platelets were incubated with 100 mg/mL fibrinogen and ADP (10 μM) for 30 minutes at 37° to create platelet aggregates. Excess reagents were removed by washing in PBS and the number of platelets/aggregate counted. For collagen-induced aggregates, 1 μg/mL collagen (Helena Lab, Lubbock, TX) was incubated for 1 hour at 37°C with intermittent shaking, followed by gravity sedimentation at room temperature for 30 minutes. The top 50% volume was removed and the remainder of the platelet aggregate suspension was added directly into Tyrode buffer with testing reagents. Anti–GPIIIa49-66 Ab (20 μg/mL) or rADAMTS-18 (35 μg/mL) was added for various time intervals and the remaining platelets/aggregate were enumerated.
Thrombin cleavage assay.
35S-methionine–labeled ADAMTS-18 was translated from pBudCe4.1/ADAMTS-18-C-myc construct using an in vitro translation kit (TNT Coupled Reticulocyte Lysate Systems; Promega, Madison, WI) following the protocol provided by the manufacturer. The protein of 35S-ADAMTS-18 was then digested by thrombin at room temperature for one hour with/without inhibition with hirudin. The reaction products were then resolved on SDS-PAGE gel and the developing film was exposed to the dried gel at 4°C overnight.
Bleeding time.
The mouse tail vein was severed 2 mm from its tip and blotted every 30 seconds on a circular sheet of filter paper to obtain an objective measurement. Termination of the bleeding time was recorded after absence of blood on the filter paper. Bleeding time differences were recorded by an unbiased observer and confirmed by 2 other observers.
Carotid artery thrombus formation.
FeCI3-induced arterial injury was performed in 6- to 8-week-old Swiss Webster mice. A 2-mm2 strip of no. 1 Whatman filter paper (Fisher Scientific, Pittsburgh, PA) soaked in 10% FeCI3 was dried and prepared for use. The right common carotid artery was exposed by blunt dissection, and a 4 × 8-mm paraffin strip was placed around the artery to isolate it from surrounding tissue. A Doppler flow probe (Model BPM2 Blood Perfusion Monitor; Vasamedics, St Paul, MN) was positioned around the artery, 1-mm proximal to the bifurcation of the external carotid artery. The strip of filter paper was applied to the adventitial surface of the artery, 5 mm from probe for 20 minutes. The filter paper was removed, the field was flushed with saline, and the blood flow was continuously monitored and recorded at base line, start injury, and each minute for 35 minutes.
Postischemic stroke model.
A 3 × 0.2-mm polyethylene thread attached to a 9-mm 7/0 suture was inserted into the right internal carotid artery and advanced to the bifurcation of the middle cerebral artery. The obstruction was removed at 60 to 90 minutes. Mice were killed at 48 hours or 8 days. Mouse brains were dissected into 8 coronal planes at 1-mm intervals. Each section was stained red with triphenyltetrazolium chloride, fixed in 10% formalin, and scanned into a commercial photo image-editing program, and the data were stored in a disk. The unstained area of infarct was computer calculated using an automated image analysis program (Image J 1.32; National Institutes of Health [NIH], Bethesda, MD).
Neurologic test battery.
Animals were evaluated on the following behavioral tests, 8 days after stroke onset, by methods previously described in detail.33 For the postural reflex test, mice are held by the tail above the table and slowly lowered to the table edge to evaluate forelimb flexion. Neurologically normal mice extend both forelimbs toward the edge. Mice with ischemic damage consistently flex the forelimb contralateral to the damaged hemisphere. Mice with mild deficit were given the score of 1, a score of 2 was assigned for moderate deficit, and a score of 3 was given when the deficit was marked. For rotor rod, an accelerating rotor rod was used to measure the mouse's balance and motor coordination after operation. Each mouse was given 3 trials 15 minutes apart, with a 30-second adaptation at the beginning of the first trial. The rotation dial reading is increased from 1.5 to 10 with an accelerating rate of 0.5 every 15 seconds. The dial reading is recorded as the mouse falls. The time at falling from the rod is averaged for the three trials. For transverse beam, motor behavior was evaluated using the transverse beam test adapted for mice. The apparatus consists of a wooden beam positioned above the bench top. At one end of the beam is an enclosed darkened goal box. A doorway provides access to the box from the beam. At the other end of the beam, a 300-W halogen lamp is mounted. Mice are trained to escape from the bright light by running to the opposite end and entering the darkened goal box. Performance was scored using the following scale: 1: mouse is unable to balance on the beam; 2: mouse is able to balance but cannot place limbs on the beam; 3: mouse traverses beam by dragging a limb; 4: mouse places limbs on the beam at least once during traverse; 5: mouse places limbs on the beam during 50% of the time; 6: mouse uses limbs during more than 50% of the time; 7: mouse traverses beam with no more than 2-foot slips. Each mouse is given a total of 5 trials.
Results
Oxidative platelet fragmentation is independent of platelet cyclic AMP or Gαq−/−
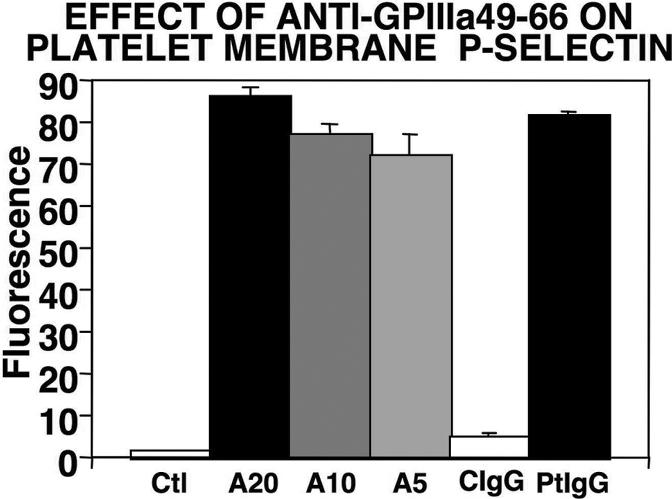
A unique mechanism of Ab-induced oxidative platelet fragmentation has been described.3,4,6 Ligation of GPIIIa49-66 induces the activation of platelet NADPH oxidase and 12-LO with release of platelet ROSs. This oxidative platelet fragmentation requires Ca2+ flux6 and is associated with P-selectin activation (Figure S1, available on the Blood website; see the Supplemental Materials link at the top of the online article). However, it occurs in the presence of PGE-1 and dibutryl cyclic AMP (Figure S2), or absence of Gαq (Figure S3), conditions that inhibit classic thrombin-induced platelet aggregation induced by outside-in signaling. Thus a new mechanism of platelet activation and death is described.
ADAMTS-18 C-terminal peptide mimics the effect of Ab-induced platelet oxidative fragmentation
In searching for a natural ligand to induce oxidative platelet fragmentation, we used the platelet GPIIIa49-66 peptide as “bait.” We then screened a phage surface display 7-mer peptide library using the GPIIIa49-66 peptide as bait in 3 rounds of washes, elutions, and amplification. Twenty clones were isolated. One of these clones (VHCVQLY) had 70% identity with ADAMTS-18 (1148-1152). An 18-mer peptide of ADAMTS-18 was therefore synthesized from the C-terminal TSR motif and conjugated to biotin, Bio-VQTRSVHCVQQGRPSSSC-OH. The peptide alone had no effect on platelet oxidative fragmentation. However, platelet fragmentation was obtained with the 18-mer biotin-peptide plus an antibiotin Ab or with streptavidin beads, not with the biotin peptide alone (Figure 1A). No effect was noted with a biotin-labeled scrambled peptide Bio-VQTRSVQVHCQGRPSSSC-OH plus antibiotin Ab or streptavidin beads. Oxidation is shown in Figure 1B. These data suggest that β3 clustering is required and Fc receptor plays no role.
Figure 1.

Effect of rADAMTS-18 on oxidative platelet fragmentation and binding to platelets. (A) Fragmentation with 18-mer peptide. Gel-filtered platelets were treated with biotin-peptide (bp) as well as bp + streptavidin (str) and scrambled biotinylated ADAMTS-18 peptide (scp) to induce integrin clustering (n = 6). Platelet fragmentation was assessed by fluorescence-activated cell sorting (FACS) as described in “Methods.” (B) Platelet oxidation. CT (control IgG), Ab (patient IgG). Eighteen-mer biotin-peptide was incubated with an antibiotin Ab, 1:200 dilution ( ) or irrelevant IgG (□; n = 6). Oxidation was assessed as discussed in “Methods.” (C) ADAMTS-18 or control protein was synthesized by ribosomal translation, using 35S-methionine. 35S-ADAMTS-18 (131 mCi/mg) was added to gel-filtered platelets overnight at 4°C, using doubling concentrations. Ctl refers to 35S-ribosomal translation of luciferase. AD-CTD refers to 35S-ribosomal translation of ADAMTS-18 with absent C-terminal 157 amino acids (13% deleted). Note absence of binding with C-terminal–deficient ADAMTS-18. Eighteen-mer refers to 10 μM nonradioactive 18-mer peptide added with the 35S-ADAMTS-18 incubation. Note inhibition of binding with C-terminal 18-mer peptide. Error bars indicate SEM.
) or irrelevant IgG (□; n = 6). Oxidation was assessed as discussed in “Methods.” (C) ADAMTS-18 or control protein was synthesized by ribosomal translation, using 35S-methionine. 35S-ADAMTS-18 (131 mCi/mg) was added to gel-filtered platelets overnight at 4°C, using doubling concentrations. Ctl refers to 35S-ribosomal translation of luciferase. AD-CTD refers to 35S-ribosomal translation of ADAMTS-18 with absent C-terminal 157 amino acids (13% deleted). Note absence of binding with C-terminal–deficient ADAMTS-18. Eighteen-mer refers to 10 μM nonradioactive 18-mer peptide added with the 35S-ADAMTS-18 incubation. Note inhibition of binding with C-terminal 18-mer peptide. Error bars indicate SEM.
Binding of ADAMTS-18 to platelets
Formal proof of ADAMTS-18 or anti–GPIIIa49-66 Ab reactivity against GPIIIa was obtained using platelets of GPIIIa−/− knockout mice. Neither ADAMTS-18 nor anti–GPIIIa49-66 Ab fragmented GPIIIa−/− platelets (n = 2, data not shown). Further proof of binding to platelets was obtained with 35S-ADAMTS-18 binding studies. Figure 1C demonstrates saturation binding of ADAMTS-18, inhibited by the 18-mer peptide. No binding was noted with a control 35S-luciferase protein or with a C-terminal truncated 35S-ADAMTS-18. Similar results were obtained with an ADAMTS-18 385-AA fragment.
Thrombin cleaves full-length ADAMTS-18
Since thrombin is generated during the formation of platelet thrombus formation, we examined whether thrombin could cleave ADAMTS-18 and release a fragment of the C-terminal region containing the 18-mer peptide. 35S-methionine–labeled ADAMTS-18 was synthesized with an in vitro translation system (using the expression vector pBudce 4.1 ADAMTS-18). This revealed 2 dominant bands of 135 and 75 kDa as well as other faint bands. The 135-kDa radioactive band represents intact ADAMTS-18, since the empty vector did not transcribe the 135-kDa band but did have a nonspecific 75-kDa band. Figure 2A demonstrates that after incubation with thrombin at room temperature for one hour, significant amounts of an ADAMTS-18 fragment was observed with ∼MW at 85 kDa and 45 kDa. The cleavage activity can be inhibited by hirudin indicating specific digestion by thrombin. Further immunoprecipitation studies with an Ab against the C-terminal myc tag on 35S-ADAMTS-18 revealed a 45-kDa fragment. Thus thrombin can cleave ADAMTS-18 to produce a C-terminal 45-kDa active fragment.
Figure 2.

Thrombin cleaves ADAMTS-18. (A) (Left panel) S35-methionine labeled ADAMTS-18 was incubated with different concentrations of thrombin with and without hirudin, at room temperature for 1 hour and resolved with SDS-PAGE. The sample loading lanes are (1) S35-ADAMTS-18 control (5 units/mL hirudin alone); (2) S35-ADAMTS-18 + 5 units/mL thrombin + hirudin; (3) S35-ADAMTS-18 + 5 units/mL thrombin; (4) S35-ADAMTS-18 + 2.5 units/mL thrombin + hirudin; (5) S35-ADAMTS-18 + 2.5 units/mL thrombin. Arrows point to thrombin cleaved bands. (Right panel) Immunoprecipitation with anti-Myc Ab following thrombin cleavage, demonstrating C-terminal 45-kDa fragment. (B) Graphic of truncated ADAMTS-18 constructs. The GPIIIa49-66 binding site is depicted. Three C-terminal recombinant peptide fragments were tested for their ability to induce platelet fragmentation and oxidation. These included the larger fragment of 385 amino acids (AAs) from the C-terminal end, an intermediate fragment of 188 AAs, and a smaller fragment of 66 AAs. (C) Effect of truncation on platelet fragmentation. Ctls refer to buffer and control IgG. Pt is patient IgG. Dilutions of the 3 fragments: recombinant peptides were made at neat (n), 1:2, and 1:4 in buffer. The initial concentration of the large 385-AA fragment was 1.3 μM, whereas the initial concentrations of the 188-AA and 66-AA peptides were 19 and 85 μM, respectively (n = 6). (D) Fragmentation induced by 385-AA fragment is inhibited by DPI (nM) and catalase (units/mL). Ctl refers to control buffer; 385-AA fragment refers to 1.3 μM 385-AA fragment. DPI 180, 140, 120, and 10 refer to nM DPI + 385-AA fragment. CAT 4000, 1000, and 500 refer to catalase units + 385-AA fragment. (E) Oxidation induced by 385-AA fragment inhibited by DPI and catalase. The designation is the same as that used as in panel D. (C-E) Where shown, error bars indicate SEM.
Effect of truncated rADAMTS-18 on platelet fragmentation
We reasoned that a C-terminal fragment of ADAMTS-18 might be the operational end for platelet oxidative fragmentation because ADAMTS-18 binds to GPIIIa49-66 by its 18-mer moiety located in the C-terminal region, ADAMTS-18 oxidative activity can be inhibited by an scFv Ab directed against the 18-mer, and thrombin cleavage could expose a C-terminal functional fragment. Accordingly, 3 truncated fragments within the C-terminal 4 TSR motifs were cloned in an E coli expression vector pGEX-4T-2 (Figures 2B and S4). Figure 2C demonstrates that the rADAMTS-385 amino acid (AA) fragment (837-1221) encompassing the 4 TSR motifs (containing the GPIIIa binding site) fragmented platelets at various dilutions, starting at n(neat) = 1.3 μM. The rADAMTS-188–amino acid structure (1034-1221) was considerably less potent with no platelet fragmentation at 1.3 μM and partial activity at n = 19 μM. The small rADAMTS-66–amino acid structure (1156-1221) not containing the GPIIIa binding site was inactive at n = 85 μM. Both fragmentation and oxidation induced by the 385-AA fragment were inhibited by DPI and catalase (Figure 2D,E), as previously reported for anti–GPIIIa49-66 Ab.3,4 Similar results were obtained with apocynin for fragmentation and oxidation with an ∼IC50 of 30 μM, n = 3.
Proof of ADAMTS-18 induced platelet fragmentation was further obtained by measurement of LDH release into the supernatant at 0 to 240 minutes. LDH at 0 time was 10 ± 1 units/L, which increased 83-fold at 60 minutes and 241- and 260-fold at 120 and 240 minutes, respectively. Similar results were obtained with anti–GPIIIa49-66 Ab, confirming our previous report.4
HUVECs contain ADAMTS-18
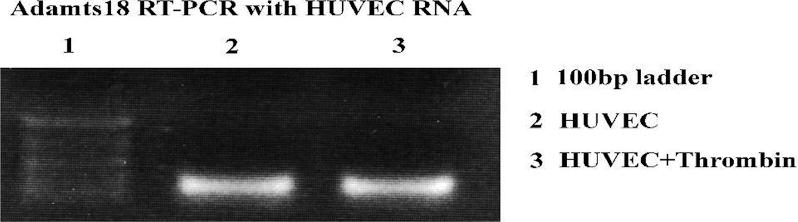
We next considered the possibility that endothelial cells could be a source of secreted ADAMTS-18, since ADAMTS-18 has been reported to be present in endothelial cells.21 We therefore performed reverse-transcription (RT)–PCR on HUVECs, which confirmed the presence of ADAMTS-18 mRNA (Figure S5). We also performed immunocytochemistry in human tissues and found ADAMTS-18 to be present on endothelium of arterial blood vessels as well as umbilical veins (Figure 3).
Figure 3.

Immunohistochemistry of arteries and veins for ADAMTS-18. (A,B) Umbilical artery and vein, respectively. (C,E,G) Pulmonary vein and (D,F,H) artery, obtained from 3 different patient archival pathology specimens.  depict positively stained endothelial cells for ADAMTS-18.
depict positively stained endothelial cells for ADAMTS-18.
Protease activation of HUVEC ADAMTS-18
Since all the ADAMTSs are secretory proteins with an N-terminal signal sequence, we assumed that it would be present in HUVEC conditioned media. Concentrated conditioned media from HUVECs contained limited to no oxidative platelet fragmentation activity. Since thrombin is generated during the formation of platelet thrombus formation and thrombin can generate a C-terminal 45-kDa fragment, we next examined whether thrombin could induce the oxidative platelet fragmentation function of C-terminal endothelial cell ADAMTS-18. Figure 4A demonstrates a time course for thrombin-induced activation of oxidatively inactive ADAMTS-18 in concentrated HUVEC conditioned media, indicating constitutive secretion of inactive ADAMTS-18. Thrombin-treated conditioned media were neutralized with hirudin at various time points and then used for platelet fragmentation studies at 4 hours. Initially, 50% functional activity was noted at approximately 15 minutes with full activity at 60 minutes. Similar results were obtained with brain microvascular endothelial cells (BMECs; Figure S6). Figure 4B demonstrates parallel platelet fragmentation of oxidatively active HUVEC media compared with anti–GPIIIa49-66 Ab.
Figure 4.

Thrombin activation and secretion of ADAMTS-18 in vitro and in vivo. (A) Kinetics of thrombin activation of concentrated HUVEC-conditioned media. HUVEC- conditioned media were treated with thrombin (0.5 units/mL) for various time intervals, followed by neutralization with hirudin. Aliquots were then used to induce platelet fragmentation. Incubation of thrombin and hirudin for 240 minutes prior to incubation had no effect on fragmentation (n = 3). (B) Comparison of anti–GPIIIa49-66 Ab with thrombin-activated HUVEC ADAMTS-18. Thrombin-treated media were neutralized with hirudin prior to incubation with gel-filtered platelets. Hirudin alone had no effect. Note similar kinetic response. (C) In vitro secretion and activation. HUVECs were treated with 0.5 units/mL thrombin for 15 to 30 minutes followed by washing and further incubation for 2 and 4 hours. The supernatant was assayed for ADAMTS-18 by ELISA. (D) Supernatant assay for platelet fragmentation (n = 3-4). (E,F) In vivo secretion and activation. (E) Mice were injected intravenously with various concentrations of thrombin in 100-μL volume and their plasma tested at 1 hour for ADAMTS-18 by ELISA. (F) Plasma from thrombin-stimulated mice was incubated with gel-filtered platelets and platelet fragmentation monitored as described in “Methods.” T-Plasma (plasma from thrombin-stimulated animals at various thrombin concentrations); T-Plasma + Hir (treatment with hirudin to neutralize any residual thrombin); + ADIgG (+ anti-ADAMTS IgG Ab) for 15-minute preincubation prior to platelet incubation with T-Plasma + hirudin; + Ctl IgG (control IgG for + ADIgG). (G) Inhibition of HUVEC-ADAMTS-18 induced platelet fragmentation with anti–ADAMTS-18 Ab or 18-mer peptide. (i) Anti–GPIIIa49-66 Ab was added to gel-filtered platelets in the presence and absence of the 18-mer peptide. (ii) HUVEC-ADAMTS-18 was incubated with control or anti-ADAMTS Ab or 18-mer peptide for 60 minutes at 37°C, prior to incubation with gel-filtered platelets for 4 hours at 37°C (n = 3). (i) Positive control with Buff (buffer), CtIgG (control IgG), PtIgG (patient IgG), 18m (18-mer peptide), or irrel (irrelevant peptide). (ii) AD (HUVEC conditioned media), CtIgG (control IgG), + ADAb (+anti–ADAMTS-18 Ab), 18m, or irrel (n = 6). Where shown, error bars indicate SEM.
Effect of endothelial cell activation on ADAMTS-18 secretion and activation from HUVECs
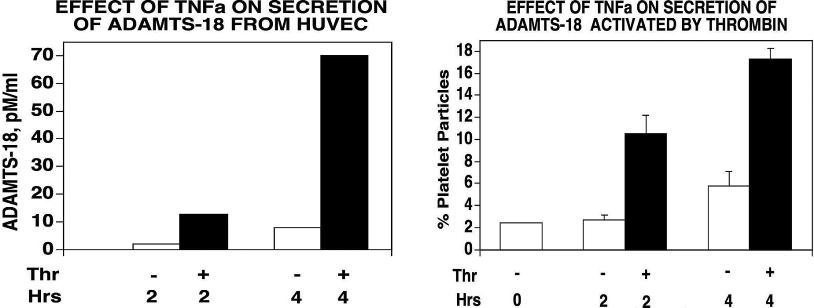
Limited to no ADAMTS-18 was present in unconcentrated HUVEC media, as assayed immunologically (enzyme-linked immunosorbent assay [ELISA] capable of detecting 10 ng). We therefore examined whether thrombin could enhance the secretion and oxidative platelet activation function of ADAMTS-18 from HUVECs. We reasoned that activation of HUVECs with endothelial cell activators, thrombin, or TNFα might enhance ADAMTS-18 secretion. Figure 4C demonstrates that thrombin (0.5 units/mL) incubated with HUVECs for 15 to 30 minutes (followed by washing to remove thrombin) followed by an additional 2- to 4-hour incubation enhanced ADAMTS-18 secretion. This was more than 90% inhibited by brefeldin A, a Golgi transport inhibitor that inhibits cell secretion. Similar results were obtained with 10 ng/mL TNFα (Figure S7). Thrombin also functionally activated HUVEC ADAMTS-18 to induce platelet fragmentation (Figure 4D).
Thrombin-induced in vivo generation of platelet-reactive ADAMTS-18
Mice have no immunologically detectable ADAMTS-18 in their plasma. However, in vivo stimulation of mice with intravenous thrombin at 4 to 16 units/mL led to ADAMTS-18 detection in the plasma, assayed 1 hour after thrombin injection (Figure 4E), which oxidatively fragmented platelets and was inhibited by anti-ADAMTS Ab (Figure 4F). These data indicate in vivo activation of endothelial cells by thrombin promotes ADAMTS-18 secretion and platelet oxidative fragmentation function.
Binding of HUVEC ADAMTS-18 to platelets is inhibited by anti–ADAMTS-18 Ab as well as 18-mer C-terminal peptide
We confirmed the presence of secreted and activated HUVEC ADAMTS-18 and its binding to platelets using an Ab raised against the C-terminal 18-mer peptide as possible inhibitor. Both the Ab as well as the 18-mer peptide inhibited HUVEC ADAMTS-18–induced fragmentation by approximately 70% at optimum concentration (Figure 4Gii). A second rabbit Ab against the N-terminal domain of ADAMTS-18 was inactive (data not shown). The 18-mer ADAMTS-18 peptide also inhibits anti–GPIIIa49-66–induced platelet fragmentation, which would be predicted if HUVEC ADAMTS-18 and the Ab bind to GPIIIa49-66 (Figure 4Gi). These data support the binding of the 18-mer as well as HUVEC ADAMTS-18 to platelet GPIIIa49-66, suggesting that the C-terminal portion of ADAMTS-18 may contain its functional properties.
Recombinant ADAMTS-18 385-AA fragment destroys platelet aggregates
In considering a possible mechanism for C-terminal ADAMTS-18–induced platelet oxidative fragmentation, we hypothesized that this could represent a physiologic clearing agent for the dissolution of platelet thrombi on activated endothelium. To test these hypotheses, we prepared collagen as well as ADP-induced platelet aggregates in the presence of fibrinogen and obtained platelet-fibrinogen aggregates of approximately 30 and approximately 10 platelets/aggregates, respectively. Figure 5A demonstrates that both anti–GPIIIa49-66 Ab and the ADAMTS-18 385-AA fragment disaggregate collagen-induced platelet clumps to the same extent, with nadir at 2 hours, whereas the scrambled ADAMTS-18 385-AA or the 66-AA fragment did not. Similar results were obtained with ADP aggregates with nadir at 4 to 8 hours. To determine whether decreased aggregate size was a function of disaggregation or platelet destruction or both, we assayed LDH release into the media. As could be predicted from previous experiments,4 which described Ab-induced oxidative platelet fragmentation in vitro with LDH release, similar results were obtained with ADAMTS-18 measurements of LDH release as an indicator of platelet lysis. With collagen-induced platelet aggregates, LDH release increased approximately 15-fold at 1 hour (n = 2) above background. Figure 5B demonstrates a photomicrograph of these events. Thus, the ADAMTS-18 385-AA fragment induces platelet aggregate destruction.
Figure 5.

Effect of polyclonal anti–GPIIIa49-66 or rADAMTS-18 385-AA fragment (AD-18F) on disaggregation and destruction of ex vivo platelet aggregates and anti–ADAMTS-18 Ab on in vivo bleeding time. (A) Collagen platelet aggregate. Ctl (scrambled [sc] ADAMTS-18F or 66-AA fragments), PtIgG (patient anti–GPIIIa49-66), media, and AD-18F were incubated with aggregates for 0.5, 1, 2, 4, and 8 hours at 37°C. The numbers of platelets/aggregate were then counted. Note that both AD-18F and anti–GPIIIa49-66 have a similar kinetic effect, representative of 3 experiments. (B) Photomicrograph of platelet collagen aggregates before and after treatment with anti–GPIIIa49-66 or control (scAD-18F or 66-AA fragment). (C) Effect of prestimulated platelet PAR-1 on induction of platelet fragmentation with AD-18F. Gel-filtered platelets were treated with the PAR-1 agonist TFLLRN (200 μM) for 30 minutes at 37°C and then followed with various AD-18F concentrations (50-6 μg/mL) for 4 hours. and ■ are without and with TFLLRN (T) prestimulation, respectively. Note increased sensitivity of activated platelets to AD-18F at low AD-18F concentration. Error bars indicate SEM. (D) Effect of polyclonal anti–ADAMTS-18 Ab on mouse bleeding time. BALB/C mice were injected intravenously with 10 μg anti–ADAMTS-18 IgG (AD-IgG) or preimmune IgG (PIS-IgG) and their bleeding time was monitored 60 minutes later (n = 10).
Recombinant ADAMTS-18 385-AA fragment reacts more strongly with TFLLRN-activated platelets
Since activated platelets have more GPIIb-IIIa reactive receptors on their surface, we reasoned that the 385-AA fragment might react more avidly with platelets pretreated with the thrombin receptor against TFLLRN. Figure 5C demonstrates this to be the case, with TFLLRN-activated platelets particularly at low 385-AA concentration (∼ 2-fold greater sensitivity). Similar results were obtained with thrombin as well as anti–GPIIIa49-66 Ab (data not shown).
Anti–ADAMTS-18 Ab shortens the bleeding time
Since the ADAMTS-18 385-AA fragment appears to be a negative regulator of platelet aggregate stability, we reasoned that an Ab against ADAMTS-18 18-mer peptide would shorten the bleeding time. Figure 5D demonstrates a dramatic 4.5-fold shortening of the mouse tail vein bleeding time 60 minutes after intravenous injection of 10 μg of rabbit anti–ADAMTS-18 IgG. Preimmune IgG had no effect, (3.6 ± 0.65 vs 0.8 ± 0.04 minutes, respectively), P = .002, n = 10. These data support the hypothesis that C-terminal ADAMTS-18 is a negative regulator of platelet aggregate stability.
Effect of ADAMTS-18 385-AA fragment on carotid artery platelet thrombus formation
We next designed an in vivo experiment to more directly assess the effect of ADAMTS-18 385-AA fragment on a classic arterial platelet thrombus induced with FeCI3. Figure 6A demonstrates an approximately 3-fold prevention of carotid artery platelet thrombus formation as measured by carotid artery blood flow. Figure 6B depicts the SEM at 5 minute intervals. Differences are statistically significant at 20 to 35 minutes, P less than .05, Student t test.
Figure 6.

Effect of ADAMTS-18 385-AA fragment on inhibition of FeCI3-induced carotid artery platelet thrombus formation. Swiss-Webster mice were injected intravenously with 35 μg active ADAMTS-18 385-AA fragment or inactive scrambled (sc) AD-18F or 66-AA fragment. The right carotid artery was exposed and an FeCI3 filter paper patch applied 2 hours after intravenous injection. Blood flow (mL/min) was measured with a Doppler flow meter. (A) Mean continuous flow was measured in 6 control (inactive 66-AA fragment), 2 control scAD-18F, and 6 experimental (active AD-18F) mice over a period of 38 to 43 minutes. (B) Same experiment with SEM given every 5 minutes as well as a single saline control experiment. *Statistically significant differences (P < .05) by Student t test (n = 5-6).
Effect of ADAMTS-18 385-AA fragment on postischemic cerebral stroke
We next examined an experimental cerebral stroke model induced by ischemia of the middle cerebral artery. Table 1 demonstrates approximately 50% protection of infarction and functional neurologic damage by ADAMTS-18 385–amino acid fragment. Protection was obtained if ADAMTS-18 fragment (or Ab) was given 2 hours before or 2 hours after 90-minute obstruction followed by release of the middle cerebral artery. No bleeding was observed. Platelet counts remained within 90% of normal range at 4 hours following ADAMTS-18 385-AA fragment versus control injection (1.0 × 106 vs 0.9 × 106 μL, respectively, n = 4). ADP, collagen, or TFLLRN-induced platelet aggregation in vitro was unaffected. These data indicate that ADAMTS-18 can regulate ischemic cerebral stroke without bleeding.
Table 1.
ADAMTS-18 385-AA fragment given 2 hours before or after obstruction of right middle cerebral artery and examined 8 days after removal of obstruction
| No. | % Infarct | Postural reflex* | Rotor rod† | Transverse beam† | |
|---|---|---|---|---|---|
| 385AA | 13 | 23.9 ± 4.0 | 1.02 ± 0.17 | 4.62 ± 0.58 | 5.97 ± 0.48 |
| 66AA+ | 8 | 43.8 ± 7.3 | 2.87 ± 0.13 | 3.23 ± 0.45 | 3.87 ± 0.90 |
| Saline | 14 | 43.0 ± 4.8 | 2.31 ± 0.25 | 3.19 ± 0.34 | 4.52 ± 0.46 |
| P§ | < .001 | < .002 | < .04 | < .01 |
Higher number reflects greater neurologic damage.
Lower number reflects greater neurologic damage.
Student t test comparison of intravenous injections of saline vehicle versus 35 μg ADAMTS-18 385-AA fragment versus functionally inactive ADAMTS-18 66-AA fragment (control). Since differences were not noted between pre– and post–ADAMTS-18 385-AA fragment infusion, both groups were averaged.
Discussion
These data demonstrate that the C-terminal fragment of a natural endothelial cell–secreted product, ADAMTS-18, can induce oxidative platelet fragmentation and platelet aggregate/thrombus dissolution after thrombin cleavage. Our data support a possible physiologic role for ADAMTS-18 in hemostasis. We propose the following hypothesis. Thrombin induces platelet thrombus formation and endothelial cell activation. Platelet thrombus formation is a dynamic process in which platelets are deposited and released from the endothelial surface depending upon an equilibrium between endothelial injury and thrombus dissolution (regulation). Thrombin-induced endothelial cell activation stimulates the secretion of HUVEC ADAMTS-18 that binds to platelet GPIIIa49-66. Thrombin cleaves ADAMTS-18 to produce the active 45-kDa C-terminal fragment that clusters β3. Clustering could be due to interchain disulfide bridging, since this fragment is enriched with cysteine. Clustering induces 12(S)-HETE production that regulates thrombus size by inducing oxidative fragmentation. Neutralization of thrombin by natural inhibitors halts thrombin-induced ADAMTS-18 release, preventing further platelet destruction. The degree of thrombus/platelet destruction is dependent upon the local platelet-endothelial concentration of ADAMTS-18 as well as the half-life of 12(S)-HETE and the C-terminal ADAMTS-18 in vivo. It is unlikely that the local ADAMTS-18 secreted by HUVECs would have any appreciable effect on circulating platelets or their function, since a 60-fold higher concentration used for mouse clinical studies had no effect on platelet count. It is likely that ADAMTS-18 has a greater effect on thrombin-activated platelets in vivo (within the platelet thrombus) than unactivated platelets in the circulation.
Our data also support the clinical use of ADAMTS-18 385-AA fragment. The 385-AA fragment can prevent the induction of experimental carotid artery thrombosis induced by FeCI3 damage. Doppler flow experiments clearly demonstrated a 3-fold protection of flow in this vessel as well as reversal of the partially occluded vessel. Recent unpublished studies (Y.-S.L., W.Z., and S.K., 2008) revealed the ability of an anti–GPIIIa49-66 scFv MoAb to reverse FeCI3-induced occlusion of the artery 2 hours after cessation of blood flow. Similar protection was noted in a postischemic middle cerebral artery stroke model in which ADAMTS-18 385-AA fragment significantly prevented and protected against infarction and neurologic damage by approximately 50%.
This report focuses on the C-terminal thrombin-cleavable domain of ADAMTS-18 that oxidatively fragments platelets and their thrombi. The N-terminal protease function remains to be established. Despite the similarity shared by ADAMTS family members, most differences among them are found in the C-terminal domains of the protein, suggesting that the C-terminal domains of ADAMTS may determine their in vivo location and substrate specificity.22,34,35 In addition, an important function of many ADAMTSs is their ability to bind to extracellular matrix presumably through thrombospondin (TSR) repeats in the C-terminal end of the protein.34,35 Of interest is a report that a sequence of ADAMTS-1 (CSVTCG) in TSR repeats can interact with CD36 on the surface of endothelial cells.36,37 It has been suggested that C-terminal processing regulates the activity of ADAMTS following autocatalytic release.26,38 Indeed, previous reports have shown that the TSR repeats of ADAMTS-4 determine the specificity of its substrate, GAG aggrecan.39 It is therefore conceivable that a better understanding of the function of the C-terminal TSR repeats of ADAMTS will shed light on the biologic function of these important proteins.
Some ADAMTS proteins including ADAMTS-18 are “orphan” ADAMTSs with no known function. Here we report an ADAMTS-18 protein with a function, which can operate in the absence of its metalloproteinase domain, but rather through a C-terminal 18-mer fragment binding site. Various recombinant truncated constructs of rADAMTS-18 were synthesized at its C-terminal region. One such C-terminal fragment containing 385 amino acids (AAs) and 4 TSR motifs was capable of inducing the same platelet oxidative-fragmentation reaction, as substantiated by its inhibition with catalase and DPI.
Thrombin is a master regulator of many proteins (factor XIII, factor XI, factor VIII, factor V, protein C, and fibrinogen) as well as its receptor (PAR-1) in which proteolytic cleavage is required to activate the factor or induce its function. Our studies provide a new proteolytic cleavage function for thrombin, in which a cleaved peptide becomes activated, rather than a larger protein. It is of interest in this regard that ADAMTS-13 has recently been shown to be inactivated by thrombin, contributing to the loss of ADAMTS-13 VWF cleavage function.40 It is also of interest that ADAMTS-13 has been reported to limit platelet thrombus formation in a shear rate–dependent platelet thrombus model on collagen, by its cleavage of ultralarge VWF.41
There is a precedent for functionally active peptides induced by enzymes (protein-peptide-enzyme), prothrombin-prothrombin kringle 2-factor-Xa,42 antithrombin-antithrombin fragment-thrombin, elastase,43 plasminogen-angiostatin-MMP-2,44 collagen IVα3-tumastatin-MMP-9,45 collagen VIII-endostatin-elastin,46 fibrinogen-alphastatin-plasminogen,47 fibrinogen-fibrin fragment E-MMP-9,48 histidine-rich glycoprotein-histidine–rich fragment,49 and high-molecular-weight kininogen-bradykinin-kallikrein.50 These peptides induce important physiologic functions regulating angiogenesis,42–48 chemotaxis,49 apoptosis,49 immune complex formation,49 fibrinogenesis,49 vasodilation, and blood pressure.51
Because ADAMTS-18 mRNA is present in endothelial cells and ADAMTS-18 has a secretory signal peptide, we hypothesized that endothelial cells might be producing this protein to regulate platelet aggregate formation and that it might be detectable in the plasma following in vivo endothelial cell activation with thrombin-treated mice. This proved to be the case with HUVECs or BMECs in culture as well as thrombin-stimulated mice in vivo. We therefore performed experiments to validate this hypothesis. rADAMTS-18 (385 AAs) was capable of (1) dissolving platelet aggregates produced ex vivo with fibrinogen and collagen or ADP; (2) regulating the in vivo bleeding time; (3) protecting against FeCI3-induced carotid artery thrombus formation; and (4) protecting against postischemic cerebral infarction. These confirmatory experiments support a possible physiologic and/or therapeutic role for ADAMTS-18 in hemostasis.
Thus we have discovered a natural ligand, ADAMTS-18, that binds to platelet integrin GPIIIa49-66 by its 18-mer C-terminal end and induces oxidative-platelet fragmentation and platelet thrombus destruction, following thrombin cleavage.
Supplementary PDF file available online.
Supplementary PDF file available online.
Supplementary PDF file available online.
Supplementary PDF file available online.
Supplementary PDF file available online.
Supplementary PDF file available online.
Supplementary PDF file available online.
Acknowledgments
This work was supported by NIH grants HL 13336 and DA 04315 (S.K.), and DA 020816 (Z.L.).
Footnotes
An Inside Blood analysis of this article appears at the front of this issue.
The online version of this article contains a data supplement.
Presented at the 49th Annual Meeting of the American Society of Hematology, Atlanta, GA, December 9, 2007.52
The publication costs of this article were defrayed in part by page charge payment. Therefore, and solely to indicate this fact, this article is hereby marked “advertisement” in accordance with 18 USC section 1734.
Authorship
Contribution: Z.L. performed the initial phage library screening, cDNA cloning, protein purification, and testing of ADAMTS-18; M.A.N. performed the platelet oxidative fragmentation studies; Y.-S.L. performed the mouse microsurgery experiments; R.P. performed the HUVEC work and bleeding times; W.Z. performed the 35S-ribosomal experiments; S.D. produced the scrambled ADAMTS-18 385-AA fragment; H.Y. performed the immunochemistry; D.Q. developed the neurologic studies; S.J. helped interpret the neurologic experiments; and S.K. designed and orchestrated the various experiments and wrote the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
Correspondence: Simon Karpatkin, New York University School of Medicine, Department of Medicine, 550 First Ave, New York, NY 10016; e-mail: simon.karpatkin@med.nyu.edu.
References
- 1.Morris L, Distenfeld A, Amorosi E, Karpatkin S. Autoimmune thrombocytopenic purpura in homosexual men. Ann Intern Med. 1982;96:714–717. doi: 10.7326/0003-4819-96-6-714. [DOI] [PubMed] [Google Scholar]
- 2.Zhang W, Nardi MA, Li Z, Borkowsky W, Karpatkin S. Role of molecular mimickry of hepatitis C-virus (HCV) protein with platelet GPIIIa in hepatitis C-related immunologic thrombocytopenia. Blood. 2009;113:4086–4093. doi: 10.1182/blood-2008-09-181073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nardi M, Feinmark SJ, Hu L, Li Z, Karpatkin S. Complement-independent Ab-induced peroxide lysis of platelets requires 12-lipoxygenase and a platelet NADPH oxidase pathway. J Clin Invest. 2004;113:973–980. doi: 10.1172/JCI20726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nardi M, Tomlinson S, Greco M, Karpatkin S. Complement-independent, peroxide induced antibody lysis of platelets in HIV-1-related immune thrombocytopenia. Cell. 2001;106:551–561. doi: 10.1016/s0092-8674(01)00477-9. [DOI] [PubMed] [Google Scholar]
- 5.Nardi M, Liu L-X. Karpatkin S GPIIIa (49-66) is a major pathophysiologically-relevant antigenic determinant for anti-platelet GPIIIa of HIV-1-related immunologic thrombocytopenia (HIV-1-ITP). Proc Natl Acad Sci U S A. 1997;94:7589–7594. doi: 10.1073/pnas.94.14.7589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nardi MA, Gor Y, Feinmark SJ, Xu F, Karpatkin S. Platelet particle formation by anti GPIIIa49-66 Ab, Ca2+ ionophore A23187, and phorbol myristate acetate is induced by reactive oxygen species and inhibited by dexamethasone blockade of platelet phospholipase A2, 12-lipoxygenase, and NADPH oxidase. Blood. 2007;110:1989–1996. doi: 10.1182/blood-2006-10-054064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li Z, Nardi MA, Karpatkin S. Role of molecular mimicry to HIV-1 peptides in HIV-1-related immunologic thrombocytopenia. Blood. 2005;106:572–576. doi: 10.1182/blood-2005-01-0243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Colige A, Beschin A, Samyn B, et al. Characterization and partial amino acid sequencing of a 107-kDa procollagen I N-proteinase purified by affinity chromatography on immobilized type XIV collagen. J Biol Chem. 1995;270:16724–16730. doi: 10.1074/jbc.270.28.16724. [DOI] [PubMed] [Google Scholar]
- 9.Colige A, Li SW, Sieron AL, et al. cDNA cloning and expression of bovine procollagen I N-proteinase: a new member of the superfamily of zinc-metalloproteinases with binding sites for cells and other matrix components. Proc Natl Acad Sci U S A. 1997;94:2374–2379. doi: 10.1073/pnas.94.6.2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Colige A, Vandenberghe I, Thiry M, et al. Cloning and characterization of ADAMTS-14, a novel ADAMTS displaying high homology with ADAMTS-2 and ADAMTS-3. J Biol Chem. 2002;277:5756–5766. doi: 10.1074/jbc.M105601200. [DOI] [PubMed] [Google Scholar]
- 11.Fernandes RJ, Hirohata S, Engle JM, et al. Procollagen II amino propeptide processing by ADAMTS-3: insights on dermatosparaxis. J Biol Chem. 2001;276:31502–31509. doi: 10.1074/jbc.M103466200. [DOI] [PubMed] [Google Scholar]
- 12.Li SW, Arita M, Fertala A, et al. Transgenic mice with inactive alleles for procollagen N-proteinase (ADAMTS-2) develop fragile skin and male sterility. Biochem J. 2001;355:271–278. doi: 10.1042/0264-6021:3550271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Georgiadis KE, Hirohata S, Seldin MF, Apte SS. ADAM-TS8, a novel metalloprotease of the ADAM-TS family located on mouse chromosome 9 and human chromosome 11. Genomics. 1999;62:312–315. doi: 10.1006/geno.1999.6014. [DOI] [PubMed] [Google Scholar]
- 14.Clark ME, Kelner GS, Turbeville LA, et al. ADAMTS9, a novel member of the ADAM-TS/metallospondin gene family. Genomics. 2000;67:343–350. doi: 10.1006/geno.2000.6246. [DOI] [PubMed] [Google Scholar]
- 15.Robker RL, Russell DL, Espey LL, et al. Progesterone-regulated genes in the ovulation process: ADAMTS-1 and cathepsin L proteases. Proc Natl Acad Sci U S A. 2000;97:4689–4694. doi: 10.1073/pnas.080073497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kuno K, Okada Y, Kawashima H, et al. ADAMTS-1 cleaves a cartilage proteoglycan, aggrecan. FEBS Lett. 2000;478:241–245. doi: 10.1016/s0014-5793(00)01854-8. [DOI] [PubMed] [Google Scholar]
- 17.Tortorella MD, Burn TC, Pratta MA, et al. Purification and cloning of aggrecanase-1: a member of the ADAMTS family of proteins. Science. 1999;284:1664–1666. doi: 10.1126/science.284.5420.1664. [DOI] [PubMed] [Google Scholar]
- 18.Abbaszade I, Liu RQ, Yang F, et al. Cloning and characterization of ADAMTS11, an aggrecanase from the ADAMTS family. J Biol Chem. 1999;274:23443–23450. doi: 10.1074/jbc.274.33.23443. [DOI] [PubMed] [Google Scholar]
- 19.Somerville RP, Longpre JM, Jungers KA, et al. Characterization of ADAMTS-9 and ADAMTS-20 as a distinct ADAMTS subfamily related to Caenorhabditis elegans GON-1. J Biol Chem. 2003;278:9503–9513. doi: 10.1074/jbc.M211009200. [DOI] [PubMed] [Google Scholar]
- 20.Bergeron F, Leduc R, Day R. Subtilase-like pro-protein convertases: from molecular specificity to therapeutic applications. J Mol Endocrinol. 2000;24:1–22. doi: 10.1677/jme.0.0240001. [DOI] [PubMed] [Google Scholar]
- 21.Cal S, Obaya AJ, Llamazares M, et al. Cloning, expression analysis, and structural characterization of seven novel human ADAMTSs, a family of metalloproteinases with disintegrin and thrombospondin-1 domains. Gene. 2002;283:49–62. doi: 10.1016/s0378-1119(01)00861-7. [DOI] [PubMed] [Google Scholar]
- 22.Porter S, Clark IM, Kevorkian L, Edwards DR. The ADAMTS metalloproteinases. Biochem J. 2005;386:15–27. doi: 10.1042/BJ20040424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vazquez F, Hastings G, Ortega MA, et al. METH-1, a human ortholog of ADAMTS-1, and METH-2 are members of a new family of proteins with angio-inhibitory activity. J Biol Chem. 1999;274:23349–23357. doi: 10.1074/jbc.274.33.23349. [DOI] [PubMed] [Google Scholar]
- 24.Gao G, Westling J, Thompson VP, et al. Activation of the proteolytic activity of ADAMTS4 (aggrecanase-1) by C-terminal truncation. J Biol Chem. 2002;277:11034–11041. doi: 10.1074/jbc.M107443200. [DOI] [PubMed] [Google Scholar]
- 25.Rodriguez-Manzaneque JC, Milchanowski AB, Dufour EK, Leduc R, Iruela-Arispe ML. Characterization of METH-1/ADAMTS1 processing reveals two distinct active forms. J Biol Chem. 2000;275:33471–33479. doi: 10.1074/jbc.M002599200. [DOI] [PubMed] [Google Scholar]
- 26.Flannery CR, Zeng W, Corcoran C, et al. Autocatalytic cleavage of ADAMTS-4 (aggrecanase-1) reveals multiple glycosaminoglycan-binding sites. J Biol Chem. 2002;277:42775–42780. doi: 10.1074/jbc.M205309200. [DOI] [PubMed] [Google Scholar]
- 27.Jin H, Wang X, Ying J, et al. Epigenetic identification of ADAMTS18 as a novel 16q23.1 tumor suppressor frequently silenced in esophageal, nasopharyngeal and multiple other carcinomas. Oncogene. 2007;26:7490–7498. doi: 10.1038/sj.onc.1210559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sjoblom T, Jones S, Wood LD, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 29.Li Z, Nardi MA, Wu J, et al. Platelet fragmentation requires a specific structural conformation of human monoclonal antibody against beta3 integrin. J Biol Chem. 2008;283:3224–3230. doi: 10.1074/jbc.M705902200. [DOI] [PubMed] [Google Scholar]
- 30.Nardi M, Karpatkin S. Antiidiotype antibody against platelet anti-GPIIIa contributes to the regulation of thrombocytopenia in HIV-1-ITP patients. J Exp Med. 2000;191:2093–2100. doi: 10.1084/jem.191.12.2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Karpatkin S, Nardi MA, Hymes KB. Sequestration of anti-platelet GPIIIa antibody in rheumatoid factor immune complexes of human immunodeficiency virus 1 thrombocytopenic patients. Proc Natl Acad Sci U S A. 1995;92:2263–2267. doi: 10.1073/pnas.92.6.2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.National Center for Biotechnology Information Basic Local Alignment Search Tool (BLAST). [Accessed February 2009]; http://www.ncbi.nlm.nih.gov/blast/Blast.cgi.
- 33.Quartermain D, Li YS, Jonas S. Acute enoxaparin treatment widens the therapeutic window for tPA in a mouse model of embolic stroke. Neurol Res. 2007;29:469–475. doi: 10.1179/016164107X164102. [DOI] [PubMed] [Google Scholar]
- 34.Kuno K, Matsushima K. ADAMTS-1 protein anchors at the extracellular matrix through the thrombospondin type I motifs and its spacing region. J Biol Chem. 1998;273:13912–13917. doi: 10.1074/jbc.273.22.13912. [DOI] [PubMed] [Google Scholar]
- 35.Kashiwagi M, Enghild JJ, Gendron C, et al. Altered proteolytic activities of ADAMTS-4 expressed by C-terminal processing. J Biol Chem. 2004;279:10109–10119. doi: 10.1074/jbc.M312123200. [DOI] [PubMed] [Google Scholar]
- 36.Iruela-Arispe ML, Lombardo M, Krutzsch HC, Lawler J, Roberts DD. Inhibition of angiogenesis by thrombospondin-1 is mediated by 2 independent regions within the type 1 repeats. Circulation. 1999;100:1423–1431. doi: 10.1161/01.cir.100.13.1423. [DOI] [PubMed] [Google Scholar]
- 37.Lawler J. The functions of thrombospondin-1 and-2. Curr Opin Cell Biol. 2000;12:634–640. doi: 10.1016/s0955-0674(00)00143-5. [DOI] [PubMed] [Google Scholar]
- 38.Georgiadis KE, Crawford T, Tomkinson K, et al. ADAMTS-5 is autocatalytic at a E753-G754 site in the spacer domain [abstract]. Trans Orthop Res Soc. 2002;27:167A. [Google Scholar]
- 39.Tortorella M, Pratta M, Liu RQ, et al. The thrombospondin motif of aggrecanase-1 (ADAMTS-4) is critical for aggrecan substrate recognition and cleavage. J Biol Chem. 2000;275:25791–25797. doi: 10.1074/jbc.M001065200. [DOI] [PubMed] [Google Scholar]
- 40.Lam JK, Chion CK, Zanardelli S, Lane DA, Crawley JT. Further characterization of ADAMTS-13 inactivation by thrombin. J Thromb Haemost. 2007;5:1010–1018. doi: 10.1111/j.1538-7836.2007.02514.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shida Y, Nishio K, Sugimoto M, et al. Functional imaging of shear-dependent activity of ADAMTS13 in regulating mural thrombus growth under whole blood flow conditions. Blood. 2008;111:1295–1298. doi: 10.1182/blood-2007-09-110700. [DOI] [PubMed] [Google Scholar]
- 42.Lee TH, Rhim T, Kim SS. Prothrombin kringle-2 domain has a growth inhibitory activity against basic fibroblast growth factor-stimulated capillary endothelial cells. J Biol Chem. 1998;273:28805–28812. doi: 10.1074/jbc.273.44.28805. [DOI] [PubMed] [Google Scholar]
- 43.O'Reilly MS, Pirie-Shepherd S, Lane WS, Folkman J. Antiangiogenic activity of the cleaved conformation of the serpin antithrombin. Science. 1999;285:1926–1928. doi: 10.1126/science.285.5435.1926. [DOI] [PubMed] [Google Scholar]
- 44.O'Reilly MS, Wiederschain D, Stetler-Stevenson WG, Folkman J, Moses MA. Regulation of angiostatin production by matrix metalloproteinase-2 in a model of concomitant resistance. J Biol Chem. 1999;274:29568–29571. doi: 10.1074/jbc.274.41.29568. [DOI] [PubMed] [Google Scholar]
- 45.Hamano Y, Zeisberg M, Sugimoto H, et al. Physiological levels of tumstatin, a fragment of collagen IV alpha3 chain, are generated by MMP-9 proteolysis and suppress angiogenesis via alphaV beta3 integrin. Cancer Cell. 2003;3:589–601. doi: 10.1016/s1535-6108(03)00133-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wen W, Moses MA, Wiederschain D, Arbiser JL, Folkman J. The generation of endostatin is mediated by elastase. Cancer Res. 1999;59:6052–6056. [PubMed] [Google Scholar]
- 47.Staton CA, Brown NJ, Rodgers GR, et al. Alphastatin, a 24-amino acid fragment of human fibrinogen, is a potent new inhibitor of activated endothelial cells in vitro and in vivo. Blood. 2004;103:601–606. doi: 10.1182/blood-2003-07-2192. [DOI] [PubMed] [Google Scholar]
- 48.Bootle-Wilbraham CA, Tazzyman S, Thompson WD, Stirk CM, Lewis CE. Fibrin fragment E stimulates the proliferation, migration and differentiation of human microvascular endothelial cells in vitro. Angiogenesis. 2001;4:269–275. doi: 10.1023/a:1016076121918. [DOI] [PubMed] [Google Scholar]
- 49.Olsson AK, Larsson H, Dixelius J, et al. A fragment of histidine-rich glycoprotein is a potent inhibitor of tumor vascularization. Cancer Res. 2004;64:599–605. doi: 10.1158/0008-5472.can-03-1941. [DOI] [PubMed] [Google Scholar]
- 50.Mori K, Nagasawa S. Studies on human high molecular weight (HMW) kininogen: II, structural change of HMW kininogen by the action of human plasma kallikrein. J Biochem. 1981;89:1465–1473. doi: 10.1093/oxfordjournals.jbchem.a133339. [DOI] [PubMed] [Google Scholar]
- 51.Rocha E, Silva M, Beraldo WT, Rosenfeld G. Bradykinin, a hypotensive and smooth muscle stimulating factor released from plasma globulin by snake venoms and by trypsin. Am J Physiol. 1949;156:261–273. doi: 10.1152/ajplegacy.1949.156.2.261. [DOI] [PubMed] [Google Scholar]
- 52.Li Z, Nardi MA, Li Y-S, et al. C-terminal ADAMTS-18 fragment induces oxidative platelet fragmentation, dissolves platelet aggregates, and protects against carotid artery occlusion and cerebral stroke.. Blood; Paper presented at the Annual Meeting of the American Society of Hematology; December 9. 2007; Atlanta, GA. 2007. p. 113A. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary PDF file available online.
Supplementary PDF file available online.
Supplementary PDF file available online.
Supplementary PDF file available online.
Supplementary PDF file available online.
Supplementary PDF file available online.
Supplementary PDF file available online.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}