

Abstract
We used replica exchange molecular dynamics (REMD) simulations to evaluate four different AMBER force fields and three different implicit solvent models. Our aim was to determine if these physics-based models captured the correct secondary structures of two α-helical and two β-peptides: the 14-mer EK helix of Baldwin and co-workers, the C-terminal helix of ribonuclease, the 16-mer C-terminal hairpin of protein G, and the trpzip2 miniprotein. The different models gave different results, but generally we found that AMBER ff96 plus the implicit solvent model of Onufriev, Bashford, and Case gave reasonable structures, and is fairly well-balanced between helix and sheet. We also observed differences in the strength of ion pairing in the solvent models, we but found that the native secondary structures were retained even when salt bridges were prevented in the conformational sampling. Overall, this work indicates that some of these all-atom physics-based force fields may be good starting points for protein folding and protein structure prediction.
I. Are Physics-Based Force Fields Accurate Enough for Protein Structure Predictions?
All-atom force fields, such as CHARMM,1 AMBER,2,3 and OPLS,4 are widely used for computer simulations of the properties of proteins and small peptides. However, such force fields are not yet commonly used to predict native protein structures, in part because of computational limitations in the conformational sampling of systems with many degrees of freedom, as in proteins, but also in part because of concerns that the force fields themselves might not be good enough. It has been difficult to distinguish whether physics-based protein structure prediction has been hindered by insufficient sampling or by limitations in the force fields.
However, a few tests on protein structure prediction have been possible so far because of mechanistically focused sampling or large dedicated computer resources. There are some notable successes indicating that modern physics-based force fields might be accurate enough to predict the native structures of proteins. Using supercomputer resources, Duan and Kollman performed a milestone microsecond molecular dynamics simulation of the 36-residue villin headpiece in explicit solvent starting from an unfolded conformation.5 Scheraga’s group, starting from a random configuration, folded the 46-residue protein A to within 3.5 Å root-mean-square deviation (rmsd) using Monte Carlo dynamics with an implicit solvation model.6 Another milestone was the folding of villin by the Pande group on Folding@Home, a distributed grid computing system running on the screen savers of volunteer computers worldwide.7
Recently, higher accuracies have also been achieved. High-resolution structures of villin have recently been reached by Pande et al.8 and Duan et al.9, 10 In addition, three groups have folded the 20-residue Trp-cage peptide to ~ 1 Å: Simmerling et al.,11 the IBM Blue Gene group of Pitera and Swope,12 and Duan et al.13 Recently, Lei and Duan folded the albumin binding domain, a 47-residue, three-helix bundle, to 2.0 Å.14 Furthermore, our group has found that AMBER ff96 in combination with a generalized Born/surface area implicit solvation model does well in identifying structures within about 3 Å from native for eight small single-domain globular proteins less than 75 amino acids in length.15 Such studies are encouraging for the application of physics-based potentials to the structure prediction problem.
Studies on shorter model peptides have enabled a more detailed look at differences among and potential deficiencies of force fields. Gnanakaran and Garcia performed 10 ns replica exchange molecular dynamics simulations of short alanine peptides with explicit water and a modified AMBER force field, and found that obtaining correct secondary structures was sensitive to backbone torsional energies.16 Okamoto and coworkers performed 10 ns replica exchange molecular dynamics (REMD) simulations on the C-terminal hairpin of protein G and the helical C peptide of ribonuclease A using six popular force fields with water molecules included explicitly.17,18 They found that AMBER ff94 and ff99 were too helical, GROMOS96 favored extended structures, and AMBER ff96, CHARMM22, and OPLS-AA were more balanced but did not reach a high level of accuracy in either peptide. Sakae and Okamoto also explored a method to optimize partial charge assignments and backbone torsional energies in these force fields using Protein Data Bank (PDB) structures, finding improved performance in simulations of short peptides.19–21
Much work has also focused on force fields used with implicit solvation, such as the generalized Born surface area (GBSA) method, as implicit models tend to be orders of magnitude faster for large protein simulations. Zhou22 compared the results of 3 ns explicit and implicit REMD simulations for the protein G hairpin, considering the AMBER ff94, ff96, ff99, and OPLS-AA force fields in combination with explicit (TIP3P for AMBER, SPC for OPLS) or implicit (GBSA for AMBER, surface generalized Born for OPLS) solvents. That study found that implicit solvation tended to destabilize the native structure, most prominently in the AMBER94 and AMBER99 force fields, which showed strong helical biases. Performing better in implicit solvent was AMBER ff96. More recently, Levy and co-workers examined the G hairpin and C peptide using OPLS-AA and a newer generalized Born model with a nonpolar solvation term;23 they found good agreement between experiment and their 4 ns REMD simulations for both peptides. Both the Zhou and Levy studies noted the overstabilized ion pairing they found in implicit solvent; non-native salt bridges significantly slowed conformational sampling or destabilized the native structure altogether. In contrast to the generalized Born approach, Luo and coworkers used a Poisson–Boltzmann implicit solvent to fold the G hairpin using AMBER ff94, ff03, and several variants of ff99, and found that ff03 was among the best performers.24 Interestingly, their simulations omitted the nonpolar surface area term, which they suggest can lead to non-native ion-pairing interactions.25
These detailed force field tests show that, despite their successes, two key issues remain unresolved in physics-based potentials: balance in backbone conformational preferences and implicit solvation models, particularly in ion-pairing interactions. We believe these are key to improving protein structure prediction with physical models. In this work, our aim is to perform an unbiased test of a family of modern force fields with implicit solvation, in order to assess the state of the art. We ask a very simple question: are these models capable of producing both basic α-helical and β-sheet structural motifs, where appropriate? In the interest of an unbiased performance assessment, here we do not evaluate based on the theoretical validity of the models or the manner in which they were derived.
We test several AMBER force fields (ff96, ff99, ff99SB, and ff03) and implicit solvation models (denoted here as igb1, igb5, igb7) for their abilities to stabilize under extensive sampling the small C-terminal protein G β-hairpin26 and the synthetic EK α-helix.27 For the best-performing force field/solvation pair, we then did additional tests on two other peptides: the helical C peptide of ribonuclease A28 and the synthetic trpzip2 hairpin.29 We believe the present simulations are extensive, with three independent 10 ns REMD trials for each peptide/force field/solvent combination. The goal of these simulations is to assess the stability of the native structures during the sampling procedure, in terms of resistance to unfolding during the finite simulation length. In addition, we perform a simple test of the role of ion pairs in the simulations by modifying the sampling so that they cannot form. We find that strong ion pairs in the implicit solvent models do not contribute significantly to stability, as the native secondary structures are still recovered when ion pairing is prevented.
II. Background
A. Force Fields
The latest release of the AMBER package, version 9, contains several all-atom force fields. Each of these comprises a collection of parameters for bonded potential energy terms (stretching, bending, and torsion) and nonbonded interactions (charge and van der Waals). In the present study, we examine four AMBER force fields: ff96, ff99, ff99SB, and ff03. The original ff94 parameter set developed by the Kollman group30 is one of the earlier force fields adopted by many researchers. The atomic charges in ff94 were derived from Hartree–Fock calculations; nonelectrostatic parameters such as torsion and van der Waals interactions were fitted first to small molecule data and then to larger organic compounds. Because ff94 was found to overemphasize helical conformations, ff96 was developed using quantum calculations to improve on peptide backbone ϕ–ψ torsion parameters.31
Kollman and co-workers later attempted a broader systematic refinement of many of the torsional parameters in ff94/ff96.32 They used RESP charges derived from high-level ab initio calculations to parametrize energies in an 82-molecule training set. The subsequent force field is denoted ff99, and is intended for use both with and without polarization effects. A number of groups, however, have since noticed an inaccurate representation of glycine and a strong α-helical emphasis in ff99. Several modifications have been proposed; among them, Hornak et al. performed a careful reparametrization of backbone torsional terms using alanine and glycine tetrapeptides.33 That new version, called ff99SB, was designed to provide better balance between extended and helical conformations. In ff03, Duan et al. refitted the charges in ff99 altogether, using quantum calculations with a continuum dielectric, to better reproduce the effects of solvent.34
B. Models for Implicit Solvation
Implicit solvation methods are designed to reproduce the solvation free energy as a function of protein composition and conformation, in a term that can be added to the intramolecular Hamiltonian for the protein. Implicit models represent the solvent as a uniform dielectric medium, neglecting the geometry and detailed bonding patterns of the solvent, such as water’s hydrogen bonds. Short- and long-range components of this free energy are treated separately: short-range interactions include van der Waals attractions and the formation of the protein cavity, and are typically modeled as being proportional to the solvent-accessible surface area. This simple treatment has been the standard approach since Eisenberg and McLachlan’s work in 1986,35 although it has been suggested that an additional cavity volume term may be important as well.36
The long-range electrostatic component of the solvation free energy has been the subject of much work in the past two decades. Numerical solution of Poisson’s equation for the protein charge distribution provides a straightforward approach, but is impractical for molecular dynamics simulations of modest lengths. Thus, a number of researchers have focused on the development of fast, approximate solutions to the electrostatic equations that recover exact results in ideal limits. The generalized Born model of Still and co-workers forms the basis for a number of implicit solvation models.37 It provides an approximation to the electrostatic free energy when a charge distribution is created in a cavity of low dielectric (the protein, ε = 1) embedded in a uniform dielectric medium (water, εW = 80):
| (1) |
where the sum is over pairs of atoms i and j, qi is the charge on atom i, and Ri is the effective Born radius of atom i. Equation 1 can also be modified with a Debye–Hückel term to incorporate salt effects, although we do not consider that case in our work. The function f provides an effective interaction distance between charge pairs. The main approximation of Still and co-workers is the following functionality for f:
| (2) |
This particular form yields the correct asymptotic free energies for point charges in both limits rij →∞(isolated ions) and rij → 0 (point dipole), while remaining smoothly differentiable at intermediate distances.
The effective Born radius for each atom, Ri, is crucial to the calculation of the solvation free energy.38 In a broad physical sense, Ri measures the distance from a charge in the protein to the boundary with the solvent. Approximating the electric field in the solvated case with the Coulombic expression (the so-called Coulomb-field approximation39,40) gives, for Ri
| (3) |
Here ρi is the intrinsic radius of atom i and the integral I is performed over the entire protein cavity minus the sphere of radius ρi surrounding atom i. For deeply buried atoms I is large, while for surface atoms it is small and Ri is close to the intrinsic radius, ρi. The computation of I is particularly challenging since the integration domain is highly irregular and the integral must be performed for each atom. Moreover, this computation is an expensive part of MD simulations and must be updated every one or two time steps to avoid numerical instabilities. Importantly, the Born radii appear to be key to the accuracy of implicit solvation: a study by Onufriev, Case, and Bashford in 2002 found that when the “perfect” Poisson–Boltzmann-determined radii were used in GB models, the remaining GB free energies were an excellent approximation to the Poisson–Boltzmann model.38 Implicit solvation models in AMBER differ mainly in the manner in which I is calculated.
In this work, we use three solvation models: igb1, igb5, and igb7, in the AMBER terminology. The solvation model called igb1 comes from Hawkins, Cramer, and Truhlar (HCT), who suggested that the integration domain for I be parsed into the atomic spheres composing the protein itself, so that the integral could be calculated by summing the analytical pairwise contributions to I of an atom with every other solute atom.41 This neglects the contributions to I of interstitial regions between the atomic spheres, and it overcounts regions of atomic overlap. HCT addressed the latter problem by scaling down the intrinsic radii used to calculate I by an element-dependent empirical factor; they determined the scaling factors by fitting predictions to a test set of small organic compounds. HCT parameters suitable for biomolecules were later developed by Tsui and Case,42 and are incorporated with the generalized Born equations into AMBER under the option igb=1.
While the HCT approach works well for smaller molecules, it was found to consistently underestimate the Born radii of buried atoms in macromolecules, due to the neglect of the atomic interstitial regions in I.39 Furthermore, the HCT equations can have singularities in the calculated Born radii when I → ρ−1, which can lead to numerical instabilities. Onufriev, Bashford, and Case (OBC)43 suggested a correction to these problems in which the Born radii are scaled by the extent of their burial, measured by the dimensionless quantity Ψ = ρ̃iI. Here I is determined by the usual HCT approach and the tilde indicates the scaled intrinsic radii. OBC chose a simple, three-parameter scaling function to modify eq 3:
| (4) |
The parameters α, β, and γ were optimized to reproduce Poisson–Boltzmann model predictions for a myoglobin unfolding trajectory. This approach largely eliminated the systematic errors for large molecules, while retaining good accuracy for smaller ones. Moreover, in a meta-study of implicit solvation models by Feig and co-workers in 2003,44 the OBC model yielded the best agreement with Poisson–Boltzmann calculations among pairwise GB models and was suggested as the best high-speed choice among the 23 methods and parametrizations studied. The OBC model is incorporated in AMBER under the option igb=5.
Mongan et al. recently attempted to improve the treatment of interstitials by including pairwise “neck” regions formed by the solvent-inaccessible space between two atoms fixed in space.45 Their model, called GBn, adds to the HCT value for I an additional summation over such neck regions for atom pairs. In these interstitials, the value of the integral is approximated by a Padé-like expression fitted to numerical solutions for test cases. Then, the Born radii are determined using the OBC methodology (eq 4) with a new set of values for α, β, and γ to accommodate the neck terms they have included. Using REMD simulations, Mongan et al. computed backbone free energies for the alanine decamer and found better agreement with explicit water simulations for GBn than for the older OBC model. The GBn model is incorporated in AMBER as the option igb=7.
III. Methods
We tested the performance of force fields and solvation models on four peptides. The peptides were chosen both because experiments show that they have substantial structure, and because they are short enough that we can do extensive conformational sampling. (1) We studied the 16-mer C-terminal hairpin of protein G, which has a near-native β-hairpin structure, when excised from the wild-type protein.26 NMR measurements estimate the population of the hairpin conformation to be about 40% in solution.26 The hairpin is stabilized by hydrophobic pairing of tyrosine/phenylalanine and tryptophan/valine on one side of the sheet, and contains four potential salt bridges, one of which does not disrupt the native strand pairing. (2) We examined the synthetic EK peptide of Baldwin and co-workers,27 which contains repeats of AEAAKA that tend to form α-helical structure due to pairing of the polar, oppositely charged glutamate and lysine residues along one side of the helix. We studied the 14-mer two-repeat version of this peptide; it has about 40% helicity according to circular dichroism measurements.
In addition, for the best-performing force fields and solvent models, we simulated two additional peptides. (3) We studied the Baldwin C peptide, a 13-residue helix excised from ribonuclease A that achieves partial helix formation around 0 °C and, based on the sensitivity of helicity to pH, appears to be at least partially stabilized by salt-bridge interactions.28 (4) Finally, we examined the 12-mer trpzip2 miniprotein engineered by Cochran, Skelton, and Starovasnik.29 trpzip2 forms a β-hairpin structure due to π-stacking interactions between four tryptophan residues on one side of the sheet. In addition, a Glu5–Lys8 salt bridge appears to stabilize the turn region of this peptide.
We tested four AMBER force fields: ff96, ff99, ff99SB, and ff03. In various combinations, we tested three solvation models: igb1 (HCT with Tsui–Case parameters), igb5 (OBC), and igb7 (GBn). Each of these GBSA models included the default surface area term specified in AMBER. The ionic radii used are the amber6, mbondi2, and bondi sets for igb1, igb5, and igb7, respectively; these follow the suggestions of the AMBER9 manual. We did not test igb1 with ff99 and ff03 because our preliminary comparisons showed that they would not differ much from igb5 in these cases (data not shown). The force fields and solvation models were selected using the tleap and sander programs of the AMBER package. Peptides were capped with ACE and NME blocking groups, for consistency with previous work18,22,23 and to minimize the possibility of salt-bridge traps that might prevent equilibration in the chosen simulation length.22,23
For all calculations, we simulated with replica exchange molecular dynamics (REMD).46 REMD enhances sampling by allowing configurations to heat up and cool down while maintaining proper Boltzmann distributions, in effect letting a conformational search process at higher temperatures inform that at lower ones. Each of our REMD simulations consisted of 20 replicas with temperatures exponentially spaced between 270 and 700 K. We used an implementation similar to that described in ref 47. All trajectories were generated using the sander program in the AMBER 9 package, while replica swaps and information collection was managed by a custom Python wrapper. Each peptide was simulated for a total of 10 ns, and analysis was performed on the last 1 ns using 1000 trajectory snapshots spaced every 1 ps.
We performed three REMD runs on each peptide to test the stability of the presumed native structure. Initial velocities for all three runs were different, and data from the three repeats for each initial structure were averaged to obtain the final results. We initiated the runs from a presumed native structure, taken from the PDB coordinates for the hairpins and the C peptide (1GB1, 1LE1, and 1FS3) and from the ideal helix for the EK peptide.
Root-mean-square-deviation (rmsd) and clustering analyses were performed on the 270 K trajectory from each REMD run. We also investigated results from the 300 K trajectories (not reported here), and found a uniform decrease in secondary structure content but otherwise no qualitative difference in the results. Secondary structures were assigned based on hydrogen bonding patterns and dihedral angles. Backbone rmsd values were computed for each analysis frame, taking reference configurations identical to the initial structures used. The rmsd trajectory tool for the VMD visualization program was used to perform these calculations.48 A Python-based clustering algorithm was also used to identify dominant configurations from the analysis frames. Multiple histogram analysis49 on all trajectories for T < 350 K was used to compute potential of mean force (PMF) curves at 270 K, using the final 2 ns of the 10 ns runs. A bin size of 0.2 Å was used, and the PMF curves were normalized such that when multiplied by −1/kBT, exponentiated, and integrated over their corresponding degree of freedom, the resulting value was unity.
IV. Results
A. Performance of Force Fields and Solvent Models
Our assessment of each of the force fields is based on the extent to which they maintain correct basic hairpin and helical tendencies, under extensive conformational sampling. Because experimental measurements on these short peptides are difficult to convert directly into structural ensembles, we do not believe a more detailed analysis–comparing the fractional stabilities, for example–is particularly informative here. Instead, we focus primarily on the degree of balance between helical and β motifs, by examining the dominant structures produced in each simulation and comparing to a single, presumed native structure. Though certainly the peptides experience fluctuations away from these presumed structures in reality, these conformations still serve as a useful milepost for indicating proximity to the correct topology.
Our main results are shown in Figures 1–3 and in Table 1. Figure 1 shows the dominant conformations of the EK peptide after REMD simulation. These conformations come from the most populous cluster after the clustering algorithm is performed on the trajectory; of the three run repeats for each condition, the one whose top cluster contained the most secondary structure is shown. The simulations with ff96, ff99, and ff99SB with igb1 or igb5 perform well, all retaining a near-perfect helix. The strong helical tendency of ff99 has been seen before.45 On the other hand, whereas ff96 was previously found to give poor helices in explicit solvent,17,50 here we find that ff96 works well with these implicit solvent models. The difference may be due to the fact that the implicit models increase the helical propensity of ff96, relative to explicit water. Another difference may be greater conformational sampling in the 10 ns simulations using implicit solvent, which lacks viscosity as compared to previous explicit water simulations.
Figure 1.
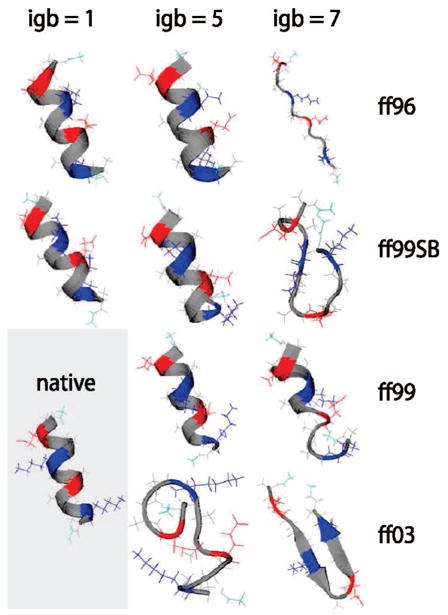
Top cluster conformations for the EK peptide from replica exchange simulations. The conformation with the most secondary structure among the dominant clusters of the three repeat runs for each force field/solvent combination is shown. Colors are residue-specific: gray, hydrophobic; green, polar; red, negatively charged; blue, positively charged. The presumed native configuration is shown at the bottom left. The solvent model igb1 was not tested with ff99 and ff03.
Figure 3.

(left) Double-error plot of average backbone rmsd for the EK peptide and G hairpin for the last 1 ns of the 10 ns REMD simulations. The points represent the average over three independent REMD runs; the error bars show the maximum and minimum averaged rmsd’s among the three runs. Colors denote the solvent model (blue, igb1; red, igb5; green, igb7) and symbols indicate the force field (diamonds, ff96; triangles, ff99; squares, ff99SB; circles, ff03). The hollow diamond indicates the result for the modified ff96/igb5 runs using the salt bridge prevention sampling protocol (see text). Gray lines serve as a guide to the eye. (right) Comparison of average rmsd values to the average fraction of native backbone hydrogen bonds formed. For clarity, bars giving the standard deviations over the data set of the calculation are placed above the curves.
TABLE 1.
Summary of Simulation Data Calculated over the Final 1 ns of Each REMD Simulationa
| peptide | force field | solvent | mean rmsd | min rmsd | max rmsd | avg secondary structure |
|---|---|---|---|---|---|---|
| EK helix | ff96 | igb1 | 2.8 | 0.6 | 4.5 | – –HHHHHHHHHHH– – |
| EK helix | ff96 | igb5 | 2.3 | 2.0 | 2.6 | – –HHHHHHHHHHH– – |
| EK helix | ff96 | igb7 | 5.6 | 5.4 | 5.7 | – – – – – – – – – – – – – – |
| EK helix | ff96 | igb5-R | 1.0 | 0.7 | 1.2 | – –HHHHHHHHHHH– |
| EK helix | ff99 | igb5 | 2.3 | 2.1 | 2.8 | – – – – –HHHH– – – – – |
| EK helix | ff99 | igb7 | 2.8 | 2.7 | 3.0 | – – – – – – – – – – – – – – |
| EK helix | ff99SB | igb1 | 1.9 | 1.3 | 2.3 | – – –HHHHHH– – – – – |
| EK helix | ff99SB | igb5 | 2.1 | 1.6 | 2.7 | – – –HHHHHHH– – – – |
| EK helix | ff99SB | igb7 | 4.3 | 3.2 | 5.1 | – – – – – – – – – – – – – – |
| EK helix | ff03 | igb5 | 4.4 | 3.7 | 5.3 | – – – – – – – – – – – – – – |
| EK helix | ff03 | igb7 | 4.9 | 4.2 | 5.7 | – – – – – – – – – – – – – – |
| G hairpin | ff96 | igb1 | 4.3 | 3.6 | 5.0 | – – – – – – – – – – – – – – |
| G hairpin | ff96 | igb5 | 4.4 | 3.7 | 5.1 | – – – – – – – – – – –HH–E– – – |
| G hairpin | ff96 | igb7 | 2.0 | 1.1 | 2.6 | – –EEEEE– – – –EEEEE– – |
| G hairpin | ff96 | igb5-R | 3.9 | 1.7 | 6.7 | – – – EEE– – – – –EEEE– – – |
| G hairpin | ff99 | igb5 | 5.9 | 5.6 | 6.1 | – – – – – – – – – – – – – – – – – – |
| G hairpin | ff99 | igb7 | 5.6 | 5.2 | 6.1 | – – – – – – – – – – – – – – – – – – |
| G hairpin | ff99SB | igb1 | 4.8 | 4.4 | 5.3 | – – – – – – – – – – – – – – – – – – |
| G hairpin | ff99SB | igb5 | 5.3 | 5.0 | 5.4 | – – – – – – – – – – – – – – – – – – |
| G hairpin | ff99SB | igb7 | 5.6 | 5.0 | 6.1 | – – – – – – – – – – – – – – – – – – |
| G hairpin | ff03 | igb5 | 2.7 | 1.7 | 3.3 | – – – –EEE– – – –EEE– – – – |
| G hairpin | ff03 | igb7 | 4.2 | 3.0 | 5.3 | – – – – – – – – – – – – – – – – – – |
| C helix | ff96 | igb5 | 2.4 | 2.0 | 2.9 | – – –HHHHHHHHHHH– |
| C helix | ff96 | igb5-R | 3.1 | 2.8 | 3.3 | – –HHHHHHHHHHHH– |
| trpzip2 | ff96 | igb5 | 1.5 | 0.8 | 2.6 | – –EEE– – –EEEE– – |
| trpzip2 | ff96 | igb5-R | 3.6 | 2.5 | 4.7 | – –EE– – – –EE– – – – |
The mean rmsd is an average of the set of three trials for each run condition. The minimum and maximum averaged rmsd values of the three trials are also shown; note that these do not refer to the minimum and maximum values over the course of the trajectories, but instead over the trial averages. The average secondary structure shows the most frequently appearing motif of helix (H), extended (E), or other (–) for each residue. The “igb5-R” solvent indicates igb5 with the salt bridge repulsion. All rmsd values are in units of angstroms.
Other force field/solvent combinations are problematic. The ff03 force field unfolds the helix completely, forming a coil-dominated structure with igb5 and a hairpin with igb7. These results are consistent among all trials, indicating at least minor difficulties in achieving α-helical structures in ff03 with implicit solvent. A second weakness appears to be the igb7 solvent model. The results for igb7 consistently show significant deviations from helix: starting from the native structure, the helix essentially unfolds when igb7 is used with all force fields except ff99SB, where it partially unfolds. The variety of structures produced with different force fields suggests that igb7 does not adequately stabilize helical structures.
Figure 2 shows our tests on the protein G hairpin. Here, successful predictions appear to depend more on the force field than on the solvation model. It is clear that ff99 erroneously tends toward helical conformations, as previous researchers have seen. ff99SB gives slightly better structures than ff99, although the hairpin never fully develops in the former, and some helical tendency is still evident for igb5. However, ff96 performs well for all three solvent models, consistent with findings of Okamoto et al.17 The ff03 parameters also fare well; the particular combination of ff03 and igb5 generates an extremely stable hairpin with near-perfect alignment. Compared to its poor performance on the helical EK peptide, this might indicate that ff03’s dihedral torsions are slightly biased toward β-structure when used with implicit solvent.
Figure 2.
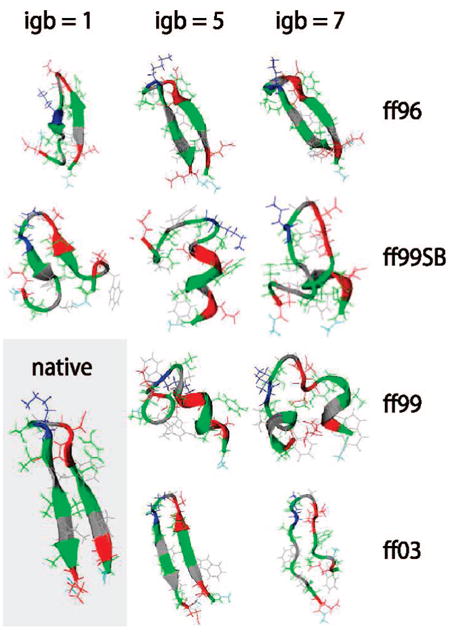
Top cluster conformations for the protein G hairpin from replica exchange simulations. See Figure 1 for further details.
In these visual assessments of structure, force field ff96 appears the most consistent and balanced. We find that igb5 performs well with it, and that igb1 is almost as good, but igb7 is problematic. Figure 3a presents a more quantitative comparison using a type of plot that we have found to be informative. This double-error plot shows the rmsd error in predicting the helix vs the rmsd error in predicting the β-hairpin. The rmsd is time-averaged over the last 1 ns of the simulation trajectories. This kind of diagnostic does not give detailed information about the ensemble of structures sampled in each simulation, but we believe it is at least a good indication of the basic extent to which helical and hairpin structures are sampled. Therefore, the plot in Figure 3a gives two simple types of information. First, points closer to the main diagonal, the y = x line, indicate that a force field is well-balanced, which is to say that errors in predicting β-hairpins are roughly equal to errors in predicting α-helical structures. Second, points closer to the origin indicate smaller overall errors of the model relative to the presumed native structures compared to points more distant from the origin. Hence the best force fields and solvent models give points on this graph that are closest to the main diagonal and closest to the origin.
By these criteria, the best overall force field/solvation pairs are ff96 + igb1, ff96 + igb5, and ff03 + igb5. We note that it is difficult to discriminate further, using small differences in average rmsd values (~1 Å), due to both the imperfect nature of the rmsd measurement to a presumed native structure and the run-to-run variations. However, we find that globally the most consistent force field, with multiple solvents, appears to be ff96. Similarly, we find igb5 to be the most consistent solvent when paired with multiple force fields. Therefore, we consider the choice of ff96 + igb5 to be a reasonable overall choice, with rmsd’s centered around 2.9 Å for the helix and 3.9 Å for the hairpin.
The error bars in Figure 3a show the minimum and maximum rmsd values of the three run repeats. Much of this variance is likely due to structural fluctuations around the stable conformation in each case, on the order of ~ 1–2 Å. On the other hand, two runs in particular exhibit fairly pronounced fluctuations: those involving ff96/igb1 and a modified version of ff96/igb5 that is described later in the paper. For those two cases, there is significant run-to-run variation among the three trials, and examining these further, we also found that each individual run could be characterized by significant structural fluctuations as the simulations progressed with time. This kind of behavior is typical of a rough folding landscape.
As a reference comparison, Figure 3b shows the correlation between averaged rmsd (as reported above) and the time-averaged fraction of native backbone hydrogen bonds formed in each peptide, compiled over all simulation test cases in our study. Both the EK peptide and protein G hairpin were reported to remain only partially stable in solution. Though a detailed comparison of structural ensembles with experiment is difficult (since it is not clear whether the reported fractional stabilities correspond to partial unfolding or temporal fluctuations), we can ask what kinds of fluctuations emerge from partially stable structures in the simulations. For the instructive case in which 50% of the native hydrogen bonding is present, Figure 3b suggests rmsd averages of 2.0 Å for the EK helix and 3.0 Å for the G hairpin. Thus, it is not unreasonable to expect that partially stable peptides experience rmsd fluctuations around a native structure that are on the order 2–3 Å, and that the fluctuations might be greater for the hairpin than the helix.
Due to the finite length of these simulations, it is indeed possible that equilibrium convergence is not fully attained. However, we believe that the dramatic differences among the systems studied in the 10 ns REMD runs provides a useful force field diagnostic. To illustrate some of these differences, Figure 4 shows some of the convergence properties of the simulations, showing a well- and poor-performing force field/solvent pair for each of the two peptides. A time progression of the rmsd from presumed native structure shows that the poor-performing pairs quickly depart the native basin, in under 1 ns of REMD simulation time. For these, the populations of the dominant clusters increase with time, indicating that the peptides find increasingly stable alternate conformations, away from the native. In contrast, the best-performing pairs largely remain a short distance from the native basin, although with time it appears that the rmsd fluctuations for these slightly increase. Moreover, in the well-performing simulations, the dominant cluster populations decrease during the run relative to the initial 2 ns. This could indicate that the systems locate slightly more diverse conformational ensembles than the native structure alone. On the time scale of the run, it appears these conformational populations start stabilizing around the 8 ns time point.
Figure 4.
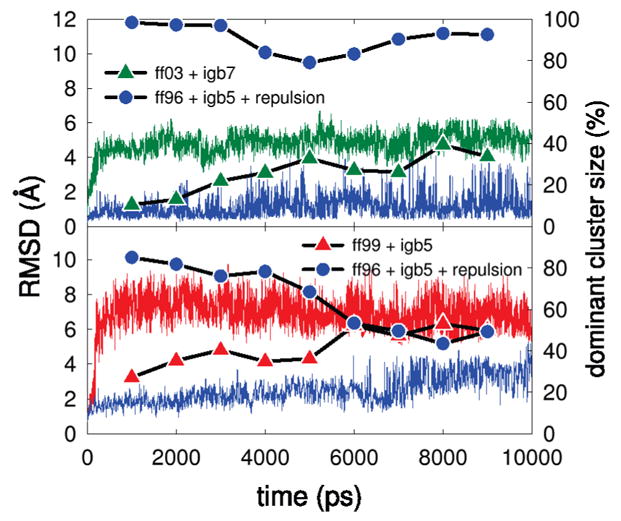
Comparison of simulation progress between the best- and worst-performing force fields. The top panel corresponds to the EK peptide and the bottom to the protein G hairpin. Continuous thin lines depict the rmsd to the presumed native structure, whereas points and thick lines indicate the populations of the dominant configurational clusters in a 2000 ns window centered around the indicated time.
B. Salt-Bridge Effects on Peptide Stability
Several research groups have previously found that implicit solvent models give non-native salt-bridge-trap conformations.51 We looked for salt-bridge traps in our simulations. The G hairpin contains a single lysine residue that can pair with either of two aspartate or two glutamate residues. One of these four potential salt bridges, Lys7 and Asp10, does not disrupt the native hairpin conformation. Figure 5 compares the potential of mean force (PMF) for this native salt bridge with different solvent models for ff96. These PMF curves were extracted from the last 2 ns of each simulation, and averaged over three runs. In the strictest sense, these curves must be taken as approximate given the finite simulation durations and the limitations of the 2 ns data window, and are best taken as a measure of probabilities during the sampling procedure. However, error bars measured from differences among the independent runs do give an indication as to the variation of these computations, and they show that there are some statistically distinct features in these measurements for different force fields. Note that each force field/solvent pair produces a minimum around 2.8 Å corresponding to the formation of the salt bridge and a shallower minimum around 4.5 Å due to the second charged aspartate oxygen.
Figure 5.
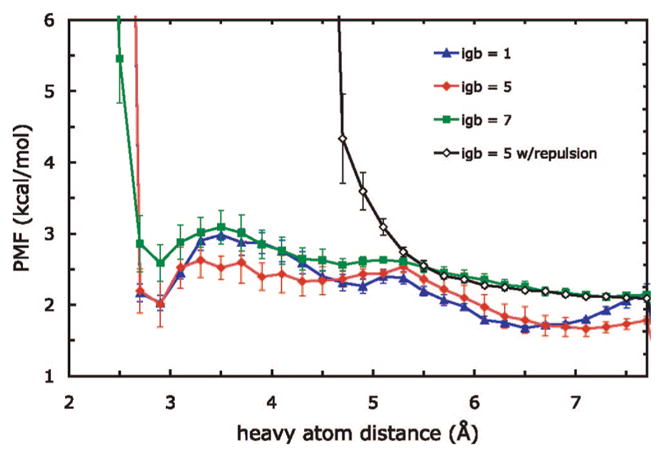
Potential of mean force between native ion-pairing side-chain atoms in the protein G hairpin. The distance is calculated between the charged N in Lys7 and one charged O in Asp10; the two PMFs for each of the aspartate oxygens are averaged. The PMFs were aligned such that the exponential of −1/kBT times their value is proportional to the fraction of simulation time spent in the corresponding distance bin. Error bars shown are computed over the two aspartate oxygens and three run trials.
An analysis of the PMF curves shows the relative populations of ion pairs in each of the three solvents tested. The main minima in the PMFs in igb1 and igb5 are roughly equal in depth, indicating that the salt bridge is formed roughly the same fraction of time in both solvents. The heights of the barriers between igb1 and igb5 are different, however, which suggests that salt bridges in igb1 less readily dissociate once formed. In the igb7 case, the higher absolute position of the minimum means that salt bridges are less populated overall. Qualitatively, our simulations show a ranking of igb1, igb5, and igb7 from strongest to weakest salt-bridge stabilization in the protein G hairpin, in terms of populations of different salt-bridge states.
It is interesting to consider these results in light of the recent work of Lwin and Luo24 that found that salt-bridge populations are also significantly affected by the choice of force field. Indeed, our results show that the other force fields, beyond ff96, alter the strength of salt-bridge formation; in many of these cases, however, the effect is indirect, through a major altering of the secondary structure preference of the peptide. Still, it is challenging to rigorously separate force field and implicit solvent effects on ion pairing in these simulations. Lwin and Luo also suggest that the nonpolar surface area term contributes significantly to ion pairing.25 Our simulations clearly show an effect of the electrostatic part alone, since each of the three GB models is paired with the same surface area term. A useful future study might examine the effect of turning off the surface area component of the implicit models.
We find in the ff96 + igb5 REMD trajectories that non-native salt bridges are convergence traps, persisting for 100–1000 ps in run time at the lowest temperature replica, but not stable over longer times. In the protein G hairpin, the most frequently seen such decoy configuration entailed a one-residue shift in the strand alignment to accommodate the non-native Lys7–Asp11 pairing. This shifted alignment was more frequent in the ff96 + igb1 case, consistent with the relative salt-bridge stabilization of igb1 and igb5. For the EK peptide, we found non-native salt-bridge pairing occasionally led to a hairpin conformation from which it was difficult to reach the native helical state.
We performed a simple test to establish whether the salt bridges contributed significantly to the stability of the native structures in the force field. We excluded the possibility of salt-bridge formation during the simulations by removing the associated configurations from the sampling space. In our molecular dynamics runs, we introduced a repulsive potential felt only upon close approach between any two ion-pairing atoms, steep enough to prevent salt bridges but not so steep that it introduced numerical instabilities. The form of the potential is shown in Figure 6, chosen because it is an option in the AMBER 9 package. We chose parameters to create a steep repulsion that would mimic the effects of a hard overlap term: the potential is zero at large distances, rises harmonically from 6 to 4 Å with a force constant of 0.5 kcal/mol/Å2, and is continuously linear thereafter. For comparison, salt bridges typically form when the pairwise distance is less than 4 Å. We stress that this modification is not intended to be a reparametrization of the force field, but simply an easy way to exclude those portions of configuration space that correspond to salt-bridge formation. We have also verified that increasing the steepness of the repulsion does not have a statistically significant effect (data not shown).
Figure 6.
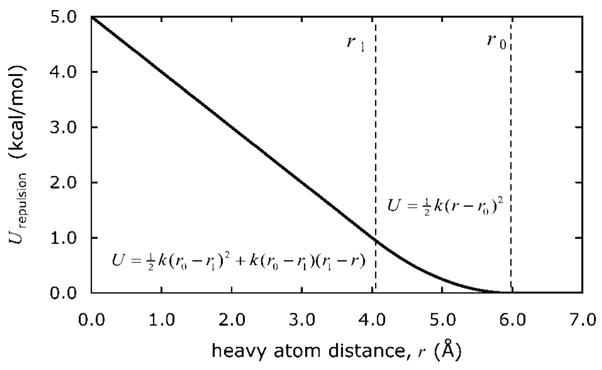
Repulsive energy function used between potential ion-pairing heavy atoms to prevent salt-bridge formation. Decreasing from large distances, the potential is zero until r0, harmonic with a force constant k until r1, and continuously linear thereafter. We used r0 = 6 Å, r1 = 4 Å, and k = 0.5 kcal/mol.
We used the salt-bridge-free sampling on the best-performing force field, ff96 + igb5. Figure 7 shows the effect of removing the possibility of salt bridges on the PMF calculations for the protein G hairpin. The modification has two effects: as expected, it pushes the potential ion-pairing heavy atoms apart, but interestingly, it also increases the probability that the native contact is formed relative to the non-native salt bridge, as shown by the lower PMF for the former.
Figure 7.
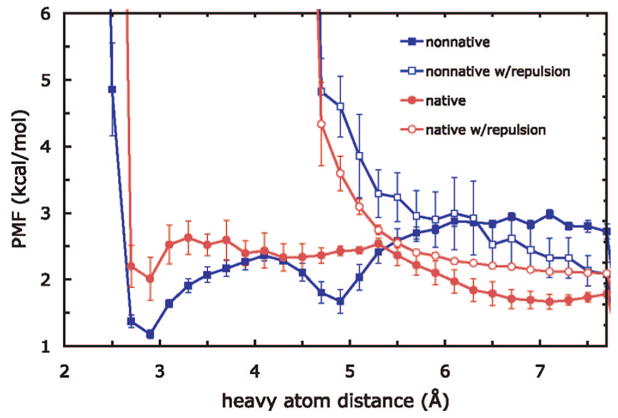
Comparison of salt-bridge interactions in the protein G hairpin for ff96 + igb5 with and without the salt-bridge-free sampling (using a repulsive potential to prevent salt bridges). The native interaction is Lys7–Asp10, while the non-native is Lys7–Asp11.
In all of the peptides we examined, we found that removing the possibility of salt bridges from the sampling did not significantly destabilize the native secondary structures. Figure 8 shows a comparison of the structures for the EK helix, protein G hairpin, Baldwin’s C peptide, and the trpzip2 mini protein. All four peptides contain potential salt bridges. Even without the repulsion, the ff96 + igb5 force field/solvent model combination yields reasonable results across all four systems. With the salt-bridge sampling, similar cluster structures were obtained, and in some cases, we found that convergence was accelerated. One minor exception is the case of the protein G hairpin, which exhibited a large run-to-run variation in the kinds of structures sampled when the salt bridge was not allowed to form (Figure 3a): this could be an indication that the native basin was broadened by the removal of salt bridges, since it resulted in both structures closer to and farther from the normal ff96/igb5 case. Overall, however, the fact that the conformations of the most populated cluster for each peptide did not change significantly during salt-bridge-free sampling suggests that the very close, strong association between charged ions is not critical for stability in these peptides.
Figure 8.
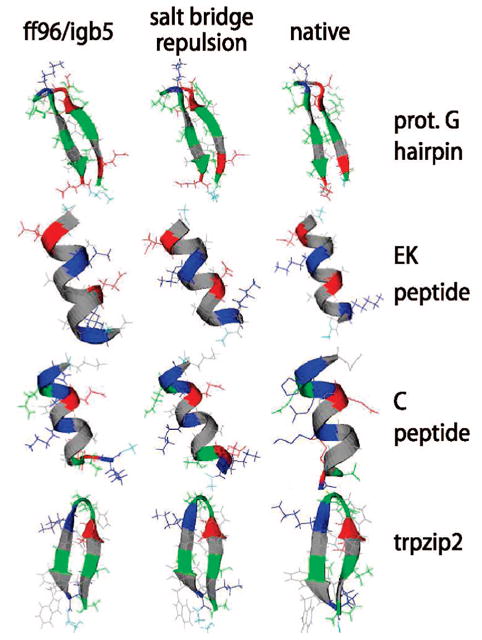
Top cluster conformations for four peptides from replica exchange simulations using the best-performing force field/solvent pair, 96 + igb5. The conformation with the most secondary structure among the dominant clusters of the three repeat runs for each scenario is shown. Colors are identical to those in previous figures.
We do caution that this salt-bridge-free sampling is a diagnostic, and not a general fix for ion-pairing overstabilization. In particular, it is possible that proteins with buried salt bridges, not investigated here, could be significantly destabilized by removal of salt-bridge interactions. Here, due to the small size of the systems studied, essentially every residue is exposed to solvent, and hence, all possible ion-pairing interactions are surface interactions. In contrast to this work, Simmerling and co-workers have attempted to address the ion-pairing overstabilization problem by rebalancing the force field, reducing the intrinsic (low-dieletric) radii of hydrogen atoms bound to charged nitrogen atoms.52
We also conducted a brief test of salt-bridge-free sampling on three larger proteins with surface salt-bridge pairs, to ensure it did not lead to unphysical artifacts in larger systems than peptides. We performed REMD simulations of three larger proteins, the 56-residue α/β protein G (PDB code 1GB1), 56-residue all-β src SH3 (1SRL), and the 46-residue all-α protein A (1BDD). Two simulations were performed for each, one with and one without the salt-bridge repulsion, and each run was initialized from the experimental native structure. We found that the rmsd-to-native of each protein after 10 ns of REMD remained the same in the salt-bridge-free case. The backbone rmsd’s for the final dominant cluster configurations in the normal/repulsion-applied cases were 1.2 Å/1.2 Å for protein G, 2.7 Å/2.6 Å for src SH3, and 2.2 Å/2.0 Å for protein A. Although such short REMD simulations for proteins of this size certainly do not reach equilibrium, these results do show that the added repulsion does not destabilize the native structure on this time scale.
V. Conclusions
We performed extensive REMD simulations of a series of candidate peptides to test four AMBER force fields with three generalized Born solvation models. We found that the combination of the AMBER ff96 force field with the Onufriev, Bashford, and Case solvent (igb5) was the most consistent in capturing the behavior of all four test peptides, with balance between strand and helical conformations. The combinations of ff03 with igb5 and ff96 with the older igb1 model of Hawkins, Cramer, and Truhlar were also consistent. In other cases, we found significant backbone torsional biases in some of the force fields when paired with GB models: ff99 had a strong tendency toward helical conformations, and while ff99SB was better, it still failed to predict the correct secondary structure of one peptide. These biases are likely due to a combination of force field and implicit solvation effects.
We also found that solvent models differed in the strength of ion-pairing interactions. Furthermore, even in the best-performing cases, we found that salt bridges could stabilize non-native contacts in the protein G C-terminal hairpin and the helical EK peptide. We introduced “salt-bridge-free sampling” to assess ion-pairing contributions to stability. We found that without these interactions the native peptide structures largely remained stable. Thus, the ion-pairing interactions in these peptides are not critical to maintaining the stability of the native structure, at least in the finite simulation time used.
Force fields and solvent models have well-known flaws. Is there a cancellation of errors in the combination of ff96 with igb5, whereby β-propensity in the force field is compensated by α-propensity in the implicit solvent? This is indeed a possibility. Since the focus of our study is force fields paired with implicit solvents, we have not performed comparisons with explicit simulations, and thus cannot directly comment on the corresponding performance of each force field in particular explicit water models. In the context of implicit models exclusively, however, there are some consistent features. For example, igb7 almost always destabilized the structures of each force field, while ff99 had a strong helical bias despite the solvation model used. On the other hand, igb5 and ff96 consistently found native or near-native structure when paired with multiple force fields or solvents, respectively. It is also interesting to note that other studies have shown balance in ff96: Okamoto et al. found ff96 to be balanced among the secondary structure types, using a different solvent model,17 and Zhou found that ff96 best reproduces the free energy surface of the protein G hairpin in comparing implicit to explicit solvation.22
In summary, we find that the AMBER ff96 force field and the implicit solvation model of Onufriev, Bashford, and Case best captures the correct balance of helical and β-hairpin tendencies of a few small peptides, of the AMBER force fields and implicit solvation models. We believe it may provide a starting point for predicting native protein structures and other properties of peptides and small globular proteins.
Acknowledgments
We thank Albert Wu for helpful discussions and for sharing with us his earlier force field tests, and Vincent Voelz and Banu Ozkan for helpful suggestions and ideas. We also thank John Chodera for providing references and helpful commentary. We appreciate the support of NIH Grants GM34993 and GM63592, and of the Air Force Materials Research Lab at Wright-Patterson Air Force Base, USAF-5408-04-SC-0003. This investigation was supported in part by the National Institutes of Health under the Ruth L. Kirschstein National Research Service Award GM08284. Its contents are solely the responsibility of the authors and do not necessarily represent the official view of the NIH.
Contributor Information
M. Scott Shell, Department of Chemical Engineering, University of California Santa Barbara, Santa Barbara, California 93106-5080.
Ryan Ritterson, Graduate Group in Biophysics, University of California San Francisco, San Francisco, California 94143.
Ken A. Dill, Department of Pharmaceutical Chemistry, University of California San Francisco, San Francisco, California 94143
References and Notes
- 1.Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, Karplus M. J Comput Chem. 1983;4:187. [Google Scholar]
- 2.Pearlman DA, Case DA, Caldwell JW, Ross WS, Cheatham TE, III, Debolt S, Ferguson D, Seibel G, Kollman P. Comput Phys Commun. 1995;91:1. [Google Scholar]
- 3.Case DA, Cheatham TE, Darden T, Gohlke H, Luo R, Merz KM, Onufriev A, Simmerling C, Wang B, Woods RJ. J Comput Chem. 2005;26:1668. doi: 10.1002/jcc.20290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jorgensen WL, Tirado-Rives J. J Am Chem Soc. 1988;110:1657. doi: 10.1021/ja00214a001. [DOI] [PubMed] [Google Scholar]
- 5.Duan Y, Kollman PA. Science. 1998;282:740. doi: 10.1126/science.282.5389.740. [DOI] [PubMed] [Google Scholar]
- 6.Vila JA, Ripoll DR, Scheraga HA. Proc Natl Acad Sci USA. 2003;100:14812. doi: 10.1073/pnas.2436463100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zagrovic B, Snow CD, Shirts MR, Pande VS. J Mol Biol. 2002;323:927. doi: 10.1016/s0022-2836(02)00997-x. [DOI] [PubMed] [Google Scholar]
- 8.Jayachandran G, Vishal V, Pande VS. J Chem Phys. 2006;124:164902. doi: 10.1063/1.2186317. [DOI] [PubMed] [Google Scholar]
- 9.Lei H, Duan Y. J Mol Biol. 2007;370:196. doi: 10.1016/j.jmb.2007.04.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lei H, Wu C, Liu H, Duan Y. Proc Natl Acad Sci USA. 2007;104:4925. doi: 10.1073/pnas.0608432104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Simmerling C, Strockbine B, Roitberg AE. J Am Chem Soc. 2002;124:11258. doi: 10.1021/ja0273851. [DOI] [PubMed] [Google Scholar]
- 12.Pitera JW, Swope W. Proc Natl Acad Sci USA. 2003;100:7587. doi: 10.1073/pnas.1330954100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chowdhury S, Lee MC, Xiong G, Duan Y. J Mol Biol. 2003;327:711. doi: 10.1016/s0022-2836(03)00177-3. [DOI] [PubMed] [Google Scholar]
- 14.Lei H, Duan Y. J Phys Chem B. 2007;111:5458. doi: 10.1021/jp0704867. [DOI] [PubMed] [Google Scholar]
- 15.Ozkan SB, Wu GHA, Chodera JD, Dill KA. Proc Natl Acad Sci USA. 2007;104:11987. doi: 10.1073/pnas.0703700104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gnanakaran S, Garcia AE. J Phys Chem B. 2003;107:12555. [Google Scholar]
- 17.Yoda T, Sugita Y, Okamoto Y. Chem Phys Lett. 2004;386:460. [Google Scholar]
- 18.Yoda T, Sugita Y, Okamoto Y. Chem Phys. 2004;307:269. [Google Scholar]
- 19.Sakae Y, Okamoto Y. Chem Phys Lett. 2003;382:626. [Google Scholar]
- 20.Sakae Y, Okamoto Y. J Theor Comput Chem. 2004;3:339. [Google Scholar]
- 21.Sakae Y, Okamoto Y. J Theor Comput Chem. 2004;3:359. [Google Scholar]
- 22.Zhou R. Proteins. 2003;53:148. doi: 10.1002/prot.10483. [DOI] [PubMed] [Google Scholar]
- 23.Felts AK, Harano Y, Gallicchio E, Levy RM. Proteins. 2004;56:310. doi: 10.1002/prot.20104. [DOI] [PubMed] [Google Scholar]
- 24.Lwin TZ, Luo R. Protein Sci. 2006;15:2642. doi: 10.1110/ps.062438006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lwin TZ, Zhong R, Luo R. J Chem Phys. 2006;124:034902. doi: 10.1063/1.2161202. [DOI] [PubMed] [Google Scholar]
- 26.Blanco FJ, Rivas G, Serrano L. Nat Struct Biol. 1994;1:584. doi: 10.1038/nsb0994-584. [DOI] [PubMed] [Google Scholar]
- 27.Scholtz JM, Barrick D, York EJ, Stewart JM, Baldwin RL. Proc Natl Acad Sci USA. 1995;92:185. doi: 10.1073/pnas.92.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bierzynski A, Kim PS, Baldwin RL. Proc Natl Acad Sci USA. 1982;79:2470. doi: 10.1073/pnas.79.8.2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cochran AG, Skelton NJ, Starovasnik MA. Proc Natl Acad Sci USA. 2001;98:5578. doi: 10.1073/pnas.091100898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cornell WD, Cieplak P, Bayly CI, Gould IR, Merz KM, Ferguson DM, Spellmeyer DC, Fox T, Caldwell JW, Kollman PA. J Am Chem Soc. 1995;117:5179. [Google Scholar]
- 31.Kollman PA, Dixon R, Cornell W, Fox T, Chipot C, Pohorille A. The development/application of “minimalist” organic/biochemical molecular mechanic force field using a combination of ab initio calculations and experimental data. In: Van Gunsteren WF, Weiner PK, editors. Computer Simulation of Biomolecular Systems. Escom; Dodrecht, Netherlands: 1997. pp. 83–96. [Google Scholar]
- 32.Wang J, Cieplak P, Kollman PA. J Comput Chem. 2000;21:1049. [Google Scholar]
- 33.Hornak V, Abel R, Okur A, Strockbine B, Roitberg A, Simmerling C. Proteins. 2006;65:712. doi: 10.1002/prot.21123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Duan Y, Wu C, Chowdhury S, Lee MC, Xiong G, Zhang W, Yang R, Cieplak P, Luo R, Lee T. J Comput Chem. 2003;24:1999. doi: 10.1002/jcc.10349. [DOI] [PubMed] [Google Scholar]
- 35.Eisenberg D, McLachlan AD. Nature. 1986;319:199. doi: 10.1038/319199a0. [DOI] [PubMed] [Google Scholar]
- 36.Wagoner JA, Baker NA. Proc Natl Acad Sci USA. 2006;103:8331. doi: 10.1073/pnas.0600118103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Still WC, Tempczyk A, Hawley RC, Hendrickson T. J Am Chem Soc. 1990;112:6127. [Google Scholar]
- 38.Onufriev A, Case DA, Bashford D. J Comput Chem. 2002;23:1297. doi: 10.1002/jcc.10126. [DOI] [PubMed] [Google Scholar]
- 39.Onufriev A, Bashford D, Case DA. J Phys Chem B. 2000;104:37112. [Google Scholar]
- 40.Bashford D, Case DA. Annu Rev Phys Chem. 2000;51:129. doi: 10.1146/annurev.physchem.51.1.129. [DOI] [PubMed] [Google Scholar]
- 41.Hawkins GD, Cramer CJ, Truhlar DG. Chem Phys Lett. 1995;246:122. [Google Scholar]
- 42.Tsui V, Case DA. Biopolymers. 2001;56:275. doi: 10.1002/1097-0282(2000)56:4<275::AID-BIP10024>3.0.CO;2-E. [DOI] [PubMed] [Google Scholar]
- 43.Onufriev A, Bashford D, Case DA. Proteins. 2004;55:383. doi: 10.1002/prot.20033. [DOI] [PubMed] [Google Scholar]
- 44.Feig M, Onufriev A, Lee MS, Im W, Case DA, Brooks CL. J Comput Chem. 2004;25:265. doi: 10.1002/jcc.10378. [DOI] [PubMed] [Google Scholar]
- 45.Mongan J, Simmerling CL, McCammon JA, Case DA, Onufriev A. J Chem Theory Comput. 2007;3:156. doi: 10.1021/ct600085e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sugita Y, Okamoto Y. Chem Phys Lett. 1999;314:141. [Google Scholar]
- 47.Chodera J, Swope WC, Pitera JW, Dill KA. Multiscale Model Simul. 2006;5:1214. [Google Scholar]
- 48.Humphrey W, Dalke A, Schulten K. J Mol Graphics. 1996;14:33. doi: 10.1016/0263-7855(96)00018-5. [DOI] [PubMed] [Google Scholar]
- 49.Kumar S, Bouzida D, Swendsen RH, Kollman PA, Rosenberg JM. J Comput Chem. 1992;13:1011. [Google Scholar]
- 50.Garcia AE, Sanbonmatsu KY. Proc Natl Acad Sci USA. 2002;99:2782. doi: 10.1073/pnas.042496899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou R, Berne BJ. Proc Natl Acad Sci USA. 2002;99:12777. doi: 10.1073/pnas.142430099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Geney R, Layten M, Gomperts R, Hornak V, Simmerling C. J Chem Theory Comput. 2006;2:115. doi: 10.1021/ct050183l. [DOI] [PubMed] [Google Scholar]