

Abstract
Protonated versions of N-heterocyclic carbenes (NHC,H+) are classically prepared by closing the ring through the introduction of the CH+ fragment. He we report a totally different synthetic approach, which can be viewed as the addition of a 1,3-diazaallyl anion to a compound featuring two leaving groups (hereafter named “di-electrophile”). Using 1,3- and 1,4-dibromides, six and seven membered NHC,H+s have been prepared in good yields. Similarly, with 1,3,2-dioxathiolane-2,2-dioxide as a di-electrophile, imidazolidinium salts were obtained. To illustrate its broad scope of application, this synthetic route has been expanded to the preparation of protonated cyclic amino alkyl carbenes (CAACs) and amino thio carbenes, using 1-aza-allyl and 1,3-azathio-allyl anions, respectively.
1. Introduction
The chemistry of N-heterocyclic carbenes (NHCs) has become a major area of research as these stable carbenes [1] have proven to be outstanding ligands for transition metals [2], as well as potent nucleophilic organic catalysts [3]. In this field, the conjugate acids (NHC,H+s) play a very important role. Indeed, they are by far the most frequently used precursors of NHCs, and oxidative addition of the CH bond to transition metal centers can even directly afford the carbene complexes [4]. Moreover, NHC,H+s, have found numerous applications as ionic liquids [5], which are important components of “Green” chemistry. Imidazolium A, imidazolidinium B, tetrahydropyrimidinium C, and related compounds are usually prepared by closing the ring through the introduction of the CH+ fragment, as shown by the disconnection (a) [1–3] (Scheme 1).
Scheme 1.

Recently, we have reported the synthesis of P-heterocyclic carbenes (PHCs) [6] and cyclic alkyl amino carbenes (CAACs) [7] by deprotonation of the corresponding conjugate acids D and E (Schemes 2 and 3), respectively. These cationic heterocycles were prepared using retrosynthetic approaches that are significantly different from those classically used for NHC precursors A–C. In the case of PHCs, we performed a formal [3+2]-cycloaddition involving a 1,3-diphosphaallyl cation and a dipolarophile (Scheme 2).
Scheme 2.
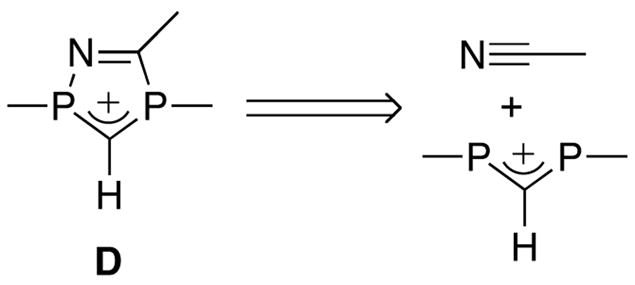
Scheme 3.

The applicability of this route is rather limited, since very few hetero-allyl cations are available. Potentially much broader in scope is the pathway used for CAAC precursors E, in which the heterocycles are formed by addition of an aza-allyl anion to an epoxide, followed by the transformation of the resulting alkoxide into a leaving group, and subsequent ring closure by intramolecular nucleophilic substitution (Scheme 3). Indeed, this approach can be viewed as the addition of a (di)hetero-allyl anion to a compound featuring two leaving groups (hereafter named “di-electrophile”) (Scheme 3). Here we report the synthesis of a variety of protonated NHCs and related compounds, which illustrates the broad scope of application of this synthetic approach.
2. Results and discussion
We first extrapolated our method to the synthesis of new CAAC precursors. Aldimines 1 featuring a secondary alkyl substituent at carbon are readily available from aldehydes and amines. Deprotonation leads to the corresponding aza-allyl anions, which readily react at room temperature with 1,3-dibromopropane to directly afford the desired cationic heterocycles 3a–c. If 1,3-dibromobutane is used as a di-electrophile, the primary formed neutral acyclic adducts 2d,e are observed, and can even be isolated. Heating at 110°C for one hour is necessary to induce the ring closure and cleanly obtain cyclic salts 3d,e, presumably due to steric hindrance (Scheme 4).
Scheme 4.

We then investigated the possibility of preparing classical NHC,H+s. We chose to use a single formamidine, namely the dimesityl formamidine [8a], since the substitution pattern on this fragment should have no influence on the fate of the reaction; note that formamidines are readily available in large quantities [8]. On the other hand, to illustrate the versatility of our synthetic approach, we tested several different di-electrophiles (Schemes 5 and 6). Neat 1,3-dibromopropane was added to a tetrahydrofuran solution of the lithium salt of the formamidine, and after stirring overnight at room temperature, the tetrahydropyrimidinium bromide 4, was isolated in 50% yield (without optimization). Note that Herrmann et al. reported a 23% yield when this compound was prepared by the classical route [9]. The reaction is not limited to 1,3-dibromides, 1,4-dibromides such as α,α′-dibromo-o-xylene can be used. In this case, the seven-membered cationic heterocycle 5 was isolated in 93% yield. Since examples of medium size, cationic heterocycles are quite rare, a single crystal X-ray diffraction study was carried out. The ORTEP view of the molecule, along with selected geometric parameters, is shown in Figure 1.
Scheme 5.

Scheme 6.

Figure 1.

Molecular view of the crystal structure of 5. Selected bond lengths [Å] and angles [°]: C1-N1 1.314(2), N1-C2 1.498(2), C2-C3 1.498(3), C3-C8 1.400(2), C8-C9 1.509(2), C9-N2 1.494(2), N2-C1 1.325(2); N1-C1-N2 127.50(14).
Among the most popular NHCs are certainly those derived from imidazolidinium salts. These five-membered heterocycles are not available by addition of 1,2-dibromides to diaza-allyl anions. Indeed, either HBr-elimination reactions occur, or in the case of per-substituted 1,2-dibromide derivatives, the steric hindrance prevents the substitution reaction. However, the 1,3,2-dioxathiolane-2,2-dioxide readily acts as the desired di-electrophile. At room temperature, the addition of the lithium salt of the formamidine to the cyclic sulfate cleanly led after 16 hours to adduct 6, which has been characterized by NMR spectroscopy. The cyclization reaction to 7 required an additional 24 hours in refluxing THF. After work up, imidazolium salt 7 was isolated in 93% yield (Scheme 6). The counteranion can be easily exchanged using, for example, tetraphenyl borate.
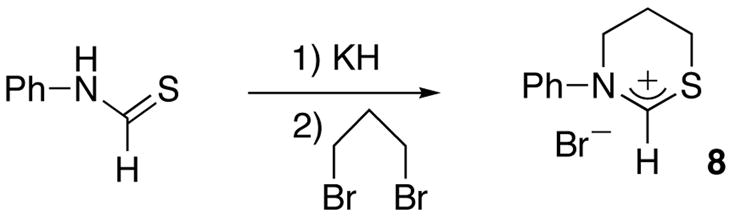
To demonstrate even further the broad applicability of our synthetic approach, we reacted the potassium salt of N-Phenyl thioamide [10] with 1,3-dibromopropane. Here again, the cyclization occurred at room temperature and after work up and recrystallization, heterocycle 8 was isolated in 39% yield (Scheme 7).
Scheme 7.
3. Conclusions
The synthetic strategy described here appears to be very general. Not only can the substituents of the starting mono- and dihetero allyl anions be modified at will, but a variety of di-electrophiles are available, including enantiomerically pure variants. Importantly, the synthesis of almost all ring-size, protonated heterocyclic carbenes should be reachable, which would allow for significant tuning of the carbene bond angle, and other stereoelectronic parameters of importance for the catalytic activity of the ensuing transition metal complexes.
4. Experimental section
General
All manipulations were performed under an inert atmosphere of argon using standard Schlenk techniques. Dry, oxygen-free solvents were employed. 1H, and 13C NMR spectra were recorded on Varian Inova 300, 500 and Bruker Avance 300 spectrometers.
Synthesis of heterocyclic salts 3a–e
1.02 eq. of the desired imine 1[11] was added at room temperature to a diethyl ether solution of LDA (10 mL of ether for every gram of LDA). After stirring for 2 h at room temperature the solvent and diisopropylamine were removed under vacuum. The lithium salts of the aza-allyl anions were obtained as thermally stable viscous oils, and used for the next step without further purification. The salts were dissolved in diethyl ether and 1.02 eq. of 1,3-dibromopropane was added via syringe at room temperature. After stirring for 16 h at room temperature and 2h at 40°C, the solvent was removed under vacuum. After extraction with CH2Cl2, and evaporation of the solvent, the residue was washed with ether and then hexane to give cyclic iminium salts 3a–c as white solids. A similar procedure was carried out for derivatives 3d,e, except that 1,3-dibromobutane was used as the di-electrophile, and high temperature (110°C, 1 hour) was required for cyclization. Derivatives 3d,e were also obtained as white solids. 3a: (73% yield); 1H NMR (CDCl3, 25°C, 300 MHz) δ = 8.96 (s, 1H, NCH), 3.93 (m, 2H, CH2), 1.98 (m, 2H, CH2), 1.74 (m, 2H, CH2), 1.64 (s, 9H, CH3), 1.44 (s, 6H, CH3); 13C{1H} NMR (CD3CN, 25°C, 75 MHz) δ = 181.39 (NCH), 71.00 (C), 48.79 (CH2), 38.52 (C), 31.35 (CH2), 27.97 (CH3), 26.15 (CH3), 20.05 (CH2). 3b: (65% yield); 1H NMR (CDCl3, 25°C, 300 MHz) δ = 9.54 (s, 1H, NCH), 4.80 (sept, J = 6.9, 1H, CH), 3.73 (m, 2H, CH2), 1.98 (m, 2H, CH2), 1.71 (m, 2H, CH2), 1.49 (d, J = 6.9, 6H, CH3), 1.39 (s, 6H, CH3); 13C{1H} NMR (CDCl3, 25°C, 75 MHz) δ = 182.93 (NCH), 64.54 (CH), 47.39 (CH2), 37.16 (C), 31.01 (CH2), 26.56 (CH3), 20.78 (CH3), 18.89 (CH2). 3c: (70% yield); 1H NMR (CD3CN, 25°C, 300 MHz) δ = 9.18 (s, 1H, NCH), 7.14 (s, 2H, Ph), 4.00 (m, 2H, CH2), 2.36 (s, 3H, p-CH3), 2.28 (s, 6H, o-CH3), 2.19 (m, 2H, CH2), 1.91 (m, 2H, CH2), 1.51 (s, 6H, CH3); 13C{1H} NMR (CD3CN, 25°C, 75 MHz) δ = 190.72 (NCH), 141.97 (C), 140.15 (C), 132.38 (C), 130.87 (CH), 66.18 (C), 55.34 (CH2), 30.30 (CH2), 25.85 (CH3), 21.02 (CH3), 19.32 (CH2) 17.44 (CH3). 3d: (61% yield); 1H NMR (CDCl3, 25°C, 300 MHz) δ = 9.41 (s, 1H, NCH), 4.49 (m, 1H, CH), 2.00-1.60 (m, 4H, CH2), 1.66 (s, 9H, CH3), 1.49 (s, 3H, CH3), 1.39 (d, J = 6.6, 3H, CH3), 1.37 (s, 3H, CH3); 13C{1H} NMR (CDCl3, 25°C, 75 MHz) δ = 183.33 (NCH), 70.30 (C), 53.23 (CH), 38.19 (C), 29.50 (CH3), 26.94 (CH2), 26.88 (CH2), 26.74 (CH3), 26.31 (CH3), 21.46 (CH3). 3e: (70% yield) 1H NMR (CDCl3, 25°C, 300 MHz) δ = 9.54 (s, 1H, NCH), 7.30 (t, 1H, J = 7.8, Ph), 7.13-7.10 (2 d, J = 7.8, 2H, Ph), 4.09 (m, 1H, CH), 2.47 (sept, J = 6.7, 1H, CH), 2.33 (sept, J = 6.7, 1H, CH), 2.26-1.65 (m, 4H, CH2), 1.45 (s, 3H, CH3), 1.44 (s, 3H, CH3), 1.25-1.00 (m, 15H); 13C{1H} NMR (CDCl3, 25°C, 75 MHz) δ = 190.85 (NCH), 142.68 (C), 142.10 (C), 136.24 (C), 131.56 (CH), 125.29 (CH), 124.97 (CH), 62.63 (CH), 38.49 (C), 29.04, 28.99, 28.50 (CH2), 25.97 (CH2), 25.77, 25.60, 25.30, 23.04, 22.56, 17.80.
Synthesis of six-membered heterocyclic salts 4
n-BuLi (2.5 M in hexane, 2.90 mL, 7.25 mmol) was added at room temperature to a THF solution (80 mL) of dimesityl formamidine (2.00 g, 7.13 mmol). The resulting yellow solution was stirred at room temperature for 20 min before 1,3-dibromopropane (1.08 mL, 1.5 eq.) in THF (80 mL) was added dropwise at room temperature. The pale brownish solution was stirred at room temperature overnight. After evaporation of the solvent under vacuum, the resulting viscous oil was washed with hexane (60 mL) and Et2O (60 mL). The residue was dissolved in CH2Cl2 (80 mL), and filtered. After evaporation of the solvent 4(Br−) was obtained as a white powder (1.43 g, 50%): 1H NMR (CDCl3, 25°C, 300 MHz) δ = 7.55 (s, 1H, NCH), 6.97 (m, 4H, Ph), 4.30 (br t, J = 5.7, 4H, NCH2), 2.62 (m, J = 5.7, 2H, CH2), 2.38 (s, 12H, o-CH3), 2.30 (s, 6H, p-CH3). A mixture of 4(Br−) (1.34 g, 3.33 mmol) and NaBPh4 (1.20 g, 3.50 mmol) in CH2Cl2 (30 mL) was stirred overnight. After filtration through celite, the solvent was removed to afford a pale brown solid. Recrystallization from CH2Cl2/Et2O yielded 4(BPh4−) as a white solid (1.95 g, 91%): 1H NMR (CDCl3, 25°C, 300 MHz) δ = 7.41 (m, 8H, BPh4), 7.06 (s, 1H, NCH), 6.93 (m, 12H, BPh4), 6.82 (m, 4H, Ph), 2.69 (t, J = 5.6, 4H, NCH2), 2.31 (s, 6H, p-CH3), 2.07 (s, 12H, o-CH3), 1.42 (m, J = 5.6, 2H, CH2); 13C{1H} NMR (THF-D8, 25°C, 75 MHz) δ = 165.35 (q, J = 49.4, ipso-BPh4), 156.95 (NCH), 141.18, 137.83, 137.18, 135.42, 130.75, 125.84, 121.96, 46.72, 21.06, 19.93, 17.71. Mass(FAB) m/z: 321 (M-BPh4)+.
Synthesis of seven-membered heterocyclic salt 5
n-BuLi (2.5 M in hexane, 1.50 mL, 3.75 mmol) was added at room temperature to a THF solution (50 mL) of dimesityl formamidine (1.00 g, 3.57 mmol). The resulting clear yellow solution was stirred at room temperature for 20 min before adding dropwise a THF solution (50 mL) of α,α′-dibromo-o-xylene (1.41 g, 1.5 eq.). The pale yellow solution was stirred overnight at room temperature. The solvent was removed under vacuum and the residue was washed with hexane (80 mL) and Et2O (60 mL). Recrystallization from a CH2Cl2/Et2O/hexane solution afforded 5 as colorless crystals (1.54 g, 93%): 1H NMR (CD3CN, 25°C, 300 MHz) δ = 7.58 (s, 1H, NCH), 7.52 (m, 2H, Ph), 7.45 (m, 2H, Ph), 7.03 (s, 4H, Ph), 5.41 (m, 4H, NCH2), 2.28 (s, 6H, p-CH3), 2.24 (s, 12H, o-CH3). 13C{1H} NMR (CD3CN, 25°C, 75 MHz) δ = 158.84 (NCH), 141.56, 141.37, 136.15, 135.75, 131.59, 131.09, 130.69, 57.31 (CH2), 21.38 (p-CH3), 18.97 (o-CH3); Mass(FAB) m/z: 383(M-Br)+.
Synthesis of imidazolidinium salts 7
n-BuLi (2.5 M in hexane, 0.57 mL, 1.44 mmol) was added at room temperature to a THF solution (8 mL) of dimesityl formamidine (0.40 g, 1.42 mmol) and the yellow solution was stirred for 20 min. Then, 1,3,2-dioxathiolane 2,2-dioxide (0.18 g, 1.43 mmol) in THF (5 mL) was added dropwise at room temperature and the solution turned colorless. The reaction mixture was stirred overnight and NMR indicated the clean formation of 6. 13C{1H} NMR (THF-D8, 25°C, 75 MHz) δ = 153.49 (NCH), 130.21 (CH), 129.14 (CH), 65.28 (SO4CH2), 49.26 (NCH2), 19.31 (CH3), 21.04 (CH3), 19.31 (CH3), 18.80 (CH3). The reaction mixture was dissolved in 10 mL of THF and refluxed for 24 h. Removal of the solvent under vacuum and washing with hexane (30 mL) and Et2O (30 mL) afforded 7(LiSO4−) as a white solid (0.54 g, 93%). 1H NMR (CDCl3, 25°C, 300 MHz) δ = 8.17 (s, 1H, NCH), 6.92 (s, 4H, Ph), 4.48 (s, 4H, NCH2), 2.31 (s, 12H, o-CH3), 2.28 (s, 6H, p-CH3); 13C{1H} (THF-D8, 25°C, 75 MHz) δ = 159.54 (NCH), 140.76, 136.80, 135.28, 130.22, 52.04 (NCH2), 21.23 (CH3), 17.89 (CH3). Anion exchange: A suspension of 7(LiSO4−) (0.54 g, 1.32 mmol) and NaBPh4 (0.45 g, 1.32 mmol) in CH2Cl2 (35 mL) was stirred overnight at room temperature. Solids were removed by filtration through celite to give a pale yellow solution. Removal of the solvent afforded 7(BPh4−) as a pale yellow solid (0.69 g, 77%). 1H NMR (CDCl3, 25°C, 300MHz) δ = 7.22 (m, 8H, BPh4), 6.94 (s, 4H, Ph), 6.83 (m, 12H, BPh4), 6.21 (s, 1H, NCH), 3.01 (s, 4H, NCH2), 2.35 (s, 6H, p-CH3), 2.03 (s, 12H, o-CH3); 13C{1H} NMR (CDCl3, 25°C, 75 MHz) δ = 164.10 (q, J = 49.5, ipso-BPh4), 157.60 (NCH), 140.94, 135.95, 134.48, 130.07, 129.56, 125.54, 121.70, 50.86 (NCH2), 21.14 (p-CH3), 17.55 (o-CH3). Mass(FAB) m/z: 307 (M-BPh4)+.
Synthesis of heterocyclic salt 8
A THF solution (25 mL) of phenyl thioamide (0.40 g, 2.91 mmol) was added dropwise at room temperature to a suspension of KH (0.12 g, 1.05 eq.) in THF (5 ml). The pale yellow suspension was stirred for 1.5 h and 1,3-dibromopropane (0.65 g, 3.21 mmol) was added. After stirring for 2 days, the solvent was removed under vacuum. The yellow residue was washed with hexane (30 ml) and ether (20 ml), and then filtered through celite and recrystallized from a CH2Cl2/Et2O solution, yielding 8 as white crystals (0.29 g, 39 %). 1H NMR (CDCl3, 25°C, 300 MHz) δ = 10.35 (s, 1H, NCH), 7.69 (m, 2H, Ph), 7.48 (m, 3H, Ph), 4.55 (t, J = 5.6, 2H, NCH2), 3.62 (t, J = 5.6, 2H, SCH2), 2.54 (m, 2H, CH2); 13C{1H} NMR (CDCl3, 25°C, 75 MHz) δ = 175.43 (NCH), 145.17, 130.66, 130.54, 122.84, 51.81 (NCH2), 26.36 (SCH2), 20.51 (CH2); Mass (FAB) m/z: 178 (M-Br)+.
Crystal structure determination of compound 5
The Bruker X8-APEX [12a] X-ray diffraction instrument with Mo-radiation was used for data collection of the compound 5. All data frames were collected at low temperatures (T = 100 K) using an ω ϕ-scan mode, and integrated using a Bruker SAINTPLUS software package [12b]. The intensity data were corrected for Lorentzian polarization. Absorption corrections were performed using the SADABS program. The Bruker SHELXTL software package [12c] was used for direct methods of phase determination and structure refinement. Atomic coordinates, isotropic and anisotropic displacement parameters of all the non-hydrogen atoms of two compounds were refined by means of a full matrix least-squares procedure on F2. All H-atoms were included in the refinement in calculated positions riding on the atoms to which they were attached. Drawing was generated using Ortep3 [12d]. Crystal and structure parameters of 5: size 0.54 × 0.40 × 0.08 mm3, orthorhombic, space group Pca2(1), a = 16.4966(18) Å, b = 11.6143(13) Å, c = 14.1552(15) Å, α = β = γ = 90°, V = 2712.1(5)Å3, ρcalcd = 1.343 g/cm3, 2θmax = 57.20°, Mo-radiation (λ = 0.71073 Å), low temperature = 100(2)° K, reflections collected = 19811, independent reflections = 6156 (Rint = 0.0246, Rsig = 0.0385), 5625 (91.4%) reflections were greater than 2σ(I), index ranges −21<=h<=19, −15<=k<=15, −18<=l<=18, absorption coefficient μ = 1.731 mm−1; max/min transmission = 0.8739 and 0.4549, 304 parameters were refined and converged R1 = 0.0247, wR2 = 0.0614, with intensity I>2σ(I), the final difference map was 0.331 and −0.242 e.Å−3. Structural data for compound 5 have been deposited in the Cambridge Crystallographic Data Center under CCDC 603563, and can be obtained free of charge at www.ccdc.cam.ac.uk/conts/retrieving.html.
Acknowledgments
Thanks are due to the NIH (R01 GM 68825) and RHODIA for financial support of this work.
References
- 1.For reviews on stable singlet carbenes: a) Hahn FE. Angew Chem Int Ed. 2006;45:1348. doi: 10.1002/anie.200503858.Kirmse W. Angew Chem Int Ed. 2004;43:1767. doi: 10.1002/anie.200301729.Alder RW, Blake ME, Chaker ME, Harvey JN, Paolini F, Schütz J. Angew Chem Int Ed. 2004;43:5896. doi: 10.1002/anie.200400654.Canac Y, Soleilhavoup M, Conejero S, Bertrand G. J Organomet Chem. 2004;689:3857.Bourissou D, Guerret O, Gabbaï FP, Bertrand G. Chem Rev. 2000;100:39. doi: 10.1021/cr940472u.
- 2.For reviews on stable carbenes as ligands for transition metal catalysts: a) Scott NM, Nolan SP. Eur J Inorg Chem. 2005:1815.Peris E, Crabtree RH. Coord Chem Rev. 2004;248:2239.Crudden CM, Allen DP. Coord Chem Rev. 2004;248:2247.César V, Bellemin-Laponnaz S, Gade LH. Chem Soc Rev. 2004;33:619. doi: 10.1039/b406802p.Herrmann WA. Angew Chem Int Ed. 2002;41:1290. doi: 10.1002/1521-3773(20020415)41:8<1290::aid-anie1290>3.0.co;2-y.
- 3.For recent reviews on stable carbenes as organic catalysts: a) Enders D, Balensiefer T. Acc Chem Res. 2004;37:534. doi: 10.1021/ar030050j.Nair V, Bindu S, Sreekumar V. Angew Chem Int Ed. 2004;43:5130. doi: 10.1002/anie.200301714.Johnson JS. Angew Chem Int Ed. 2004;43:1326. doi: 10.1002/anie.200301702.
- 4.For recent examples for the formation of metal–carbene complexes by oxidative insertion into the CH bond of imidazolium salts see: a) Grundemann S, Albrecht M, Kovacevic A, Faller JW, Crabtree RH. J Chem Soc, Dalton Trans. 2002:2163. doi: 10.1021/ja026735g.McGuiness DS, Cavell KJ, Yates BF, Skelton BW, White AH. J Am Chem Soc. 2001;123:8317. doi: 10.1021/ja010628p.Ho VM, Watson LA, Huffmann JC, Caulton KG. New J Chem. 2003;27:1446.Clement ND, Cavell KJ, Jones C, Elsevier CJ. Angew Chem Int Ed. 2004;43:1277. doi: 10.1002/anie.200353409.Duin MA, Clement ND, Cavell KJ, Elsevier CJ. Chem Commun. 2003:400. doi: 10.1039/b211235c.Cavell KJ, McGuinness DS. Coord Chem Rev. 2004;248:671.
- 5.a) Fei ZF, Geldbach TJ, Zhao DB, Dyson PJ. Chem Eur J. 2006;12:2123. [Google Scholar]; b) Rangits G, Kollar L. J Mol Cat A. 2006;246:59. [Google Scholar]; c) Lin IJB, Vasam CS. J Organomet Chem. 2004;690:3498. [Google Scholar]
- 6.Martin D, Baceiredo A, Gornitzka H, Schoeller WW, Bertrand G. Angew Chem Int Ed. 2005;44:1700. doi: 10.1002/anie.200462239. [DOI] [PubMed] [Google Scholar]
- 7.a) Lavallo V, Mafhouz J, Canac Y, Donnadieu B, Schoeller WW, Bertrand G. J Am Chem Soc. 2004;126:8670. doi: 10.1021/ja047503f. [DOI] [PubMed] [Google Scholar]; b) Lavallo V, Canac Y, Prasang C, Donnadieu B, Bertrand G. G Angew Chem Int Ed. 2005;44:5705. doi: 10.1002/anie.200501841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.a) Tamaoki A, Yamomoto K, Maeda K. JP 55049353 Jpn Kokai Tokkyo Koho. 1980; b) Roberts RM. J Org Chem. 1949;14:277. doi: 10.1021/jo01154a013. [DOI] [PubMed] [Google Scholar]; c) Cotton FA, Haefner SC, Matonic JH, Wang X, Murillo CA. Polyhedron. 1997;16:541. [Google Scholar]
- 9.Herrmann WA, Schneider SK, Öfele K, Sakamoto M, Herdtweck E. J Organomet Chem. 2004;689:2441. [Google Scholar]
- 10.Perrin CL, Lollo CP, Hahn CS. J Org Chem. 1985;50:1405. [Google Scholar]
- 11.a) Stevens CV, Peristeropoulou M, De Kimpe N. Tetrahedron. 2001;57:7865. [Google Scholar]; b) Daugulis O, Brookhart M. Organometallics. 2002;21:5926. [Google Scholar]
- 12.a) Bruker. APEX 2 version 1.0–22. Bruker AXS Inc.; Madison, Wisconsin, U.S.A.: 2004. [Google Scholar]; b) Bruker. SAINT version V7.06A. Bruker AXS Inc.; Madison, Wisconsin, USA: 2003. [Google Scholar]; c) SHELXTL, version 6.14, Bruker. Bruker AXS Inc.; Madison, Wisconsin, USA: 2003. [Google Scholar]; ORTEP3 for Windows. Farrugia LJ. J Appl Crystallogr. 1997;30:565. [Google Scholar]