

1. Introduction
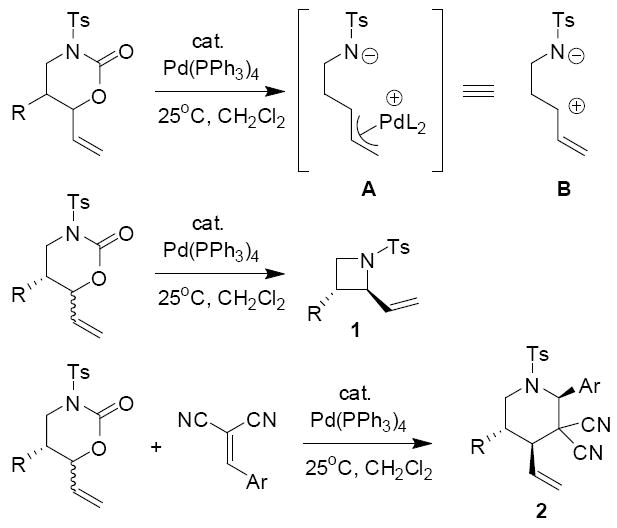
Previously we have shown that palladium catalysts effect the decarboxylation of vinyl oxazinones to form zwitterionic π-allyl palladium intermediates (A, Scheme 1).1 Thus, vinyl oxazinones can be equated with zwitterionic synthons (B). In the absence of other electrophiles, the zwitterionic intermediate cyclizes to form vinyl azetidines with high diastereoselectivity. Moreover, in the presence of suitably electrophilic Michael acceptors,2 the intermediates undergo diastereoselective cycloadditions to generate highly substituted piperidine derivatives.3
Scheme 1.
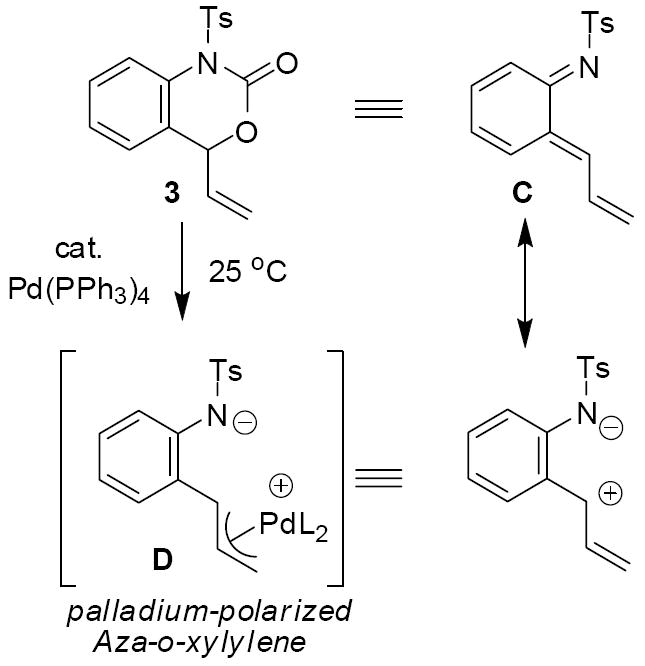
The related vinyl benzoxazinones (3) undergo similar palladium-induced decarboxylation (Scheme 2).4 However, these substrates give rise to aza-ortho-xylylene synthons (C).5 The decarboxylation of vinyl benzoxazinones under conditions of palladium catalysis occurs at 25 °C and is thus significantly milder than thermal decarboxylation of benzoxazinones which typically requires temperatures near 200 °C.6 In addition, the aza-ortho-xylylene equivalents generated in the presence of palladium are more polarized than standard aza-ortho-xylylenes. Thus, while standard aza-ortho-xylylenes preferentially react with electron rich olefins, palladium-polarized aza-ortho-xylylenes (D) prefer to react with electron deficient olefins. Herein, we further describe the cyclization and cycloaddition chemistry of these unique intermediates.
Scheme 2.
2. Decarboxylative macrocycloadditions
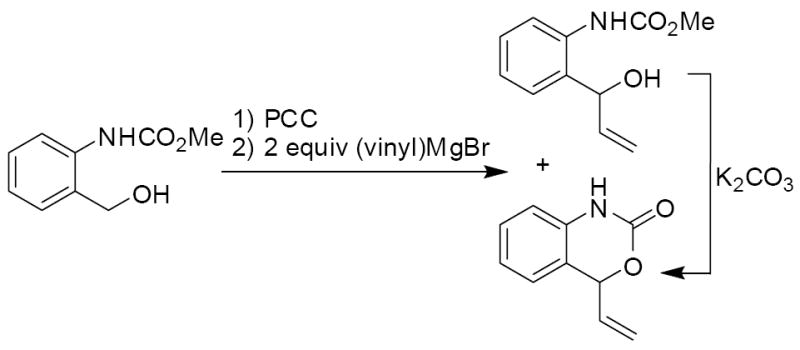
To begin, vinyl benzoxazinone was synthesized from ortho-hydroxymethyl phenylcarbamate (Scheme 3).4 Specifically, the alcohol was oxidized to the aldehyde with PCC on alumina,7 followed by addition of a vinyl Grignard reagent. This procedure often produced the desired benzoxazinone, however the ratio of cyclized and uncyclized carbamate was not always reproducible. Thus, the product of Grignard addition was treated with potassium carbonate to ensure high-yielding cyclization.8 Finally, N-tosylation under standard conditions gave the desired starting material.9 Importantly, this process could be carried out without chromatographic purification of any intermediates.
Scheme 3.
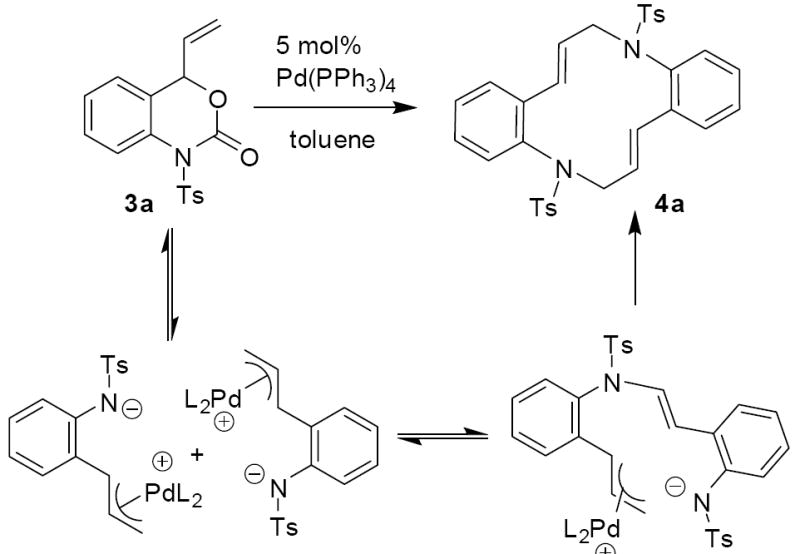
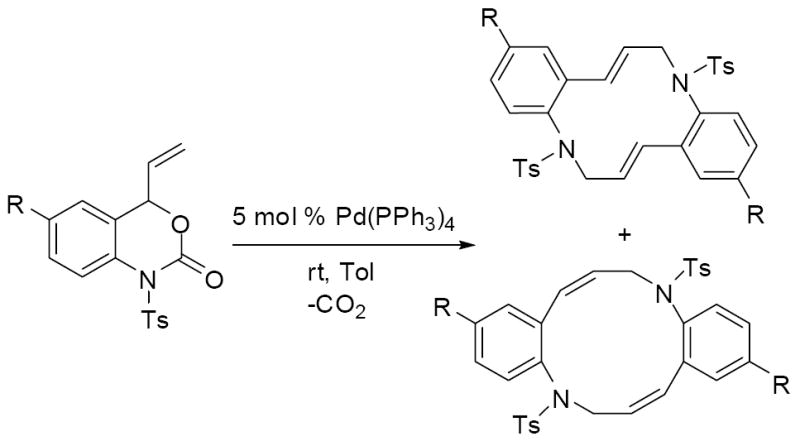
Next, vinyl carbamate 3a, was treated with Pd(PPh3)4 in toluene at room temperature. After 10 minutes, 1H NMR spectroscopic analysis indicated the clean formation of the 12-membered macrocycle 4a, however the product was isolated in only 36% yield (Scheme 4).10 The low isolated yield led us to believe that the zwitterionic intermediate may also form oligomers or polymers under the reaction conditions. It is also important to note that, in contrast to saturated vinyl oxazinones which rapidly form vinyl azetidines under these conditions (Scheme 1),1 the formation of vinyl azetidines is never observed in the reactions of vinyl benzoxazinones.
Scheme 4.
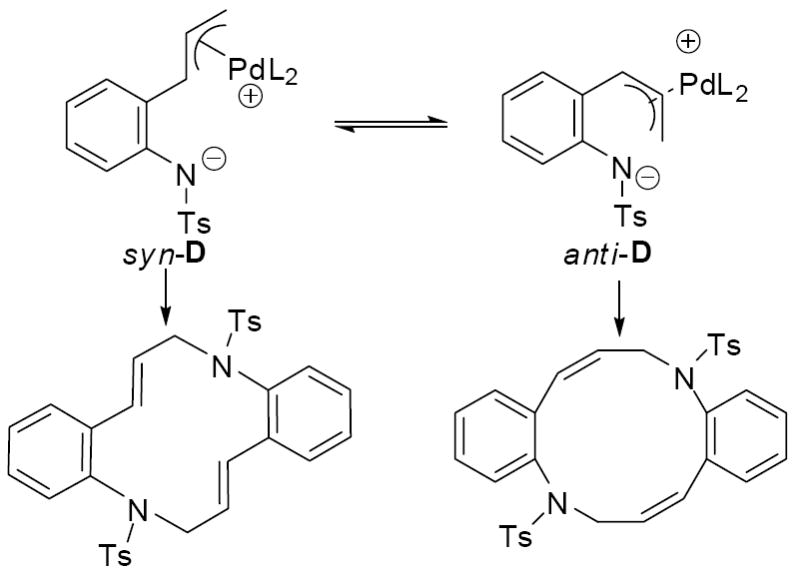
Given the discovery of a rapid, formal [6+6] cycloaddition, we briefly examined the generality of this macrocycloaddition. As can be seen in table 1, addition of groups to the position para to the tosylamide significantly improved the isolated yields of products. To explain this observation, we speculate that the group is blocking electrophilic aromatic substitution that can take place with the protio derivative. Surprisingly, the fluorine-containing derivative did not form any isolable products when allowed to react under analogous conditions in toluene at room temperature. However, switching the solvent to dichloroethane and performing the reaction at 60 °C did allow for the isolation of a high yield of the fluorinated macrocycle. Interestingly, the coupling constant for the olefinic protons (J = 9.8 Hz) of the macrocyclic product (4d) indicated that the product had an exclusively cis-cis geometry whereas the macrocycles obtained from toluene (4a-c) were clearly the trans-trans geometry (J = 16.2 Hz). These results suggested that the conformation of the zwitterionic π-allyl intermediate may be different in different solvents, with the syn-allyl complex being favored in toluene and the anti-allyl complex being more accessible in dichloroethane (Scheme 5).
Table 1.
Formation of macrocycles.
 | |||||
|---|---|---|---|---|---|
| entry | R | conditions | vinyl JHH (Hz) | product | yield% |
| 1 | H | toluene, 25 °C | 16.2 | trans-4a | 36 |
| 2 | Me | toluene, 25 °C | 16.2 | trans-4b | 87 |
| 3 | OMe | toluene, 25 °C | 16.2 | trans-4c | 94 |
| 4 | F | ClCH2CH2Cl, 60 °C | 9.8 | cis-4d | 92 |
Scheme 5.
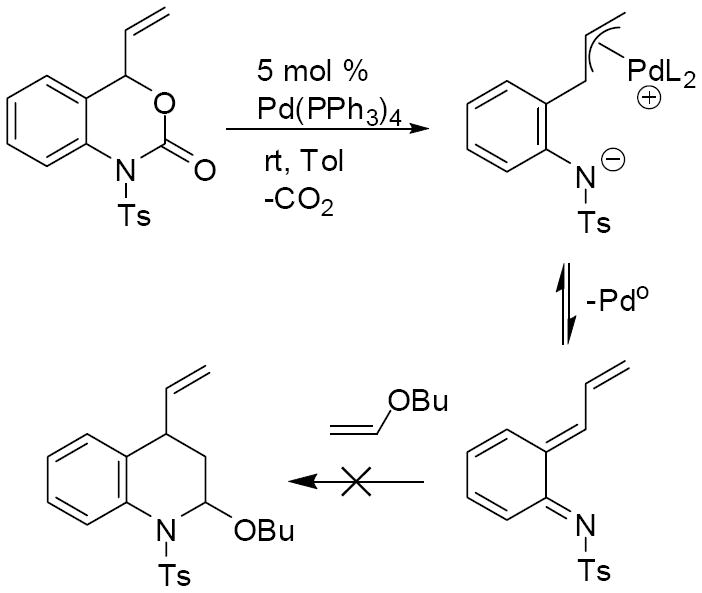
Next, in an attempt to test for the presence of free aza-ortho-xylylenes in this reaction media, the same reaction was conducted in the presence of butyl vinyl ether. It was reasoned that, if a free aza-ortho-xylylene was being formed, then the electron-rich olefin should trap the intermediate via [4+2] cycloaddition (Scheme 6). Since, no cycloaddition product was observed, we conclude that free aza-ortho-xylylene intermediates are not formed to any appreciable extent under our reaction conditions.
Scheme 6.
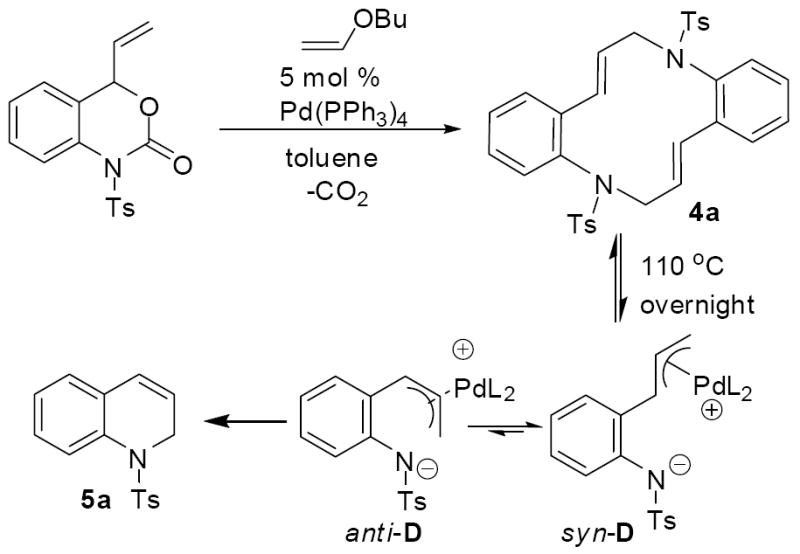
Conducting the same trapping experiment at 100 °C led to an interesting result. While the macrocycle (4a) was formed cleanly as the kinetic product, it quantitatively formed the hydroquinoline (5a) upon heating overnight (Scheme 7).11 With the knowledge that hydroquinolines are the thermodynamic product, we set forth to develop a decarboxylative cyclization that would give rise to hydroquinolines. Here, it proved important to consider the conformations of the π-allyl complex that give rise to the macrocycle vs. hydroquinoline. While the trans macrocycle is assuredly formed from the thermodynamically more stable syn conformation of the π-allyl complex (syn-D), the hydroquinoline must be formed from the less stable anti-conformation (anti-D, Scheme 7). Thus, in most cases, the trans-macrocycles are probably the kinetic product because they are derived from the more stable conformation.
Scheme 7.
3. Decarboxylative cyclizations
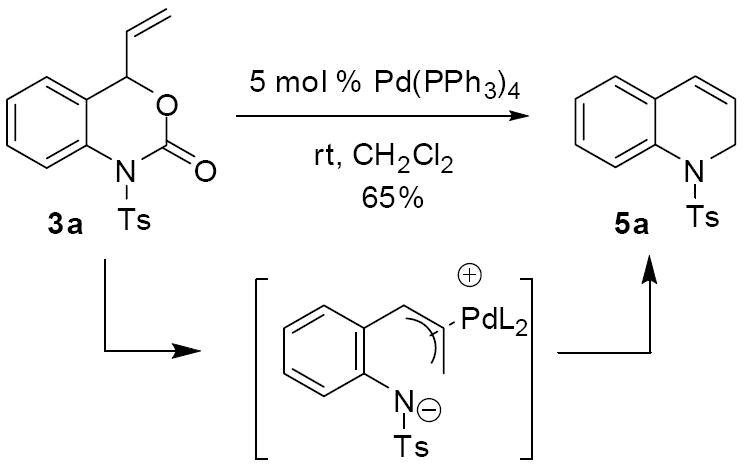
With the goal of biasing the conformation such that the anti-conformation of the π-allyl complex would be viable, our attention turned to chlorinated solvents. Our previous results suggested that the trans macrocycle was favored in toluene, but in ClCH2CH2Cl we were able to access a cis-macrocycle. These results indicated that the anti-conformation of zwitterionic intermediate may be more accessible in dichloroethane. Investigation of several solvents quickly led to the conclusion that dichloromethane was the ideal solvent for decarboxylative cyclization. Ultimately, treatment of the vinyl benzoxazinone 3a with 5 mol% Pd(PPh3)4 in CH2Cl2 at room temperature for 4 hours effected complete conversion of the starting material and product 5a was isolated in 65% yield (Scheme 8). Importantly, monitoring the reaction by 1H NMR spectroscopy showed that macrocycle 4a was not an intermediate in the reaction. Furthermore, the same analysis showed that vinyl azetidines are not intermediates in this transformation.1,12 Therefore, it appears that the dihydroquinoline is the kinetic product in dichloromethane solvent.
Scheme 8.
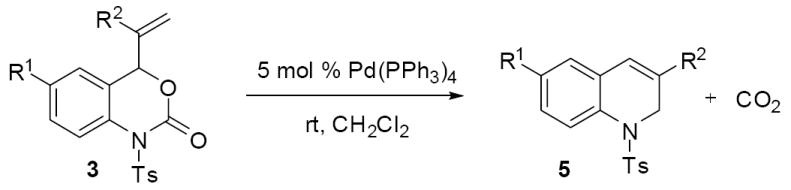
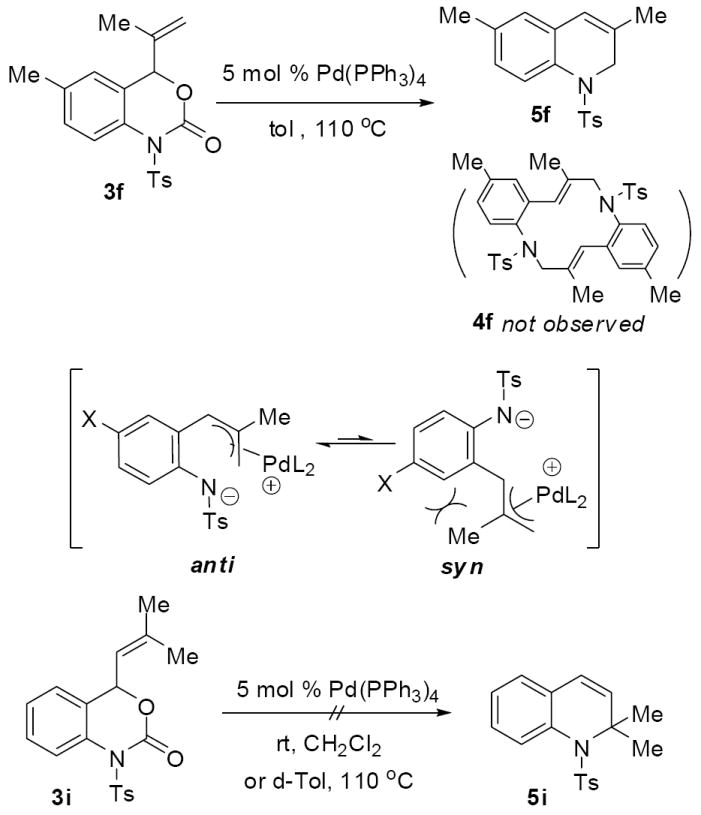
Other vinylbenzoxazinones reacted similarly to 3a. Thus, subjecting a variety of vinyl benzoxazinones to catalytic Pd(PPh3)4 in dichloromethane at room temperature produced the desired dihydroquinolines in good to excellent yields (table 2). While most yields were satisfactory, the yield of the fluorinated product 5d was somewhat low at 51%. Careful investigation of this crude reaction mixture showed that the mass balance was made up by macrocycle 4d. Notably, the 2-methallyl derivative of the fluorinated carbamate produced cyclized product 5h in substantially higher yield (82%) and the macrocycle was not observed in the crude reaction mixture. Thus, the methallyl group (R2 = CH3) appears to sterically preclude formation of macrocycles which leads to high product yields in each case where the 2-substituted allyl groups were utilized (5e-h). In fact, performing the reaction of methallyl reactant 3f in toluene, a solvent that typically favors macrocycloaddition, lead to exclusive formation of the dihydroquinoline product with no evidence of macrocycle formation (Scheme 9). While the methyl group may sterically slow intermolecular macrocycloaddition, it may also bias the conformation of the π-allyl complex by disfavoring the syn-π-allyl complex. Although steric bulk is tolerated in the 2-position of the allyl group, there is a limit on sterics that are tolerated in the cyclization; a substrate (3i) with a terminally disubstituted vinyl group does not undergo any reaction, even under forcing conditions (110 °C) and only starting material is present after 12 h. Since decarboxylation after formation of a π-allyl palladium complex is facile and generally leads to irreversible transformation of the reactant, this experiment suggests that substrate (3i) is unreactive toward formation of the requisite π-allyl palladium complex.
Table 2.
 | ||||
|---|---|---|---|---|
| entry | R1 | R2 | product | yield% |
| 1 | H | H | 5a | 65 |
| 2 | p-Me | H | 5b | 50 |
| 3 | p-MeO | H | 5c | 80 |
| 4 | p-F | H | 5d | 51 |
| 5 | H | Me | 5e | 77 |
| 6 | p-Me | Me | 5f | 94 |
| 7 | p-MeO | Me | 5g | 92 |
| 8 | p-F | Me | 5h | 82 |
Scheme 9.
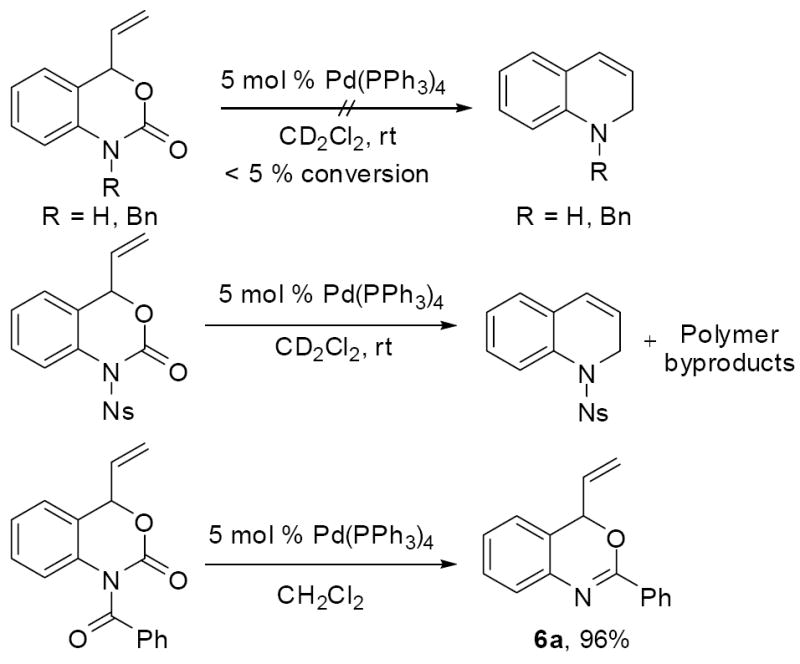
Another current limitation of the decarboxylative cyclization to form dihydroquinolines is the inability to use protecting groups other than tosyl. For example, unprotected and benzyl protected vinyl oxazinones do not react under any conditions (Scheme 10). Presumably, these substrates can form π-allyl palladium complexes, but decarboxylation to form the relatively unstable aniline anion is unfavorable. Use of the more tractable nosyl protecting group did lead to substantial decarboxylative cyclization,13 but the formation of a large amount of oligomeric/polymeric material limits the utility of that protecting group in our chemistry. Lastly, the benzoyl protecting group does facilitate decarboxylative cyclization, however the benzoyl group is non-innocent and takes part in the cyclization to form benzoxazine 6a in high yield. This transformation is expected on the basis of Cook’s precedent that oxazoles can be formed in a similar manner.14
Scheme 10.
4. Decarboxylative cycloaddition of olefins
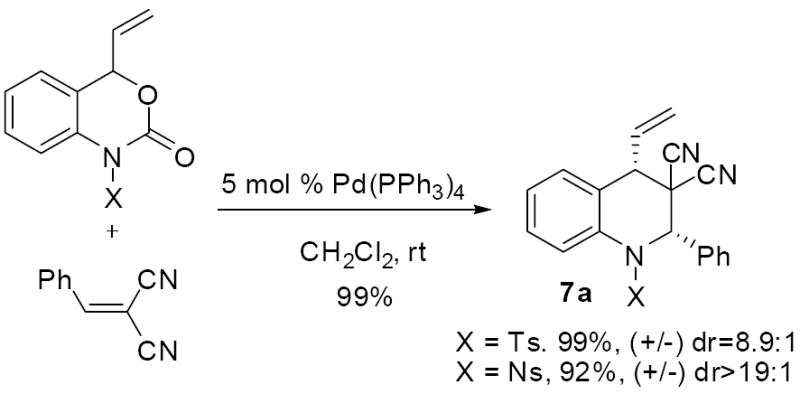
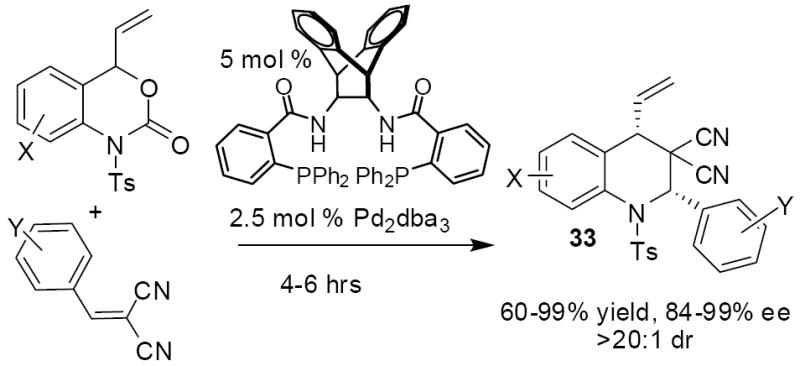
While attempts to intercept aza-ortho-xylylene intermediates with electron rich olefins such as butyl(vinyl) ether were unsuccessful, coordination of palladium is expected to polarize the aza-ortho-xylylene toward reaction with electron deficient olefins. Thus, allowing the vinyl benzoxazinone (3a, X = Ts) to react with one equivalent of benzylidene malononitrile produced the cycloaddition product 7a in excellent yield with good diastereoselectivity. Here, the nosyl protecting group also produced a high yield of product, however 5 equivalents of Michael acceptor were required to achieve high yields. Thus, we proceeded to use the tosyl-protected benzoxazinones in development of a highly asymmetric cycloaddition which utilizes a variant of the well-known Trost ligand.4,15
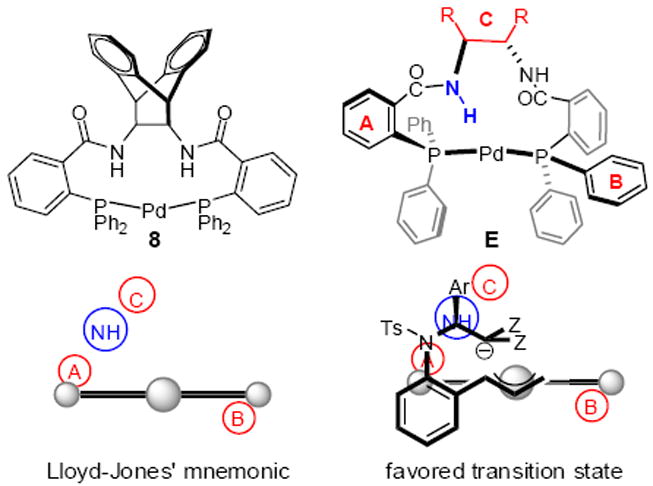
In our communication of the asymmetric decarboxylative cycloaddition of electron-deficient olefins, we provided a stereochemical rationale based on Lloyd-Jones’ mnemonic for the C1-symmetric complex of the Trost ligand with Pd(0).16 Since that time, Lloyd-Jones has refined his model to incorporate a critical H-bonding interaction between the incoming nucleophile and the amide NH in the ligand backbone.17 In doing so, he has developed a new mnemonic device that nicely predicts our observed stereochemistry. Specifically, they found that the Trost ligand binds to palladium to produce 8, which exists in a conformation represented as E. Simplifying this further, leads to a predictive model that has sterically hindering groups represented as A, B, and C, while there is an attractive interaction with the NH (Figure 1). Thus, the nucleophile prefers to align on the left side of the palladium complex to maximize H-bonding with the NH group, while the aryl (Ar) group directs itself up and away from a steric interaction with ligand backbone (C). Such an H-bond-directed attack of the nucleophile on the π-allyl complex accounts for our observed stereochemistry.
Figure 1.
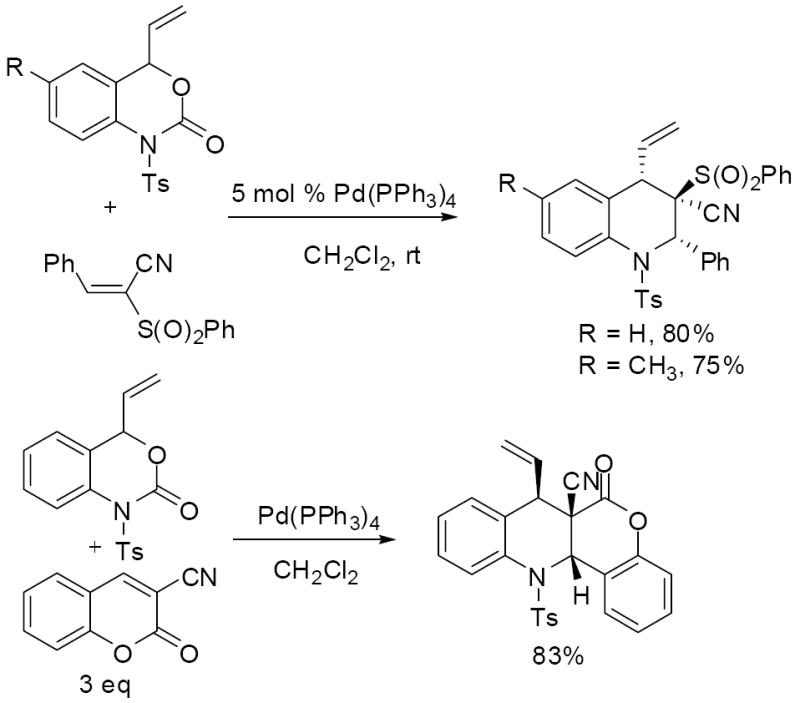
In addition to benzylidene malononitriles, a variety of other electrophiles were investigated for their ability to intercept the polarized aza-o-xylylene intermediates. Simple enones, like methyl vinyl ketone, are not electrophilic enough to out-compete intramolecular cyclization, thus dihydroquinoline is the only product. Other acrylonitrile derivatives are, however, electrophilic enough to promote cycloaddition rather than cyclization. For instance, phenylsulfonyl acrylonitrile provides the products in good yield using just one equivalent of electrophile (Scheme 13). Moreover, the products are formed as single diastereomers; presumably the stereochemistry is governed by a cyclization where all of the large groups occupy pseudo-equatorial positions about the 6-membered ring. Cyanocoumarins are also adequate partners in the cycloaddition chemistry, however, higher concentrations of cyanocoumarin are required to achieve similar yields to the sulfonyl acrylonitrile. Investigation of the reaction by 1H NMR spectroscopy shows that product is formed as a 7:1 mixture along with dihydroquinoline produced via intramolecular cyclization.
Scheme 13.
5. Decarboxylative cycloaddition of tosyl isocyanate
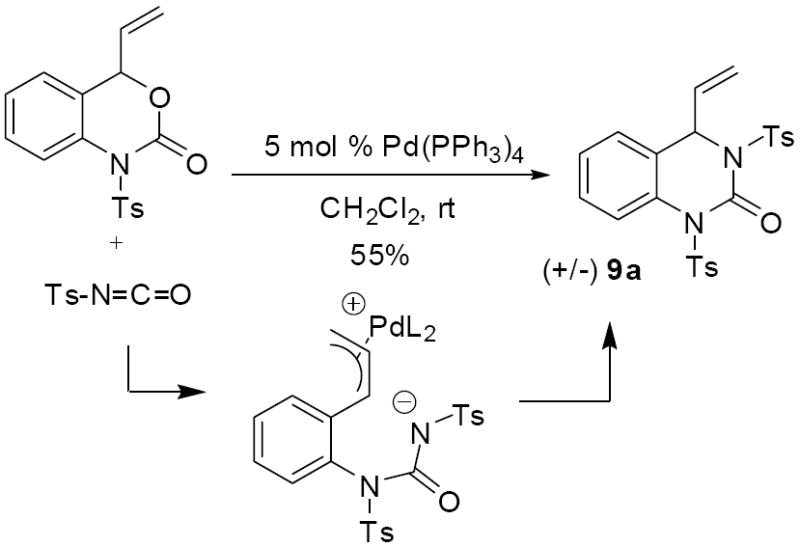
Electrophiles other than Michael acceptors are also viable partners for decarboxylative cycloaddition. For example, treatment of the vinyl benzoxazinone 3a with tosyl isocyanate produced the dihydroquinazolinone (9a, Scheme 14)).18 Unfortunately, the same reaction performed with a chiral Trost ligand that was successful in cycloaddition of Michael acceptors led to only low enantioselectivity in the formation of 9a (24% ee).
Scheme 14.
6. Decarboxylative aminations
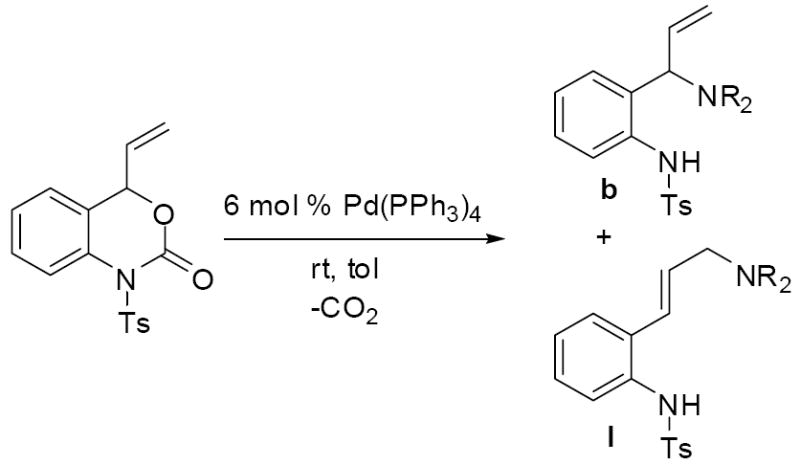
The reaction of tosyl isocyanate with the vinyl oxazinone amounts to the amination of the more hindered π-allyl terminus, where the amination is clearly directed so a 6-membered ring is formed. Next, we became curious whether free amine nucleophiles would react with the aza-ortho-xylylene intermediates with the standard regiochemical preference for reaction at the less hindered allyl terminus.19 Toward this end, vinyl benzoxazinone 3a was treated with pyrrolidine in the presence of catalytic Pd(PPh3)4. Interestingly, the pyrrolidine reacted exclusively at the more hindered allyl terminus to provide the aminated product 10a in excellent yield (97%) and piperidine reacts similarly (table 3, entries 1 and 2). In addition primary benzylamines react to give clean monoallylation with the same regiochemical preference for nucleophilic attack at the more hindered allyl terminus. While it is relatively unusual to observe attack at the more hindered allyl terminus of palladium π-allyl complexes,19 our current hypothesis is that the basic tosylamide acts to direct the addition via a 6-membered transition state (Scheme 15). Such a hypothesis is consistent with Cooks’ detailed studies on similar systems.20
Table 3.
Decarboxylative aminations
 | ||||
|---|---|---|---|---|
| entry | amine | product | b:l | yield (%) |
| 1 | 10a | >19:1 | 97 | |
| 2 | 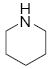 |
10b | >19:1 | 96 |
| 3 | 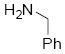 |
10c | >19:1 | 98 |
| 4 | 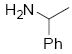 |
10d | >19:1 | 82a |
| 5 | 10e | - | <5 | |
| 6 | 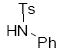 |
10f | <1:19 | 65 |
obtained as a 1.2:1 mixture of diastereomers
Scheme 15.
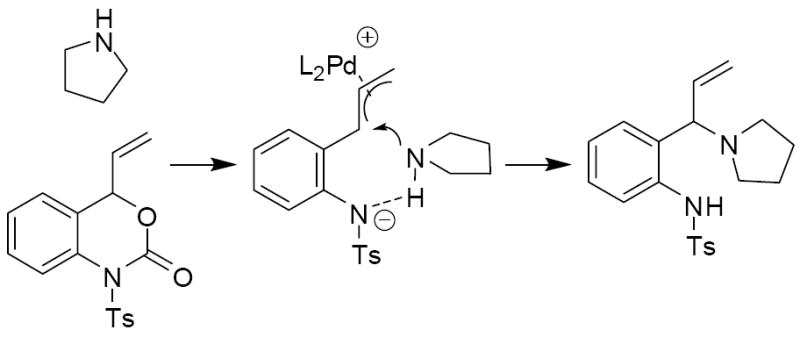
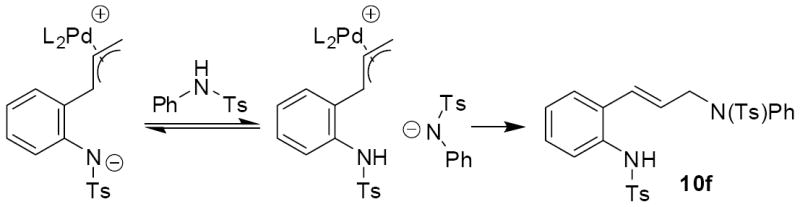
Analogous treatment of the benzoxazinone 3a with aniline failed to give any product, presumably due to the lower nucleophilicity of aniline relative to aliphatic amines. However, an activated aniline derivative does participate in the reaction (entry 6, table 3). Here, a near thermoneutral proton transfer can produce the anionic sulfonamide nucleophile which is substantially more reactive than aniline (Scheme 16). Another interesting feature of the reaction with N-tosyl aniline is the regiochemistry of the isolated product. While the more basic amines produced the branched product with high selectivity, N-tosyl aniline produced the linear product exclusively.21 Once again, we attribute this divergent behavior to the generation of the sulfonamide anion prior to C—N bond formation.
Scheme 16.
7. Conclusions
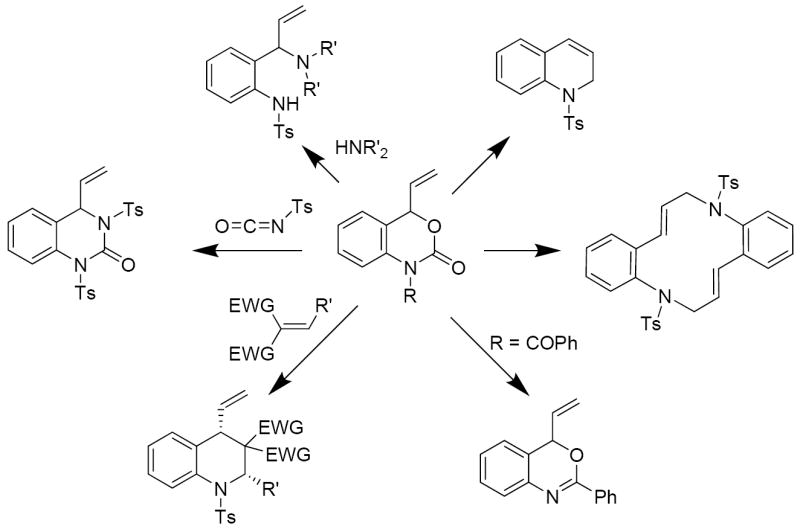
In conclusion, palladium-polarized aza-ortho-xylylenes are versatile intermediates that can be generated under mild conditions via decarboxylation of vinyl benzoxazinones. These intermediates undergo dimerization reactions to produce macrocycles, cyclizations to form dihydroquinolines, cycloadditions with electrophilic π-bonds and nucleophilic attack by amines. Thus, a wide array of structurally unique aniline derivatives can be accessed (Scheme 17).
Scheme 17.
8. Experimental
8.1. General
Toluene and methylene chloride were dried over activated alumina. Commercially available reagents were used without additional purification unless otherwise stated. Products were purified on silica gel from Sorbent Technologies (230×400 mesh, 60 Å porosity, pH 6.5-7.5). 1H and 13C NMR spectra were obtained on a Bruker Avance 400 or Bruker Avance 500 DRX spectrometer and referenced to residual protio solvent signals. Structural assignments are based on 1H, 13C, DEPT-135, COSY, and HMQC spectroscopies, and by comparison with literature data.11 High resolution mass spectrometry was performed on an AUTOSPEC-Q tandem hybrid mass spectrometer (VG Analytical Ltd, Manchester, UK). FTIR spectra were acquired on a Shimadzu FTIR-8400S spectrometer. HPLC analysis was performed on a Shimadzu SCL-10A VP instrument.
8.2. General procedure for catalytic decarboxylative macrocycloaddition to form 4
In a Schlenk tube under argon, Pd(PPh3)4 (0.05 mmol) and vinyl benzoxazinanone 1 (1 mmol) were dissolved in 5 mL of toluene. The resulting yellow solution was stirred at ambient temperature under Ar until reaction completion was indicated by TLC (ca. 10 min). Following solvent evaporation under reduced pressure, the crude product was purified via flash chromatography (SiO2, 2:1 hexane:ethyl acetate).
8.2.1. (7E,15E)-5,13-ditosyl-5,6,13,14-tetrahydrodibenzo[b,h] [1,7]diazacyclododecine (4a)
1H NMR (400 MHz, CDCl3) δ 2.47 (6 H, s: CH3Ts), 3.62 (2 H, dd, J=13.2, 5.9 Hz: CH2), 3.73 (6 H, s: CH3), 4.66 (2 H, ddd, J=13.2, 5.9, 1.3 Hz: CH2), 5.46 (2 H, dt, J=16.1, 5.9 Hz: =CHCH2), 6.94 (2 H, d, J=7.8 Hz: Ar CH), 7.15-7.25 (4 H, m: Ar CH), 7.28 (2 H, d, J=8.6 Hz: Ar CH), 7.41 (4 H, d, J=8.6 Hz: Ar CH), 7.50 (2 H, d, J=16.1 Hz: =CH), 7.83 (4 H, d, J=8.1 Hz: Ar CH). 13C NMR (100 MHz, CDCl3) δ 21.7 (CH3Ts), 50.9 (CH2), 124.6 (=CHCH2), 127.0 (Ar CH), 128.2 (Ar CH), 128.4 (Ar CH), 128.5 (Ar CH), 129.0 (Ar CH), 129.6 (quat. Ar C), 130.0 (Ar CH), 136.0 (quat. Ar C), 137.3 (=CH), 139.0 (quat. Ar C), 143.9 (quat. Ar C); FTIR (CH2Cl2):νmax 3053, 2926, 1346, 1271, 1161, 760. HRMS calcd for C32H34N3O4S2 [M+NH4] 588.1991, found 588.1990.
8.2.2 (7Z,15Z)-2,10-difluoro-5,13-ditosyl-5,6,13,14-tetrahydrodibenzo[1,7] diazacyclododecine (4d)
1H NMR (400 MHz, CDCl3) δ 2.36 (6 H, s: CH3Ts), 4.46 (4 H, dd, J=3.73, 1.39 Hz: CH2), 5.67 (2 H, dt, J=9.8 Hz, 4.26 Hz: =CHCH2), 6.29 (2 H, d, J=9.8 Hz: =CH), 6.91 (2 H, t, J=8.75 Hz: Ar CH), 7.11 (4 H, d, J=7.89 Hz: Ar CH), 7.19 - 7.26 (2 H, m: Ar CH), 7.33 (4 H, d, J=8.2 Hz: Ar CH), 7.53 (2 H, d, J=8.2 Hz: Ar CH).13C NMR (100 MHz, CDCl3) δ 21.6 (CH3Ts), 45.1 (CH2), 113.0 (Ar CH), 117.8 (quat. Ar C), 118.6 (=CHCH2), 122.3 (Ar CH), 124.3 (=CH), 127.2 (Ar CH), 128.2 (Ar CH), 129.2 (Ar CH), 136.1 (quat. Ar C), 143.8 (quat. Ar C), 156.8 (quat. Ar C), 158.8 (quat. Ar C); FTIR (CH2Cl2): νmax 3065, 2926, 1612, 1472, 1263, 1167, 750. HRMS calcd. for C32H28F2N2O4S2Na [M+Na] 629.1356, found 629.1359.
8.3. General procedure for catalytic decarboxylative dihydroquinoline synthesis from vinyl benzoxazinanones
In a Schlenk tube under argon, Pd(PPh3)4 (0.05 mmol) and vinyl benzoxazinanone 1a (1 mmol) were dissolved in methylene chloride (5 mL). The resulting yellow solution was stirred at ambient temperature under Ar until the completion of the reaction was indicated by TLC (generally 1h-4h). Following solvent evaporation under reduced pressure, the crude product was purified via flash chromatography (SiO2, 7:1 hexane: ethyl acetate).
8.3.1 6-methyl-1-tosyl-1,2-dihydroquinoline (5b)
1H NMR (400 MHz, CDCl3) δ 2.32 (3 H, s: overlapping CH3Ts, CH3), 2.35 (3 H, s: overlapping CH3Ts, CH3), 4.41 (2 H, dd, J=4.1, 1.6 Hz: CH2), 5.56 (1 H, dt, J=9.6, 4.1 Hz: =CHCH2), 5.98 (1 H, d, J=9.6 Hz: =CH), 6.75 (1 H, s: Ar CH), 7.08 (3 H, d, J=8.1 Hz: Ar CH), 7.31 (2 H, d, J=8.3 Hz: Ar CH), 7.59 (1 H, d, J=8.1 Hz: Ar CH). 13C NMR (100 MHz, CDCl3) δ 21.0 (overlapping CH3, CH3Ts), 21.6 (overlapping CH3, CH3Ts), 45.5 (CH2), 123.8 (=CHCH2), 126.0 (=CH), 126.7 (Ar CH), 127.0 (Ar CH), 127.3 (Ar CH), 128.7 (Ar CH), 129.0 (Ar CH), 129.3 (quat. Ar C), 132.4 (quat. Ar C), 136.4 (quat. Ar C), 136.5 (quat. Ar C), 143.3 (quat. Ar C); FTIR (CH2Cl2): νmax 3053, 2986, 1489, 1421, 1271, 1258, 895, HRMS calcd for C17H18NO2S [M+] 300.1058, found 300.1059.
8.3.2 6-methoxy-3-methyl-1-tosyl-1,2-dihydroquinoline (5g)
1H NMR (400 MHz, CDCl3) δ 1.65 (3 H, s: CH3), 2.36 (3 H, s: CH3Ts), 3.81 (3 H, s: OCH3), 4.23 (2 H, s: CH2), 5.66 (1 H, s: =CH), 6.41 (1 H, d, J=2.8 Hz: Ar CH), 6.79 (1 H, dd, J=8.8, 2.8 Hz: Ar CH), 7.09 (2 H, d, J=8.1 Hz: Ar CH), 7.25 (2 H, d, J=8.1 Hz: Ar CH), 7.61 (1 H, d, J=8.8 Hz: Ar CH).13C NMR (100 MHz, CDCl3) δ 20.8 (CH3), 21.6 (CH3Ts), 49.6 (CH2), 55.5 (OCH3), 110.8 (Ar CH), 112.0 (Ar CH), 121.0 (=CH), 126.5 (quat. Ar C), 126.9 (Ar CH), 128.2 (Ar CH), 128.9 (Ar CH), 131.8 (quat. Ar C), 134.4 (=C), 135.8 (quat. Ar C), 143.3 (quat. Ar C), 158.3 (quat. Ar C); FTIR (CH2Cl2): νmax 3052, 2929, 1346, 1259, 1165. HRMS calcd for C18H19NO3SNa [M+Na] 352.0984, found 352.0982.
8.4. General procedure for decarboxylative cycloadditions
In a Schlenk tube under argon, Pd(PPh3)4 (0.05 mmol), vinyl benzoxazinanone 1 (1 mmol) and the electrophile (1-1.1 mmol) were dissolved in 5 mL of methylene chloride. The resulting yellow solution was stirred at ambient temperature under Ar until reaction completion was indicated by TLC (generally 4-6h). Following solvent evaporation under reduced pressure, the crude product was purified via flash chromatography (SiO2, 5:1 Hexane: Ethyl acetate).
8.4.1 2-phenyl-1-tosyl-4-vinyl-1,2-dihydroquinoline-3,3(4H)-dicarbonitrile (7a)
1H NMR (400 MHz, CDCl3) δ ppm 2.47 (3 H, s: CH3Ts), 2.56 (1 H, d, J=9.6 Hz: CHCH=), 5.09 (1 H, d, J=16.8 Hz: CH=CH(H)trans), 5.61 (1 H, dd, J=10.1, 1.0 Hz: CH=CH(H)cis), 5.77 (1 H, s: CHPh), 5.91 (1 H, dt, J=16.8, 10.1 Hz: CH=CH2), 7.17 (1 H, d, J=7.7 Hz: Ar CH), 7.29 (2 H, d, J=7.7 Hz: Ar CH), 7.39 - 7.51 (5 H, m: Ar CH), 7.51 - 7.67 (4 H, m: Ar CH), 7.89 (1 H, dd, J=7.99, 1.04 Hz: Ar CH).13C NMR (75 MHz, CDCl3) δ 21.6 (CH3Ts), 49.1 (CHCH=), 50.0 (CCN2), 65.8 (CHPh), 111.2 (CN), 113.9 (CN), 125.0 (=CH2), 126.8 (Ar CH), 127.0 (Ar CH), 127.3 (Ar CH), 128.2 (Ar CH), 128.6 (Ar CH), 129.2 (Ar CH), 129.3 (CH=CH2), 129.6 (Ar CH), 130.0 (Ar CH), 130.2 (Ar CH), 131.6 (quat. Ar C), 134.5 (quat. Ar C), 135.1 (quat. Ar C), 136.7 (quat. Ar C), 145.1 (quat. Ar C); FTIR (CDCl3): νmax 3053, 2986, 2305, 1597, 1483, 1421, 1262, 895. HRMS calcd for C26H21N3O2SNa [M+Na] 462.1252, found 462.1251. HPLC, Diacel Chiralpak AD-H column (96% Hexane/IPA, 1.0 mL/min), tr (S,S) = 22.8 min, tr (R,R) = 32.1 min.; N-nosyl derivative: 1-(4-nitrophenylsulfonyl)-2-phenyl-4-vinyl-1,2-dihydroquinoline-3,3(4H)-dicarbonitrile. 1H NMR (400 MHz, CDCl3) δ ppm 3.02 (1H, d, J=9.5 Hz: CHCH=), 5.33 (1H, d, J=16.8 Hz: CH=CH(H)trans), 5.69 (1 H, d, J=10.2 Hz: CH=CH(H)cis), 5.86 (1 H, s: CHPh), 5.95 (1H, ddd, J=16.6, 10.2, 9.5 Hz: CH=CH2), 6.97-8.26 (overlapping Ar CH).
8.5. General procedure for decarboxylative amination
Pd(PPh3)4 (0.012 mmol), vinyl benzoxazinanone 3a (0.2 mmol) and the amine (0.3 mmol) were dissolved in methylene chloride (1.0 mL) in a well-sealed reaction vial under argon. The resulting solution was stirred at ambient temperature under Ar until reaction completion was indicated by TLC (generally 30 min to 1 h). Following solvent evaporation under reduced pressure, the crude product was purified via flash chromatography (SiO2, 4:1 Hexane: Ethyl acetate).
8.5.1 4-methyl-N-(2-(1-(pyrrolidin-1-yl) allyl)phenyl)benzenesulfonamide (10a)
NMR (400 MHz, CDCl3) δ ppm 1.85 (4H, s), 2.39 (3H, s: CH3Ts and CH2, overlapping), 2.55 (2H, br s), 3.69 (1H, d, J = 8.84 Hz), 4.96 (1H, d, J = 9.92 Hz), 5.09 (1H, d, J = 16.84 Hz), 5.87-5.78 (1H, m), 6.93 (1H, d, J = 7.16), 6.99 (1H, d, J = 7.16 Hz), 7.19-7.15 (1H, m), 7.28-7.25 (1H, m), 7.53 (1H, d, J = 8.04 Hz), 7.80 (1H, d, J = 8.04 Hz), 11.87 (1H, br s); 13C NMR (75 MHz, CDCl3) δ 21.5, 23.6, 51.8, 74.3. 117.2, 118.0, 123.0, 127.0, 128.2, 128.7, 128.8, 129.5, 136.4, 137.1, 137.8, 143.4; FTIR (CH2Cl2):νmax 2935, 1494, 1338, 1151, 756, 655; HRMS calcd for C20H24N2O2SNa [M+Na] 379.1456, found 393.1451.
8.5.2 4-methyl-N-(2-(1-(piperidin-1-yl)allyl)phenyl)benzenesulfonamide (10b)
NMR (400 MHz, CDCl3) δ ppm 1.49 (2 H, br s), 1.68 (4H, m), 2.40 (3H, s: CH3Ts), 2.49 (4H, br s), 3.62 (1H, d, J = 9.60 Hz), 5.08 (2H, dd, J = 18.50, 13.50), 5.76 (1H, dt, J = 16.9, 9.9, 1H), 6.99-6.95 (2H, m),7.20- 7.16 (1H, m), 7.28-7.25 (2H, m), 7.47 (1H, d, J = 8.00 Hz), 7.77 (2H, d, J = 7.77 Hz), 12.18 (1H, br s); 13C NMR (75 MHz, CDCl3) δ 21.5, 24.3, 26.0, 51.1, 74.1, 118.9, 119.4, 123.4, 126.9, 128.1, 128.6, 129.0, 129.6, 134.4, 137.5, 138.2, 143.3; FTIR (CH2Cl2):νmax 2935, 1492, 1340, 1150, 754; HRMS calcd for C21H26N2NaO2S [M+Na] 393.1613, found 393.1611.
8.5.3 N-(2-(1-(benzylamino)allyl)phenyl)-4-methylbenzenesulfonamide (10c)
NMR (400 MHz, CDCl3) δ ppm 2.37 (3H, s: CH3Ts), 3.65 (1H, d, J = 13.04), 3.74 (1H, d, J = 13.04 Hz), 4.14 (1H, d, J = 6.76, Hz), 5.05 (2H, t, J = 10.40 Hz), 5.84-5.76 (1H, m), 7.02-6.97 (2H, m), 7.21-716 (3H, m), 7.35 (3H, m), 7.40 (2H, d, J = 7.60 Hz), 7.60 (1H, d, J = 7.60 Hz), 7.66 (2H, d, J = 7.60 Hz), 11.31 (1H, br s); 13C NMR (75 MHz, CDCl3) δ 21.4, 50.9, 65.1, 117.1, 119.3, 123.4, 127.1, 127.5, 127.8, 128.3, 128.5, 128.7, 129.4, 136.9, 137.5, 138.4, 143.2; FTIR (CH2Cl2):νmax 3028, 1494, 1150, 933, 756; HRMS calcd for C23H25N2O2SNa [M+H] 393.1637, found 393.1630.
8.5.4 (E)-4-methyl-N-(3-(2-(4-methylphenylsulfonamido)phenyl)allyl)-N-phenylbenzenesulfonamide
NMR (500 MHz, CDCl3) δ ppm 2.41 (3 H, s : CH3Ts), 2.47 (3H, s: CH3Ts), 4.23 (2H, d, J = 6.5 Hz), 5.83 (1H, dt, J = 15.7, 6.4 Hz), 6.10 (1H, br s), 6.21 (1H, d, J = 15.70 Hz), 7.10-7.06 (3H, m), 7.18-7.13 (2H, m), 7.29-7.22 (3H, m), 7.30 (1H, br s), 7.37-7.32 (2H, m), 7.52 (2H, d, J = 8.25 Hz), 7.56 (2H, d, J = 8.25 Hz); 13C NMR (100 MHz, CDCl3) δ 21.6 (CH3Ts), 21.6 (CH3Ts), 52.9, 124.5, 126.3, 127.2, 127.3, 127.8, 128.1, 128.6, 128.6, 128.7, 128.8, 129.1, 129.5, 129.7, 131.4, 133.2, 135.19, 136.4, 139.2, 143.7, 143.9. HRMS calcd for C29H28N2O4S2Na [M+Na] 555.1388, found 555.1402.
Scheme 11.
Scheme 12.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wang C, Tunge JA. Org Lett. 2006;8:3211–3214. doi: 10.1021/ol0610744. [DOI] [PubMed] [Google Scholar]
- 2.(a) Kuhn O, Mayr H. Angew Chem Int Ed. 1999;38:343–346. doi: 10.1002/(SICI)1521-3773(19990201)38:3<343::AID-ANIE343>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]; (b) Lemek T, Mayr H. J Org Chem. 2003;68:6880–6886. doi: 10.1021/jo0344182. [DOI] [PubMed] [Google Scholar]
- 3.For related cycloadditions see: Aoyagi K, Nakamura H, Yamamoto Y. J Org Chem. 2002;67:5977. doi: 10.1021/jo025747h.Sekido M, Aoyagi K, Nakamura H, Kabuto C, Yamamoto Y. J Org Chem. 2001;66:7142. doi: 10.1021/jo0158332.
- 4.Wang C, Tunge JA. J Am Chem Soc. 2008;130:8118–8119. doi: 10.1021/ja801742h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wojciechowski K. Eur J Org Chem. 2001:3587–3605. [Google Scholar]
- 6.Consonni R, Dalla Croce P, Ferraccioli R, La Rosa C. J Chem Soc, Perkin Trans. 1996;1:1809–1814. [Google Scholar]
- 7.Chong PY, Janicki SZ, Petillo PA. J Org Chem. 1998;63:8515–8521. [Google Scholar]
- 8.Rauno G, Luis J, Concepcion P, Jesus HR. Tetrahedron. 1989;45:203–214. [Google Scholar]
- 9.Consonni R, Croce PD, Ferraccioli R, Rosa CL. J Chem Soc, Perkin Trans. 1996;1:1809–1814. [Google Scholar]
- 10.For a related macrocyclic triflamide see: Satake A, Ishii H, Shimizu I, Inoue Y, Hasegawa H, Yamamoto A. Tetrahedron. 1995;51:5331–5340.
- 11.(a) Arisawa M, Terada Y, Takahashi K, Nakagawa M, Nishida A. J Org Chem. 2006;71:4255–4261. doi: 10.1021/jo060308u. [DOI] [PubMed] [Google Scholar]; Larock RC, Hightower TR, Hasvold LA, Peterson KP. J Org Chem. 1996;61:3584–3585. doi: 10.1021/jo952088i. [DOI] [PubMed] [Google Scholar]; (c) Weissman A, Muren JF. J Med Chem. 1971;14:49–53. doi: 10.1021/jm00283a013. [DOI] [PubMed] [Google Scholar]
- 12.Fugami K, Miura K, Morizawa Y, Oshima K, Utimoto K, Nozaki H. Tetrahedron. 1989;45:3089–98. [Google Scholar]
- 13.(a) Fukuyama T, Jow CK, Cheung M. Tetrahedron Lett. 1995;36:6373. [Google Scholar]; (b) Maligres PE, See MM, Askin D, Reider PJ. Tetrahedron Lett. 1997;38:5253. [Google Scholar]
- 14.(a) Cook GR, Shanker PS, Pararajasingham K. Angew Chem Int Ed. 1999;38:110–113. [Google Scholar]; (b) Cook GR, Sun L. Org Lett. 2004;6:2481–2484. doi: 10.1021/ol049087+. [DOI] [PubMed] [Google Scholar]
- 15.Trost BM, Van Vranken DL, Bingel C. J Am Chem Soc. 1992;114:9327–9343. [Google Scholar]
- 16.Lloyd-Jones GC, Stephen SC, Fairlamb IJS, Martorell A, Dominguez B, Tomlin PM, Murray M, Fernandez JM, Jeffery JC, Riis-Johannessen T, Guerziz T. Pure Appl Chem. 2004;76:589–601. [Google Scholar]
- 17.Personal communication of a manuscript that is submitted for publication Butts CP, Filali E, Lloyd-Jones GC, Norrby P-O, Sale DA, Schramm Y. 2009 Dec 22; doi: 10.1021/ja8099757.
- 18.Trost BM, Fandrick DR. J Am Chem Soc. 2003;125:11836–7. doi: 10.1021/ja037450m. [DOI] [PubMed] [Google Scholar]
- 19.Trost BM, Van Vranken DL. Chem Rev. 1996;96:395–422. doi: 10.1021/cr9409804. [DOI] [PubMed] [Google Scholar]
- 20.Cook GR, Yu H, Sankaranarayanan S, Shanker PS. J Am Chem Soc. 2003;125:5115–5120. doi: 10.1021/ja028426w. [DOI] [PubMed] [Google Scholar]
- 21.Investigation of the early stages of the reaction by 1H NMR spectroscopy does not show any branched product, thus the linear product is suspected to be the kinetic product.