

Abstract
Integrins link the inside of a cell with its outside environment and in doing so regulate a wide variety of cell behaviors. Integrins are well known for their roles in angiogenesis and cell migration but their functions in bone formation are less clear. The majority of integrin signaling proceeds through focal adhesion kinase (FAK), an essential component of the focal adhesion complex. We generated transgenic mice in which FAK was deleted in osteoblasts and uncovered a previously unknown role in osteoblast differentiation associated with bone healing. FAK mutant cells migrated to the site of skeletal injury and angiogenesis was unaffected yet the transgenic mice still exhibited numerous defects in reparative bone formation. Osteoblast differentiation itself was unperturbed by the loss of FAK, whereas the attachment of osteoclasts to bone matrix was disrupted in vivo. We postulate that defective bi-directional integrin signaling affects the organization of the collagen matrix. Finally, we present a compensatory candidate molecule, Pyk2, which localized to the focal adhesions in osteoblasts that were lacking FAK.
Keywords: osteoblasts, focal adhesion kinase (FAK), skeletal regeneration, osteoclasts, Pyk2
Introduction
Integrins function as cell-substrate adhesion molecules, serving as receptors for major extracellular matrix molecules including collagens and fibronectin [1-4]. In addition to their adhesive functions, integrins mediate bi-directional signaling between the extracellular matrix and the cell. Upon activation of integrin molecules, cytoskeletal and signaling molecules are recruited into focal adhesion structures [5-7].
Although integrin molecules provide a platform for the signaling complex, they do not have intrinsic enzymatic activities in their cytoplasmic domains [8]. Therefore, transmission of downstream signals has to be mediated by non-receptor tyrosine kinases, such as FAK or Src family kinases [9]. Integrin mediated attachment activates FAK by autophosphorylation at tyrosine 397, which provides a binding site for Src [10, 11]. In turn, FAK binds to a number of signaling molecules. For example, the N-terminal domain of FAK binds to Shc [12]. The C-terminal domain of FAK binds to Talin and Paxillin [13, 14], which link the integrin-FAK signaling complex to the actin cytoskeleton [15, 16]. Furthermore, FAK is required for the phosphorylation of Src and other downstream targets [17, 18]. Since several different integrins transduce signals via FAK [19, 20], the deletion of FAK is one mechanism to cause a major reduction in integrin signaling within a cell population.
The role of FAK in bone formation, remodeling and repair in vivo is unknown because deletion of FAK in the mouse germline results in embryonic death at e8.5-9.0 [21], long before the skeleton forms. Our goal was to understand in greater detail the role of FAK in these essential stages of osteogenesis in vivo. To that end, we generated a conditional deletion of FAK using a Cre-loxP approach by crossing mice, in which the Cre recombinase is driven by 2.3 Kb of the collagen type I(α)I promoter, with mice carrying a floxed FAK allele [22, 23]. In this study, we first established immortalized calvarial osteoblast and bone marrow cell lines which are devoid of FAK, and assayed them for their ability to differentiate into osteoblasts. Second, we investigated the function of FAK in adult bone regeneration. Lastly, we explored a possible compensatory role for the closely related non-receptor type kinase, Pyk2, in osteoblast differentiation when FAK is not present.
Materials and Methods
Mice and genotyping
All the procedures for animal breeding and surgeries followed the approved protocols and guidelines from the Administrative Panel for Laboratory Animal Care (APLAC) at Stanford University and the Laboratory Animal Resource Center (LARC) at UCSF. Heterozygote transgenic mice containing loxP sequences (floxed FAK) in the flanking introns of the second kinase domain of fak were generated [23] and crossed with FAK heterozygote mice (FAK+/−) (Ilic, 1995 #10059) to obtain FAKfl/− and FAKfl/fl genotypes. Transgenic mice carrying Cre recombinase driven by the 2.3 Kb collagen type I(α)I promoter (Cre+/−) (Dacquin, 2002 #8151) were crossed with FAKfl/− and FAKfl/fl mice. Floxed fak and the recombined allele resulting from Cre recombinase activity were differentiated using P1 (5′-gagaatccagctttggctgttg -3′) and P2 (5′-gaatgctacaggaaccaaataac-3′) PCR primers. Genotyping was performed by tail biopsy followed by PCR.
Antibodies
Anti-mouse FAK antibody was obtained from Transduction Laboratories (San Jose, CA). Rabbit antibody against phophospecific Pyk2-402P was purchased from Biosource (Camarillo, CA). β-actin antibody was purchased from Sigma (AC-15). Biotinylated-monoclonal Cre antibody was obtained from Abcam (ab24580; Cambridge, MA). Antibodies were used at 1:1000 and 1:100 for Western blot. Secondary antibodies were purchased from Jackson ImmunoResearch (West Grove, PA).
Calvarial osteoblast and bone marrow cell isolation
In order to generate cell lines in which the fak allele was deleted, two different transgenic mouse lines were generated. Mice carrying the genotypes of Cre+;FAKfl/fl and Cre+;FAKfl/− were bred with p53−/− mice to produce animals that lacked functional FAK and whose cells, when isolated, were immortalized [21]. Control calvarial osteoblasts were prepared from Cre−;FAKfl/fl;p53−/− or Cre−;FAKfl/−;p53−/− mice. Calvarial osteoblast isolation and bone marrow cell culture were performed as described [24, 25].
Clonal cell line establishment and in vitro differentiation
Primary cells from calvaria and bone marrow were grown in 15% FBS α-MEM, which consisted of minimum medium with α-modification, supplemented with 15% fetal calf serum (Hyclone, Logan, Utah) and 1% penicillin/streptomycin (Gibco BRL, Gaithersburg, MD). Cells were infected at a MOI 10 with adenovirus carrying cre and gfp under regulation of the CMV promoter (Ad-cre) [23]. Single cell clones that maintained cre expression were identified using primers of cre1 (5′-cctggaaaatgcttctgtcctttgcc-3′) and cre2 (5′-gagttgatagctggctggtggcagatg-3′). These clones were removed from the study. Single cell cloning was performed using a limited dilution and individual clones were established as cell lines. Control FAKfl/fl calvarial and bone marrow cells that were not treated with adeno-cre were handled in a similar manner. The differentiation of skeletal progenitor cells into osteoblasts was induced with ascorbic acid (56mM) and β-glycerophosphate (5mM) in vitro. The media were changed every 48h for 2 weeks. Osteoblast differentiation was determined by 0.2% Alizarin Red staining.
Western blot and Immunofluorescence
Detailed procedures were described previously [26]. Protein concentration was determined by BCA method [27]. One microgram of total protein was loaded on each lane for Western blot analysis. Proteins were transferred onto nitrocellulose membranes and blotted with the appropriate primary antibodies. Bands were visualized using an enhanced chemiluminescent kit (Amersham, Piscataway, NJ). β-actin was used to confirm equal amount protein loading. For immunofluorescence, fluorescein-conjugated donkey IgG was used to detect signals under an epifluorescence microscope (Zeiss, Germany). Nuclei were stained with Hoechst 33342 dye (Invitrogen, Carlsbad, CA) prior to mounting.
Histology, immunohistochemistry and whole mount skeletal staining
Tissues were harvested at different time points and embedded in paraffin. Sections were cut at 7 μm thickness. For histology, sections were stained using Movat's pentachrome [28]. Tartrate resistant acid phosphatase [29] staining procedure was previously described [30]. For Cre immunohistochemistry, tissue sections were permeabilized with cold acetone for 10 min followed by overnight incubation with the primary antibody. After Streptavidin-HRP incubation (1:2000), signal was detected using a DAB kit (Vector lab, Burlingame, CA). Whole mount skeletal staining with Alizarin Red and Alcian Blue was performed according to previous reports [31]. Proliferating cell nuclear antigen (PCNA) staining followed the manufacturer's instruction (Zymed, San Francisco, CA). Nuclei were stained with Hoechst 33342 dye (Invitrogen, Carlsbad, CA) prior to mounting.
Generation of fak probes and in situ hybridization
The N-terminal FAK probe was made by digesting mouse FAK cDNA with Cla I and Xho I and ligating the resulting fragment into pBluescript. A Sma I and Xba I fragment of FAK cDNA was ligated into pBluescript to generate the C-terminal region of the FAK probe. Probe sequences were verified by DNA sequencing. Digoxigenin-labeled probes for both the N- and C-terminal portions of FAK were transcribed with T3 RNA polymerase and used for in situ hybridization [32].
Surgical procedures
Previously, we described the healing process following pinhole injury in mouse tibiae [33]. Briefly, mice were anaesthetized and the anterior-proximal tibia was exposed. Injuries were made using a Dremel® drill with a 1 mm core drill bit (Racine, WI). Wounds were closed and animals received Buprenorphine for analgesia. Animals were allowed to ambulate freely after recovery. For the bone chip experiment, murine tibiae were isolated and snap-frozen in liquid nitrogen followed by trituration with a mortar and pestle. Then, the bone chips were transplanted into the tibial pinhole injury and the wound was closed. Animals were sacrificed at post-surgical day 7, 14, 21 and 28.
Results
FAK null cells can differentiate into osteoblasts
Extracellular matrix signals are required for osteoblast differentiation [34-36], and one class of molecules that might mediate this extracellular signaling are the integrins. Our first objective was to determine if integrin signaling, specifically via FAK, was essential for osteoblast differentiation. Along with other investigators, we have shown that function-blocking antibodies to integrins resulted in a significant reduction in formation of mineralized nodules in vitro [35-37]. These findings suggest that integrin signaling is necessary for the differentiation of progenitors into osteoblasts. There are, however, caveats to these types of in vitro experiments. The most obvious limitations are related to the specificity and/or efficacy of the function-blocking antibodies [38] and the inability to directly assess integrin activity. One read-out of integrin function is the state of ERK phosphorylation [39], but it is an indirect measure and therefore may not be the most accurate indicator of integrin activity.
To circumvent these limitations, we employed a genetic strategy for significantly reducing integrin signaling, in which FAK was inactivated by crossing FAKfl/− and FAKfl/fl mice with p53 null mice to generate FAKfl/fl;p53−/− and FAKfl/−;p53−/− double transgenic mice. Cells were then isolated from neonatal calvaria or from adult bone marrow of these mice, and transduced with an adenovirus carrying cre to eliminate functional FAK gene expression. The adenovirus construct contained gfp under the control of an internal ribosomal entry site, which allowed us to evaluate the infection efficiency using GFP expression. We found that Cre protein levels gradually increased during the first 72h and then tapered off (Fig. 1A). Concomitant with Cre expression was a gradual loss of FAK, which was substantially reduced by d5 (Fig. 1A). FAK null clones (n=35 from calvarial osteoblasts, and n=22 from bone marrow) were then established and the absence of FAK was verified using Western blot (Fig. 1B). The inactivation of p53 ensured that these cell lines were genetically immortalized [21]. Clones from FAKfl/fl;p53−/− served as positive controls (n=28 from calvarial osteoblasts, and 33 from bone marrow), and any FAK null clones that contained chromosomal cre were identified by PCR and then removed from further study.
Figure 1.
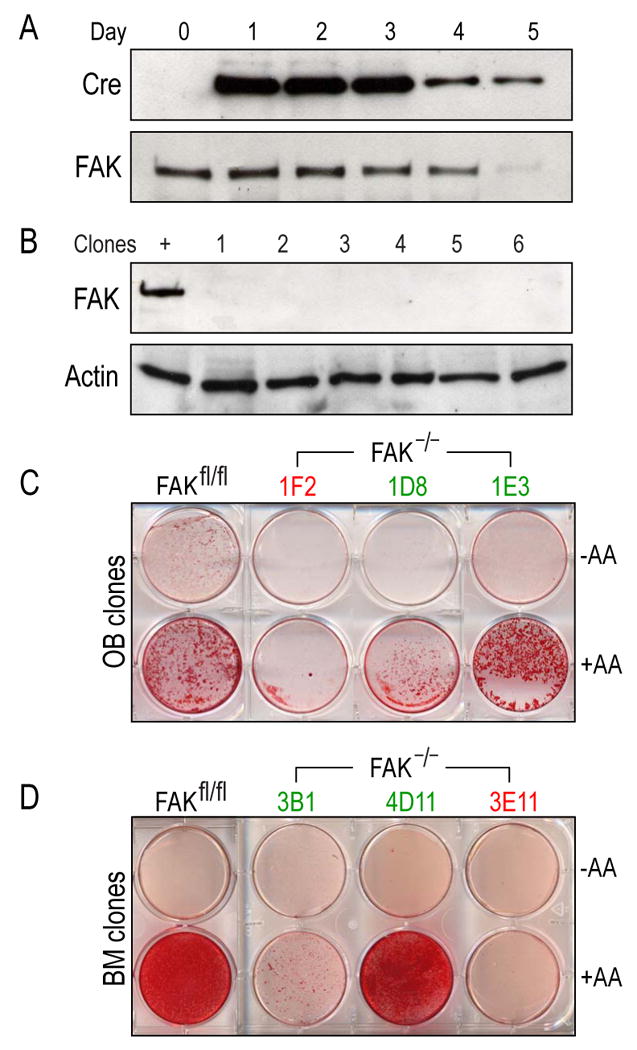
Establishment of FAK null osteoblast and bone marrow cell lines. (A) In vitro knockout of fak in osteoblasts. Cells from floxed animals (FAKfl/fl;p53−/−) were treated with adenovirus carrying cre. One microgram of total protein lysates was analyzed by Western blot using a monoclonal antibody against Cre. Signal was detected using enhanced chemiluminescence. After 5 days, the Cre protein level was tapered off. The same lysates were subjected to Western blot using a monoclonal antibody against FAK. FAK level was significantly reduced after 5 days of infection. (B) Establishment of FAK null osteoblast clonal lines. One microgram of total protein from immortalized calvarial osteoblasts was analyzed using monoclonal antibody against FAK. β-actin was used to ensure an equal amount of protein loading. +; positive control (FAKfl/fl;p53−/−), lane 1-6; FAK null osteoblast clonal lysates. (C,D) Calvarial osteoblast (C) and bone marrow cell line (D) differentiation in vitro. Immortalized calvarial FAK null clones were induced to differentiate in vitro. Cells were grown to confluency and differentiation was induced using ascorbic acid and β-glycerophosphate. After 2 weeks, cells were stained with 0.2% Alizarin Red to detect mineralization. A portion of FAK null clones (30%) differentiated in vitro. Mineralized and non-mineralized clones were labeled in red and green respectively. FAKfl/fl; parental cell line (FAKfl/fl;p53−/−), −AA; uninduced, +AA: induced by ascorbic acid and β -glycerophosphate; OB: osteoblast; BM; bone marrow.
With these reagents in hand we undertook a series of assays to test whether or not FAK null clones could differentiate into osteoblasts. Using clones derived from calvarial osteoblasts and from the bone marrow, we found that a majority of the clones (14/20, 70%) from both sources could not differentiate into osteoblasts (Fig. 1C,D, green labels). Conversely, 30% of FAK null clones could differentiate into osteoblasts when grown in osteogenic media (6/20; Fig. 1C,D, red labels). When wild type clones were challenged to differentiate, 8 out of 12 clones could differentiate (data not shown). These results suggest that while the majority of FAK null cells could not complete their differentiation into osteoblasts, a minority of FAK mutant cells could. Furthermore, results were comparable whether the source of the osteoprogenitor cells was the calvaria or the bone marrow (Fig. 1 and data not shown). This discrepancy raised a question whether FAK, and therefore integrin signaling, was essential for osteoblast differentiation. An abundant literature, however, implicates integrin signaling in the program of bone formation [40-43]. We decided to take another tactic that used the well characterized collagen type I promoter [44] to drive Cre expression and thus block FAK activity in developing embryos.
FAK inactivation in osteoprogenitor cells did not disrupt fetal skeletogenesis
Collagen type I is the predominant extracellular matrix protein in bone [45]. The 2.3 Kb fragment of the collagen type I(α)I promoter specifically drives expression in mature osteoblasts, and by crossing floxed FAK mice with Cre mice containing the 2.3 Kb promoter of collagen type I(α)I we generated mice carrying Cre+;FAKfl/fl and Cre+;FAKfl/− genotypes.
We confirmed that Cre-mediated recombination resulted in FAK inactivation by optimizing a PCR that amplified the intervening sequence between the two loxP sites. When Cre recombinase was active, the distance between the two PCR primers P1 and P2 was shortened (Fig. 2A). A typical genotyping PCR result showed the wild type allele (fl, 1.6 Kb) compared to the recombined allele (rec; 327bp; Fig. 2B). Hereafter, we refer to these mice as FAK mutants.
Figure 2.
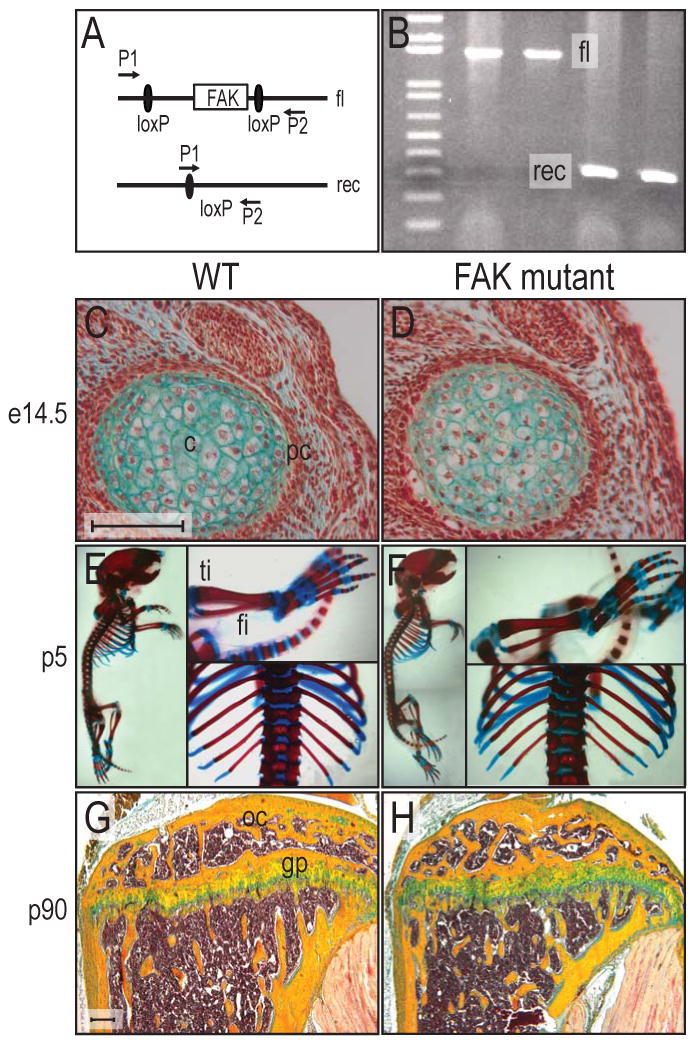
Cre-mediated recombination of fak and skeletal pattern in FAK mutants. (A) Genomic structure of floxed fak mouse. When the floxed gene is recombined by Cre recombinase, the distance between the P1 and P2 primer is shortened. Floxed and recombined alleles generate 1.6kbp and 327bp PCR products respectively. (B) PCR analysis of Cre-mediated recombination in the animals. Typical screening gel from tail clips. fl; floxed allele, rec; recombined allele by Cre recombinase. (C,D) Cross section of humeri at e14.5 of wild type mouse (C) and FAK mutant (D) were collected and processed for Movat's pentachrome staining. Both wild type and FAK mutant showed the same pattern of chondrogenesis in limb development. (E,F) Whole mount bone and cartilage staining of wild type and FAK mutant. Wild type P5 mouse (E) and FAK mutant (F) were stained with Alizarin Red and Alcian Blue. Skeletal system in FAK mutant was not affected. (G,H) Longitudinal sections of mouse tibiae from wild type (G) and FAK mutant (H). Tibiae from three month old wild type and FAK mutant showed well developed growth plates. fi: fibula, ti: tibia, gp: growth plate, oc: ossification center. Bar: 100 μm.
Given in vitro evidence describing an essential role for integrin signaling in osteoblast differentiation, we were surprised to find that FAK mutant embryos had an intact skeleton. We examined mice at a variety of embryonic stages (e.g., from e13.5 through post-natal and adult life) and despite careful histological staining, whole mount analyses, X-ray, and micro CT, we did not detect differences in patterning of the skeleton nor in the onset, rate, or extent of bone formation. For example, we inspected the fetal skeleton for discrepancies in the commitment or allocation of cells to chondrogenic and osteogenic lineages, and failed to detect any differences (Fig. 2C,D). The patterning and overall growth of the skeleton was indistinguishable between wild type and FAK mutants (Fig. 2E,F). We also monitored the growth of the juvenile and adult skeletons, as well as the architecture of the growth plates, and found no differences between wild type and FAK mutants (Fig. 2G,H). In addition, we examined bones that form through intramembranous ossification and failed to uncover any detectable alterations (data not shown). These analyses suggested that the loss of FAK is not critical for osteoblast differentiation in vivo. An alternative possibility is that FAK activity may only be transiently required and that other signaling pathways replace its function during fetal skeletogenesis. To separate out this potential redundancy we turned to an examination of the adult FAK mutant skeleton, reasoning that a subtle defect in osteoblast differentiation might manifest itself as a disruption in skeletal repair.
Acute injury in adult skeleton initiated FAK signaling
Adult bone formation is an integral component of skeletal remodeling and adult bone formation is greatly enhanced in response to a skeletal injury. We therefore generated small, mono-cortical tibial injuries in adult mutant mice in order to test whether the absence of FAK adversely affected bone regeneration.
Since fak has multiple splicing forms in vivo [46, 47] we used in situ hybridization to show where FAK transcripts were originally expressed in the adult skeleton, and where the gene had been deleted. cDNA probes were generated against the N- and C-terminal portions of FAK (Fig. 3A) to show that the Cre mediated recombination effectively deleted fak in collagen type I-expressing osteocytes (Fig. 3B,C). The C-terminal probe, however, recognized a truncated FAK gene in osteocytes (Fig. 3D,E), which generates the FAK related non-kinase protein, FRNK [48, 49]. FRNK lacks the kinase domains of FAK which mediate most of signaling functions of FAK [50].
Figure 3.
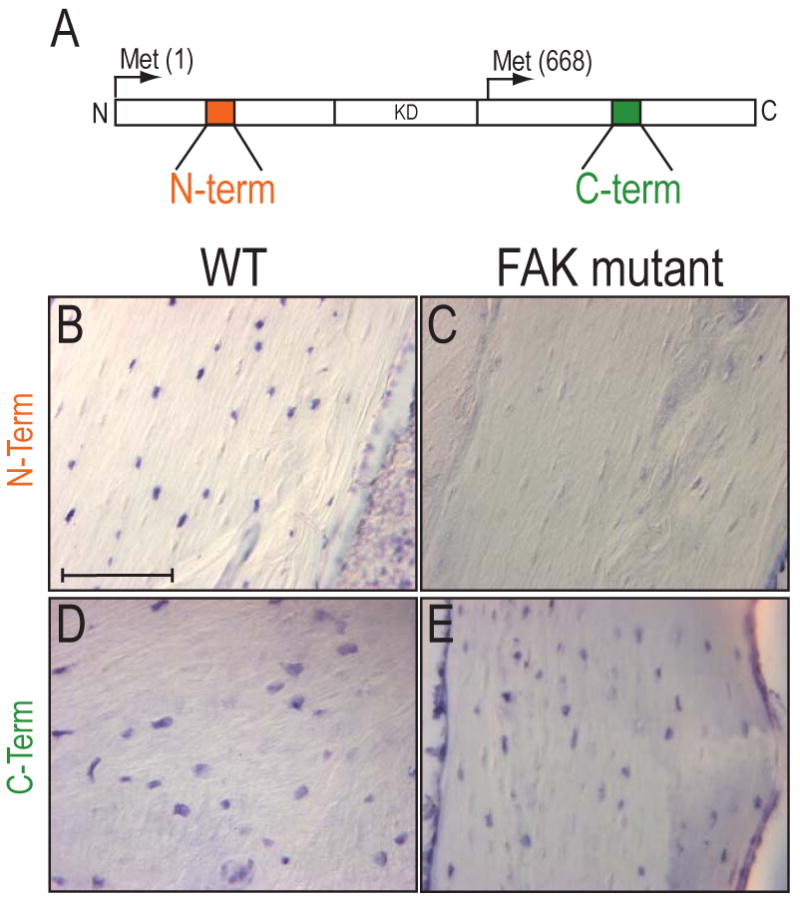
In situ hybridization for FAK in bone tissues. (A) Diagram of FAK protein and cDNA probe regions. FRNK is initiated at a methionine 668 and results in a truncated FAK protein. (B-E) In situ hybridization for N-terminal (B,C) and C-terminal of FAK transcripts (D,E) in wildtype and FAK mutant tibiae. FAK mutants were void of the transcripts for the N-terminal of FAK (C), while they expressed transcripts for the C-terminal of FAK transcripts (E). Bar: 100 μm.
Loss of FAK results in delayed bone regeneration
We confirmed that in FAK mutants collagen type I (col I), a molecular marker of osteoprogenitor cells [51, 52] continued to be expressed in the intact adult skeleton (Fig. 4A) and that the Cre protein co-localized with col I expression in the periosteum (Fig. 4B), and in col I-expressing osteocytes. The co-expression of runx2 and sox9 in the periosteum (Fig. 4C,D) indicated that at least some of the cells in which FAK was inactivated were osteoprogenitor cells [53-55].
Figure 4.
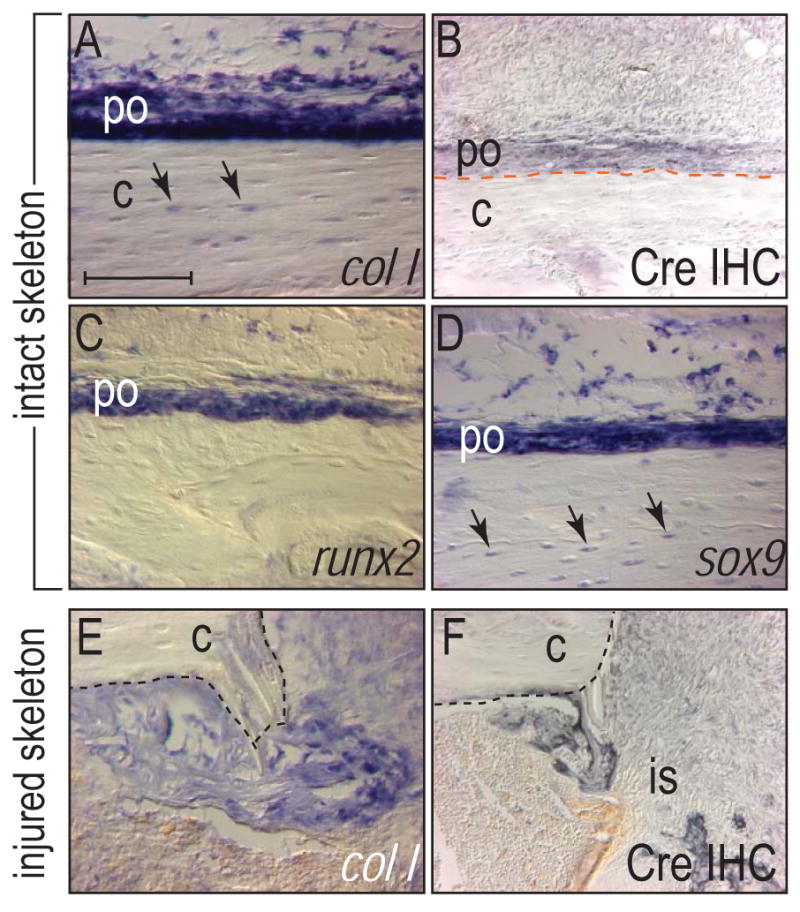
Molecular analyses of osteogenic genes in wild type animals and Cre expression in the Col1-Cre mouse. (A) In situ hybridization of wild type tibia for collagen type I. Col I was expressed in osteocytes (arrows) and in the cambial layer of the periosteum. (B) Immunohistochemistry for Cre in the tibia of the Col1-Cre mouse. Cre protein was detected in the periosteum. (C,D) In situ hybridization for runx2 (C) and sox9 (D) in wild type tibia. Sox9 was expressed in both periosteum and osteocytes (arrows). (E,F) Collagen type I (col I) in situ hybridization and Cre immunohistochemistry in the pinhole injury sites from FAK mutants at d7. Col I and Cre expression was colocalized in the injury site. (E). c: cortex, is: injury site, po: periosteum. Bar: 100 μm.
We generated mono-cortical defects in the tibiae of FAK mutants and wild type mice and then examined the healing response over a protracted time course. We first confirmed that FAK was inactivated in cells that contributed to bone repair. In FAK mutants, col I was strongly expressed in cells occupying the injury site (Fig. 4E) and this expression coincided with Cre immunostaining in the injury site (Fig. 4F). Thus the injury site was populated by osteoprogenitor cells deficient in FAK.
We next evaluated how FAK deletion affected skeletal repair over the course of healing. On post-surgical d7, wild type cells in the injury site had differentiated into osteoblasts and deposited a bony matrix intermingled with new blood vessels (n=4; Fig. 5A and data not shown). In FAK mutants, however, there was no evidence of a bony matrix (n=4; Fig. 5B).
Figure 5.
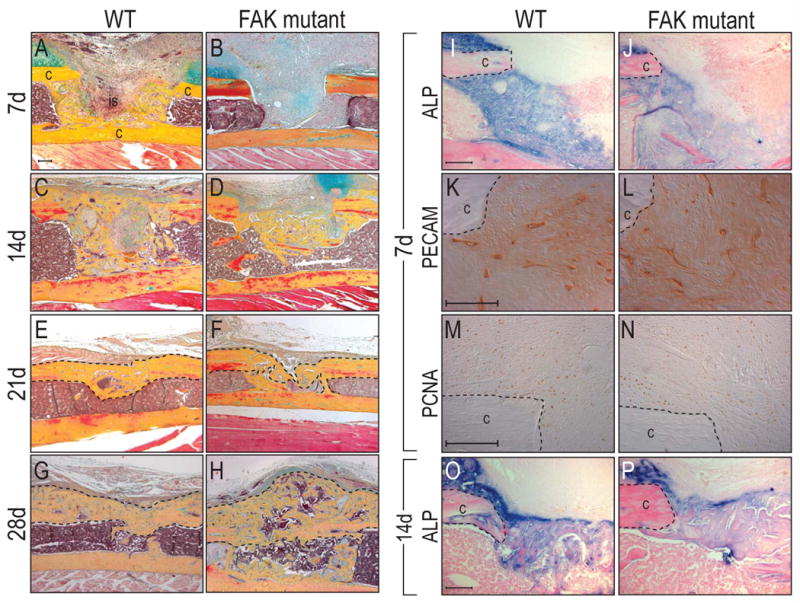
Delayed bone healing in FAK mutants. Mono-cortical tibial injuries were induced in wild type and FAK mutants. (A-H) Histology of wild type and FAK mutant injury at post-surgical d7, 14, 21 and 28. FAK mutants showed delayed skeletal healing compared to wild type animals. At 28 days after surgery, FAK mutants developed exuberant calluses compared to wild type counterparts. (I,J) Alkaline phosphatase activity in the injury sites at post-surgical d7. FAK mutants (J) showed less enzyme activity in the injury site compared to wild type animals (I). (K,L) PECAM staining at post-surgical d7 showed equivalent number of endothelial cells in both wild type (K) and FAK mutant (L). (M,N) PCNA staining revealed a similar proliferative activity in wild type and FAK mutant. c: cortex, is: injury site. Bar: 100 μm.
By post-surgical d14, a bony bridge spanned the cut cortices in wild type mice. In FAK mutants a bony matrix was evident but was insufficient to bridge the defect (n=3 for both wild type and mutants; Fig. 5C,D). At post-surgical d21, the amount of bone in the injury site was roughly equivalent between wild type and FAK mutants, indicating that FAK deletion delayed, but ultimately did not prevent, the deposition of a matrix that underwent mineralization (n=3; Fig. 5E,F). At post-surgical d28, wild type injury sites had undergone extensive bone remodeling (n=4 for both wild type and mutants; Fig. 5G). FAK mutants, on the other hand, developed large bony calluses compared to wild type counterparts and retained new bone in the marrow cavity (Fig. 5H).
To identify the mechanisms for delayed bone healing in FAK mutants, we performed a series of experiments. First, we examined alkaline phosphatase activity in the injury sites at post-surgical d7. Wild type animals showed significantly higher alkaline phosphatase activity than FAK mutants in the injury sites (Fig. 5I,J). Next, immunostaining for PECAM in the injury sites showed no obvious difference in wild type and FAK mutants (Fig. 5K,L). Since FAK is involved in cell proliferation [56], a proliferative defect might be responsible for fewer osteoblasts in the injury site. Therefore, we performed PCNA immunostaining of the injury sites at post-surgical d7. Both wild type and FAK mutant mice showed a similar proliferative activity. (Fig. 5M,N). By post-surgical d14, however, both wild type animals and FAK mutants showed comparable alkaline phosphatase in the injury sites (Fig. 5O,P) suggesting that osteoblast differentiation in FAK mutant mice was recovered. Together, these analyses demonstrate that the initial lack of a bony matrix was more likely due to a delay in osteoblast differentiation.
Bone matrix deposited by FAK mutant osteoblasts is defective
Almost as soon as new bone is deposited in an injury site, osteoclasts begin to remodel the matrix. We found evidence of abundant TRAP activity in wild type injury sites, in keeping with the exuberant remodeling that is characteristic of repair (Fig. 6A). In FAK mutant injury sites, osteoclast activity was low at post-surgical d7 (Fig. 6B), which further supported our conclusion that new bone deposition was delayed in the mutants. At all time points examined (i.e., post-surgical d7, 14, 21, and 28) we observed diminished TRAP activity (Fig. 6C,D).
Figure 6.
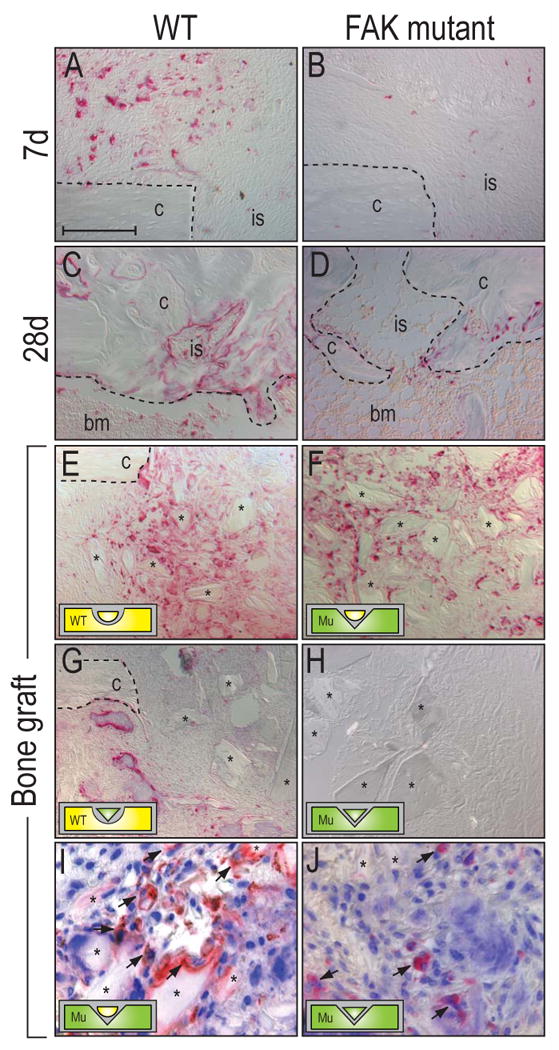
Defective osteoclast activity in FAK mutant mice. (A-D) TRAP staining of the injury sites in wild type (A,C) and FAK mutants (B,D) at post-surgical d7 and 28. Wild type animals showed higher TRAP activity throughout the healing process than FAK mutants. (E-F) Remodeling of bone chips [75] in wild type and FAK mutants at 7 days post-surgery. (E) Wild type bone chips in a wild type injury site. (F) Wild type bone chips in a FAK mutant. (G) FAK mutant bone chips in a wild type animal. (H) FAK mutant bone chips into a FAK mutant. Osteoclast activity in the animals that received FAK mutant bone chips did not activate bone remodeling. (I, J) Double staining of TRAP activity and nuclei in the bone grafted injury sites. (I) Wild type bone chips in a FAK mutant injury site. (J) Mutant bone chips in a FAK mutant injury site. Arrows indicate multi-nucleated osteoclasts that attached to the transplanted bone chips. bm: bone marrow, c: cortex, is: injury site. Bar: 100 μm.
Thus, our analyses demonstrate that FAK inactivation hampers the program of bone regeneration and remodeling. Initially, loss of FAK impedes the deposition of a mineralized matrix; with time, this defect is overcome, but a delay in osteoclast remodeling of the mutant extracellular matrix then hinders the latter stages of bone regeneration. Our genetic strategy, however, drives FAK inactivation in collagen type I-expressing osteoblasts and not in osteoclast precursors (Fig. 2). Was there some mechanism by which osteoclasts became defective in the adult animal? Or was the defect in bone regeneration due to perturbations in the deposition or organization of the extracellular matrix in the FAK mutant? We devised another method to independently assess the in vivo activity of osteoblasts and osteoclasts in another adult repair scenario.
We first tested whether osteoclasts from FAK mutant mice were defective in their ability to attach and remodel bone matrix. In a previous study, we demonstrated that bone matrix implanted into a mono-cortical defect serves as an excellent scaffold for osteoblasts to attach and deposit a mineralized matrix. Concomitant with this exuberant osteoblast activity is enhanced osteoclast remodeling of the implanted matrix [33]. Therefore, we prepared bone chips from wild type and mutant tibiae using a preparation that kills osteocytes and osteoblasts in the bone chips but leaves the mineralized matrix intact [57]. Next, mono-cortical defects, identical to those generated in our earlier experiments, were prepared in wild type and FAK mutant mice. Wild type bone chips were introduced into wild type injury sites (n=3; Fig. 6E), and wild type bone chips were placed into FAK mutant injury sites (n=4; Fig. 6F). Likewise, FAK mutant bone chips were introduced into wild type injury sites (n=3; Fig. 6G), and FAK mutant bone chips were placed into FAK mutant injury sites (n=3; Fig. 6H). The response was then examined at post-surgical d7.
Abundant TRAP activity was evident around wild type bone chips implanted in wild type injury sites (Fig. 6E), confirming the exuberant bone remodeling that characterizes this assay. When wild type bone chips were introduced into FAK mutants, we also noted an equivalent TRAP activity (Fig. 6F), indicating that the intrinsic activity of osteoclasts in mutant animals was not affected by the loss of FAK. In contrast, when attempting to remodel FAK mutant bone chips, wild type osteoclasts as well as osteoclasts from FAK mutant mice showed diminished activity (Fig. 6G,H), demonstrating that the matrix in FAK mutant animals was not suitable for remodeling by osteoclasts from either source. These data support the conclusion that the organization of the mineralized matrix and not the osteoclasts were defective in FAK mutant mice. To confirm that the TRAP positive cells were osteoclasts, we performed a double staining for TRAP and nuclei. Our data showed that TRAP positive cells in the injury site were multi-nucleated, a typical feature of osteoclasts. In addition, the results showed that osteoclasts in the FAK mutant attached to wild type bone chips, whereas osteoclasts failed to adhere to mutant bone chips in vivo (Fig. 6I,J, arrows).
When considered together, our data indicate that FAK mutant osteoblasts fail to secrete a bony matrix in a timely fashion. Even though the FAK mutant matrix eventually becomes mineralized, it still does not serve as a favorable substrate for osteoclast attachment and remodeling. Consequently, there is a delay in remodeling the FAK mutant matrix. Collectively, these defects culminate in delayed bone regeneration.
Pyk2 may substitute for FAK in vitro for osteoblast differentiation
By examining the program of skeletal repair in FAK mutant mice we gained insight into one compensatory mechanism that might compensate for the loss of FAK during osteoblast differentiation. One candidate molecule that may replace FAK function is Pyk2 kinase, which shares ∼45% sequence homology with FAK [58]. Functionally, Pyk2 can partially replace FAK in cell migration assays [41] and spreading [59]. We returned to the FAK null cell lines we had generated (Fig. 1) and evaluated a subset of them for evidence of altered Pyk2 activation of localization.
When Pyk2 is activated, Tyrosine 402 of Pyk2 is phosphorylated and this form of the protein can be detected with a phospho-specific antibody [60]. Western analyses showed that the level of total Pyk2 was decreased in FAK null cells (Fig. 7A, lanes 2 and 3). The level of activated Pyk2, however, was increased in these cells (Fig. 7B, lanes 2 and 3).
Figure 7.
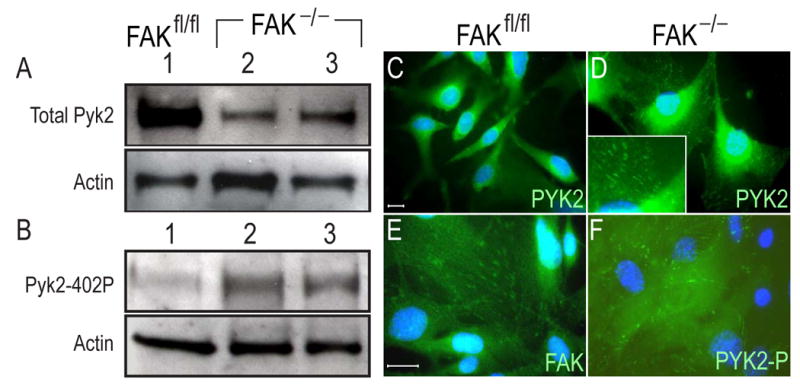
Western and Immunofluorescence analyses of FAK and Pyk2 in FAK null osteoblast cell lines. (A) Western blot of total Pyk2 protein in FAK null osteoblast clones (FAK−/−;p53−/−). A total of 2 μg of cell lysate was loaded in each lane. Membrane was blotted with Pyk2 antibody (A) and phosphospecific Pyk2 antibody (B). β-actin was used for a loading control. The activated form of Pyk2 was increased in FAK null cells. (C,E) Immunofluorescence of total Pyk2 (C) and FAK (E) in wild type (FAKfl/fl;p53−/−) osteoblast cells. Pyk2 was found in the perinuclear region in wild type cells. (D,F) Immunofluorescence analyses of total Pyk2 (D) and phosphospecific Pyk2 (F) in FAK null osteoblast cells (FAK−/−;p53−/−). Inset in panel D; high magnification of focal adhesions. When FAK was deleted, Pyk2 was redistributed into focal adhesions in FAK null cells. Active form of Pyk2 was found in focal adhesions in FAK null cells. Bar; 10 μm.
We then used cytohistochemistry to compare localization of Pyk2 in wild type cells. Pyk2 was localized in perinuclear region in wild type cells. In contrast, in FAK null cells Pyk2 was redistributed. The punctated pattern of Pyk2 staining suggested that in the absence of FAK, Pyk2 relocated to the focal adhesions (Fig. 7C,D). The same punctated staining pattern was observed using a FAK antibody in wild type cells (Fig. 7E), lending further support to our hypothesis that Pky2 protein occupied a new cellular location when FAK was deleted. Finally, we used an antibody against phospho-Pyk2 to show that activated Pyk2 was present at focal adhesion sites in FAK null osteoblasts (Fig. 7F). Taken together, these in vitro data indicated that functional Pyk2 relocated to the focal adhesions when FAK was deleted, where it could functionally substitute, at least in part, deleted FAK.
Discussion
FAK is not required for osteoblast differentiation in vitro
An abundant literature implicates integrin signaling in osteoblast differentiation but most of these data are obtained from in vitro assays. For example, a variety of integrins are expressed on the osteoblast cell surface and function-blocking antibodies to these integrins reduce osteoblast differentiation in vitro [34-36, 61, 62]. Chemicals that block potential downstream targets of integrin signaling, such as ERK and RAF, inhibit osteoblast differentiation in vitro [42, 63-65].
Our in vitro results with immortalized FAK null osteoblasts (i.e., FAKfl/fl;p53−/− calvarial and bone marrow osteoblasts), however, produced contradictory results. While the majority of FAK null cells did not differentiate into osteoblasts, 30% of them did. Our first thought was that these cells might have lost their potential to differentiate after prolonged culturing. We found, however, that freshly isolated calvarial osteoblasts from FAK mutant mice could also differentiate into osteoblasts. These data demonstrate that FAK may not be critical for osteoblast differentiation.
Generation of FAK conditional knockout mice in mature osteoblasts
Previous genetic studies have failed to shed light on the role of integrins in osteogenesis because most integrin mutants either die before osteoblast differentiation ensues (i.e., β1 and α5 integrin null mutants, (Stephens, 1995 #13347; Yang, 1993 #21776)), or the mutants are born and exhibit no skeletal phenotype (i.e., α1β1 or α2β1 integrin mutant, (Chen, 2002 #11877; Gardner, 1996 #13512)).
We undertook an alternative approach that circumvented early lethality and targeted significant disruption of integrin signaling to osteoblasts; the cells that deposit the bony matrix of the skeleton. We exploited the fact that when integrins bind to the extracellular matrix FAK is activated [66]. Therefore, perturbation of FAK in collagen type I expressing cells appeared to be an effective method to inhibit most, if not all, integrin signaling involved in osteoblast differentiation.
We used a Cre-loxP system to generate a conditional knockout of FAK, using the well characterized collagen type I(α)I promoter. Three fragments of the promoter have been used to generate transgenic mice [22, 44, 67]; among these, the 2.3 Kb Col I promoter fragment is specific for bone cells, although it is activated after osteoblast differentiation is initiated [51].
Using floxed FAK mice and 2.3 Kb collagen I-Cre mice, we generated mutants in which FAK is silenced in osteoblasts. FAK mutants did not express full length fak transcripts but did express a truncated form of FAK, FRNK (Fig. 3). FRNK in FAK mutants, however, is defective in many aspects of integrin signaling because it lacks the kinase domain and auto-phosphorylation site which are essential for its activation and subsequent binding of Src [48]. We could not rule out the possibility that even after gene deletion pre-existing fak transcripts remained functional in the osteoblasts of Cre+;FAKfl/fl mutants [68, 69]. To minimize any potential contribution from residual FAK and to achieve efficient Cre recombination, we introduced a fak null allele into the colony and generated a second line of transgenics (Cre+;FAKfl/−). Detailed histological analyses of FAK mutants from embryonic stages to adulthood in both lines showed the same normal skeletal anatomy and morphology (Fig. 2).
FAK mutants show delayed bone healing and remodeling
Because FAK is involved in both cell cycle progression and migration, adult skeletal repair represented a particularly attractive model in which to re-assess the role of FAK in osteoblast differentiation and skeletal progenitor migration. We evaluated the role of FAK in skeletal regeneration by generating small skeletal injuries in FAK mutants and their wild type counterparts.
The first and most obvious observation we made was that FAK mutant mice healed their skeletal injuries by generating new bone (Fig. 5). This confirmed the ability of cells to differentiate into osteoblasts in the absence of FAK-mediated integrin signaling. There was, however, a defect in this bone regeneration in FAK mutants. They were slow in depositing the extracellular matrix that could undergo normal mineralization (Fig. 5). In addition, FAK mutants generated larger calluses than wild type animals during the process of skeletal healing. These observations prompted us to investigate the ability of bone remodeling in FAK mutants.
Bi-directional integrin signaling is disrupted in FAK mutants
To identify the defects in bone remodeling in FAK mutants, we undertook a second assay in which bone chips were implanted into injury sites (Fig. 6). In this preparation of bone chips any contribution from living cells is nullified during the freezing step; however, the procedure leaves the bone matrix intact. This second assay confirmed that osteoclasts in FAK mutants are not defective since they could remodel the bone chips from wild type animals. On the contrary, both wild type and FAK mutant osteoclasts could not bind and remodel FAK mutant collagen matrix as efficiently as they could bind to and remodel wild type matrix (Fig. 6). These data strongly suggest that bone matrix in FAK mutant is not favorable for osteoclast binding in vivo.
To date, there is little evidence showing that integrin signaling can modulate collagen matrix organization in osteoblasts. The present study supports that idea and suggests that FAK-dependent bidirectional signaling between the inside osteoblast and the outside affects extracellular matrix organization. Thus, we propose that a loss of FAK in osteoblasts affects their ability to properly organize the collagen-rich matrix they secrete; consequently, osteoclasts can not properly attach to and remodel the defective matrix as efficiently as they do with wild type matrix.
Pyk2 may compensate for FAK in osteoblast differentiation
FAK does not appear to be critical in the late stages of osteoblast differentiation, but the possibility exists that other closely related molecules, such as Pyk2, compensate and thus obscure the requirement for FAK. For example, upon integrin activation FAK is recruited to focal adhesions in cells, a step that is mediated by the focal adhesion targeting (FAT) domain in the C-terminus of FAK [70]. FAK contains binding sites for other signaling molecules, which stabilize the entire signaling complex in the focal adhesion [4]. Targeting FAK into different locations in the cells resulted in destabilization of the signaling complex mediated by integrin and FAK [50, 71]. Therefore, the localization of FAK in the focal adhesion is an important factor for its full activation [72].
Pyk2 is localized to the perinuclear region [70, 73, 74], and over-expression of Pyk2 causes its translocation to focal adhesions with low efficiency [70]. In FAK null osteoblasts, however, Pyk2 spontaneously localizes to the focal adhesions, where it is present in its active form (Fig. 7). Pyk2 at the focal adhesion may subsume some of the functions of the deleted FAK and thus mask a further requirement for FAK in the process of osteoblast differentiation.
In sum, our data underscore the complex interactions between cells and their extracellular matrix that are integral to the process of differentiation and tissue repair. These data indicate that FAK is not a critical requirement for the differentiation of mesenchymal precursor cells into osteoblasts. However, once osteoblasts secrete their collagen type I, FAK dependent integrin signaling appears to be critical for the structural protein to become organized into a fibrillar network. If the osteoid matrix is defective, then remodeling of the matrix may also be disrupted. Such is the case in the FAK mutants here, where osteoclasts do not appear to be able to bind and remodel the matrix as efficiently as they remodel wild type bone matrix.
Acknowledgments
This work was supported by: Northern California Arthritis Foundation Chapter Grant, NIH RO1 (AR45989) (J.B.K.), NIH COHRCD grant (P60 DE13058) (C.H.D.), NIH R01 DE 012462 and Air Force FA9550-04-1-0075 (J.A.H). A portion of this work was conducted in a facility constructed with support from Research Facilities Improvement Grant # C06-RR16490, from NCRR-NIH.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Greenberg RS, Bernstein AM, Benezra M, Gelman IH, Taliana L, Masur SK. FAK-dependent regulation of myofibroblast differentiation. Faseb J. 2006;20:1006–8. doi: 10.1096/fj.05-4838fje. [DOI] [PubMed] [Google Scholar]
- 2.Yeo MG, Partridge MA, Ezratty EJ, Shen Q, Gundersen GG, Marcantonio EE. Src SH2 arginine 175 is required for cell motility: specific focal adhesion kinase targeting and focal adhesion assembly function. Mol Cell Biol. 2006;26:4399–409. doi: 10.1128/MCB.01147-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Damsky CH, Ilic D. Integrin signaling: it's where the action is. Curr Opin Cell Biol. 2002;14:594–602. doi: 10.1016/s0955-0674(02)00368-x. [DOI] [PubMed] [Google Scholar]
- 4.Sieg DJ, Hauck CR, Schlaepfer DD. Required role of focal adhesion kinase (FAK) for integrin-stimulated cell migration. J Cell Sci. 1999;112(Pt 16):2677–91. doi: 10.1242/jcs.112.16.2677. [DOI] [PubMed] [Google Scholar]
- 5.Schoenwaelder SM, Burridge K. Bidirectional signaling between the cytoskeleton and integrins. Curr Opin Cell Biol. 1999;11:274–86. doi: 10.1016/s0955-0674(99)80037-4. [DOI] [PubMed] [Google Scholar]
- 6.Danen EH, Yamada KM. Fibronectin, integrins, and growth control. J Cell Physiol. 2001;189:1–13. doi: 10.1002/jcp.1137. [DOI] [PubMed] [Google Scholar]
- 7.Schwartz MA. Integrin signaling revisited. Trends in Cell Biology. 2001;11:466–470. doi: 10.1016/s0962-8924(01)02152-3. [DOI] [PubMed] [Google Scholar]
- 8.Liu S, Calderwood DA, Ginsberg MH. Integrin cytoplasmic domain-binding proteins. J Cell Sci. 2000;113(Pt 20):3563–71. doi: 10.1242/jcs.113.20.3563. [DOI] [PubMed] [Google Scholar]
- 9.Schaller MD, Borgman CA, Cobb BS, Vines RR, Reynolds AB, Parsons JT. pp125FAK a structurally distinctive protein-tyrosine kinase associated with focal adhesions. Proc Natl Acad Sci U S A. 1992;89:5192–6. doi: 10.1073/pnas.89.11.5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chan PY, Kanner SB, Whitney G, Aruffo A. A transmembrane-anchored chimeric focal adhesion kinase is constitutively activated and phosphorylated at tyrosine residues identical to pp125FAK. J Biol Chem. 1994;269:20567–74. [PubMed] [Google Scholar]
- 11.Schlaepfer DD, Hauck CR, Sieg DJ. Signaling through focal adhesion kinase. Prog Biophys Mol Biol. 1999;71:435–78. doi: 10.1016/s0079-6107(98)00052-2. [DOI] [PubMed] [Google Scholar]
- 12.Klemke RL, Cai S, Giannini AL, Gallagher PJ, de Lanerolle P, Cheresh DA. Regulation of cell motility by mitogen-activated protein kinase. J Cell Biol. 1997;137:481–92. doi: 10.1083/jcb.137.2.481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen HC, Appeddu PA, Parsons JT, Hildebrand JD, Schaller MD, Guan JL. Interaction of focal adhesion kinase with cytoskeletal protein talin. J Biol Chem. 1995;270:16995–9. doi: 10.1074/jbc.270.28.16995. [DOI] [PubMed] [Google Scholar]
- 14.Brown MC, Perrotta JA, Turner CE. Identification of LIM3 as the principal determinant of paxillin focal adhesion localization and characterization of a novel motif on paxillin directing vinculin and focal adhesion kinase binding. J Cell Biol. 1996;135:1109–23. doi: 10.1083/jcb.135.4.1109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Smilenov LB, Mikhailov A, Pelham RJ, Marcantonio EE, Gundersen GG. Focal adhesion motility revealed in stationary fibroblasts. Science. 1999;286:1172–4. doi: 10.1126/science.286.5442.1172. [DOI] [PubMed] [Google Scholar]
- 16.Otey CA, Pavalko FM, Burridge K. An interaction between alpha-actinin and the beta 1 integrin subunit in vitro. J Cell Biol. 1990;111:721–9. doi: 10.1083/jcb.111.2.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Schaller MD, Hildebrand JD, Shannon JD, Fox JW, Vines RR, Parsons JT. Autophosphorylation of the focal adhesion kinase, pp125FAK, directs SH2-dependent binding of pp60src. Mol Cell Biol. 1994;14:1680–8. doi: 10.1128/mcb.14.3.1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schlaepfer DD, Jones KC, Hunter T. Multiple Grb2-mediated integrin-stimulated signaling pathways to ERK2/mitogen-activated protein kinase: summation of both c-Src- and focal adhesion kinase-initiated tyrosine phosphorylation events. Mol Cell Biol. 1998;18:2571–85. doi: 10.1128/mcb.18.5.2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bai R, Chen S, Liu Y, Fu J, Li J, Yu H, Chang Y, Ruan G. beta1 Integrin Dysfunction in Adult Chronic Myeloid Leukemia Bone Marrow Cells. Zhongguo Shi Yan Xue Ye Xue Za Zhi. 2000;8:85–89. [PubMed] [Google Scholar]
- 20.Tahiliani PD, Singh L, Auer KL, LaFlamme SE. The role of conserved amino acid motifs within the integrin beta3 cytoplasmic domain in triggering focal adhesion kinase phosphorylation. J Biol Chem. 1997;272:7892–8. doi: 10.1074/jbc.272.12.7892. [DOI] [PubMed] [Google Scholar]
- 21.Ilic D, Furuta Y, Kanazawa S, Takeda N, Sobue K, Nakatsuji N, Nomura S, Fujimoto J, Okada M, Yamamoto T. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature. 1995;377:539–44. doi: 10.1038/377539a0. [DOI] [PubMed] [Google Scholar]
- 22.Dacquin R, Starbuck M, Schinke T, Karsenty G. Mouse alpha1(I)-collagen promoter is the best known promoter to drive efficient Cre recombinase expression in osteoblast. Dev Dyn. 2002;224:245–51. doi: 10.1002/dvdy.10100. [DOI] [PubMed] [Google Scholar]
- 23.Beggs HE, Schahin-Reed D, Zang K, Goebbels S, Nave KA, Gorski J, Jones KR, Sretavan D, Reichardt LF. FAK deficiency in cells contributing to the basal lamina results in cortical abnormalities resembling congenital muscular dystrophies. Neuron. 2003;40:501–14. doi: 10.1016/s0896-6273(03)00666-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krause DS, Theise ND, Collector MI, Henegariu O, Hwang S, Gardner R, Neutzel S, Sharkis SJ. Multi-organ, multi-lineage engraftment by a single bone marrow-derived stem cell. Cell. 2001;105:369–77. doi: 10.1016/s0092-8674(01)00328-2. [DOI] [PubMed] [Google Scholar]
- 25.Zhang R, Ducy P, Karsenty G. 1,25-dihydroxyvitamin D3 inhibits Osteocalcin expression in mouse through an indirect mechanism. J Biol Chem. 1997;272:110–6. doi: 10.1074/jbc.272.1.110. [DOI] [PubMed] [Google Scholar]
- 26.Kim JB, Islam S, Kim YJ, Prudoff RS, Sass KM, Wheelock MJ, Johnson KR. N-Cadherin extracellular repeat 4 mediates epithelial to mesenchymal transition and increased motility. J Cell Biol. 2000;151:1193–206. doi: 10.1083/jcb.151.6.1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. Measurement of protein using bicinchoninic acid. Anal Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- 28.Sheehan DC, Hrapchak BB. Columbus. Second. Ohio: Batelle Press; 1980. Theory and practice of Histotechnology; pp. 103–104. [Google Scholar]
- 29.Pera IL, Iuliano R, Florio T, Susini C, Trapasso F, Santoro M, Chiariotti L, Schettini G, Viglietto G, Fusco A. The rat tyrosine phosphatase eta increases cell adhesion by activating c-Src through dephosphorylation of its inhibitory phosphotyrosine residue. Oncogene. 2005;24:3187–95. doi: 10.1038/sj.onc.1208510. [DOI] [PubMed] [Google Scholar]
- 30.Colnot C, Thompson Z, Miclau T, Werb Z, Helms JA. Altered fracture repair in the absence of MMP9. Development. 2003;130:4123–33. doi: 10.1242/dev.00559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.McLeod MJ. Differential staining of cartilage and bone in whole mouse fetuses by alcian blue and alizarin red S. Teratology. 1980;22:299–301. doi: 10.1002/tera.1420220306. [DOI] [PubMed] [Google Scholar]
- 32.Albrecht UEG, Helms JA, Lin H. Visualization of gene expression patterns by in situ hybridization. In: Daston GP, editor. Molecular and cellular methods in developmental toxicology. CRC Press; Boca Raton, FL: 1997. pp. 23–48. [Google Scholar]
- 33.Colnot C, Romero DM, Huang S, Helms JA. Mechanisms of action of demineralized bone matrix in the repair of cortical bone defects. Clin Orthop Relat Res. 2005:69–78. doi: 10.1097/00003086-200506000-00012. [DOI] [PubMed] [Google Scholar]
- 34.Globus RK, Moursi A, Zimmerman D, Lull J, Damsky C. Integrin-extracellular matrix interactions in connective tissue remodeling and osteoblast differentiation. ASGSB Bull. 1995;8:19–28. [PubMed] [Google Scholar]
- 35.Moursi AM, Damsky CH, Lull J, Zimmerman D, Doty SB, Aota S, Globus RK. Fibronectin regulates calvarial osteoblast differentiation. J Cell Sci. 1996;109(Pt 6):1369–80. doi: 10.1242/jcs.109.6.1369. [DOI] [PubMed] [Google Scholar]
- 36.Jikko A, Harris SE, Chen D, Mendrick DL, Damsky CH. Collagen integrin receptors regulate early osteoblast differentiation induced by BMP-2. J Bone Miner Res. 1999;14:1075–83. doi: 10.1359/jbmr.1999.14.7.1075. [DOI] [PubMed] [Google Scholar]
- 37.Salasznyk RM, Klees RF, Hughlock MK, Plopper GE. ERK signaling pathways regulate the osteogenic differentiation of human mesenchymal stem cells on collagen I and vitronectin. Cell Commun Adhes. 2004;11:137–53. doi: 10.1080/15419060500242836. [DOI] [PubMed] [Google Scholar]
- 38.Gronthos S, Stewart K, Graves SE, Hay S, Simmons PJ. Integrin expression and function on human osteoblast-like cells. J Bone Miner Res. 1997;12:1189–97. doi: 10.1359/jbmr.1997.12.8.1189. [DOI] [PubMed] [Google Scholar]
- 39.Takeuchi Y, Suzawa M, Kikuchi T, Nishida E, Fujita T, Matsumoto T. Differentiation and transforming growth factor-beta receptor down-regulation by collagen-alpha2beta1 integrin interaction is mediated by focal adhesion kinase and its downstream signals in murine osteoblastic cells. J Biol Chem. 1997;272:29309–16. doi: 10.1074/jbc.272.46.29309. [DOI] [PubMed] [Google Scholar]
- 40.Tamura Y, Takeuchi Y, Suzawa M, Fukumoto S, Kato M, Miyazono K, Fujita T. Focal adhesion kinase activity is required for bone morphogenetic protein--Smad1 signaling and osteoblastic differentiation in murine MC3T3-E1 cells. J Bone Miner Res. 2001;16:1772–9. doi: 10.1359/jbmr.2001.16.10.1772. [DOI] [PubMed] [Google Scholar]
- 41.Sieg DJ, Ilic D, Jones KC, Damsky CH, Hunter T, Schlaepfer DD. Pyk2 and Src-family protein-tyrosine kinases compensate for the loss of FAK in fibronectin-stimulated signaling events but Pyk2 does not fully function to enhance FAK- cell migration. Embo J. 1998;17:5933–47. doi: 10.1093/emboj/17.20.5933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lai CF, Cheng SL. Alphavbeta integrins play an essential role in BMP-2 induction of osteoblast differentiation. J Bone Miner Res. 2005;20:330–40. doi: 10.1359/JBMR.041013. [DOI] [PubMed] [Google Scholar]
- 43.Reyes CD, Garcia AJ. Alpha2beta1 integrin-specific collagen-mimetic surfaces supporting osteoblastic differentiation. J Biomed Mater Res A. 2004;69:591–600. doi: 10.1002/jbm.a.30034. [DOI] [PubMed] [Google Scholar]
- 44.Rossert J, Eberspaecher H, de Crombrugghe B. Separate cis-acting DNA elements of the mouse pro-alpha 1(I) collagen promoter direct expression of reporter genes to different type I collagen-producing cells in transgenic mice. J Cell Biol. 1995;129:1421–32. doi: 10.1083/jcb.129.5.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Robinson RAa, W ML. Collagen-crystal relationships in bone as seen in the electron microscope. The anatomical record. 1952;114:383–409. doi: 10.1002/ar.1091140302. [DOI] [PubMed] [Google Scholar]
- 46.Burgaya F, Toutant M, Studler JM, Costa A, Le Bert M, Gelman M, Girault JA. Alternatively spliced focal adhesion kinase in rat brain with increased autophosphorylation activity. J Biol Chem. 1997;272:28720–5. doi: 10.1074/jbc.272.45.28720. [DOI] [PubMed] [Google Scholar]
- 47.Toutant M, Costa A, Studler JM, Kadare G, Carnaud M, Girault JA. Alternative splicing controls the mechanisms of FAK autophosphorylation. Mol Cell Biol. 2002;22:7731–43. doi: 10.1128/MCB.22.22.7731-7743.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schaller MD, Borgman CA, Parsons JT. Autonomous expression of a noncatalytic domain of the focal adhesion-associated protein tyrosine kinase pp125FAK. Mol Cell Biol. 1993;13:785–91. doi: 10.1128/mcb.13.2.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nolan K, Lacoste J, Parsons JT. Regulated expression of focal adhesion kinase-related nonkinase, the autonomously expressed C-terminal domain of focal adhesion kinase. Mol Cell Biol. 1999;19:6120–9. doi: 10.1128/mcb.19.9.6120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Richardson A, Parsons T. A mechanism for regulation of the adhesion-associated proteintyrosine kinase pp125FAK. Nature. 1996;380:538–40. doi: 10.1038/380538a0. [DOI] [PubMed] [Google Scholar]
- 51.Kalajzic I, Kalajzic Z, Kaliterna M, Gronowicz G, Clark SH, Lichtler AC, Rowe D. Use of type I collagen green fluorescent protein transgenes to identify subpopulations of cells at different stages of the osteoblast lineage. J Bone Miner Res. 2002;17:15–25. doi: 10.1359/jbmr.2002.17.1.15. [DOI] [PubMed] [Google Scholar]
- 52.Candeliere GA, Rao Y, Floh A, Sandler SD, Aubin JE. cDNA fingerprinting of osteoprogenitor cells to isolate differentiation stage-specific genes. Nucleic Acids Res. 1999;27:1079–83. doi: 10.1093/nar/27.4.1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Allen MR, Hock JM, Burr DB. Periosteum: biology, regulation, and response to osteoporosis therapies. Bone. 2004;35:1003–12. doi: 10.1016/j.bone.2004.07.014. [DOI] [PubMed] [Google Scholar]
- 54.Akiyama Ddagger H, Kim Ddagger JE, Nakashima K, Balmes G, Iwai N, Deng JM, Zhang Z, Martin JF, Behringer RR, Nakamura T, de Crombrugghe B. Osteo-chondroprogenitor cells are derived from Sox9 expressing precursors. Proc Natl Acad Sci U S A. 2005;102:14665–70. doi: 10.1073/pnas.0504750102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Komori T, Yagi H, Nomura S, Yamaguchi A, Sasaki K, Deguchi K, Shimizu Y, Bronson RT, Gao YH, Inada M, Sato M, Okamoto R, Kitamura Y, Yoshiki S, Kishimoto T. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell. 1997;89:755–64. doi: 10.1016/s0092-8674(00)80258-5. [DOI] [PubMed] [Google Scholar]
- 56.Taylor JM, Mack CP, Nolan K, Regan CP, Owens GK, Parsons JT. Selective expression of an endogenous inhibitor of FAK regulates proliferation and migration of vascular smooth muscle cells. Mol Cell Biol. 2001;21:1565–72. doi: 10.1128/MCB.21.5.1565-1572.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Etienne G, Ragland PS, Mont MA. Use of cancellous bone chips and demineralized bone matrix in the treatment of acetabular osteolysis: preliminary 2-year follow-up. Orthopedics. 2004;27:s123–6. doi: 10.3928/0147-7447-20040102-08. [DOI] [PubMed] [Google Scholar]
- 58.Schlaepfer DD, Broome MA, Hunter T. Fibronectin-stimulated signaling from a focal adhesion kinase-c-Src complex: involvement of the Grb2, p130cas, and Nck adaptor proteins. Mol Cell Biol. 1997;17:1702–13. doi: 10.1128/mcb.17.3.1702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Owen JD, Ruest PJ, Fry DW, Hanks SK. Induced focal adhesion kinase (FAK) expression in FAK-null cells enhances cell spreading and migration requiring both auto- and activation loop phosphorylation sites and inhibits adhesion-dependent tyrosine phosphorylation of Pyk2. Mol Cell Biol. 1999;19:4806–18. doi: 10.1128/mcb.19.7.4806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ivankovic-Dikic I, Gronroos E, Blaukat A, Barth BU, Dikic I. Pyk2 and FAK regulate neurite outgrowth induced by growth factors and integrins. Nat Cell Biol. 2000;2:574–81. doi: 10.1038/35023515. [DOI] [PubMed] [Google Scholar]
- 61.Schneider GB, Zaharias R, Stanford C. Osteoblast integrin adhesion and signaling regulate mineralization. J Dent Res. 2001;80:1540–4. doi: 10.1177/00220345010800061201. [DOI] [PubMed] [Google Scholar]
- 62.Sinha RK, Tuan RS. Regulation of human osteoblast integrin expression by orthopedic implant materials. Bone. 1996;18:451–7. doi: 10.1016/8756-3282(96)00044-0. [DOI] [PubMed] [Google Scholar]
- 63.Kim HH, Chung WJ, Lee SW, Chung PJ, You JW, Kwon HJ, Tanaka S, Lee ZH. Association of sustained ERK activity with integrin beta3 induction during receptor activator of nuclear factor kappaB ligand (RANKL)-directed osteoclast differentiation. Exp Cell Res. 2003;289:368–77. doi: 10.1016/s0014-4827(03)00288-x. [DOI] [PubMed] [Google Scholar]
- 64.Nakayamada S, Okada Y, Saito K, Tamura M, Tanaka Y. Beta1 integrin/focal adhesion kinase-mediated signaling induces intercellular adhesion molecule 1 and receptor activator of nuclear factor kappaB ligand on osteoblasts and osteoclast maturation. J Biol Chem. 2003;278:45368–74. doi: 10.1074/jbc.M308786200. [DOI] [PubMed] [Google Scholar]
- 65.Gallea S, Lallemand F, Atfi A, Rawadi G, Ramez V, Spinella-Jaegle S, Kawai S, Faucheu C, Huet L, Baron R, Roman-Roman S. Activation of mitogen-activated protein kinase cascades is involved in regulation of bone morphogenetic protein-2-induced osteoblast differentiation in pluripotent C2C12 cells. Bone. 2001;28:491–8. doi: 10.1016/s8756-3282(01)00415-x. [DOI] [PubMed] [Google Scholar]
- 66.Calalb MB, Polte TR, Hanks SK. Tyrosine phosphorylation of focal adhesion kinase at sites in the catalytic domain regulates kinase activity: a role for Src family kinases. Mol Cell Biol. 1995;15:954–63. doi: 10.1128/mcb.15.2.954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Dacic S, Kalajzic I, Visnjic D, Lichtler AC, Rowe DW. Col1a1-driven transgenic markers of osteoblast lineage progression. J Bone Miner Res. 2001;16:1228–36. doi: 10.1359/jbmr.2001.16.7.1228. [DOI] [PubMed] [Google Scholar]
- 68.Ochel HJ, Schulte TW, Nguyen P, Trepel J, Neckers L. The benzoquinone ansamycin geldanamycin stimulates proteolytic degradation of focal adhesion kinase. Mol Genet Metab. 1999;66:24–30. doi: 10.1006/mgme.1998.2774. [DOI] [PubMed] [Google Scholar]
- 69.Kornberg L, Earp HS, Parsons JT, Schaller M, Juliano RL. Cell adhesion or integrin clustering increases phosphorylation of a focal adhesion-associated tyrosine kinase. J Biol Chem. 1992;267:23439–42. [PubMed] [Google Scholar]
- 70.Schaller MD, Sasaki T. Differential signaling by the focal adhesion kinase and cell adhesion kinase beta. J Biol Chem. 1997;272:25319–25. doi: 10.1074/jbc.272.40.25319. [DOI] [PubMed] [Google Scholar]
- 71.Gilmore AP, Romer LH. Inhibition of focal adhesion kinase (FAK) signaling in focal adhesions decreases cell motility and proliferation. Mol Biol Cell. 1996;7:1209–24. doi: 10.1091/mbc.7.8.1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Igishi T, Fukuhara S, Patel V, Katz BZ, Yamada KM, Gutkind JS. Divergent signaling pathways link focal adhesion kinase to mitogen-activated protein kinase cascades. Evidence for a role of paxillin in c-Jun NH(2)-terminal kinase activation. J Biol Chem. 1999;274:30738–46. doi: 10.1074/jbc.274.43.30738. [DOI] [PubMed] [Google Scholar]
- 73.Duong LT, Rodan GA. PYK2 is an adhesion kinase in macrophages, localized in podosomes and activated by beta(2)-integrin ligation. Cell Motil Cytoskeleton. 2000;47:174–88. doi: 10.1002/1097-0169(200011)47:3<174::AID-CM2>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- 74.Williams LM, Ridley AJ. Lipopolysaccharide induces actin reorganization and tyrosine phosphorylation of Pyk2 and paxillin in monocytes and macrophages. J Immunol. 2000;164:2028–36. doi: 10.4049/jimmunol.164.4.2028. [DOI] [PubMed] [Google Scholar]
- 75.Rális ZA, Rális HM. A simple method for demonstration of osteoid in paraffin sections. Medical Laboratory Technology. 1975;32:203–13. [PubMed] [Google Scholar]