

Abstract
Water, acting as a rogue nucleophile, can disrupt transesterification steps of important phosphoryl transfer reactions in DNA and RNA. We have unveiled this risk, and identified safeguards instituted against it, during strand cleavage and joining by the tyrosine site-specific recombinase Flp. Strand joining is threatened by a latent Flp endonuclease activity (type I) towards the 3′-phosphotyrosyl intermediate resulting from strand cleavage. This risk is not alleviated by phosphate electrostatics; neutralizing the negative charge on the scissile phosphate through methylphosphonate (MeP) substitution does not stimulate type I endonuclease. Rather, protection derives from the architecture of the recombination synapse and conformational dynamics within it. Strand cleavage is protected against water by active site electrostatics. Replacement of the catalytic Arg-308 of Flp by alanine, along with MeP substitution, elicits a second Flp endonuclease activity (type II) that directly targets the scissile phosphodiester bond in DNA. MeP substitution, combined with appropriate active site mutations, will be useful in revealing anti-hydrolytic mechanisms engendered by systems that mediate DNA relaxation, DNA transposition, site-specific recombination, telomere resolution, RNA splicing and retrohoming of mobile introns.
Keywords: active site electrostatics, methylphosphonate, type I Flp endonuclease, type II Flp endonuclease, tyrosine recombinases
Introduction
Transesterification reactions at phosphodiester targets in nucleic acids are ubiquitous and copious in biological systems. They include DNA relaxation by type I and type II topoisomerases, conservative site-specific recombination, DNA transposition, telomere resolution in spirochetes, RNA splicing and retrohoming by mobile introns (Cech, 1983; Mizuuchi, 1997; Belfort et al, 2002; Craig, 2002; Chaconas, 2005). The active nucleophiles in these phosphoryl transfer steps are derived from the topoisomerase, recombinase or telomere resolvase or from the nucleic acid: an active site tyrosine or serine or 5′, 3′ or 2′ hydroxyl groups. These nucleophiles face the challenge of performing their function in the presence of an overwhelming excess of water, approximately 56 M. The vast abundance of water, combined with its overall reactivity, makes it a potential rogue nucleophile that can defeat the native nucleophiles by hydrolyzing an activated phosphodiester target. What electrostatic, chemical or conformational designs do phosphoryl transfer systems incorporate to protect themselves against the contrary action of water? Methylphosphonate (MeP) substituted DNA substrates in reactions carried out by the Flp site-specific recombinase of Saccharomyces cerevisiae provide insights into this issue with far reaching implications. This work was inspired by Shuman and colleagues who elegantly showed a critical role for phosphate electrostatics in warding off the untoward action of water during DNA relaxation by vaccinia type IB topoisomerase (Tian et al, 2003, 2005).
Flp is a eukaryotic representative of the tyrosine family of recombinases (Jayaram et al, 2002, 2004). Its physiological role is in mediating DNA amplification of the S. cerevisiae plasmid 2 μm circle by an appropriately timed recombination event that switches bi-directional replication into uni-directional ‘double-rolling-circle' replication (Futcher, 1986; Volkert and Broach, 1986; Reynolds et al, 1987). A copy number drop in the plasmid resulting from rare missegregation events can thus be corrected, contributing to the high copy propagation of this selfish DNA element. In conformity with the rest of the tyrosine family members, Flp follows the chemistry of type IB topoisomerase reaction. Attack of the scissile phosphodiester bond by the active site nucleophile Tyr-343 of Flp results in strand cleavage with concomitant formation of a 3′-phosphotyrosyl intermediate (Rice, 2002). The 5′-hydroxyl group generated at the cleavage step serves as the nucleophile for strand joining. Strand exchange ensues when the 5′-hydroxyl formed within a DNA substrate attacks the 3′-phosphotyrosyl bond formed within its partner. Recombination takes place within a synaptic arrangement containing four recombinase monomers and two DNA target sites. The reaction is completed in two steps of single strand exchanges, a Holliday junction being an obligatory intermediate in the pathway (Figure 1A). For comparison, one round of DNA relaxation by a type IB topoisomerase is diagrammed in Figure 1B.
Figure 1.

Reaction pathways and active site configurations for tyrosine site-specific recombinase and type IB topoisomerase reactions. (A) Recombination by Flp site-specific recombinase proceeds through the following steps: (a) single strand cleavages; (b) strand exchange and formation of a Holliday junction intermediate; (c) isomerization of the Holliday junction; (d, e) resolution of the junction into recombinant products. Two Flp monomers (green) activate the scissile phosphodiester bonds in cis, and the other two (magenta) donate the tyrosine nucleophile in trans. (B) A round of DNA relaxation by vaccinia topoisomerase I proceeds through (a) single strand cleavage, (b) strand rotation and (c) strand religation. Activation of the scissile phosphodiester and strand cleavage is mediated in cis by a single topoisomerase monomer. (C) In the Flp active site (Chen et al, 2000), the catalytic pentad of Arg-191, Lys-223, His-305, Arg-308 and Trp-330 are derived from one Flp monomer (green), and Tyr-343 poised to attack the scissile phosphate (gold sphere) is supplied by the neighbouring Flp monomer (magenta). (D) The active site of the small pox virus topoisomerase enzyme shown here (Perry et al, 2006) is functionally related to that of vaccinia topoisomerase (Cheng et al, 1998). It is characterized by a catalytic pentad of Arg-130, Lys-167, Lys-220, Arg-223 and His-265 plus Tyr-274, all derived from a single topoisomerase monomer.
In addition to Tyr-343, the Flp active site is characterized by a catalytic pentad comprising Arg-191, Lys-223, His-305, Arg-308 and Trp-330 (Chen et al, 2000; Rice, 2002). In the majority of tyrosine recombinases, the pentad contains a histidine at the position corresponding to Trp-330 of Flp (Esposito and Scocca, 1997; Nunes-Duby et al, 1998). The type IB topoisomerases also display a similar constellation of catalytic residues, except that His-305 and Trp-330 positions are occupied by lysine and histidine, respectively (Redinbo et al, 1998; Krogh and Shuman, 2000; Van Duyne, 2002; Perry et al, 2006) (Figure 1D). The assembly of the Flp active site is rather unusual in that it is shared between two neighbouring recombinase monomers, one of which provides the pentad and the other the tyrosine nucleophile (Chen et al, 2000; Chen and Rice, 2003). As a result, strand cleavage by Flp occurs in a trans fashion, that is, across the crossover region (or spacer) from where the tyrosine donating Flp monomer is bound (Figure 1A and C). In the other well characterized tyrosine recombinases, lambda Int, HP1 Int, Cre and XerC/XerD, the entire active site is housed within a single monomer, and strand cleavage takes place in cis (Nunes-Duby et al, 1994; Arciszewska and Sherratt, 1995; Guo et al, 1997; Hickman et al, 1997; Biswas et al, 2005).
In the catalytically active dimer of Flp, Arg-191, Lys-223, His-305 and Arg-308 of the pentad contact the scissile phosphate, with Lys-223 also contacting O2 of the adjacent cytosine (Chen et al, 2000). Trp-330 helps orient Tyr-343 primarily through hydrophobic and van der Walls interactions with residues of the adjacent Flp monomer (Chen and Rice, 2003). In addition, it appears to play a subsidiary role in activating the scissile phosphate and facilitating leaving group departure during strand cleavage (Ma et al, 2007). His-305 fits the role of a general acid/base catalyst during recombination (Whiteson et al, 2007). The arrangement of the active site residues accommodates an acid/base-assisted SN2 in-line nucleophilic substitution, through a penta-coordinated transition state, for the strand cleavage and joining steps of recombination (Mizuuchi, 1997). The array of hydrogen bond donors surrounding the scissile phosphate helps to stabilize the transition state, and imparts a significant electrophilic character to the recombination reaction.
The importance of complementary contributions of phosphate electrostatics and active site configuration during phosphoryl transfer have come to light from two pertinent observations by Shuman and colleagues on vaccinia topoisomerase (Tian et al, 2003, 2005). Substituting the scissile phosphate (P) by MeP unveils a potent endonuclease activity of topoisomerase that hydrolyzes the covalent MeP-enzyme intermediate under physiological pH. Furthermore, this nuclease reaction is independent of Arg-223, which corresponds to Arg-308 of Flp. Two insights emerge from these findings with general implications for biologically important phosphoryl transfer reactions. First, the negative charge on the phosphate helps keep the water nucleophile at bay, and preserves the integrity of the phosphotyrosyl intermediate for productive 5′-hydroxyl attack and restoration of the DNA chain. Second, when the negative charge in the transition state is partly neutralized, the requirement of a key positively charged side chain in the active site is alleviated in the normal cleavage reaction, and is dispensed with in the nuclease reaction.
We have now addressed the generality of the vaccinia topoisomerase results by substituting SP or RP MeP at the cleavage position in Flp reactions. The outcomes are distinct from those of the vaccinia system and therefore mechanistically revealing. In contrast to the vaccinia enzyme, the intrinsic endonuclease activity of Flp that hydrolyzes the cleaved tyrosyl intermediate is not enhanced by substitution of the scissile phosphate by MeP. However, the same substrate elicits a second endonuclease activity in Flp(R308A) that is mechanistically distinct from the first and the equivalent endonuclease of topoisomerase or its R223A derivative, the Flp(R308A) counterpart. The latter reaction does not involve the tyrosyl intermediate, but directly targets the scissile MeP in DNA. These mechanistic differences between Flp and vaccinia topoisomerase likely reflect the characteristic active site–substrate interactions of each enzyme as well as the distinct conformational dynamics of the individual reactions they catalyse. The two systems accomplish, through separate strategies, the common goal of effectively promoting transesterification and strongly deterring hydrolysis. In doing so, they also prevent damage to the genomes on which they act. Electrostatic, chemical and/or conformational designs to expel water must, of necessity, be an intrinsic feature of transesterification reactions catalysed by type I and type II topoisomerases and site-specific recombinases, as well as enzyme systems responsible for DNA transposition, telomere resolution, RNA splicing and retrohoming of mobile introns. As shown here for the Flp recombinase, these protective mechanisms can be explored by suitable substrate design, MeP substitution and active site mutations.
Results
Experimental layout for assaying Flp activity on MeP substituted substrates
By substituting one of the non-bridging oxygen atoms of the scissile phosphate by the uncharged methyl group, the nominal negative charges in the ground and transition states would be reduced from −1 to 0 and −2 to −1, respectively. The methyl substitution introduces chirality at the phosphate centre, RP or SP (Figure 2A). According to conventional nomenclature, replacement of the pro-R oxygen yields the SP form MeP; that of the pro-S oxygen results in the RP form MeP. Although a mixture of the two stereoisomers served as the substrate in the majority of reactions, a subset of reactions was carried out with stereochemically pure RP or SP form. All reactions used near neutral pH (8.0), buffered by 25 mM Tris–HCl.
Figure 2.

Flp reactions using half-site substrates containing a phosphodiester or methylphosphonate at the scissile position. (A) The methyl substitutions in the RP and SP stereoisomers of methylphosphonate are indicated with reference to the achiral phosphodiester. (B) In the schematic representation of a half-site substrate, the horizontal arrows represent the Flp-binding element with its terminal bp abutting the spacer segment shown in bold. The scissile phosphodiester is indicated as ‘P'. Strand cleavage in a half-site traps the tyrosyl intermediate, as the trinucleotide product (5′HO-TTT3′) diffuses away. The 5′ end of the bottom strand is blocked by phosphorylation (denoted by ‘P' in parentheses) from acting as the nucleophile in a joining reaction. (C) A free 5′-OH group on the bottom strand permits the conversion of the tyrosyl intermediate into a hairpin recombinant. Although not shown here, attack by the 5′-OH from a second half-site is also possible. The joined strand would be identical in sequence to the hairpin. (D) Flp-assisted hydrolysis could potentially target the phosphotyrosyl intermediate formed by strand cleavage (top) or the scissile phosphodiester in DNA (bottom).The former activity is called type I endonuclease, the latter, type II.
P- or MeP-'half-site' substrates contained one Flp-binding element and one cleavage site (Figure 2B and C). Because of the shared active site configuration of Flp, strand scission could occur only in the context of at least two associated half-sites, each bound by a Flp monomer. Cleavage of a half-site by Tyr-343, with formation of Flp-linked DNA, was assayed by SDS–polyacrylamide gel electrophoresis (PAGE). Hydrolysis in a half-site, with either the DNA or the tyrosyl intermediate as target for water attack, was assayed by denaturing urea–PAGE. In most reactions, the 5′-OH on the bottom strand of the half-site was blocked by phosphorylation (Figure 2B) to prevent a strand joining reaction. In one set of reactions, this hydroxyl was free (Figure 2C), so that it could compete with water in nucleophilic attack of the tyrosyl intermediate. Strand joining in the context of a half-site (intra-half-site) would produce a hairpin recombinant. Strand joining across two half-sites (inter-half-site) would produce the equivalent of a full-site with spacers containing mismatches (not shown), referred to as a ‘pseudo-full-site' under Supplementary Figure S2. The intra- and inter-half-site reaction products would be identical in sequence, and would migrate as one band during electrophoresis under denaturing conditions. The potential Flp-assisted hydrolytic reactions and their possible outcomes are indicated in Figure 2D.
‘Wild-type' Flp used in this study was Flpe, a more thermostable version of Flp that differs from native Flp at four amino acid positions (Buchholz et al, 1998). The active site mutants were also derived from Flpe. Reactions run in parallel with native Flp and Flpe indicated that they behaved almost identically within minor experimental variations.
Flp exhibits an intrinsic site-specific endonuclease activity on native and MeP half-sites that targets the Flp-linked DNA intermediate
The P- or MeP-half-site, the latter a raecemic mixture of the RP and SP stereoisomers, was treated with wild-type Flp under conditions used for standard recombination reactions at pH 8.0 (Chen et al, 1992). The 5′ end-labelled reporter strand of the half-site was 26 nucleotides long, and harboured the scissile phosphate three nucleotides upstream of the 3′ end. Samples were divided in two and analysed for the DNA–protein covalent adduct and the 23-mer hydrolysis product formed from the strand cleavage and endonuclease activities of Flp, respectively (Figure 3A and B). Note that, here and elsewhere, ‘strand cleavage' refers exclusively to the reaction catalysed by Tyr-343 of Flp to distinguish it from strand breakage mediated by alternative non-protein nucleophiles including water. With either substrate, there was a buildup of the cleaved covalent adduct in the early part of the reaction (up to 8 h for the P-half-site and 16 h for the MeP-half-site) followed by its decline at later time points. Formation of the 23-mer followed slower kinetics, and its accumulation paralleled the disappearance of the covalent adduct. Qualitatively, the reactions of the P- and MeP-half-sites were similar, although the latter was clearly weaker in its reactivity. The extent of transesterification by Flp, estimated as the sum of the covalent adduct plus the 23-mer, at the 72 h time point was >90% for the P-half-site and ∼10% for the MeP-half-site.
Figure 3.

Kinetics of Flp strand cleavage activity and type I Flp endonuclease activity in half-site substrates. In the schematic representations of half-site substrates, the asterisk indicates 32P-label at the 5′-end, ‘P' the scissile phosphate and ‘mp' the scissile methylphosphonate. Reactions were analysed by PAGE for covalent Flp-DNA adduct (line ending in a circular knob) formed by strand cleavage (A) and by denaturing PAGE for hydrolysis (23-mer). (B) The hydrolysis product from the P-half-site runs as a doublet band (left panel in B), whereas that from the MeP-half-site (right panel in B) runs almost exclusively as a single band. The end-labelled half-site (A) or its labelled strand (B) is denoted by ‘S'. The plot below represents mean values from three separate experiments.
The observed reciprocity between the tyrosyl intermediate and the 23-mer is consistent with a precursor–product relationship. This was further verified by performing the MeP-half-site reactions with two Flp mutants lacking the active site tyrosine, Flp(Y343F) and Flp(Y343G). Consistent with their inability to mediate strand cleavage, neither was capable of the nuclease reaction (Supplementary Figure S1, Supplementary data; data not shown). In overexposed gels, weak formation of the 23-mer was detected, especially late in the reaction. This product likely results from a Tyr-343 independent endonuclease activity of Flp, which will be discussed in further detail (see below). The Y343F or Y343G Flp mutants were not affected in their functional interaction with the MeP-half-site. It is known that they can use an exogenous nucleophile such as hydrogen peroxide to mediate strand nicking in a native half-site (Kimball et al, 1993; Lee and Jayaram, 1993). Indeed, the peroxidolysis reaction was detected with the MeP-half-site in presence of Flp(Y343F) or Flp(Y343G) (Supplementary Figure S2A; data not shown). Note that the 23-mer band from reactions with the P-half-site (Figure 3A) migrated as a doublet, the slower migrating species being more prominent. Further characterization of these two products (Figure 4) revealed that they differ at their 3′-termini, harbouring either a hydroxyl or a phosphate group.
Figure 4.

Stereoisomer preference of Flp in the MeP-half-site reaction; characterization of the hydrolysis products from P- and MeP-half-site substrates. (A) The hydrolysis products from stereochemically pure SP and RP forms of the MeP-half-site were analysed by denaturing PAGE. (B) The closely migrating hydrolysis products from the P-half-site (2 and 24 h reactions in lanes 2 and 3, respectively, were characterized for the chemical nature of their 3′ ends. ‘MG' refers to a Maxam–Gilbert sequencing ladder obtained by chemical modification at C plus T positions followed by strand scission (lane 1). Lane 1 is duplicated to its left at higher intensity and contrast for better visibility of individual bands. Their sizes and the bases present at 3′-ends are indicated. A peroxidolysis reaction performed on the half-site using Flp(Y343F) was run in lane 4. ‘M' stands for a synthetic oligonucleotide marker harbouring a 5′-OH end. ‘EP' refers to the extension product formed in a Klenow polymerase reaction. (C) The hydrolysis product from a raecemic mixture of the MeP-half-site (lane 2) was characterized as in (B) by comparison to the C plus T sequencing ladder (lane 1 and its higher intensity duplicate to its left) and the product of Flp(Y343F)-assisted peroxydolysis (lane 3). The MeP substitution significantly enhanced strand scission to yield elevated levels of the 23-mer during the chemical steps of sequencing. The slow migrating band well above the doublet in lane 4 is the labelled top strand of the half-site, whose 3′-OH could prime DNA elongation by the Klenow enzyme.
These results show that Flp houses an endonuclease activity, referred to from here on as type I Flp endonuclease that targets the cleaved tyrosyl-DNA intermediate formed in half-site substrates. When the 5′-OH on the bottom strand of a half-site was not blocked, formation of the hairpin product by strand joining was dominant over hydrolysis at earlier times for the P-half-site, although the latter became significant at later times (Supplementary Figure S2B). For the MeP-half-site, the hydrolysis and strand joining reactions were nearly equally competitive (Figure 2C). However, in reactions with full-site substrates, the hydrolysis reaction was undetected or weak even after prolonged incubation (previous work and Supplementary Figure S3). These findings suggest that water becomes effective in attacking the tyrosyl-DNA intermediate only when the integrity of the normal recombination complex is compromised by switching from a full-site to a half-site reaction. The dimer interface formed by adjacent Flp subunits in half-site complexes may be more pliant than in full-site complexes, and thus more yielding to water entry (see also ‘Discussion').
Unlike the potent stimulation of endonuclease activity of vaccinia topoisomerase by MeP substitution (Tian et al, 2003), there is no such enhancement of Flp type I endonuclease by a similar substitution. The first-order rate constants for the hydrolysis of the P- and MeP-tyrosyl intermediates by Flp were 6.4 × 10−5 s−1 and 1.3 × 10−5 s−1, respectively (Table I). For comparison, the corresponding values for topoisomerase were 2.2 × 10−7 s−1 and 6 × 10−3 s−1, or a rate stimulation of four orders of magnitude by MeP substitution (Tian et al, 2003, 2005) (Table I). Phosphate electrostatics does not appear to play a role in protecting the tyrosyl intermediate from attack by water during Flp recombination.
Table 1.
First-order rate constants for the type I endonuclease activity of Flp and the type II endonuclease activities of Flp(R308A) and Flp(R308A, Y343G)
| Recombinase/topoisomerase | k (s−1) | ||
|---|---|---|---|
| P | MeP | ||
| Endonuclease activity | |||
| Type I | |||
| Flp | kcl(I) | 1.8 × 10−1 | 2.4 × 10−4 |
| khydrol(I) | 6.4 × 10−5 | 1.3 × 10−5 | |
| Vaccinia topoisomerse | kcl | 4.0 × 10−1 | 1.4 × 10−3 |
| khydrol | 2.2 × 10−7 | 7.0 × 10−3 | |
| Vaccinia topoisomerase | kcl | 4.0 × 10−6 | 7.0 × 10−4 |
| (R223A) | khydrol | — | 3.5 × 10−3 |
| Type II | |||
| Flp(R308A) | khydrol(II) | — | 8.7 × 10−5 |
| Flp(R308A, Y343G) | khydrol(II) | — | 5.3 × 10−5 |
Rate constants were estimated from the data shown in Figures 3, 5 and 6 using the software package Prism5 for Windows (GraphPad Software, Inc.). The type I reaction follows path (I) and the type II reaction follows path (II). 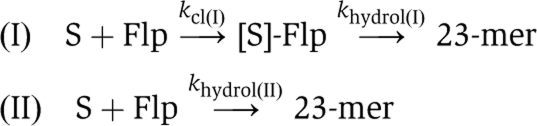 The first step of the type I reaction, strand cleavage, is assumed to be virtually irreversible. The trinucleotide product of strand cleavage, which is not stably hydrogen bonded to the complementary strand, would diffuse away and be unavailable for the back reaction. Subscripts I and II are ascribed to the rate constants to distinguish between the type I and type II reactions. The values for the vaccinia topoisomerase (Tian et al, 2005), which exhibits only the type I endonuclease, are shown for comparison with those for the corresponding Flp activity. The first step of the type I reaction, strand cleavage, is assumed to be virtually irreversible. The trinucleotide product of strand cleavage, which is not stably hydrogen bonded to the complementary strand, would diffuse away and be unavailable for the back reaction. Subscripts I and II are ascribed to the rate constants to distinguish between the type I and type II reactions. The values for the vaccinia topoisomerase (Tian et al, 2005), which exhibits only the type I endonuclease, are shown for comparison with those for the corresponding Flp activity. | |||
Flp is stereoselective for the SP form of the MeP-half-site: characterization of the 3′-termini of hydrolysis products from P- and MeP-half-sites
The Flp crystal structures reveal that both non-bridging oxygen atoms on the scissile phosphate take part in hydrogen bonding interactions with a subset of residues from the catalytic pentad (Chen et al, 2000; Conway et al, 2003). Introducing a methyl group in place of either oxygen would eliminate hydrogen bonding capability at the substituted position, which could be deleterious to the strand cleavage and/or strand joining steps of recombination. The effect of specific methyl substitution on type I Flp endonuclease was tested in reactions of wild-type Flp with stereochemically pure RP and SP forms of the MeP half-site.
The strong preference observed for the SP substrate (Figure 4A) was qualitatively the same as that reported for the vaccinia topoisomerase. The six- to eight-fold bias in 23-mer formation is contributed in part by the strand cleavage step (three- to four-fold). By inference, the hydrolysis step provides the additional bias. As strand cleavage by the Tyr-343 nucleophile is accompanied by inversion of configuration at the phosphate centre, the hydrolysis reaction prefers the MeP-tyrosyl intermediate whose chirality is opposite to that of the starting SP MeP-half-site.
The major product of type I Flp endonuclease from the P-half-site (the top band of the 23-mer doublet in Figure 3A) was determined to contain a free 3′-OH by several independent criteria (Figure 4B). It migrated in between the appropriate rungs of a Maxam–Gilbert sequencing ladder derived from the labelled strand of this half-site (Figure 4B, lanes 1–3) and slightly above the product of Flp(Y343F)-assisted strand nicking by hydrogen peroxide (Figure 4B, lane 4). Earlier work showed that peroxidolysis leaves a 3′-phosphate and a 5′-hydroxyl on either side of the break point (Kimball et al, 1993; Lee and Jayaram, 1993). Furthermore, this type I Flp endonuclease product had the same mobility as a synthetic 23-mer of the predicted sequence bearing a 3′-OH (Figure 4B, lane 5), and served as a primer in a Klenow polymerase extension reaction (Figure 4B, lane 7). The size of the fully extended product was the same as that given by the synthetic 23-mer (Figure 4B, lane 9). By contrast, the minor product of type I Flp endonuclease from the P-half-site (the weaker bottom band of the 23-mer doublet in lanes 2 and 3 of Figure 4B) was deduced to contain a 3′ phosphate terminus. It co-migrated with the expected band in the sequencing ladder (Figure 4B, lanes 1–3) as well as with the product of peroxydolysis (Figure 4B, lane 4) migrated slightly faster than the synthetic marker oligonucleotide with a 3′-OH (Figure 4B, lane 5) and could not be extended by Klenow polymerase (Figure 4B, lane 7). By similar reasoning, the 23-mer yielded by type I Flp endonuclease from the MeP-half-site (Figure 4C, lane 2) was deemed to contain a methylphosphate at its 3′-end. Note that the top band of the doublet in the Maxam–Gilbert ladder in lane 1 of Figure 4C contained a 3′-OH end, as verified by Klenow polymerase extension reaction (Figure 4C, compare lanes 4 and 5).
Although both the P-tyrosyl and MeP-tyrosyl intermediates are targets for water attack, the resulting products are distinct in the two instances. Hydrolysis of the P-tyrosyl bond leaves a 3′-OH terminus on the DNA as the primary product and a 3′-phosphate terminus in significantly lower yield. The hydrolysis product from the MeP-tyrosyl intermediate harbours almost exclusively a 3′-methylphosphate terminus. The two types of products reveal water attack from opposite orientations, causing the departure of either tyrosine or the 3′-OH as the leaving group. The transfer of the scissile phosphate from DNA to Tyr-343 of Flp, predicted by the latter reaction, is being probed by labelling the DNA substrate internally with 32P at this position as well as scouting for the signal for the predicted phosphotyrosine containing tryptic peptide from Flp in mass spectrometry. It is possible, though, that the phosphate group may be removed from tyrosine by Flp in a second hydrolytic step. The presence of the methyl group in place of oxygen at the non-bridging position appears to restrict the direction in which water approaches its scissile target. Overall, these results speak to the conformational flexibility and versatility of the Flp active site in accommodating distinct reaction mechanisms.
The formation of a 3′-hydroxyl rather than a 3′-phosphate end from hydrolysis of the P-tyrosyl intermediate offers a potential advantage in preserving DNA integrity in the physiological context of the recombination reaction. Should an abortive hydrolytic event occur, the DNA end generated would be readily amenable to repair by DNA ligase or extension by a repair polymerase. A 3′-phosphate end would require an additional dephosphorylation step, which can be mediated by Tpp1 (3′-phosphatase) of S. cerevisiae (Vance and Wilson, 2001). Regardless of the nature of the 3′ end, the 5′-hydroxyl group adjacent to the nick will have to be phosphorylated before repair by ligation. As S. cerevisiae apparently lacks a polynucleotide kinase, 5′-hydroxyl ends are thought to be processed by a cycle of base removal and re-synthesis (Vance and Wilson, 2001).
Replacement of Arg-308 by alanine in Flp induces a nuclease activity that targets MeP and is independent of the covalent tyrosyl-DNA intermediate
Shuman and colleagues noticed that the requirement of Arg-223 in vaccinia topoisomerase (which corresponds to Arg-308 in Flp) is alleviated for induction of its latent endonuclease activity towards the scissile MeP linkage (Tian et al, 2005). The mechanism of action of the mutant is the same as that of the wild-type enzyme, namely, formation of the MeP-tyrosyl intermediate followed by its hydrolysis. We therefore wished to know how lack of Arg-308 would affect the endonuclease activity of Flp.
Consistent with the requirement for Arg-308 for activation of the scissile phosphate, Flp(R308A) was virtually inactive towards the P-half-site (Figure 5A). Trace amounts of a band with the mobility of the covalent DNA–protein adduct were detected; however, its appearance was long delayed relative to that in a wild-type Flp reaction (compare Figure 5A with Figure 3A). Little or no hydrolysis product was observed even at late time points. Reactions of Flp(R308A) on the MeP-half-site revealed a strikingly different outcome. The 23-mer hydrolytic product was formed early, compared with the wild-type Flp reaction (Figure 5B; compare to Figure 3B). At these or later time points, there was no evidence for the formation of the protein-linked DNA intermediate. Thus, unlike the type I Flp endonuclease reaction, the Flp(R308A) reaction showed no obvious precursor–product relationship between the cleaved tyrosyl intermediate and the hydrolysis product. Furthermore, absence of the covalent adduct was not due to its rapid and quantitative conversion to the hydrolysis product. Tyr-343 could be completely dispensed with in this novel reaction (Figure 6). The rate of hydrolysis of the MeP-half-site by Flp(R308A, Y343G) was similar to that by Flp(R308A), within a factor of two (Table I), and the final yields of the 23-mer were comparable between the single and double mutants.
Figure 5.

Reactions of Flp(R308A) assayed on P- and MeP-half-sites. Reactions and analysis of strand cleavage (A) and hydrolysis (B) products were performed as described in Figure 3.
Figure 6.

Hydrolysis in the MeP-half-site is independent of Tyr-343-mediated strand cleavage. Reactions and product analysis were as in Figures 3 and 5. (A) strand cleavage and covalent adduct formation; (B) hydrolysis and 23-mer formation.
Direct hydrolysis by Flp(R308A) favoured the SP MeP-half-site over its RP counterpart (Supplementary Figure S4A), as did the type I Flp endonuclease. Thus, the stereochemical preference of the reaction is unaltered regardless of whether Tyr-343 or water is used for strand breakage.
Characterization of the 23-mer product from the Flp(R308A)-MeP-half-site reaction revealed a methylphosphate at its 3′-end (Supplementary Figure S4B). Formation of the same product by direct hydrolysis of the scissile MeP by Flp(R308A) or hydrolysis of the MeP-tyrosyl bond by Flp suggests that water is potentially capable of carrying out in-line attack opposite a 5′-hydroxyl or a tyrosine leaving group. The presence of Arg-308 effectively blocks the former mode of attack.
The novel endonuclease activity engendered by Flp(R308A), with its sharp mechanistic distinction from type I Flp endonuclease, is termed type II Flp endonuclease. The type II activity suggests that, in addition to its normal role in phosphate activation as a member of the catalytic pentad, Arg-308 also serves the equally important function of warding off water from the reaction centre. It is also remarkable that there is no formation of the MeP-tyrosyl intermediate during Flp(R308A) action on the MeP-half-site. The failure of Tyr-343, as opposed to the success of water, in attacking the scissile MeP suggests that Arg-308 may also assist the activation of the tyrosine nucleophile during normal recombination. Perhaps a water molecule oriented with assistance from Arg-308 may facilitate proton abstraction from Tyr-343. The point of import is that, by chemically neutralizing a negative charge on the phosphate group, the requirement of a positively charged side chain in the Flp active site can be eliminated.
The first-order rate constants for the type II endonuclease activities of Flp(R308A) and Flp(R308A, Y343G) were estimated to be 8.7 × 10−5 s−1 and 5.3 × 10−5 s−1, respectively (Table I). The type II activity is thus more robust than the type I endonuclease of wild-type Flp, whose rate constant in the MeP reaction was derived as 1.3 × 10−5 s−1 (Table I). However, it is significantly weaker than topoisomerase I(R223A) endonuclease with a hydrolysis rate constant of 3.5 × 10−3 s−1 (Tian et al, 2003) (Table I). Unlike Flp(R308A), wild-type Flp and topoisomerase I(R223A) direct their endonuclease activity against the MeP-tyrosyl intermediate, and not the scissile phosphodiester in DNA.
Discussion
As biological catalysts, enzymes must not only carry out specific biochemical reactions that they are designed for but must also often guard against errant reactive species present in the cellular environment. A salient example is transesterification events involving phosphodiester bonds in DNA and RNA. Side reactions, in particular hydrolysis, can potentially damage the integrity of the genome or interrupt the steps of information processing at the RNA level. The significance of the problem, the solutions to it devised by the Flp site-specific recombinase and their general implications are illustrated in the present studies.
Channelling the phosphotyrosyl intermediate of strand cleavage away from hydrolysis: Flp and vaccinia topoisomerase paradigms
The 3′-phosphotyrosyl intermediate formed during Flp recombination is susceptible to hydrolysis by the type I Flp endonuclease activity. Competition from this futile reaction is negligible during a normal recombination reaction but becomes apparent during a half-site reaction. In the absence of a 5′-hydroxyl terminus on the bottom strand of the half-site, the tyrosyl intermediate can be quantitatively converted into the hydrolytic product. An active 5′-hydroxyl can effectively suppress competition from water, the hydrolysis reaction becoming significant only at later times of the reaction course (Supplementary Figure S2B). The estimated rate constant for 5′-hydroxyl-mediated strand joining is 3.1 × 10−3 s−1, whereas that for hydrolysis is 9.7 × 10−5 s−1.
Flp does not follow the topoisomerase paradigm in exploiting phosphate electrostatics to protect the strand cleavage intermediate from attack by water. During type I Flp endonuclease action, the rates of hydrolysis of 3′-P-tyrosyl or 3′MeP-tyrosyl intermediates are comparable. In stark contrast, neutralizing phosphate electrostatics causes a large stimulatory effect on the corresponding endonuclease activity of vaccinia topoisomerase (Tian et al, 2003). Although MeP substitution slows down strand cleavage by topoisomerase by a factor of ∼200, it accelerates the rate of hydrolysis of the MeP-tyrosyl intermediate by a factor of ∼3–4 × 104 (see Table I). The half-life of phosphotyrosyl intermediate is approximately 36 days at physiological pH whereas that of MeP-tyrosyl intermediate is less than 2 min.
Protection of the covalent Flp-DNA intermediate is built into the compact design of the recombination synapse comprising an Flp tetramer in association with two DNA partners. In the crystal structure of the Flp-recombination complex, the interface between two Flp monomers that partake in the trans donation of tyrosine and assembly of a functional active site displays extensive protein–protein interactions that bury a large surface area of ∼1500 Å2 (Conway et al, 2003). Furthermore, invading 5′-hydroxyl groups from cleaved strands are seen poised to attack their target phosphotyrosyl bonds and mediate a pair of strand exchanges. The structures of the recombination synapses of the Flp-related recombinases Cre and λ Int also suggest that a pair of coordinated strand cleavages concomitantly orients the resulting 5′-hydroxyl groups for interpartner strand joining (Guo et al, 1997; Biswas et al, 2005). This conformational feature of tyrosine family recombination, namely the tight coupling between strand cleavage and strand joining, can effectively deny water the chance to thwart the reaction midway through completion.
The half-site Flp reaction, exploited in this study, could occur in the context of a [half-site-Flp] dimer or a [half-site-Flp] tetramer or perhaps even a [half-site-Flp] trimer (Chen et al, 1993; Lee and Jayaram, 1995, 1997; Qian and Cox, 1995; Lee et al, 1996). A [half-site-Flp] monomer is insufficient for the reaction as it takes two monomers of Flp to assemble a functional active site. Regardless of the oligomeric state of the half-site synapse, the discontinuity in the DNA apparently loosens the complex sufficiently for water to access the reaction centre.
Vulnerability of the scissile phosphodiester bond in DNA to attack by water: type II Flp endonuclease
The scissile MeP in half-site reactions becomes susceptible to direct hydrolysis in the presence of Flp(R308A) by the type II Flp endonuclease activity. Wild-type Flp does not exhibit this activity, and the phosphate containing half-site is immune to it. A corresponding activity is not detected in the R223A mutant of vaccinia topoisomerase. Conversely, Flp(R308A) is not capable of the type I endonucleolytic cleavage, whereas the vaccinia mutant is as competent as the wild-type enzyme for hydrolysis of the 3′-MeP-tyrosyl bond.
The positively charged Arg-308 side chain, rather than the negatively charged phosphate, is responsible for repelling water during the strand cleavage reaction by Flp. If phosphate were to play a critical role in this protective mechanism, the MeP half-site should become exposed to hydrolysis by wild-type Flp, and it is not. As discussed below, the role of Arg-308 in recombination is more than catalytic. Furthermore, the role of Arg-308 in excluding water is not simply steric; Flp(R308Q), similarly to Flp(R308A), is also a type II endonuclease, as are Flp variants harbouring other Arg-308 substitutions (unpublished data).
Catalytic and non-catalytic contributions of Arg-308 to recombination: activation of the scissile phosphate and repulsion of water
Consistent with its key catalytic role in recombination, the crystal structure of Flp reveals Arg-308 within hydrogen bonding distance of the scissile phosphate (Chen et al, 2000; Conway et al, 2003). Mutations at Arg-308 cause severe defects in strand cleavage as well as strand joining (Parsons et al, 1988; Chen et al, 1992, 1993; Lee and Jayaram, 1993; Jayaram et al, 2004). However, MeP activation is not impeded by lack of Arg-308, as evidenced by its efficient hydrolysis by Flp(R308A). Thus, MeP substitution reveals a dual role for Arg-308: a catalytic role of phosphate activation and a non-catalytic role of water exclusion. Eliminating the negative charge on phosphate ameliorates the requirement of Arg-308 in the activation step. The penalty, though, for lack of Arg-308 is high susceptibility to hydrolysis.
Unlike Arg-308 in Flp, Arg-223 in vaccinia topoisomerase appears to satisfy an exclusively catalytic function. Activation of the scissile phosphate for strand cleavage is dependent on Arg-223, but this requirement is largely relieved for the scissile MeP. Presence or absence of Arg-223 makes little difference to the hydrolysis of the 3′-MeP-tyrosyl linkage. Neither the wild-type topoisomerase nor the Arg-223 mutant promotes direct hydrolysis of the scissile MeP in DNA.
Flp type II endonuclease: mechanistic similarity to Flp type II RNase
The type II Flp endonuclease is mechanistically similar in some respects to the type II Flp RNase that was revealed by a ribonucleotide substitution immediately 3′ to the scissile phosphodiester target of recombination (Xu et al, 1998). In this reaction, which is independent of Tyr-343, the vicinal 2′-hydroxyl directly attacks the phosphodiester bond to cause chain breakage. The 3′ terminal phosphate left at the strand nick is presumably generated through the 2′,3′-cyclic phosphate intermediate. There are also important differences between type II Flp endonuclease and type II Flp RNase. The scissile targets for the two activities are not the same, rather they are immediate neighbours of each other. Arg-308 is essential for the type II RNase activity, whereas it is the lack of Arg-308, in combination with MeP substitution that elicits the type II endonuclease activity. These differences notwithstanding, the two activities highlight the danger from stray nucleophiles that Flp-activated phosphodiester bonds are subject to.
Recombinase and topoisomerase active sites engender distinct strategies for excluding water
Several instances in which Flp, as well as vaccinia and human type I topoisomerase, active sites can be coaxed to accept non-native nucleophiles have been encountered (for details see Appendix in Supplementary data). The results from this study demonstrate that water is indeed such a nucleophile, and threatens Flp recombination on two fronts, during strand cleavage and strand joining. The peril to vaccinia topoisomerase reaction, by contrast, is principally during the strand joining step. The risks faced by these enzymes and the solutions they have devised to overcome these risks can be rationally accommodated in terms of their active site configurations and the conformational features of the reactions they catalyse.
The topoisomerase binds to DNA and relaxes it as a monomer. Engagement of the tyrosine nucleophile occurs concomitant with DNA binding by topoisomerase, and leaves water little chance of attacking the scissile phosphodiester (Figure 7A). The cleaved strand must go through a round of rotation before joining occurs. The conformational flexibility associated with the dynamics of rotation likely opens the door for water to attack the phosphotyrosyl intermediate (Figure 7B). However, as shown by the MeP substitution, the negative charge on the phosphate prevents water from being the spoiler during the normal reaction (Tian et al, 2003, 2005).
Figure 7.

Vulnerability of the transesterification steps of DNA relaxation and site-specific recombination to hydrolysis. (A) During DNA relaxation by type I topoisomerase, the strand cleavage step is protected by the engagement of the active site in cis and the in-line orientation of the active site tyrosine with respect to the scissile phosphate. (B) The tyrosyl intermediate resulting from strand cleavage is a potential target for attack by water during the strand rotation step that precedes ligation. Protection is derived from phosphate electrostatics. (C) The association of a single Flp monomer with DNA activates the scissile phosphate but full engagement of the active site must await the binding of a second Flp monomer and donation of the active site tyrosine. Hydrolysis of the phosphodiester bond is at potential risk. Active site electrostatics comes to the rescue. (D) During normal recombination, the synaptic architecture of the Flp tetramer, with the tight polypeptide interface between active pairs of Flp monomers, barricades water from dissipating the cleaved tyrosyl intermediate. Furthermore, the conformational dynamics accompanying cleavage orients the 5′-OH groups for strand joining.
Flp binds to each of the two binding elements of a recombination target site as a monomer, but assembles a shared active site by donation of the Tyr-343 nucleophile in trans. Binding of a Flp monomer, which activates the scissile phosphate (Lee and Jayaram, 1993), and active site engagement within a Flp dimer are not coincident. The temporal separation of the two events makes the scissile phosphate vulnerable to attack by water (Figure 7C). In wild-type Flp, the positively charged Arg-308 side chain minimizes this risk by repelling or misorienting water. Hydrolysis becomes prominent when the active site lacks Arg-308 and MeP occupies the scissile position. Protection of the strand joining step of recombination derives primarily from the synaptic architecture and the post-cleavage conformational dynamics that give the 5′-hydroxyl a large advantage over a competitor nucleophile (Figure 7D).
The Flp-related tyrosine recombinases Cre, lambda Int and XerC/XerD resemble vaccinia topoisomerases in that each houses its active site within a protein monomer. Nevertheless, the full chemical competence and functional regulation of the cis active site may require trans allosteric activation by a neighbouring monomer (Guo et al, 1997; Hallet et al, 1999; Biswas et al, 2005; Hazelbaker et al, 2005). It would be interesting to test whether Cre, Int and XerC/XerD are more Flp-like or topoisomerase-like in regards to the reaction steps that are prone to hydrolysis and whether they use phosphate electrostatics or active site electrostatics to deal with this encumbrance.
Substrate and active site electrostatics as a general mechanism to repel water during phosphoryl transfer in nucleic acids
The responses of Flp and vaccinia topoisomerase, as well as their mutant derivatives lacking a conserved catalytic arginine, to neutralization of phosphate charge are illuminating with respect to both mechanism and nucleophile specificity of the relevant transesterification reactions. Indeed, appropriate pairing of the electrostatics of the substrate and of the enzyme active site is critical for activation of the target phosphodiester bond. In addition, these electrostatic attributes serve to promote the authentic reaction steps by preserving the integrity of the phosphodiester bond in the DNA substrate and/or the cleaved tyrosyl intermediate. We believe that MeP substitution, in conjunction with specific active site mutations, will be instructive in understanding protective designs used by a variety of enzyme systems that carry out phosphoryl transfer reactions in DNA and RNA.
Materials and methods
Synthetic oligonucleotides and assembly of substrates
Oligonucleotides used for the assembly of half-site and full-site substrates are listed in Supplementary Table S1 (Supplementary data). All standard oligonucleotides were purchased from IDT (Coralville, IA). The MeP-containing oligonucleotide (a raecemic mixture of the RP and SP forms) was supplied by TriLink Technologies (San Diego, CA). Hybridization of oligonucleotides was performed as described earlier (Lee et al, 1996).
Stereochemically pure RP and SP methylphosphonate containing oligonucleotides
The oligonucleotide substrates containing RP or SP form of MeP were obtained using published procedures (Wozniak et al, 2005, 2006). The first step was the synthesis of the 5′ and 3′ protected (3′-5′)-methylphosphonothioate (DMTAP(S)MeTTBDMS) followed by its desilylation to DMTAP(S)MeT using Et3N (2.5 eq) and Et3N × 3HF (5 eq). Separation of the P-diastereomers of DMTAP(S)MeT was accomplished by silica gel chromatography. Stereochemically pure RP and SP forms of the dinucleoside (3′-5′) methylphosphonothioate were then converted to the corresponding MeP (DMTAP(O)MeT) on treatment with oxone. The stereochemically pure MeP dinucleotides were phosphitylated at the 3′-OH position with 2-O-cyanoethyl tetra-N,N,N′,N′-(diisopropyl) phosphoramidite. These compounds were further incorporated (without purification) into the required oligonucleotides using standard phosphoramidite synthetic protocols, with the exception that the coupling time of 20 min was applied. Further details of individual steps of stereoisomer separation and oligonucleotide synthesis can be provided on request.
Although the scissile phosphodiester in the native Flp substrate is bordered by a C on its 5′ side, the corresponding base in the stereochemically pure RP and SP MeP substrates used in our assays was an A. This substitution was necessitated by technical reasons. Separation of the DMTCp(S)MeT stereoisomers was more challenging and less clean than that of the DMTAp(S)MeT stereoisomers. Replacement of the C–G bp by the A–T bp at the cleavage proximal position decreases the efficiency of the reaction but does not abolish it (Senecoff et al, 1988).
Purification of Flp and Flp mutants
All proteins, wild-type Flpe and mutant derivatives, were partially purified using protocols described by Lee et al (1996), and further purified to near homogeneity over a DNA-affinity column (Meyer-Leon et al, 1987).
In vitro Flp reactions
In vitro Flp reactions were carried out using 5′-end labelled substrates at 30°C under buffer conditions described by Chen et al (1992). Each reaction mixture contained 25 mM Tris–HCl, pH 8.0, 1 mM EDTA, 100 mM NaCl, 5% glycerol, 5% polyethylene glycol, approximately 0.2 pmol DNA substrate(s) and the Flp protein in a total volume of 30 μl. The stoichiometry of Flp or a Flp mutant in the reactions was approximately 4 monomers per Flp-binding element in the DNA substrate. Reactions were terminated by addition of sodium dodecyl sulfate (0.1% final concentration), and DNA was isolated by phenol–chloroform extraction and ethanol precipitation. In processing the reactions to assay Flp endonuclease activities, the proteinase K treatment that normally precedes DNA extraction was omitted. By doing so, after electrophoresis, hydrolysis products could be visualized free of the strand cleavage product carrying short Flp peptides at the 3′ end.
Characterization of hydrolysis products by Klenow polymerase extension reaction
The 32P-labelled strand of the hydrolysis product was excised from the gel after fractionation by electrophoresis, and the DNA purified by phenol extraction and ethanol precipitation. It was hybridized to a complementary strand that had a 5′ extension of additional nucleotides. The Klenow polymerase reaction was carried out in the presence of all four deoxynucleoside triphosphates, and analysed by denaturing PAGE.
Strand nicking using hydrogen peroxide
Hydrogen peroxide-mediated strand nicking was carried out in the presence of Flp or a Tyr-343 mutant of Flp as described earlier (Kimball et al, 1993).
Quantification of reaction products
Bands corresponding to substrate and products were quantified using a phosphorimager. For low-efficiency reactions, the following procedure was used to derive the fraction of substrate converted to product. Multiple serial dilutions of a reaction were fractionated alongside the undiluted reaction, and the substrate band was quantified from those that fell within the linear range of estimation. Appropriate correction factors were then applied in the calculation of product yields.
Supplementary Material
Supplementary Information
Review Process File
Acknowledgments
We thank S Shuman and A Landy for helpful comments on the paper. We are grateful to Malgorzata Bukowiecka-Matusiak for useful suggestions for synthesis of methylphosphonate dimers. Milena Sobczak provided excellent technical assistance in synthesizing methylphosphonate oligonucleotides. We acknowledge help from Y-C Tsai and KA Johnson in deriving kinetic constants. This work was supported by NIH award GM035654 to MJ. Partial support was provided by the Robert F Welch Foundation and a Faculty Research Award from the University of Texas at Austin.
References
- Arciszewska LK, Sherratt DJ (1995) Xer site-specific recombination in vitro. EMBO J 14: 2112–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belfort M, Derbyshire V, Parker MM, Cousineau B, Lambowitz AM (2002) Mobile introns: pathways and proteins. In Mobile DNA II, Craig NL, Craigie R, Gellert M, Lambowitz AM (eds), pp 761–783. Washington, DC: ASM Press [Google Scholar]
- Biswas T, Aihara H, Radman-Livaja M, Filman D, Landy A, Ellenberger T (2005) A structural basis for allosteric control of DNA recombination by lambda integrase. Nature 435: 1059–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchholz F, Angrand PO, Stewart AF (1998) Improved properties of FLP recombinase evolved by cycling mutagenesis. Nat Biotechnol 16: 657–662 [DOI] [PubMed] [Google Scholar]
- Cech TR (1983) RNA splicing: three themes with variations. Cell 34: 713–716 [DOI] [PubMed] [Google Scholar]
- Chaconas G (2005) Hairpin telomeres and genome plasticity in Borrelia: all mixed up in the end. Mol Microbiol 58: 625–635 [DOI] [PubMed] [Google Scholar]
- Chen JW, Lee J, Jayaram M (1992) DNA cleavage in trans by the active site tyrosine during Flp recombination: switching protein partners before exchanging strands. Cell 69: 647–658 [DOI] [PubMed] [Google Scholar]
- Chen JW, Yang SH, Jayaram M (1993) Tests for the fractional active-site model in Flp site-specific recombination. Assembly of a functional recombination complex in half-site and full-site strand transfer. J Biol Chem 268: 14417–14425 [PubMed] [Google Scholar]
- Chen Y, Narendra U, Iype LE, Cox MM, Rice PA (2000) Crystal structure of a Flp recombinase-Holliday junction complex: assembly of an active oligomer by helix swapping. Mol Cell 6: 885–897 [PubMed] [Google Scholar]
- Chen Y, Rice PA (2003) The role of the conserved Trp330 in Flp-mediated recombination: functional and structural analysis. J Biol Chem 278: 24800–24807 [DOI] [PubMed] [Google Scholar]
- Cheng C, Kussie P, Pavletich N, Shuman S (1998) Conservation of structure and mechanism between eukaryotic topoisomerase I and site-specific recombinases. Cell 92: 841–850 [DOI] [PubMed] [Google Scholar]
- Conway AB, Chen Y, Rice PA (2003) Structural plasticity of the Flp-Holliday junction complex. J Mol Biol 326: 425–434 [DOI] [PubMed] [Google Scholar]
- Craig NL (2002) Mobile DNA: an introduction. In Mobile DNA II, Craig NL, Craigie R, Gellert M, Lambowitz AM (eds), pp 3–11. Washington, DC: ASM Press [Google Scholar]
- Esposito D, Scocca JJ (1997) The integrase family of tyrosine recombinases: evolution of a conserved active site domain. Nucl Acids Res 25: 3605–3614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Futcher AB (1986) Copy number amplification of the 2 micron circle plasmid of Saccharomyces cerevisiae. J Theor Biol 119: 197–204 [DOI] [PubMed] [Google Scholar]
- Guo F, Gopaul DN, Van Duyne GD (1997) Structure of Cre recombinase complexed with DNA in a site-specific recombination synapse. Nature 389: 40–46 [DOI] [PubMed] [Google Scholar]
- Hallet B, Arciszewska LK, Sherratt DJ (1999) Reciprocal control of catalysis by the tyrosine recombinases XerC and XerD: an enzymatic switch in site-specific recombination. Mol Cell 4: 949–959 [DOI] [PubMed] [Google Scholar]
- Hazelbaker D, Radman-Livaja M, Landy A (2005) Receipt of the C-terminal tail from a neighboring lambda Int protomer allosterically stimulates Holliday junction resolution. J Mol Biol 351: 948–955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman AB, Waninger S, Scocca JJ, Dyda F (1997) Molecular organization in site-specific recombination: the catalytic domain of bacteriophage HP1 integrase at 2.7Å resolution. Cell 89: 227–237 [DOI] [PubMed] [Google Scholar]
- Jayaram M, Mehta S, Uzri D, Voziyanov Y, Velmurugan S (2004) Site-specific recombination and partitioning systems in the stable high copy propagation of the 2-micron yeast plasmid. In Progress in Nucleic Acid Research and Molecular Biology, Moldave K (ed), Vol. 77, pp 127–172. Academic Press [DOI] [PubMed] [Google Scholar]
- Jayaram M, Tribble G, Grainge I (2002) Site-specific recombination by the Flp protein of Saccharomyces cerevisiae. In Mobile DNA II, Craig NL, Craigie R, Gellert M, Lambowitz AM (eds), pp 192–218. Washington, DC: ASM Press [Google Scholar]
- Kimball AS, Lee J, Jayaram M, Tullius TD (1993) Sequence-specific cleavage of DNA via nucleophilic attack of hydrogen peroxide, assisted by Flp recombinase. Biochemistry 32: 4698–4701 [DOI] [PubMed] [Google Scholar]
- Krogh BO, Shuman S (2000) Catalytic mechanism of DNA topoisomerase IB. Mol Cell 5: 1035–1041 [DOI] [PubMed] [Google Scholar]
- Lee J, Jayaram M (1993) Mechanism of site-specific recombination. Logic of assembling recombinase catalytic site from fractional active sites. J Biol Chem 268: 17564–17570 [PubMed] [Google Scholar]
- Lee J, Jayaram M (1995) Role of partner homology in DNA recombination. J Biol Chem 270: 4042–4052 [DOI] [PubMed] [Google Scholar]
- Lee J, Jayaram M (1997) A tetramer of the Flp recombinase silences the trimers within it during resolution of a Holliday junction substrate. Genes Dev 11: 2438–2447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Whang I, Jayaram M (1996) Assembly and orientation of Flp recombinase active sites on two-, three- and four-armed DNA substrates: implications for a recombination mechanism. J Mol Biol 257: 532–549 [DOI] [PubMed] [Google Scholar]
- Ma CH, Kwiatek A, Bolusani S, Voziyanov Y, Jayaram M (2007) Unveiling hidden catalytic contributions of the conserved His/Trp-III in tyrosine recombinases: assembly of a novel active site in Flp recombinase harboring alanine at this position. J Mol Biol 368: 183–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer-Leon L, Gates CA, Attwood JM, Wood EA, Cox MM (1987) Purification of the FLP site-specific recombinase by affinity chromatography and re-examination of basic properties of the system. Nucleic Acids Res 15: 6469–6488 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuuchi K (1997) Polynucleotidyl transfer reactions in site-specific DNA recombination. Genes Cells 2: 1–12 [DOI] [PubMed] [Google Scholar]
- Nunes-Duby SE, Kwon HJ, Tirumalai RS, Ellenberger T, Landy A (1998) Similarities and differences among 105 members of the Int family of site-specific recombinases. Nucleic Acids Res 26: 391–406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunes-Duby SE, Tirumalai RS, Dorgai L, Yagil E, Weisberg RA, Landy A (1994) Lambda integrase cleaves DNA in cis. EMBO J 13: 4421–4430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons RL, Prasad PV, Harshey RM, Jayaram M (1988) Step-arrest mutants of FLP recombinase: implications for the catalytic mechanism of DNA recombination. Mol Cell Biol 8: 3303–3310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry K, Hwang Y, Bushman FD, Van Duyne GD (2006) Structural basis for specificity in the poxvirus topoisomerase. Mol Cell 23: 343–354 [DOI] [PubMed] [Google Scholar]
- Qian XH, Cox MM (1995) Asymmetry in active complexes of FLP recombinase. Genes Dev 9: 2053–2064 [DOI] [PubMed] [Google Scholar]
- Redinbo MR, Stewart L, Kuhn P, Champoux JJ, Hol WG (1998) Crystal structures of human topoisomerase I in covalent and noncovalent complexes with DNA. Science 279: 1504–1513 [DOI] [PubMed] [Google Scholar]
- Reynolds AE, Murray AW, Szostak JW (1987) Roles of the 2 micron gene products in stable maintenance of the 2 micron plasmid of Saccharomyces cerevisiae. Mol Cell Biol 7: 3566–3573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice PA (2002) Theme and variation in tyrosine recombinases: structure of a Flp-DNA complex. In Mobile DNA II, Craig NL, Craigie R, Gellert M, Lambowitz AM (eds), pp 219–229. Wahington, DC: ASM Press [Google Scholar]
- Senecoff JF, Rossmeissl PJ, Cox MM (1988) DNA recognition by the FLP recombinase of the yeast 2 micron plasmid. A mutational analysis of the FLP binding site. J Mol Biol 201: 405–421 [DOI] [PubMed] [Google Scholar]
- Tian L, Claeboe CD, Hecht SM, Shuman S (2003) Guarding the genome: electrostatic repulsion of water by DNA suppresses a potent nuclease activity of topoisomerase IB. Mol Cell 12: 199–208 [DOI] [PubMed] [Google Scholar]
- Tian L, Claeboe CD, Hecht SM, Shuman S (2005) Mechanistic plasticity of DNA topoisomerase IB: phosphate electrostatics dictate the need for a catalytic arginine. Structure 13: 513–520 [DOI] [PubMed] [Google Scholar]
- Van Duyne GD (2002) A structural view of tyrosine recombinase site-specific recombination. In Mobile DNA II, Craig NL, Craigie R, Gellert M, Lambowitz AM (eds), pp 93–117. Washington, DC: ASM Press [Google Scholar]
- Vance JR, Wilson TE (2001) Uncoupling of 3′-phosphatase and 5′-kinase functions in budding yeast. Characterization of Saccharomyces cerevisiae DNA 3′-phosphatase (TPP1). J Biol Chem 276: 15073–15081 [DOI] [PubMed] [Google Scholar]
- Volkert FC, Broach JR (1986) Site-specific recombination promotes plasmid amplification in yeast. Cell 46: 541–550 [DOI] [PubMed] [Google Scholar]
- Whiteson KL, Chen Y, Chopra N, Raymond AC, Rice PA (2007) Identification of a potential general acid/base in the reversible phosphoryl transfer reactions catalyzed by tyrosine recombinases: Flp H305. Chem Biol 14: 121–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wozniak LA, Janicka M, Bukowiecka-Matusiak M (2005) Consequences of P-chirality in chimeric 2′-O-methyloligoribonucleotides with stereoregular methylphosphonothioate linkages. Euro J Organic Chem 24: 5189–5197 [Google Scholar]
- Wozniak LA, Bukowiecka-Matusiak M, Gora M, Stec WJ (2006) One-Pot synthesis of dinucleoside(3′,5′)-methylphosphonothioates and their seleno congeners via the phosphonotriazolidite approach. Synlett 9: 1331–1334 [Google Scholar]
- Xu C-J, Grainge I, Lee J, Harshey RM, Jayaram M (1998) Unveiling two distinct ribonuclease activities and a topoisomerase activity in a site-specific DNA recombinase. Mol Cell 1: 729–739 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information
Review Process File