

Abstract
Aging is associated with accumulation of genomic rearrangements consistent with aberrant repair of DNA breaks. We have shown previously that DNA repair by nonhomologous end joining (NHEJ) becomes less efficient and more error-prone in senescent cells. Here we show that the levels of Ku70 and Ku80 drop approximately two fold in replicatively senescent cells. Intracellular distribution of Ku also changes. In the young cells roughly half of Ku is located in the nucleus and half in the cytoplasm. In senescent cells the nuclear levels of Ku do not change, while the cytoplasmic Ku fraction disappears. Upon treatment with gamma-irradiation, in the young cells cytoplasmic Ku moved into the nuclear and membrane fractions, while no change in the Ku distribution occurred in senescent cells. Upon treatment with UVC Ku moved out of the nucleus in the young cells, while most Ku remained nuclear in senescent cells. This suggests that the nuclear Ku in senescent cells is unable to respond to DNA damage. We hypothesize that overall decline in Ku levels, changes in Ku intracellular distribution, and the loss of appropriate response of Ku to DNA damage in senescent cells contribute to the decline of NHEJ and to age-related genomic instability.
Keywords: Ku70, Ku80, senescence, DSB repair
1. Introduction
The frequency of genomic rearrangements increases with age (reviewed in [1]). The best example of age-related genomic instability is exponential increase in cancer incidence with age [2], as genomic rearrangements are a hallmark of most tumors [3]. Increased frequency of chromosomal aberrations in non-malignant tissues has also been detected in humans and in mice [4-8]. Studies of transgenic mice carrying lacZ reporter gene show that genomic rearrangements such as deletions, inversions and translocations are a characteristic component of mutation spectra in aged animals [9, 10]. Sequence analysis of these rearrangements suggested that they arose as a result of aberrant repair of DNA double strand breaks (DSBs) via NHEJ pathway [9].
Human somatic cells have limited replicative lifespan in culture, and after approximately 60 population doublings (PDs) enter irreversible cell cycle arrest called replicative senescence [11, 12]. Senescent cells accumulate in aging tissues and may contribute to the functional decline of organ systems [13-17]. The number of replicatively senescent cells that accumulate in aging tissues and whether this number is sufficient to contribute to organismal aging has been controversial. This skepticism arose due to the low frequency of replicatively senescent cells detected in aged tissues using senescence associated β-galactosidase biomarker [18]. Contribution of replicative senescence to organismal aging has recently been reinvestigated using newly discovered biomarkers of senescence — DNA-damage induced foci on telomeres [17]. This analysis revealed that senescent cells account for more than 15% of the cell population in aged animals [17]. The presence of senescent cells at such high frequencies confirms that cellular senescence may contribute significantly to organismal aging.
We have used a replicative senescence model to study whether changes in the efficiency and fidelity of NHEJ process could account for age-related genomic instability. We found that NHEJ became less efficient and more error-prone in senescent human fibroblasts [19]. The efficiency of rejoining of linear DNA substrate was reduced up to 4.5 fold in senescent cells relative to young cells [19]. Furthermore, sequence analysis of end junctions showed that end joining in senescent cells was associated with extended deletions [19].
The goal of the present study was to identify the molecular factors that contribute to the decline of NHEJ during cellular senescence. To this end, we have analyzed the status of Ku protein in young and senescent cells. Ku is an essential component of NHEJ machinery (reviewed in [20-22]). Ku is a heterodimer composed of 70 and 86 kDa subunits (Ku70 and Ku80 respectively). The X-ray crystal structure of Ku shows that the two subunits form a ring that encircles duplex DNA [23]. When DSB occurs, Ku binds to DNA ends and recruits DNA-dependent protein kinase catalytic subunit, which is thought to phosphorylate and activate downstream targets such as Artemis [21, 24-26]. The members of DNA-PK complex are unique among other DNA repair proteins in that they not only localize in the nucleus but also in the membrane and cytoplasm [27, 28]. Cytoplasmic Ku70 subunit acts as antiapoptotic protein by sequestering Bax and preventing its translocation to mitochondria [29]. The function of the cytoplasmic Ku80 is unknown. Ku70 and Ku80 were also found on cell surface of some cell types where they have been implicated in cell adhesion and cell/microenvironment interactions (reviewed in [30]). The members of DNA-PK complex have also been found in the lipid rafts [28]. Thus, Ku is a protein with multiple functions and interaction between these functions is unclear. The role of Ku in aging has been established through mouse knockout studies. Ku80 knockout mice display multiple symptoms of premature aging and cells isolated from these mice undergo premature senescence in culture [31-33]. Ku70 knock out mice develop tumors [34, 35], and cells from these mice show premature senescence associated with accumulation of nondividing cells [34].
In this study, we show that the total levels of Ku decrease in senescent cells. Ku also changes its intracellular localization from nuclear/cytoplasmic in the young cells to strictly nuclear in senescent cells. Furthermore, we found that in the young cells Ku responds to DNA damage by changing its intracellular localization while this response is impaired in senescent cells. We hypothesize that these changes contribute to the decline of NHEJ during cellular senescence.
2. Materials and Methods
2.1 Cell culture
HCA2 neonatal foreskin fibroblasts were a kind gift of O.M. Pereira-Smith (University of Texas Health Sciences Center). IMR-90 embryonic lung fibroblasts were from Coriel Institute for Medical Research. Cells were cultured in minimal essential medium with Earle's salts (Invitrogen), L-glutamine, 0.1 mM nonessential amino acids and 1 mM Na pyruvate, 15% FBS (Invitrogen), and 1× penicillin-streptomycin (Invitrogen) at 37°C, 95% humidity, 5% CO2, and 3% O2. Subconfluent cells were passaged by using 0.25% trypsin in PBS, pH 7.4 (Invitrogen). Cells were passaged until senescence. HCA2 cells became senescent at population doubling (PD) 70, and IMR-90 cells became senescent at PD78.
2.2 DNA damage treatments
For Gamma-irradiation treatment cells were irradiated using Gammacell 1000 (Atomic energy of Canada LTD), at the dose of 50 Gy or 5 Gy at 2.8 Gy/min. The culture medium was changed immediately after irradiation. For UVC treatment cells were washed once with phosphate buffer saline (PBS) and irradiated in PBS at the dose of 400×100μJ/cm2 or 40×100μJ/cm2 using a UVC source (Stratalinker, Stratagene). Immediately after irradiation, PBS was replaced with fresh culture medium.
2.3 Subcellular fractionation
Cells were harvested, washed in PBS, and 3-5×106 cells were used for subcellular fractionation. Subcellular fractionation was performed using ProteoExtract Subcellular Fractionation kit (Calbiochem). Protein concentration in the fractions was determined by modified Lowry method (RC DC protein assay, BioRad).
2.4 Western blot
For analysis of total proteins, cells were harvested and 106 cells were resuspended in 40 μl of PBS with complete protease inhibitor cocktail (Roche), and lysed by addition of 40 μl Laemmli sample buffer with β-mercaptoethanol (BioRad) and boiled for 10 min with vortex every 5 min. Protein concentration was determined by modified Lowry method (RC DC protein assay, BioRad), that allows measuring proteins in Laemmli sample buffer. Proteins were separated on SDS-PAGE and transferred to nitrocellulose membrane. Membranes were hybridized with the following antibodies: Ku70 polyclonal antibody (Abcam, ab2629), Ku70 monoclonal antibody (Abcam, ab87), Ku80 polyclonal antibody (Abcam, ab10879), Ku80 monoclonal antibody (Abcam, ab3715), Lactate Dehydrogenase (LDH) monoclonal antibody (Sigma-Aldrich, L7016), Na+/K+ ATPase monoclonal antibody (Abcam, ab7671), histone H3 polyclonal antibody (Cell Signaling, 9715), GAPDH polyclonal antibody (Abcam, ab9485), LaminB1 polyclonal antibody (Santa Cruz, sc-20682), and Actin (C-2) HRP-conjugated monoclonal antibody (Santa Cruz, sc-8432).
2.5 Immunohistochemistry
Cells were seeded on fibronectin pretreated culture slides (BD Falcon) at 2×104 cells/slide. Two days after plating cells were fixed with 80% acetone for 4 min at room temperature, and air-dried. Cells were washed three times for 10 min with PBS at room temperature and blocked with goat serum (1:100) at room temperature for 1 hour. Then the cells were incubated with Ku80 monoclonal antibody (Abcam, ab3715) overnight at 4°C, washed three times for 10 min with PBS and incubated with FITC conjugated anti-mouse secondary antibody (1:200) for 1 hour at room temperature in the dark. Cells were washed for 10 min three times, stained with 1 μg/ml DAPI for 2 min at room temperature in the dark, washed briefly 3 times with PBS, and the slides were covered with Vectashield mounting media (Vector Laboratories). The images were taken using Leica confocal microscope TCS SP.
3. Results
3.1 Total levels of Ku decrease in senescent cells
To study whether total levels of Ku change in replicatively senescent cells we used two lines of primary human fibroblasts: embryonic lung fibroblasts IMR-90 and neonatal foreskin fibroblasts HCA2. Cells were passaged to senescence and the levels of Ku70 and Ku80 proteins in whole cell extract were compared using Western blot in the young cells (PD38-44 for HCA2 cells, and PD34-40 for IMR-90 cells) and in senescent cells (PD>70 for HCA2 cells, and PD>78 for IMR-90 cells) (Figure 1A). Equal loading of samples was verified by hybridization with Actin, GAPDH, and Lamin B1 antibodies. The amounts of both Ku subunits, Ku70 and Ku80 decreased at least two fold in senescent IMR-90 and HCA2 cells relative to the young cells (Figure 1B). Each Ku subunit was detected with two different antibodies (polyclonal and monoclonal) to confirm that the observed reduction in Ku is not caused by conformational change and the use of conformation-specific antibody. In summary, Ku protein levels decline in senescent fibroblasts. The observed effect was consistent in two fibroblast cell lines of different tissue origin and affected both subunits of Ku.
Figure 1.
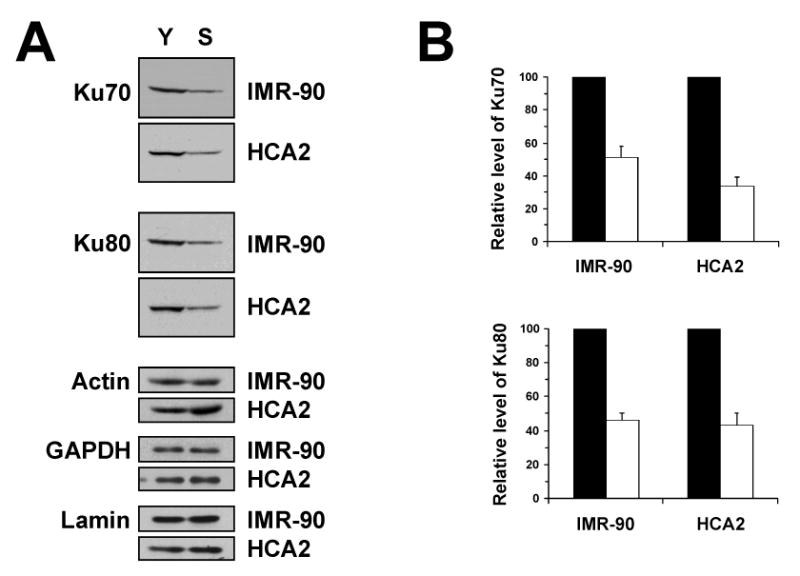
Ku levels in young and senescent cells. (A) Young and senescent IMR-90 and HCA2 cells were harvested and 30 μg of whole cell protein extract was analyzed by Western blot with Ku70 or Ku80 antibodies. Equal loading was confirmed by hybridization with actin, GAPDH, and Lamin B1 antibodies. Y, young cells; S, senescent cells. (B) The Western blots were quantified using ImageQuant software. Protein levels in senescent cells were normalized to the young cells, which were taken as 100%. Black bars represent young cells; white bars represent senescent cells. The experiments were repeated four times, and error bars show standard deviations.
3.2 Subcellular distribution of Ku in young and senescent cells
Ku is found in the nucleus, cytoplasm and membrane, therefore we aimed to test which subcellular fraction of Ku was reduced in senescent cells. Young and senescent IMR-90 and HCA2 fibroblasts were grown as described above, harvested and immediately subjected to fractionation. The fractionation procedure was based on differential solubility of cellular compartments in four sequentially applied extraction buffers. The first extraction buffer releases cytosolic proteins, the second buffer solubilizes membranes and organelles, nuclear proteins are released with the third buffer, and cytoskeleton is solubilized with the fourth buffer. The advantage of this fractionation method is that all the fractions are being extracted from a single cell sample and no protein material is discarded. Therefore, the proteins in the fractions are present in the same proportion as they are found within cells.
Following fractionation the amount of protein in each fraction and its proportion to the total protein in all four fractions was calculated. The fractions were loaded on a gel according to the proportion of total cellular protein they represent. For example, if 30 μg of total protein was loaded in all four fractions, and cytoplasm/membrane/nuclear/cytoskeleton fractions contributed 50%/19%/24%/6% of protein respectively, then 15 μg of cytoplasmic fraction, 5.7 μg of membrane fraction, 7.2 μg of nuclear fraction, and 1.8 μg of cytoskeletal fraction was loaded. Such loading shows the distribution of a given protein between subcellular fractions, keeping the same ratio between the different fractions as they are present within the cell.
Subcellular fractions were verified by hybridizing with LDH (cytoplasmic protein), Na+/K+ ATPase (membrane protein), and histone H3 (nuclear protein) (Figure 2A). Hybridization with the antibodies to the marker proteins shows that the fractionation procedure was very efficient, and there was virtually no cross-contamination between the fractions.
Figure 2.
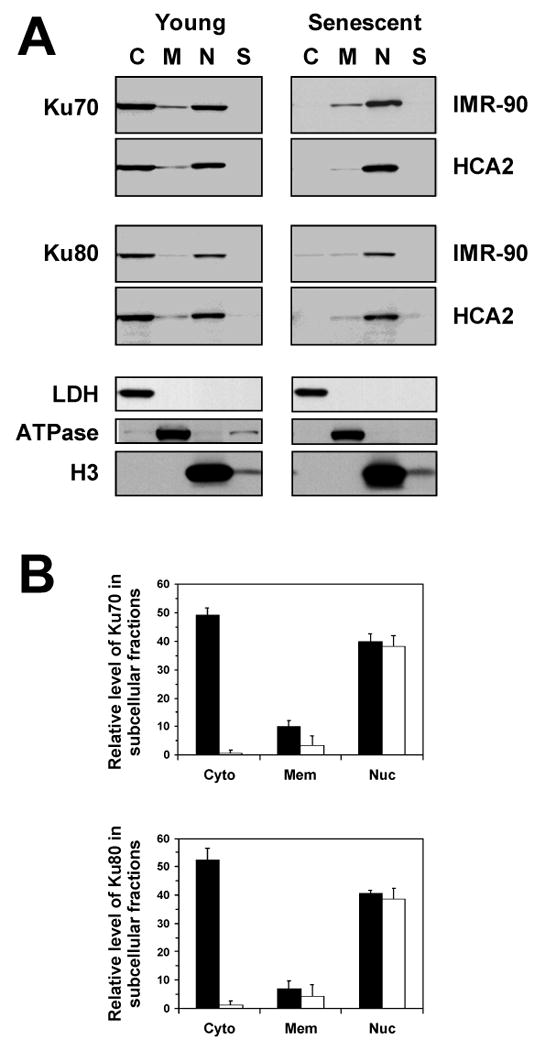
Subcellular distribution of Ku70 and Ku80 in young and senescent cells. (A) Western blot analysis of Ku in subcellullar fractions. Young and senescent IMR-90 and HCA2 cells were fractionated into cytoplasmic (C), membrane/organelle (M), nuclear (N), and cytoskeletal (S) fractions. The fractions were loaded according to the percent of total cellular protein represented by each fraction. Fractions were verified by hybridizing with LDH, Na+/K+ ATPase, and histone H3 antibodies. (B) The Western blots were quantified using ImageQuant software. The fractions were plotted according to the percent of total cellular protein represented by each fraction. The sum of the three fractions in the young cells was taken as 100%; the sum of the three fractions in senescent cells was reduced according to the data in Figure 1. Black bars represent young cells; white bars represent senescent cells. Experiments were repeated four times, and error bars show standard deviations.
Ku70 and Ku80 were localized predominantly in the cytoplasm and the nucleus in young cells (Figure 2). In senescent cells Ku was localized in the nucleus, and almost disappeared from the cytoplasmic fraction (Figure 2). A small amount of Ku was present in the membrane/organelle fraction. The results were consistent for the two cell lines HCA2 and IMR-90.
We then used immunohistochemistry to confirm Ku localization results obtained by cell fractionation. Young and senescent IMR90 and HCA2 cells were stained with Ku80 antibodies (Figure 3A). In young cells Ku was distributed evenly throughout the nucleus and cytoplasm, while in senescent cells Ku was localized in the nucleus. Thus, we show by both cell fractionation and immunohistochemical techniques that Ku is localized in the nucleus and cytoplasm in the young cells, and changes its localization to nuclear in senescent cells.
Figure 3.
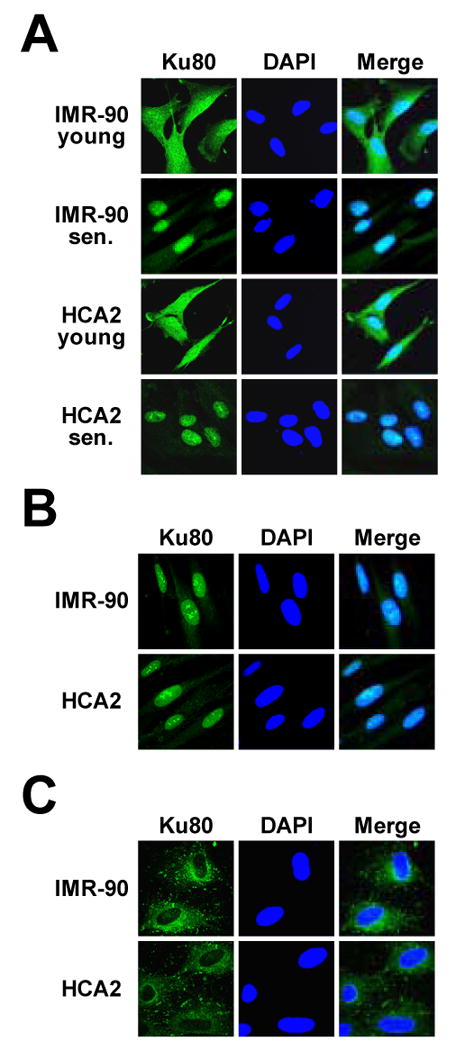
Immunohistochemical analysis of Ku localization in young and senescent human fibroblasts. Young and senescent IMR-90 and HCA2 cells were fixed and incubated with Ku80 antibodies as described in Materials and Methods. Green color is immunofluorescent staining of Ku80; blue color is DNA stained with DAPI. (A) Untreated young and senescent cells. (B) Young cells 24 hours after γ-irradiation. (C) Young cells 24 hours after UVC treatment.
3.3 Ku localization in growth-arrested cells
Replicatively senescent cells enter irreversible G0 growth arrest. We therefore tested whether the changes in Ku levels and intracellular distribution we observed in senescent cells would be found in the young growth-arrested cells. Normal human fibroblasts enter G1 arrest when cells reach confluence and remain viable on a plate, with regular media changes, for extended periods of time. Young IMR-90 and HCA2 cells were kept at confluence for 7 days and the total levels of Ku was tested by Western blot in whole cell extracts (Figure 4A). The levels of Ku70 and Ku80 remained unchanged in the G1 arrested cells. We then tested whether intracellular distribution of Ku in G1-arrested cells. Young cells were kept at confluence for 48 hours or for 7 days and Ku localization was assayed by cell fractionation (Figure 4B). After 48 hours of G1 arrest subcellular distribution of Ku was similar to that of growing young cells (Figures 2 and 4B). After extended period of arrest the distribution of Ku has changed to acquire a unique distribution where similar levels of Ku were localized in the nucleus, membrane, and cytoplasm. This distribution differs from both young and senescent cells. This result shows that the reduction of Ku levels and the changes of intracellular distribution of Ku in senescent cells are characteristic of senescent cells and not the result of the growth-arrest alone.
Figure 4.
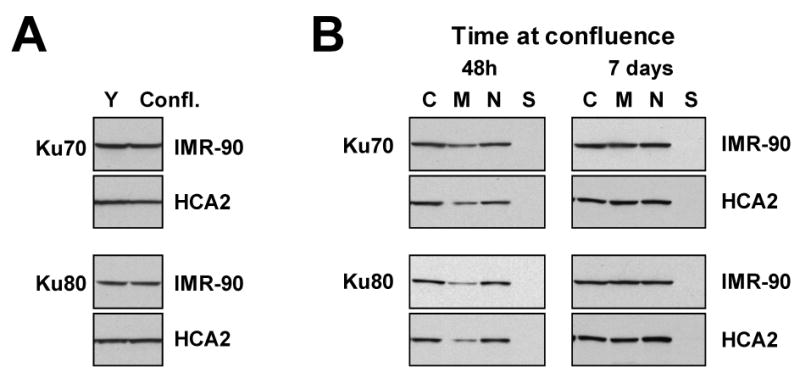
Ku levels and intracellular localization in young arrested cells. (A) Young IMR-90 and HCA2 cells were kept at confluence for 7 days. The total levels of Ku were analyzed by Western blot in whole cell extracts. (B) Cells were kept at confluence for 48 hours or for 7 days and subcellular distribution of Ku was analyzed by cell fractionation as in Figure 2. C, cytoplasmic fraction; M, membrane/organelle fraction, N, nuclear fraction; and S, cytoskeletal fraction.
3.4 Subcellular distribution of Ku changes in response to DNA damage in young but not in senescent cells
We hypothesized that nuclear translocation of Ku in senescent cells is caused by accumulation of DNA breaks and short telomeres that require Ku binding. To test this hypothesis we subjected young cells to DNA damage and examined the changes in Ku distribution. Young and senescent HCA2 and IMR-90 cells were subjected to high and low doses DNA damaging agents: 50 or 5 Gy of γ-irradiation; 400 or 40×100μJ/cm2 of UVC irradiation. γ-irradiation induces DSB breaks while UV induces primarily thymidine dimers that are not repaired by NHEJ. The cells were harvested 1 h, 5 h, and 24 hours after treatment and subjected to subcellular fractionation as described above, and subcellular fractions and whole cell extracts were analyzed by Western blot. Total levels of Ku70 and Ku80 did not change upon treatment (data not shown), while marked changes were observed in subcellular distribution of Ku. Following γ-irradiation of young cells both Ku70 and Ku80 translocated from cytoplasm into the nucleus and membrane/organelles (Figure 5). Translocation was detectable as early as 1 hour after irradiation, more than 50% of Ku left the cytoplasm by 5 hours, and almost no Ku remained in the cytoplasm by 24 hours. The high dose of γ-irradiation produced a stronger effect; only trace amounts of Ku were detectable in the cytoplasm 24 hours after treatment. The low dose also resulted in Ku moving into the nucleus and membrane/organelles, although more Ku remained in the cytoplasm and less Ku accumulated in the membrane/organelles (Figure 5C). Nuclear translocation of Ku following γ-irradiation has also been observed by immunohistochemistry (Figure 3B). It is possible that in the young cells cytoplasmic fraction of Ku serves as a reserve that is recruited to the nucleus upon DNA damage. The distribution of Ku attained by the young cells at 24 hours after γ-irradiation resembled Ku distribution in senescent cells, except the increased level of Ku in the membrane/organelle fraction of irradiated cells. In senescent cells, in contrast to the young cells, intracellular distribution of Ku did not change after γ–irradiation (Figure 5).
Figure 5.
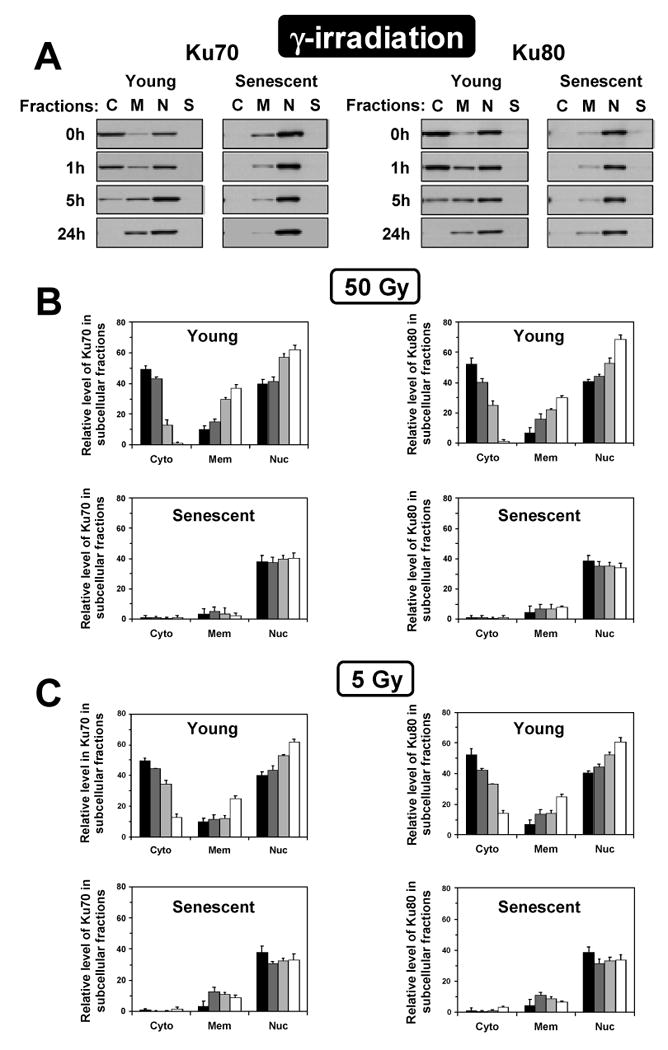
Subcellular distribution of Ku70 and Ku80 in young and senescent cells following exposure to γ-irradiation. IMR-90 and HCA2 cells were irradiated with 5 or 50 Gy of γ-irradiation, and fractionated. (A) Representative Western blots showing changes in subcellular localization of Ku70 or Ku80 after 50 Gy of γ-irradiation. The fractions were loaded according to the percent of total cellular protein represented by each fraction. C, cytoplasm; M, membranes/organelles, N, nucleus; S, cytoskeleton. (B) Quantification of the Western blots obtained after 50 Gy of γ-irradiation. Quantification was done using ImageQuant software. The results for IMR-90 and HCA2 cells were pooled. The fractions were plotted according to the percent of total cellular protein represented by each fraction. The sum of the three fractions in the young cells was taken as 100%; the sum of the three fractions in senescent cells was reduced according to the data in Figure 1. (C) Quantification of the Western blots obtained after 5 Gy of γ-irradiation. Bar's shading represent different time points after irradiation: black bars, 0 time point; dark grey bars, 1 hour after irradiation; light grey bars, 5 hours after irradiation; and white bars, 24 hours after irradiation. The experiments were repeated twice for each cell line and standard deviations are shown.
UVC-irradiation changed intracellular distribution of Ku in young cells but in a very different manner from γ-irradiation (Figure 6). In the young cells subjected to UVC-irradiation both Ku70 and Ku80 moved out of the nucleus into the cytoplasm. The high UVC dose resulted in all Ku being exported into the cytoplasm. The reduction in nuclear Ku was detectable as early as 1 hour after irradiation and almost no Ku was detectable in the nucleus by 24 h post treatment (Figure 6). Immunohistochemical staining of the UVC-irradiated young cells with Ku80 antibodies revealed “hollow” nuclei (Figure 3C), which confirms cytoplasmic translocation of Ku observed by subcellular fractionation. The lower UVC dose induced nuclear export of Ku 1 h after treatment followed by re-entry of Ku at 5 h and 24 h (Figure 6C). In senescent cells Ku response to UVC was greatly attenuated, with only a small fraction of Ku moving out of the nucleus at either high or low UVC doses. In summary this set of experiments shows that in the young cells Ku changes intracellular distribution after DNA damage. This response is impaired in senescent cells, where Ku may be permanently bound to existing DNA lesions in the nucleus and unable to participate in new transactions.
Figure 6.
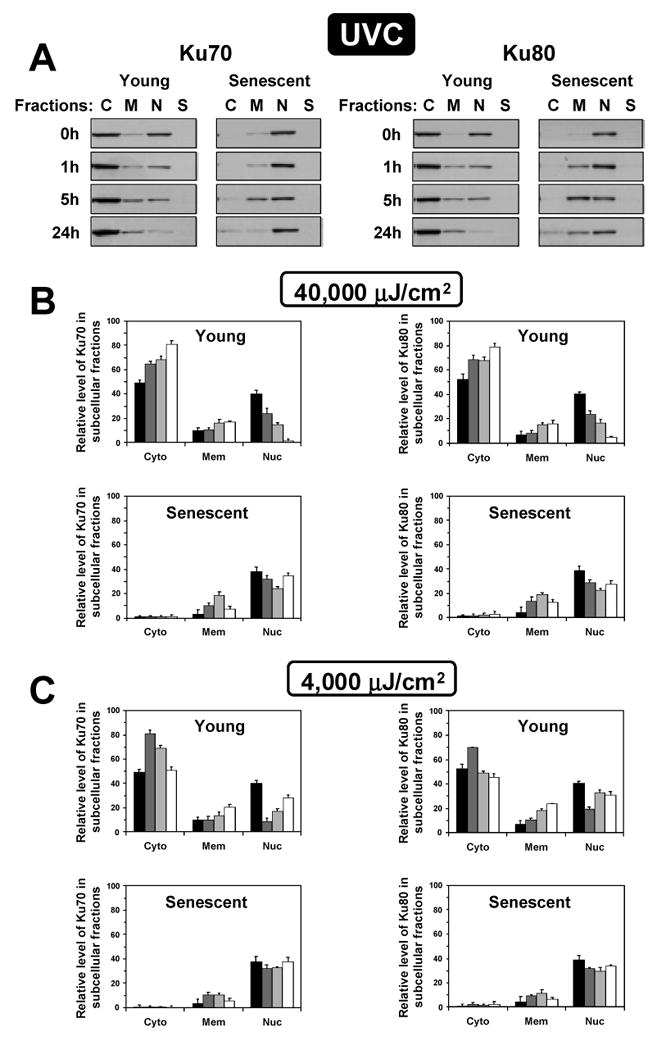
Subcellular distribution of Ku70 and Ku80 in young and senescent cells following UVC irradiation. IMR-90 and HCA2 cells were irradiated with 40×100 or 400×100 μJ/cm2 of UVC and fractionated. (A) Representative Western blots showing changes in subcellular localization of Ku70 or Ku80 after 400×100μJ/cm2 of UVC. The fractions were loaded according to the percent of the total cellular protein represented by each fraction. C, cytoplasm; M, membranes/organelles, N, nucleus; S, cytoskeleton. (B) Quantification of the Western blots obtained after the high dose (400×100μJ/cm2) of UVC. Quantification was done using ImageQuant software. The results for IMR-90 and HCA2 cells were pooled. The fractions were plotted according to the percent of total cellular protein represented by each fraction. The sum of the three fractions in the young cells was taken as 100%; the sum of the three fractions in senescent cells was reduced according to the data in Figure 1. (C) Quantification of the Western blots obtained after the low dose of UVC (40×100μJ/cm2). Bar's shading represent different time points after irradiation: black bars, 0 time point; dark grey bars, 1 hour after irradiation; light grey bars, 5 hours after irradiation; and white bars, 24 hours after irradiation. The experiments were repeated twice for each cell line and standard deviations are shown.
4. Discussion
In this study we examined the levels and intracellular localization of Ku70 and Ku80 proteins during cellular senescence. We show that the levels of both Ku70 and Ku80 in whole cell extracts drop approximately two fold in senescent cells. This effect was observed in two normal human fibroblast cell lines of different tissue origin. Ku70 has recently been proposed to be a new biomarker of aging [36], as reduced Ku levels were also reported in tissues of old rats [37] and humans [36]. We have previously shown that NHEJ in senescent cells is less efficient and more error-prone [19]. Since Ku is a major component of DSB repair machinery the low Ku levels may play a role in the decline of NHEJ in senescent cells.
The analysis of intracellular distribution of Ku by cell fractionation and immunofluorescence revealed marked differences between young and senescent cells. In the young cells Ku is present in the nucleus, cytoplasm, and a small fraction of Ku is localized to the membrane and organelles, while in senescent cells Ku is confined to the nucleus, and a very small amount is found in the membrane fraction. Treatment of young cells with γ-irradiation induced senescent-like changes in the intracellular distribution of Ku: both Ku70 and Ku80 translocated to the nucleus. UVC treatment of young cells induced translocation of Ku out of the nucleus into the cytoplasm. Senescent cells treated with either γ-irradiation or UVC did not change intracellular distribution of Ku. The lack of nuclear translocation in response to γ-irradiation may be less surprising since in senescent cells all Ku is already in the nucleus, while the absence of cytoplasmic translocation in response to UVC suggests that Ku protein in senescent cells is unable to appropriately respond to DNA damage. The differences in Ku dynamics between young and senescent cells are summarized in Figure 7.
Figure 7.

Senescence-related changes in Ku distribution and response to DNA damage. In the young cells Ku (open circles) is present predominantly in the nucleus and cytoplasm. In response to γ-irradiation Ku moves into the nucleus and membrane/organelle fraction. In response to UVC Ku moves out of the nucleus. In senescent cells the majority of Ku protein is localized in the nucleus, presumably in a DNA-bound form (crossed circles). Ku in senescent cells does not change its localization in response to γ-irradiation or UVC.
Our results suggest that Ku is regulated through its transport in and out of the nucleus. How exactly this happens and what factors and signaling pathways are involved in Ku shuttling remains to be determined. We hypothesize that the cytoplasm of young cells contains Ku that serves as a reserve that is recruited to the nucleus upon DNA damage. Senescent cells, in contrast, lack this reserve, so no additional Ku can be brought into the nucleus to repair DNA damage. Furthermore, Ku is unable to move out of the nucleus in senescent cells in response to UVC, which suggests that it is present in a bound form, possibly permanently bound to DNA breaks. It was demonstrated that senescent cells accumulate unrepairable DNA damage foci [38, 39] thus Ku may be trapped by such structures. Ku heterodimer forms a ring that is loaded on DNA strand [23], and it is unknown what takes Ku off the DNA. The release of Ku from DNA may be impaired in senescent cells. Alternatively, Ku may remain at the damage sites and shortened telomeres, which cannot be repaired by DNA repair machinery in senescent cells. In summary, we show that the level of Ku is reduced in senescent cells, senescent cells lack the cytoplasmic fraction of Ku that in the young cells is recruited to the nucleus after DNA damage, and the nuclear Ku in senescent cells is unavailable for new transactions. All these changes in Ku may contribute to the reduced efficiency and fidelity of NHEJ in senescent cells.
Acknowledgments
This work was supported by grants from National Institutes of Health, Ellison Medical Foundation, and American Federation for Aging Research to V.G.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gorbunova V, Seluanov A. Making ends meet in old age: DSB repair and aging. Mech Ageing Dev. 2005;126:621–8. doi: 10.1016/j.mad.2005.02.008. [DOI] [PubMed] [Google Scholar]
- 2.DePinho RA. The age of cancer. Nature. 2000;408:248–254. doi: 10.1038/35041694. [DOI] [PubMed] [Google Scholar]
- 3.Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194:23–28. doi: 10.1126/science.959840. [DOI] [PubMed] [Google Scholar]
- 4.Curtis H, Crowley C. Chromosome aberrations in the liver cells in relation to the somatic mutation theory of aging. Radiat Res. 1963;19:337–344. [PubMed] [Google Scholar]
- 5.Ramsey MJ, Moore DH, Briner JF, Lee DA, Olsen LA, Senft JR, Tucker JD. The effects of age and lifestyle factors on the accumulation of cytogenetic damage as measured by chromosome painting. Mutation Res. 1995;338:95–106. doi: 10.1016/0921-8734(95)00015-x. [DOI] [PubMed] [Google Scholar]
- 6.Tucker JD, Spruill MD, Ramsey MJ, Director AD, Nath J. Frequency of spontaneous chromosome aberrations in mice: effects of age. Mutat Res. 1999;425:135–141. doi: 10.1016/s0027-5107(99)00036-6. [DOI] [PubMed] [Google Scholar]
- 7.Martin GM, Smith AC, Ketterer DJ, Ogburn CE, Disteche CM. Increased chromosomal aberrations in first metaphases of cells isolated from the kidneys of aged mice. Isr J Med Sci. 1985;21:296–301. [PubMed] [Google Scholar]
- 8.Grist SA, McCarron M, Kutlaca A, Turner DR, Morley AA. In vivo human somatic mutation: frequency and spectrum with age. Mutat Res. 1992;266:189–96. doi: 10.1016/0027-5107(92)90186-6. [DOI] [PubMed] [Google Scholar]
- 9.Vijg J, Dolle MET. Large genome rearrangements as a primary cause of aging. Mech Ageing Dev. 2002;123:907–915. doi: 10.1016/s0047-6374(02)00028-3. [DOI] [PubMed] [Google Scholar]
- 10.Dolle MET, Giese H, Hopkins CL, Martus HJ, Hausdorf JM, Vijg J. Rapid accumulation of genome rearrangements in liver but not in brain of old mice. Nat Genet. 1997;17:431–434. doi: 10.1038/ng1297-431. [DOI] [PubMed] [Google Scholar]
- 11.Hayflick L. The limited in vitro lifetime of human diploid cell strains. Exp Cell Res. 1965;37:614–636. doi: 10.1016/0014-4827(65)90211-9. [DOI] [PubMed] [Google Scholar]
- 12.Hayflick L, Moorhead PS. The serial cultivation of human diploid strains. Exp Cell Res. 1961;25:585–621. doi: 10.1016/0014-4827(61)90192-6. [DOI] [PubMed] [Google Scholar]
- 13.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O, Peacocke M, Campisi J. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA. 1995;92:9363–9367. doi: 10.1073/pnas.92.20.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pendergrass WR, Lane MA, Bodkin NL, Hansen BC, Ingram DK, Roth GS, Yi L, Bin H, Wolf N. Cellular proliferation potential during aging and caloric restriction in rhesus monkeys (Macaca mulatta) J Cell Physiol. 1999;180:123–130. doi: 10.1002/(SICI)1097-4652(199907)180:1<123::AID-JCP14>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 15.Choi J, S I, Peacocke M, Peehl D, Buttyan R, Ikeguchi EF, Katz AE, Benson MC. Expression of senescence-associated beta-galactosidase in enlarged prostates from men with benign prostatic hyperplasia. Urology. 2000;56:160–166. doi: 10.1016/s0090-4295(00)00538-0. [DOI] [PubMed] [Google Scholar]
- 16.Ding G, Franki N, Kapasi AA, Reddy K, Gibbons N, Singhal PC. Tubular cell senescence and expression of TGF-beta1 and p21(WAF1/CIP1) in tubulointerstitial fibrosis of aging rats. Exp Mol Pathol. 2001;70:43–53. doi: 10.1006/exmp.2000.2346. [DOI] [PubMed] [Google Scholar]
- 17.Herbig U, Ferreira M, Condel L, Carey D, Sedivy JM. Cellular senescence in aging primates. Science. 2006;311:1257. doi: 10.1126/science.1122446. [DOI] [PubMed] [Google Scholar]
- 18.Cristofalo VJ. SA beta Gal staining: biomarker or delusion. Exp Gerontol. 2005;40:836–8. doi: 10.1016/j.exger.2005.08.005. [DOI] [PubMed] [Google Scholar]
- 19.Seluanov A, Mittelman D, Pereira-Smith OM, Wilson JH, Gorbunova V. DNA end joining becomes less efficient and more error-prone during cellular senescence. Proc Natl Acad Sci U S A. 2004;101:7624–9. doi: 10.1073/pnas.0400726101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Khanna KK, Jackson SP. DNA double-strand breaks: signaling, repair and the cancer connection. Nature Genet. 2001;27:247–254. doi: 10.1038/85798. [DOI] [PubMed] [Google Scholar]
- 21.Lieber MR, Ma Y, Pannicke U, Schwarz K. Mechanism and regulation of human nonhomologous DNA end-joining. Nat Rev Mol Cell Biol. 2003;4:712–20. doi: 10.1038/nrm1202. [DOI] [PubMed] [Google Scholar]
- 22.Collis SJ, DeWeese TL, Jeggo PA, Parker AR. The life and death of DNA-PK. Oncogene. 2005;24:949–61. doi: 10.1038/sj.onc.1208332. [DOI] [PubMed] [Google Scholar]
- 23.Walker JR, Corpina RA, Goldberg J. Structure of the Ku heterodimer bound to DNA and its implications for double-strand break repair. Nature. 2001;412:607–14. doi: 10.1038/35088000. [DOI] [PubMed] [Google Scholar]
- 24.Downs JA, Jackson SP. A means to a DNA end: the many roles of Ku. Nat Rev Mol Cell Biol. 2004;5:367–78. doi: 10.1038/nrm1367. [DOI] [PubMed] [Google Scholar]
- 25.Ma Y, Pannicke U, Schwarz K, Lieber MR. Hairpin opening and overhang processing by an Artemis/DNA-dependent protein kinase complex in nonhomologous end joining and V(D)J recombination. Cell. 2002;108:781–94. doi: 10.1016/s0092-8674(02)00671-2. [DOI] [PubMed] [Google Scholar]
- 26.Goodarzi AA, Yu Y, Riballo E, Douglas P, Walker SA, Ye R, Harer C, Marchetti C, Morrice N, Jeggo PA, Lees-Miller SP. DNA-PK autophosphorylation facilitates Artemis endonuclease activity. Embo J. 2006;25:3880–9. doi: 10.1038/sj.emboj.7601255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koike M. Dimerization, translocation and localization of Ku70 and Ku80 proteins. J Radiat Res (Tokyo) 2002;43:223–36. doi: 10.1269/jrr.43.223. [DOI] [PubMed] [Google Scholar]
- 28.Lucero H, Gae D, Taccioli GE. Novel localization of the DNA-PK complex in lipid rafts: a putative role in the signal transduction pathway of the ionizing radiation response. J Biol Chem. 2003;278:22136–43. doi: 10.1074/jbc.M301579200. [DOI] [PubMed] [Google Scholar]
- 29.Sawada M, Sun W, Hayes P, Leskov K, Boothman DA, Matsuyama S. Ku70 suppresses the apoptotic translocation of Bax to mitochondria. Nat Cell Biol. 2003;5:320–9. doi: 10.1038/ncb950. [DOI] [PubMed] [Google Scholar]
- 30.Muller C, Paupert J, Monferran S, Salles B. The double life of the Ku protein: facing the DNA breaks and the extracellular environment. Cell Cycle. 2005;4:438–41. doi: 10.4161/cc.4.3.1565. [DOI] [PubMed] [Google Scholar]
- 31.Vogel H, Lim DS, Karsenty J, Finegold M, Hasty P. Deletion of Ku 86 cases erly onset of senescence in mice. Proc Natl Acad Sci USA. 1999;96:10770–10775. doi: 10.1073/pnas.96.19.10770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hasty P, Campisi J, Hoeijmakers J, van Steeg H, Vijg J. Aging and genome maintenance: lessons from the mouse? Science. 2003;299:1355–9. doi: 10.1126/science.1079161. [DOI] [PubMed] [Google Scholar]
- 33.Lim DS, Vogel H, Willerford DM, Sands AT, Platt KA, Hasty P. Analysis of ku80-mutant mice and cells with deficient levels of p53. Mol Cell Biol. 2000;20:3772–80. doi: 10.1128/mcb.20.11.3772-3780.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gu Y, Seidl KJ, Rathbun GA, Zhu C, Manis JP, van der Stoep N, Davidson L, Cheng HL, Sekiguchi JM, Frank K, Stanhope-Baker P, Schlissel MS, Roth DB, Alt FW. Growth retardation and leaky SCID phenotype of Ku70-deficient mice. Immunity. 1997;7:653–65. doi: 10.1016/s1074-7613(00)80386-6. [DOI] [PubMed] [Google Scholar]
- 35.Li GC, Ouyang H, Li X, Nagasawa H, Little JB, Chen DJ, Ling CC, Fuks Z, Cordon-Cardo C. Ku70: a candidate tumor suppressor gene for murine T cell lymphoma. Mol Cell. 1998;2:1–8. doi: 10.1016/s1097-2765(00)80108-2. [DOI] [PubMed] [Google Scholar]
- 36.Ju YJ, Lee KH, Park JE, Yi YS, Yun MY, Ham YH, Kim TJ, Choi HM, Han GJ, Lee JH, Lee J, Han JS, Lee KM, Park GH. Decreased expression of DNA repair proteins Ku70 and Mre11 is associated with aging and may contribute to the cellular senescence. Exp Mol Med. 2006;38:686–93. doi: 10.1038/emm.2006.81. [DOI] [PubMed] [Google Scholar]
- 37.Um JH, Kim SJ, Kim DW, Ha MY, Jang JH, Chung BS, Kang CD, Kim SH. Tissue-specific changes of DNA repair protein Ku and mtHSP70 in aging rats and their retardation by caloric restriction. Mech Ageing Dev. 2003;124:967–75. doi: 10.1016/s0047-6374(03)00169-6. [DOI] [PubMed] [Google Scholar]
- 38.Sedelnikova OA, Horikawa I, Zimonjic DB, Popescu NC, Bonner WM, Barrett JC. Senescing human cells and ageing mice accumulate DNA lesions with unrepairable double-strand breaks. Nat Cell Biol. 2004;6:168–70. doi: 10.1038/ncb1095. [DOI] [PubMed] [Google Scholar]
- 39.d'Adda di Fagagna F, Reaper PM, Clay-Farrace L, Fiegler H, Carr P, Von Zglinicki T, Saretzki G, Carter NP, Jackson SP. A DNA damage checkpoint response in telomere-initiated senescence. Nature. 2003;426:194–8. doi: 10.1038/nature02118. [DOI] [PubMed] [Google Scholar]