

Abstract
We have developed theory and the computational scheme for the analysis of the kinetics of the membrane potential generated by cytochrome c oxidase upon single electron injection into the enzyme. The theory allows one to connect the charge motions inside the enzyme to the membrane potential observed in the experiments by using data from the “dielectric topography” map of the enzyme that we have created. The developed theory is applied for the analysis of the potentiometric data recently reported by the Wikström group [I. Belevich, D.A. Bloch, N. Belevich, M. Wikström and M.I. Verkhovsky, Exploring the proton pump mechanism of cytochrome c oxidase in real time, Proc. Natl. Acad. Sci. U. S. A.104 (2007) 2685–2690] on the O to E transition in Paracoccus denitrificans oxidase. Our analysis suggests, that the electron transfer to the binuclear center is coupled to a proton transfer (proton loading) to a group just “above” the binuclear center of the enzyme, from which the pumped proton is subsequently expelled by the chemical proton arriving to the binuclear center. The identity of the pump site could not be determined with certainty, but could be localized to the group of residues His326 (His291 in bovine), propionates of heme a3, Arg 473/474, and Trp164. The analysis also suggests that the dielectric distance from the P-side to Fe a is 0.4 or larger. The difficulties and pitfalls of quantitative interpretation of potentiometric data are discussed.
Keywords: Paracoccus denitrificans, Cytochrome c oxidase, Electron transfer, Proton translocation
1. Introduction
Time-resolved measurements of the membrane potential generated by proteins upon single-electron injection have been one of the most fruitful techniques in studies of charge translocation in membrane proteins such as cytochrome c oxidase (CcO) and reaction centers [1–10]. Although such measurements provide valuable data that reflect charge transfer processes in proteins, getting molecular insights from these experiments has been difficult because of lack of the theory for proper quantitative interpretation of the data. The main problem is related to the dielectric inhomogeneity of the membrane-protein system, which complicates the relationship between the observed membrane potential and the distances traveled by charges in the protein.
The goal of the present paper is to establish a connection between the measured amplitudes of the kinetic phases and charge transfer processes in the protein taking into account the actual inhomogeneous dielectric properties of the system. To this end, we have developed a continuum electrostatic model that directly relates the computed potentials of different groups of the protein to the membrane potential generated when the charges are transferred in the enzyme. The result of such calculations is what can be termed the dielectric topography map of the protein. Each residue is assigned a normalized potential, which is a measure of the dielectric depth of the residue measured from one side of the membrane, so that the difference between the corresponding values of two groups is directly proportional to the membrane potential observed in the potentiometric experiments.
We have created such a map for Paracoccus denitrificans CcO, and have applied our theory for the analysis of the potentiometric kinetic data on the O to E transition reported recently by Belevich et al. [9]. In their experiment, three protonic kinetic phases were observed. Using their data, we have attempted to establish the identity of the groups that exchange charges and generate the observed potentials. Of our particular interest is the so-called Proton Loading Site (PLS) of the pump. We will show that although the identity of PLS cannot be precisely established, this site can be localized to a small group of residues located just “above” the Binuclear Center (BNC) of the enzyme. Both the dielectric model of the enzyme and the experimental data contain uncertainties that prevent an unambiguous molecular interpretation of the potentiometric data. The limitations of the theory are discussed.
The insights obtained in the analysis are discussed in the context of other potentiometric experiments and proposed proton pumping models of CcO. This paper demonstrates the usefulness of the developed approach and with further improvements can provide a quantitative method for interpretation of potentiometric data not limited to CcO, but for other proton pumps as well.
The plan of the paper is as follows. In the next section we present theory that describes how the potentials observed in the experiment can be calculated and connected to specific groups inside the enzyme that exchange charges; the sequential kinetic model that is typically used in the analysis of potentiometric data is described next. We then present the results of the calculations on the P. denitrificans enzyme, and apply the developed theory for the analysis of the experimental data by Belevich et al. We examine the extent to which identity of the proton loading site of the pump can be determined from the data. We conclude the paper with a discussion of the results for other systems, including Rhodobacter sphaeroides mutant N139D. Limitations of the potentiometry are discussed and suggestions are made as to how to improve the accuracy of the method.
2. Theory
2.1. Relation between the measured membrane potential and calculated potentials of individual groups in the protein
For interpretation of the experimental results we need to calculate the potentials generated by moving charges in inhomogeneous dielectric medium. The system consists of a protein molecule embedded in an infinite planar hydrophobic membrane of thickness L separating two aqueous electrolyte solutions with ionic strength of I. The membrane planes together with the outer protein surfaces can be considered as two plates of a capacitor, a and b. The protein region is inhomogeneous dielectrics with a coordinate-dependent dielectric constant ε(r). In order to formulate the problem numerically, we cut on the membrane a circle of a large radius R with its center at the protein, such that ε(r) is constant outside the circle x2+y2=R2 (the z-axis is perpendicular to the membrane with its origin at the membrane center), where the potential created by a voltage difference between two plates of the capacitor has the form
| (1) |
Here, ΔVm = Vplate b − Vplate a is the potential difference between the N-side plate b (z=L/2) and the P-side plate a (z=−L/2). Boundary conditions require that potential be constant at the plates,
| (2) |
Below we will show that the normalized potential,
| (3) |
obeying the modified linearized Poisson–Boltzmann equation [11,12],
| (4) |
directly represents the required “dielectric topography” of the protein, that is, φ0(r) is the dielectric distance traveled by the electron moving from the P-side to a given point r inside the protein and 1−ϕ0(r) is the distance traveled by the proton from the N-side to r. In Eq. (4), εw is the dielectric constant of water, κ(r) is a constant equal to the Debye–Hückel screening parameter κ outside the membrane–protein system and zero inside it, and f(r) is 1 on the N-side and 0 everywhere else. The term with f(r) is introduced in order to satisfy the boundary conditions at z → ± ∞. The normalized potential ϕ0(r) ranges from 0 at z → −∞ to 1 at z → + ∞.
The voltage ΔVm is expressed in terms of the capacitance C and the total charge,
| (5) |
accumulated at each plate, so that
| (6) |
In the limit of R→∞, both Q and C are increasing, whereas their ratio remains finite. The function ϕ0(r) is independent of the cut-off radius R when R is significantly larger than the protein dimensions. The charges are inhomogeneously distributed over the plates, so that
| (7) |
Now let us consider the experimental setup. When a single electron is injected into the protein and is transferred between the redox centers, it causes translocation of protons. It is a change in the potential difference between the plates induced by a single-electron injection that is measured in experiment. Therefore, the problem is formulated differently in this case.
Let ψ(r) be the potential at point r when a single point charge q′ is placed at a point r′. If both r and r′ belong to the protein interior, ψ(r) satisfies the Poisson equation,
| (8) |
with boundary conditions (1) and (2), where now ΔVm is an unknown constant. The potential induced by q′ can be written in a general form as
| (9) |
where G(r,r′) is the potential created at r by a unit charge placed at r′. We note that this is not the Green function as defined in common textbooks (see, e.g. [13,14]) since it does not obey homogeneous boundary conditions. It is important, however, that it is symmetric with respect to its arguments. In order to prove this statement, we consider the total energy change, ΔW, when two charges, q and q′, are placed at points r and r′, respectively. It consists of three terms, ΔW=A +A′ +B, where A is the interaction energy of q with the medium, A′ is the same for q′, and B is the interaction between q and q′. The latter can be calculated either as the energy of charge q in the potential of Eq. (9), B=qq′G(r,r′), or as the energy of charge q′ in the potential created by charge q, B=q′qG(r′r). Since both equations represent the same quantity, we obtain
| (10) |
We will use this property in order to connect our calculated function ϕ0(r) with the measured trans-membrane potential generated by a single-electron injection.
Consider charge q placed at point r. According to Eq. (9), the potential generated by this charge at plate a is
| (11) |
where G(ra,r) is independent of the location of the point ra on the surface of the conductor a and is therefore designated simply as Ga(r). For the potential difference between plates a and b we obtain
| (12) |
If several charges qk move from the initial locations to the final locations , the change in the potential difference is given by
| (13) |
where nm is the number of enzymes on the membrane and summation is over charges in a single enzyme. Thus, in order to calculate the amplitudes of the kinetic phases observed in experiments, we have to relate φ(r) with ϕ0(r), the latter being obtained as a numerical solution to a different electrostatic problem discussed at the beginning of this section. This relationship can be obtained as follows.
Consider again the first problem where there are no charges, but instead a known potential difference ΔVm is maintained between the capacitor plates. The solution given by Eq. (6) can now be written in terms of function φ. First of all, the potential created by a surface charge dQa at a point r is dQaG(r,ra). Furthermore, G(r,ra)= G(ra,r) ≡ Ga (r) is independent of ra due to its symmetry, see Eq. (10). Hence, integration over surface a gives QaGa(r), and the same applies to surface b. With this and Eq. (5), we obtain that the total potential created at a given point r by both plates is Qφ(r). Comparing it with Eq. (6), we obtain the equation
| (14) |
which relates the function needed for interpretation of experiment with the one calculated numerically. It is worthwhile to note that the capacitance of our system, C, tends to infinity when the cut-off radius, R, increases infinitely. Therefore, any changes of the potential generated by a single charge must disappear, i.e., φ(r) → 0 when R → ∞, whereas the product on the left of Eq. (14) remains finite. From Eqs. (13) and (14), we obtain
| (15) |
If charges are measured in atomic units, i.e., qk = −1 for electron and +1 for proton, then the coefficient of proportionality, CV = enm/C, is the potential generated by a unit charge crossing the full membrane (from the measured value of CV =2.5 mV, see footnote to Table 2, one obtains C/nm =6.4×10−17 F). The k-th term on the right represents the relative, dimensionless dielectric distance covered by charge qk moving from to , which is equal to the potential generated due to this movement. For instance, the electron moving from the P-side (ϕ0=0) to a point r inside the protein generates the potential CVϕ0(r), and the proton moving from the N-side (ϕ0=1) to r generates CV[1 − ϕ0(r)].
Table 2.
The inverse rates (in ms) and normalized cumulative amplitudes of four kinetic phases (V4 =2) found by four-exponential fitting of the experimental kinetics of the potential generated upon single-electron injection into CcO from P. denitrificans [9]
| Set number | V1 | V2 | V3 | SD (0.1μV) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 0.020 | 0.140 | 0.636 | 2.28 | 0.336 | 1.06 | 1.62 | 5.82 | ||||
| 2 | 0.021 | 0.150 | 0.716 | 2.37 | 0.350 | 1.09 | 1.66 | 5.91 | ||||
| 3 | 0.022 | 0.155 | 0.758 | 2.41 | 0.358 | 1.11 | 1.68 | 6.00 | ||||
| 4 | 0.026 | 0.189 | 0.824 | 2.16 | 0.418 | 1.20 | 1.68 | 6.85 | ||||
| 5 | 0.010 | 0.149 | 0.769 | 2.21 | 0.259 | 1.11 | 1.68 | 7.75 | ||||
| 6 | 0.010 | 0.15 | 0.8 | 2.6 | 0.24 | 1.08 | 1.68 | 10.5 |
Sets 1–4, Our fittings of the original experimental data file provided by Belevich et al. The rate of the first phase was kept constant, other three rates and four amplitudes were optimized. The merit function is the sum of relative deviations squared. CV is the same as in set 5.
Set 5, Inverse rates, amplitudes Ui (recalculated in terms of Vi), and CV =2.477 mV are from Belevich et al. (private communication).
Thus, in order to predict the potential that a charge transfer between groups A and B inside the enzyme will generate, one needs to know the potentials ϕ0(A) and ϕ0(B) of these groups. The observed membrane potential is proportional to the difference between ϕ0(A) and ϕ0(B), as Eq. (15) states. The potentials ϕ0(gi) of different groups gi of the P. denitrificans enzyme will be calculated in the following section.
2.2. Sequential kinetic model
The observed potential generated across the measuring membrane upon single-electron injection is usually represented in the form
| (16) |
where N is the number of kinetic phases, k1 > k2 > … >kN are rate constants, Ai are the amplitudes normalized such that their sum is equal to the total number of charges translocated across the full membrane at , and CV is the potential generated by unit charge crossing the full membrane.
It will be assumed that the above potential is generated by a sequence of charge transfer processes of the form:
| (17) |
where S0 is the initial state of the enzyme before electron injection, SN is the final state when all charge translocations have occurred, and Si are intermediate states. Each intermediate state is characterized by a specific charge distribution in the enzyme and a specific potential Vi accumulated when the system reaches state Si. As the system undergoes a sequence of transitions from state S0 to state SN, the membrane potential is changing from V0 to VN.
If the potentials Vi are normalized such that VN is the total charge translocated across the membrane, the evolution of the membrane potential can be represented as follows:
| (18) |
where pn(t) are populations of states Sn. The time-dependent populations of a general sequential kinetic model, such as in Eq. (17), with N+1 states are given by
| (19) |
where ki are the rates of sequential transitions, K01 =1, and
| (20) |
(for derivation, see Supplementary information, section S1). Substituting Eq. (19) into Eq. (18) and comparing the resulting expression with the experimental fitting curve, Eq. (16), one finds the relationship between the observed apparent amplitudes of the N kinetic phases and the membrane potentials Vn that characterize the intermediate kinetic steps. The relationships are
| (21) |
It should be noticed that the observed amplitudes Ai depend not only on the potentials of the intermediate states Vn but also on the transition rates ki. It is only when the time-scales of successive kinetic phases are well separated, ki ≫ ki + 1, that the observed amplitudes Ai become equal to potential increments (Vi − Vi − 1) between the successive steps. In general, the kinetic phases are not separable —the next phase begins while the previous one is not completely finished. Hence, the kinetic overlap is critical for the correct analysis of the experimental kinetics [8,15].
Inverting the above equation, one can express the potentials Vi in terms of the observed amplitudes of the kinetic phases Ai and rates ki. A specific relation between Ai in Eq. (16) and Vi in Eq. (18) for N =4 that corresponds to one electronic and three protonic phases, as in experiment of Belevich et al., is given in section S1 of Supplementary information, see also [8,15].
Wikström and co-workers [6,9,16] used another equivalent representation for the observed potential,
| (22) |
where F1(t)=1 − p0(t), F2(t)=1 − p0(t) − p1(t),…, FN(t)= pN(t). Comparing Eqs. (18) and (22) one finds that the newly-defined “amplitudes” Ui (not to be confused with the commonly used amplitudes Ai) represent the increments of the potential between the successive phases, that is,
| (23) |
Note that both Vi and Ui are the actual potentials due to charge displacements; hence, in contrast to Ai, they are independent of the rate constants. When the rates of successive phases differ more than an order of magnitude, the overlap effect is negligible, and one has Ai = Ui.
Experimental data typically include sets of rate constants and amplitudes, either Ai [4,5,7,8] or Ui [6,9,16]. The theory of the previous section allows one to directly connect the charge transfer processes inside the enzyme with the amplitudes derived from the experimental kinetic curves.1 In order to do this, one only needs to know the potentials corresponding to different groups. We next discuss the calculations of such potentials for P. denitrificans CcO.
3. Computational methods
3.1. Electrostatic calculations
The electrostatic calculations described in section 2 were performed using a modified version of Adaptive Poisson–Boltzmann Solver (APBS), ver. 0.3.2 [17–19]. The details are given in section S2 of Supplementary information. The structure of CcO from P. denitrificans determined by Harrenga and Michel [20] (PDB code 1QLE, all redox centers oxidized) was used in the calculations. The first three subunits containing all redox and other important groups were included in the calculations (see related ref. [21]). The membrane was modeled as a planar low-dielectric slab, see Fig. 1. The orientation of the membrane with respect to the molecular frame of the protein was assumed to be the same as for bovine enzyme, for which calculations involving the membrane were performed by us earlier [22].
Fig. 1.

Schematics of CcO incorporated in the membrane and major groups involved in proton pumping activity. The photo-injection of an electron into the enzyme results in the development of the membrane potential examined in this paper.
3.2. Membrane parameters
The coordinates of 1QLE were transformed to have the best overlap with the structure of 1V55 [23](bovine CcO). The transformation was performed by the Swiss-PDB Viewer [24] best fit with the so-called structure alignment function, which minimizes RMSD of Cα atoms of homologous residues in the first two subunits. The coordinate system of 1 V55 has the xy-axes in the plane of the membrane and the z-axis perpendicular to it, directed from the P-side to the N-side. The resulting orientation of the membrane with respect to protein body can be described in a coordinate system fixed in the molecular frame of CcO. This frame was defined by three unit vectors e1, e2, and e3 connecting CuB with Fe a, Fe a3, and His326/ND1, respectively; the orientation of the membrane was then defined by a unit vector u normal to the membrane surface and aligned along the z-axis of 1 V55 structure. In the molecular frame of CcO, the normal vector u has the following form: u=0.832e1 −0.469e2 −1.178e3. Throughout the paper, we describe the membrane position, M.P., as the z-coordinate of the membrane center in the coordinate system of 1V55. Then, M.P.=194.0 Å corresponds to the membrane center located at distance 3.42 Å from CuB in the direction of vector u; hence, the z-coordinate of CuB is 190.58 Å.
The membrane width was taken to be 26 Å, which is the accepted value for the low-dielectric part of the membrane [25,26]. We also varied it between 25 and 32 Å [27,28] and found that potential variations for all key residues around the catalytic center were small, within ±0.02.
Taking into account that the membrane width=26 Å corresponds to the membrane potential=1 and that the uncertainty of the atomic positions in the X-ray structure is about 0.5 Å (from their b-value, which is around 20), we find that the uncertainty in the membrane potential due to small thermal fluctuations should be about ±0.02.
3.3. Dielectric constants
The internal water cavities in the protein were determined by an in-house geometry-based program, details are given in the Supplementary information.
Dielectric constants assigned for the membrane and the external solvent regions were εm =2 and εw =80, respectively. For the bulk protein and the internal cavities, the dielectric constants were varied in the ranges εp =4–16 and εcav =4–80, respectively, to explore various degrees of hydration and polarizability of the protein [29].
The major factor was the ratio εp:εcav, whereas the absolute value of εp was less important. Thus, changing εp at a given εp:εcav =1:2 or 1:5 shifted the potentials of CuA, PropA/a3, Arg474/HH11, and Trp272/HE by about 0.02, and less for other sites. The internal cavities that belong to the first two subunits, where all charge translocations are taking place, were included in the calculations. Temperature and ionic strength were 300 K and 0.1 M, respectively. Within the physiological range, variations in these two parameters had very little effect on the results.
4. Results
4.1. The dielectric topography map of CcO
In order to determine the potentials of different groups of CcO, the following continuum electrostatic calculations have been performed. The protein structure of CcO from P. denitrificans was incorporated into a membrane, which was modeled as a dielectric slab, see Fig. 1. The potential difference=1 was set across the membrane, and the distribution of the electric potential ϕ0(r) inside the protein was calculated by solving the modified Poisson–Boltzmann equation (Eq. (4) with appropriate boundary conditions), see [11,22] and Supplementary information. The potential ϕ0(r) varying from 0 to 1 is a measure of the “dielectric depth” inside the membrane. As shown in section 2, the experimentally observed membrane potential Vfi generated by a point charge q transferred from point r(i) to point r(f) is proportional to the difference between the electric potentials ϕ0(r) in points r(i) and r(f), see Eq. (15). In a homogeneous planar membrane, the potential Vfi would be proportional to the geometric distance between the points projected onto the normal across the membrane, i.e., the difference of the geometric depths of the two points. In a real system this is not the case; the potential is proportional instead to the difference of the dielectric depths. The potentials of the most interesting groups in CcO are shown in Table 1 (we show only a few groups of interest; the extended version of the Table, in the same format, is given in the Supplementary information).
Table 1.
Dielectric topography map of CcOa
| Membrane center (Å) |
Variation |
||||||
|---|---|---|---|---|---|---|---|
| Residue | Atom | 190.0 | 194.0 | 197.0 | 200.0 | Diel. | Width |
| Metal sites | |||||||
| CuA | (top) | 0.09 | 0.06 | 0.05 | 0.04 | ±0.01 | ±0.01 |
| (bottom) | 0.12 | 0.09 | 0.07 | 0.05 | ±0.01 | ±0.01 | |
| Heme a | Fe | 0.53 | 0.44 | 0.38 | 0.32 | ±0.01 | ±0.01 |
| Heme a3 | Fe | 0.52 | 0.43 | 0.37 | 0.31 | ±0.01 | ±0.02 |
| CuB | Cu | 0.51 | 0.42 | 0.35 | 0.29 | ±0.01 | ±0.02 |
| BNCb | 0.52 | 0.43 | 0.36 | 0.30 | ±0.01 | ±0.02 | |
| PLS candidatesa | |||||||
| His326 | HD1 | 0.38 | 0.31 | 0.25 | 0.21 | ±0.02 | ±0.02 |
| Heme a3 | HA1 | 0.32 | 0.25 | 0.21 | 0.17 | ±0.01 | ±0.01 |
| HA2 | 0.30 | 0.23 | 0.19 | 0.15 | ±0.01 | ±0.01 | |
| HD1 | 0.39 | 0.31 | 0.26 | 0.22 | ±0.01 | ±0.01 | |
| HD2 | 0.36 | 0.29 | 0.24 | 0.19 | ±0.02 | ±0.01 | |
| Arg473 | HE | 0.34 | 0.27 | 0.22 | 0.18 | ±0.01 | ±0.01 |
| Arg474 | HE | 0.29 | 0.23 | 0.19 | 0.15 | ±0.02 | ±0.01 |
| Trp164 | HE | 0.46 | 0.38 | 0.32 | 0.27 | ±0.01 | ±0.01 |
| Trp272 | HE | 0.34 | 0.26 | 0.21 | 0.17 | ±0.01 | ±0.01 |
| PLS-related sites | |||||||
| Asp399 | OD2 | 0.30 | 0.23 | 0.18 | 0.14 | ±0.01 | ±0.02 |
| Waterc | O | 0.36 | 0.29 | 0.24 | 0.19 | ±0.01 | ±0.02 |
| K-channel residuesa | |||||||
| D-channel residuesa | |||||||
The normalized potentials of the atoms shown have the meaning of the dielectric depth measured from the P-side of the membrane. The charge transfer between any of the atoms shown will produce membrane potential proportional to the difference of the potentials of the atoms. The shown data are discussed in the text.
This is an abbreviated version of the data. A more complete table is given in the Supplementary information.
BNC is defined as the midpoint between Heme a3/Fe and CuB/Cu.
The water molecule located between the two propionates of heme a3 and His326. Its position is estimated from the crystallography structure 1 V55[23].
4.1.1. Effects of dielectric inhomogeneity, membrane width, and the position of the membrane
The electrostatic computational model of the protein employed to produce the data shown in Table 1 involves several unknown parameters: the width of the membrane, the position of the membrane with respect to the protein, and the dielectric composition of the protein itself. By the width of the membrane we mean the width of the hydrophobic and low-dielectric part (εm ≈ 2) of the lipid bi-layer which affects the electrostatic calculations. Roughly, the boundaries of this part of the membrane coincide with the position of carbonyl groups of the lipids. The orientation of the membrane plane with respect to the protein was discussed in section 3, and appears to be well-defined by the structure of the protein. However, the position of the center of the low-dielectric part of the membrane is more difficult to determine from the published structural data and considered here as an unknown parameter. Finally, the dielectric composition of the protein is determined by the internal water cavities of the protein, which are filled with ill-defined number of water molecules. These regions of the protein should have higher dielectric constant. However, the exact value of the dielectric constant is not known. In this subsection, we examine how the results of our calculation depend on these parameters.
We assume that the thickness of the low-dielectric part of the membrane is in the range of 25–32 Å; for example for L-lecithin [16] the width of the low-dielectric part is close to 25–27 Å. The potential calculations were performed for various values of the membrane width; we find that the potentials of the most interesting groups located in the middle part of the membrane are not very sensitive to the membrane thickness in the range of 25–32 Å. Table 1 lists the data for membrane of 26 Å and the variation of the potential (last column) for each of the group due to uncertainty in the membrane width is reported in a separate column.
Similarly, the dependence of the results on the dielectric composition of the protein was found to be relatively unimportant within the range of reasonable variation of the dielectric constant of the protein, εp =4–16 and dielectric of the water cavities, εcav =4–20. The data shown in Table 1 were obtained for εp =4 and εcav =10, and the variations of the potentials with changing dielectric of the cavities in the range of 4–20, as well as with various dielectric constants for the protein are also shown for each group in Table 1 in the second column from the right.
The most significant parameter, however, that is crucial for the electrostatic topography of the protein was found to be the position of the membrane center, M.P. In Fig. 2, dielectric distances (difference of potentials) between some important sites of the protein calculated as functions of membrane position are shown for various values of εcav. The total dielectric distance between two sides of the membrane is set to unity. As seen from Fig. 2, dielectric inhomogeneity of the protein–membrane system has relatively minor effect on the dielectric distances. This agrees with our previous estimates [15]. The potential and distances, however, are quite sensitive to the position of the membrane center with respect to the protein. Hence, any analysis of the data on the amplitudes of various kinetic phases observed in experiment requires precise determination of the “optimal” membrane position using some additional information independent of experimental data, which would permit such determination. In the following section we will demonstrate how this can be accomplished in practice using experimental data by Belevich et al.
Fig. 2.
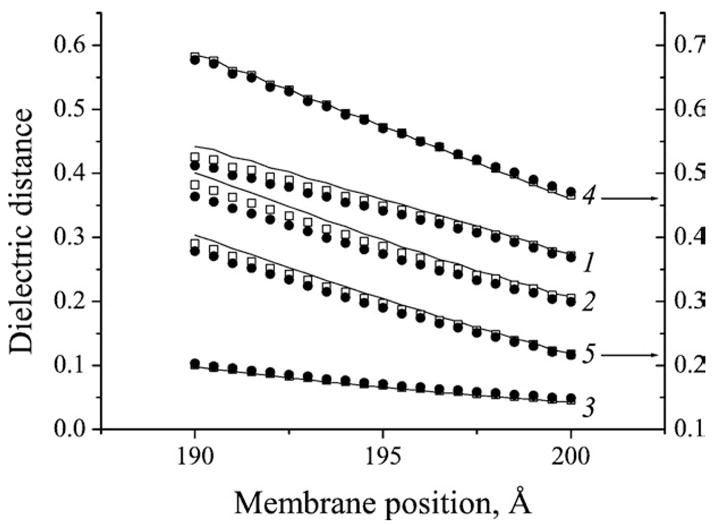
Calculated dielectric distances for P. denitrificans. Membrane width 25 Å, εp =4, εcav =4 (line), 10 (open squares), 20 (full circles). 1, from CuA to Fea; 2, from P-side to His326; 3, from P-side to CuA; 4, from P-side to Glu278; 5, from P-side to PropD1/Fea3. Left ordinate, data sets 1, 2, and 3. Right ordinate, data sets 4 and 5. For definition of the membrane position, see Methods section.
As will be shown later in the paper, the requirement of self-consistency of the model and consistency with experimental data can be utilized to reduce the degree of uncertainty in the position of the membrane, M.P. Also, there are indirect structural data that also allow one to make a reasonable estimate of the range of possible values of this important parameter. Namely, the low-dielectric part of the membrane is localized in the region roughly between the carbonyl groups of the fatty acids of the membrane lipids; some of these lipids have been recently reported by Yoshikawa and co-workers for bovine enzyme [30]. In Fig. 3, the position of some of the carbonyl groups on the bovine structure (subunits I and II) are shown, and the corresponding range of possible positions of the low-dielectric part of the membrane, assuming its width 26 Å, is schematically indicated; for these positions of the membrane, the low-dielectric part of the membrane would be localized between the carbonyl groups found in the structure. Although the interface between the membrane and the enzyme is unlikely to be identical for bacterial and bovine enzymes, the positions of redox centers in two species are amazingly similar (the z-component of the coordinates of the redox centers, normal to the membrane surface, differ only by less than 0.2 Å). If we assume that the position of the membrane center in the bovine structure is about the same as in Paracoccus structure (although the membrane width may be somewhat different), the bovine structural data can be used for an estimate of the possible positions of the membrane in the bacterial enzyme. Based on these considerations, we assume that the position of the membrane center should be expected to be between 194 and 200 Å (for the description of the coordinate system utilized in our calculations see section 3), and the position of the membrane center outside the window 194–200 Å will be considered as unlikely.
Fig. 3.
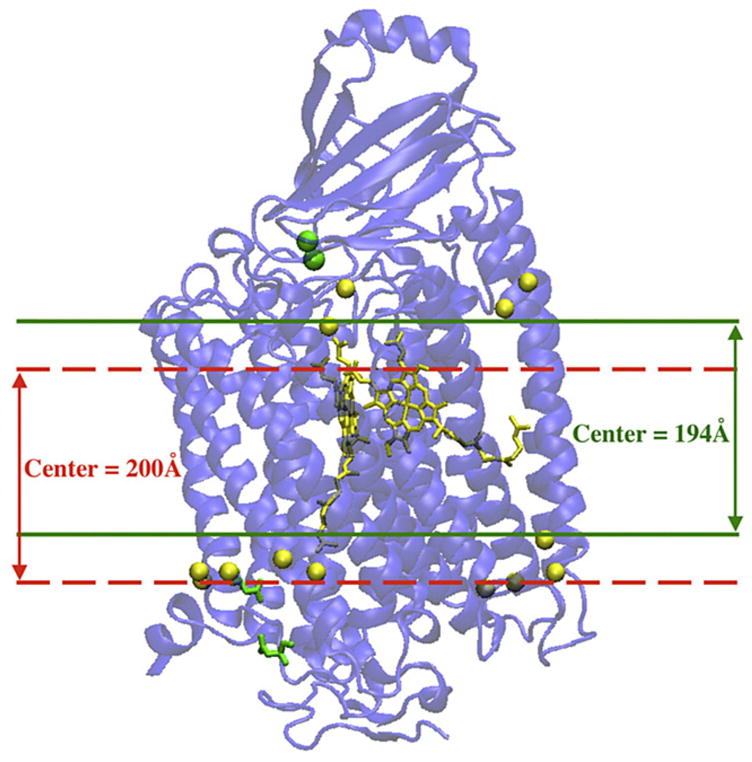
The structure of the bovine enzyme, subunits I and II [30] (PDB ID=2DYR), and possible range of membrane center positions. The coordinate system employed is described in the text. The light balls correspond to carbonyl groups of the phospholipids of the membrane, which are considered to be the boundaries of the low-dielectric (insulating) part of the membrane.
In Table 1, the potentials of CcO are shown for three possible positions of the membrane in this range, 194, 197, and 200 Å. It is noteworthy that at the membrane position 200 Å the potential of BNC is about one third of the total. We also show data for one additional position of the membrane, 190 Å, for the following reason. In the next section we will be looking for a combination of the parameters of the model including the position of the membrane M.P. that is most consistent with the potentiometric data. As we will see, the best fit to experimental data is provided for three positions of the membrane: 190, 194, and 197 Å. Based on the arguments of this section, the position of 190 Å can be discarded as it is in the conflict with structural data, however, for completeness purposes, and also to demonstrate details of our analysis these data are also included in Table 1. Thus, as we will see later, only data shown for positions 194 and 197 Å are fully consistent with structural and potentiometric data of Belevich et al. These data are shown in Table 1 in bold, and will be further discussed in the following sections of the paper.
4.1.2. Potential of CuA center
The results for the CuA center shown in Table 1 deserve special consideration. The calculated dielectric distance from CuA to the P-side of the membrane is appreciably non-vanishing in contrast to the commonly accepted opinion that it is zero because CuA does not reside within the membrane domain, but instead belongs to the part of the protein that is exposed to the aqueous solution on the P-side of the membrane. We notice, however, that CuA is buried inside the low-dielectric part of the protein and appears from the structure to be isolated from the solution, therefore there must be some non-zero potential drop from the P-side solution to CuA center. On the other hand (M. Wikström and M. Verkhovsky, private communication), when the photo-activated RubiPy donates its electron to the CuA center in less than 0.5 μs, the potential should show significant initial jump, up to about one third of the amplitude of the fast (electronic) phase, which had not been observed in the experiment. This disagreement between the theory and experiment might be due to the fact that when RubiPy complex is docking in the vicinity of the CuA center, it partially deforms the protein surface, allowing for some additional water molecules and ions to penetrate into the structure, and therefore reduces the isolation of the CuA center. In our calculations, we used the intact CcO structure since the structure of the CcO/RubiPy complex is unknown.
Allowing for possibility that the structure of the CcO/RubiPy complex involves additional “connectivity” between the P-side and CuA center, not seen in the X-ray structure of CcO, in the following analysis of the pumping mechanism of CcO, we will consider the case when the potential of CuA is set to be zero, in addition to actual data from our calculations shown in Table 1.
4.2. O to E transition in P. denitrificans. Experiment of Belevich et al. [9]
In a recent paper, Belevich et al. [9] have presented the results of the most detailed electrometric study yet on CcO from P. denitrificans, in which the kinetics of the membrane potential generated by the enzyme upon single-electron injection in the O to E transition was measured. In addition to a pure electronic kinetic phase, three protonic phases have been resolved, for which the rates ki and the corresponding amplitudes Ui were reported.
The experimental details are described in Ref. [9]. Briefly, the enzyme was incorporated into a measuring membrane. With use of RubiPy as photo-sensitized donor, an electron was injected into the enzyme prepared in the “pulsed” oxidized state O (or OH, using notation of [6]), and the membrane potential generated by the enzyme, as it made the O to E transition, was monitored with a time step of ≥1 μs. In a separate experiment, changes of the redox state of the enzyme were also monitored by optical spectroscopy. Three phases of the redox kinetics of heme a were revealed with rates k1,2,3 shown in Table 2. The kinetics of the membrane generation was analyzed in Ref. [9] with use of Eq. (22) with N=4 and rates k1,2,3 fixed at their values from the optical data whereas k4 and four amplitudes were varied to get the best fit to the experimental kinetics. It was obtained , U1 =0.24, U2 =0.84, U3 =0.60, and U4 =0.32. Instead of U1,2,3,4, we will use the cumulative amplitudes V1,2,3 of Eq. (18) shown in Table 2.
4.2.1. Qualitative interpretation
According to Ref. [9], phase 1 (10 μs) corresponds to electron transfer from CuA to heme a, see Fig. 1. Only a “fraction of electron”, β=0.70, is transferred to heme a in this phase. Further electron redistribution between CuA, heme a, and the heme a3/CuB binuclear center (BNC) is coupled to three proton transfers, which generate three additional kinetic phases. The spectroscopic data indicate that, by the end of the 150 μs phase, 60% of the electron is transferred to the heme a3/CuB site, while 40% still remains on heme a. This electron transfer is coupled to the transfer of the pumped proton from the N-side of the membrane to an unknown proton-loading site (PLS) “above” BNC. The proton transfer occurs via Glu278, which is deprotonated and reprotonated in a concerted manner since a stepwise process would require overcoming a large barrier inconsistent with the observed kinetics [31]. The next 800 μs phase corresponds to the transfer of the remaining 40% of the electron from heme a to BNC and an accompanying transfer of the chemical proton to BNC. Finally, the last 2.6 ms phase is associated with the displacement of the pumped proton from PLS to the P-side of the membrane. Presumably, this happens due to repulsion between the chemical proton arrived to BNC and the pumped proton preloaded to PLS [32]. Overall two unit charges are crossing the membrane during the transition, therefore V4 =2.
The sequence of events — 1) electron transfer to BNC, 2) loading the pumped proton to PLS via Glu278 in the D-channel, 3) transfer of the chemical proton to BNC, and 4) ejection of the pumped proton from PLS to the P-side of the membrane due to Coulomb repulsion between the pumped and chemical protons — exactly corresponds to the scheme described earlier in Ref. [33] (see also [32]), where it was proposed, based on ab initio and electrostatic calculations, that the role of PLS is played by one of the ligands of the CuB center, His326. The identity of PLS, however, has never been directly probed experimentally. Below, making use of the above data of Belevich et al., we will examine the proposed mechanism and will try to establish likely candidates for PLS.
4.2.2. Quantitative analysis
4.2.2.1. Analysis of the kinetics data. Uncertainty of exponential fittings
There is an intrinsic uncertainty in the rates and amplitudes of individual kinetic components that are found from the decomposition of the experimental signal. This is a well-known difficulty of exponential decomposition of the kinetic data. While for qualitative interpretation these uncertainties may not be of any significance, our goal is to explore the extent to which potentiometric data can be used for understanding molecular specifics of the charge transfer in the enzyme. Belevich et al. have kindly provided us with their original raw experimental data (and their own precise fitting parameters) so that we could explore various fittings. Some results obtained are shown in Table 2, Sets 1–4. Set 5 is provided by Belevich et al. in their private communication, and Set 6 is the original set of parameters from Ref. [9], we only added standard deviations (SDs). Both these fittings used a fixed rate of k1 that was determined from the optical experiment. Since the ET driving force of CuA to Fe a transition is close to zero, this transition rate k1 may be different in the enzyme incorporated in the membrane, in particular when some potential exists on the membrane. It is appropriate to stress that the details we are considering at the moment were not important for Belevich et al. in their analysis of the experimental data. For instance, small changes of k4 and V1 between Sets 5 and 6 do not affect any conclusions of Ref. [9]. On the contrary, our approach requires more quantitative evaluation of the parameters. Therefore, we explored additional fittings with different rate constants varied in the range of 10 to 26 μs. It is worth mentioning that for the rate of the electronic phase of the O to E and E to R transitions in CcO from P. denitrificans, Ruitenberg et al. [4,5] reported the values of 20 and 27 μs, respectively, which fall in the range of our values. As seen from Table 2, several different fittings could be obtained, with quite significant (for our quantitative analysis) variation of the amplitudes V1–V3.
4.2.2.2. Pumping model, identity of PLS
Our model is identical to what is qualitatively described by Belevich et al., see Fig. 4. We now provide a quantitative description of this model, and examine possible candidates for PLS. In this scheme, we have three intermediate states, S1–S3, and corresponding membrane potentials, V1–V3.
Fig. 4.
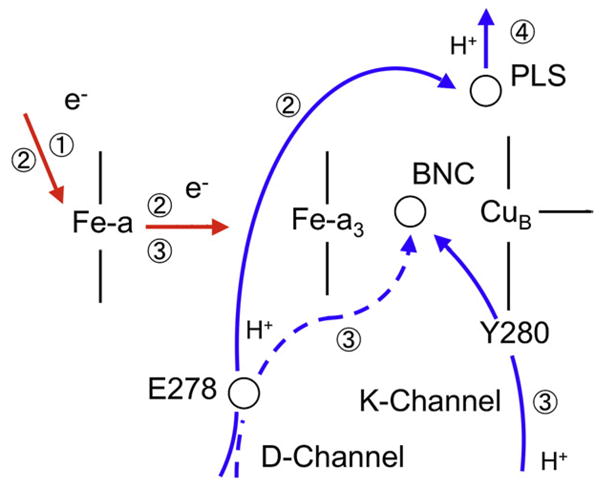
Schematics of the considered pumping model (c.f. [33]). The numbers correspond to the kinetic phase order observed in the experiment. BNC is the binuclear catalytic center, PLS is the Proton Loading Site of the pump. For O to E transition discussed in the text, the third phase presumably corresponds to proton transfer to BNC along the K-channel; in other transitions, the same transition may occur along the D-channel.
Phase 1, in which the first intermediate state S1 is formed, is associated with the transfer of the injected electron via the CuA center to heme a. As established experimentally, only a fraction of electron β=0.70 moves to heme a in this phase. Therefore, the potential generated by the enzyme when state S1 is formed can be written as
| (24) |
where VCuA and Va are normalized potentials (i.e., dielectric depths) of CuA and heme a, respectively, measured from the P-side of the membrane, obtained from our electrostatic calculations, Table 1, whereas V1 is obtained from the kinetic analysis of the experimental data, Table 2. In a similar way we obtain the relationships for other two phases.
Phase 2 corresponds to the S1 to S2 transition, where the second intermediate state S2 is formed. This phase is associated with the transfer of the pumped proton from the N-side of the membrane to an unknown group PLS, via the D-channel and Glu278, and simultaneous redistribution of the injected electron between the redox centers of the enzyme. By the end of this phase fraction (1−γ)=0.6 of the electron is localized on the a3/CuA binuclear center and fraction γ=0.4 remains on heme a. Hence, this intermediate generates the potential
| (25) |
The contribution of the proton is written in such a way as to reflect the fact that the proton moves from the N-side of the membrane, which is assigned the potential of +1, to Glu278 and further to PLS in a concerted manner [31].
Phase 3, S2 → S3, is the transfer of the chemical proton and the remaining fraction of the electron to BNC. The corresponding potential is
| (26) |
where the first term corresponds to a complete electron transfer to BNC, the second term is due to the chemical proton transfer to BNC, and the third term is due to the pumped proton from the previous phase.
The above three equations relate the potentials that we found from the kinetic analysis of the experimental data (V1,V2, and V3, Table 2) to the potentials of the specific groups of the enzyme that we calculated by solving the Poisson–Boltzmann equation, Table 1. The identities of all groups are known, except for that of PLS.
If we knew the exact values of V1–V3, and if there were no ambiguity in the electrostatic model and we could calculate the potentials of different groups unambiguously, then we could use Eq. (26) to find VPLS, and then look up the table of potentials of different groups of the protein, such as Table 1, and find the group that has the “target” value VPLS – thus, the identity of the PLS would be revealed. Unfortunately, there are ambiguities both in the membrane potentials, i.e., left-hand-sides of Eqs. (24)–(26), found from the decomposition of the experimental data (Table 2), and in the electrostatic model, which determines the potentials of the groups in the right-hand-side of Eqs. (24)–(26). Thus, the identity of PLS cannot be easily determined.
With such uncertainties both on the right- and left-hand-sides of Eqs. (24)–(26), one might think that there is no hope to find a unique solution to the problem, since there will be always a combination of parameters such that Eqs. (24)–(26) would be satisfied. It turns out, however, that the situation is not as hopeless as it appears to be.
The overall situation that we deal with resembles that in the branch of mathematics which considers so-called “ill-posed” problems, where the data are not known exactly, and there is no unique solution to the problem. In this case, one attempts to identify the “most likely” solution that best satisfies a given set of conditions. In our case, we have several independent conditions, Eqs. (24)–(26), and certain requirements on the membrane position. Therefore we proceed as follows.
We have multiple combinations of amplitudes V1–V3 that all satisfy the experimental data, some of which are shown in Table 2. Without additional conditions, one cannot choose one set of parameters over another. We consider our Eqs. (24)–(26) as these additional conditions that should be satisfied. Therefore, we will be looking for such a set of experimental parameters V1–V3, a position of the membrane, and a PLS group that best satisfy all three equations. To estimate the quality of the solution we introduce a statistical parameter χ2 as follows:
| (27) |
where Δi is the difference between left- and right-hand-side of Eqs. (24)–(26), and σi is a typical uncertainty with which the right-hand-side of Eqs. (24)–(26) could be calculated. This uncertainty is due to small variations of the membrane composition and width, as well as the accuracy with which parameters β and γ could be determined in the experiment. We assume β =0.7±0.05 and γ =0.4±0.05. For computed potentials (other than that of CuA) the maximum error (for a given membrane position) is roughly 0.02 and maximum error due to uncertainty in the atom’s position is also roughly 0.02. Overall, in our estimate the σi are 0.034, 0.049, and 0.030 for Eqs. (24), (25), and (26), respectively.
We then introduce the likelihood parameter,
| (28) |
which is evaluated for each kinetic fitting k, membrane position m, and PLS candidate p. The parameter P is a statistical measure of the quality of a given set of parameters of the model.
Table 3 lists several best combinations of kinetic parameters, membrane positions, and PLS candidates. We examined both the case when the potential of CuA is calculated from our electrostatic model (non-zero potential), and the case when the potential of CuA is set to zero, for reasons explained in section 4.1. Despite the uncertainty of the parameters of the model, we find that the number of PLS candidates turns out to be rather small. The reason for such a selection is the large number of conditions that the model should satisfy for self-consistency: three equations, Eqs. (24)–(26), and an additional condition for a specific window for the membrane position. The analysis with this method yields the following results. (The Supplementary information provides details of the analysis of PLS candidates. Table 3 gives only the summary of the results).
Table 3.
PLS candidates for six different kinetic fitting sets shown in Table 2
| Set | Most likely PLS candidates (P-value, M.P.) |
|---|---|
| A. VCuA ≠ 0 | |
| 1 | Trp164 (81% at 194.0 Å) |
| 2 | a3/D1 (52% at 193.0 Å) and His326 (39% at 193.0 Å) |
| 3 | His326 (41% at 193.5 Å) and a3/D1 (41% at 194.0 Å) |
| 4 | a3/A1 (55% at 190.5 Å), a3/A2 (45%), Trp272 (42%) |
| 5 | Trp164 (7.9% at 197.0 Å) |
| 6 | Trp164 (3.5% at 197.5 Å) |
| B. VCuA =0 | |
| 1 | Trp164 (35% at 193.5 Å) |
| 2 | a3/D1 (22% at 193.0 Å) and His326 (21% at 192.5 Å) |
| 3 | His326 (14% at 193.5 Å) and a3/D1 (13% at 193.5 Å) |
| 4 | a3/A2 (20% at 189.0 Å), a3/A1 (17%), Trp272 (12%) |
| 5 | Trp164 (20% at 197.0 Å) |
| 6 | Trp164 (14% at 197.0 Å) |
In (A), the PLS analysis was done with VCuA predicted by our electrostatic model, which in general gives non-zero potential, see Table 1; (B) same as in (A), but with the potential of CuA set to zero. P is the likelihood parameter, Eq. (28), and M.P. is the membrane position that was found for a given set of kinetic parameters.
As can be seen in Table 3, only three positions of the membrane, around 190, 194, and 197 Å are consistent with experimental data and with the pumping model considered – that is only these positions of the membrane yield any PLS groups. Only positions 194 and 197 Å however are in the window of realistic values. Hence, the PLS candidates found for the membrane position of 190 Å should be considered as unlikely. There was no PLS found for the position of the membrane at 200 Å. Some kinetic fitting sets do not provide any candidates for PLS at any position of the membrane in the interval of expected values between 194 and 200 Å.
For the membrane positions in the interval of expected values, i.e., between 194 and 200 Å, the analysis yields several PLS residues which are clustered in the region just “above” BNC. We first consider the case of VCuA ≠ 0. Here, the best candidates are: Trp164, P=0.8, Set 1; His326, PropD/heme a3, P =0.5–0.4, Set 2 and 3; naturally, the sites with close potential, such as propionate D of heme a, a water molecule between the two propionates of heme a3 and His326, and Arg 473/474, cannot be excluded as PLS candidates on the basis of potentiometry data.
In Set 4, PropA/a3 and Trp272 were identified as PLS candidates with likelihood P=0.4; however, in this case the membrane position turns out to be around 190 Å, i.e. out of the range of the expected values; hence, these candidates should be considered as unlikely. For Sets 5 and 6 (Belevich et al.) the only PLS candidate is Trp164 with a very low likelihood values, P=0.08 and 0.04. All other groups, for all kinetic parameter sets produce the likelihood parameter much smaller that what we considered above (for more quantitative details, see extended Table 3 in the Supplementary information).
When the assumption VCuA =0 is imposed, the results are as follows. PLS candidates of Sets 1–4 remain the same, however, the likelihood parameter becomes significantly smaller: Trp164 (Set 1, P=0.35), His326 and PropD/a3 (Set 2, P =0.22); PropA/a3 and Trp272 (Set 4, P =0.2) appeared here as well, but only for membrane position around 190 Å and therefore should be considered less likely candidates than others. Sets 5 and 6 now yield Trp164 as the best candidate for PLS, with P=0.2 at M.P. around 197 Å.
We see that both cases of zero and non-zero potential of CuA result in the same group of PLS candidates, which are clustered in a small region just above BNC. The likelihood parameter P allows one to qualitatively compare the candidates. It is hardly possible, however, to make a strong case for one specific candidate from this group on the basis of potentiometric data alone. Obviously, the above results are maximum that one can expect from the present theory and current data.
4.3. N139D non-pumping mutant of R. sphaeroides, Siletsky et al. [7]
While we did not make electrostatic calculations for this bacterium, data of section 4.1 can still be used for semi-quantitative discussion. In the experiments on F→O transition in N139D mutant recently reported by Siletsky et al. [7], only one electronic and one protonic phases were observed with the amplitude ratio of A2/A1 =1.3. Since the time constants of the two phases differ significantly (15 and 600 μs), the measured amplitudes directly represent the intrinsic dielectric distances, that is, A1 = U1 = V1 and A2 = U2 = V2 − V1. The total charge transferred across the full membrane is V2 =1, which gives V1 =0.435. By its physical meaning, the amplitude of the electronic phase, V1, is approximately equal to the dielectric distance from the P-side to Fe a, or equivalently from the P-side to BNC (Fe a3 or CuB), see Eq. (24) where β =0.84 [8]. For P. denitrificans, we found that for most likely M. P.=194 Å the potential of BNC is 0.43, instead of the usually assumed “geometric” value 0.33. If this is also approximately valid for R. sphaeroides, the above experimental amplitude ratio and the resulting value of V1 =0.435 can be rationalized.
It is instructive to calculate the value of V1 using geometric considerations. Inserting VCuA = 0, Va − VCuA = 0.33, and β = 0.84 into Eq. (24), we obtain V1 =0.28, which is significantly smaller than the one found above. The geometric value predicts the amplitude ratio A2/A1 =2.6, that is, two times as large as the observed value. Thus, our explanation of the experimental result implies that, “dielectrically”, heme a and catalytic center are buried deeper into CcO, closer to the N-side, than they are “geometrically”.
5. Discussion
5.1. Uncertainties, dielectric map
The electrostatic calculations presented above indicate that the major factor influencing the dielectric topography of a protein incorporated in a membrane is the relative position of the membrane center with respect to the protein body. Other factors, such as the dielectric constants for protein cavities and the width of the hydrophobic part of the membrane, have relatively minor effects. Therefore, it is important to know the actual membrane position (M.P.) with respect to the body of the protein in order to be able to calculate the dielectric distances between key sites, which could be further used to analyze experimental data on the kinetics of the potential generation. In the absence of direct structural data, here we attempted to reduce the uncertainty of the membrane position by using indirect structural data for bovine enzyme.
There is a generic uncertainty in the decomposition of a kinetic signal into individual exponential components [34]. In our case the major uncertainty of the fitting procedure arises due to the first kinetic phase. As seen from Table 2, the amplitude V1, which mainly affects the membrane position, fluctuates by about 50% among various fittings whereas other amplitudes are stable within a few percent. This is because the experimental counts are taken with a long time step of 1 μs, which results in 150 and more counts during the lifetimes of the second and subsequent phases but only 10 counts during the first phase, which is insufficient for accurate determination of V1. To reduce this uncertainty, it would be desirable that the fast part of the kinetics within, say, the initial 100 μs be measured with a short time step on the order of 0.1 μs.
It cannot be excluded, indeed it is expected, that each of the three protonic phases observed in the experiment of Belevich et al. consists of several components; by its nature, proton translocation is always a multi-step process. The fitting of a kinetic signal with larger number of exponential components is possible, but highly numerically unstable. For example, we could fit the experimental kinetic data with five exponentials instead of four; in this fit the intermediate phase with lifetime k−1 =0.57 ms splits into two components with lifetimes 0.52 and 0.71 ms. The quality of overall fitting is about the same as for four kinetic components. However, our analysis suggests that these refinements can hardly be reliable enough to be utilized for identification of the groups involved.
5.2. BNC potential 0.4
We found that the dielectric distances between the sites participating in the catalytic cycle of CcO differ significantly from the values based on the geometry. For example, the binuclear center is located at approximately one third of the membrane width from the P-side, which implies the geometric distance of 0.33, but the dielectric depth of CuB and Fe a3 from the P-side were found to be at least 0.4 (for the membrane positions that are consistent with both the structural data and kinetic potentiometry data of Belevich et al.) This result helps to rationalize the experimental data of Siletsky et al. [7] for N139D non-pumping mutant of R. sphaeroides enzyme. It is interesting to note that the “electric distance” between heme a and the P-phase had been originally estimated on the basis of equilibrium studies (effect of the imposed membrane potential on a redox equilibrium between cytochrome c and heme a in mitochondria) by P. Hinkle and P. Mitchell almost 40 years ago[35], who obtained a value of ~0.4–0.5. The present work appears to reconcile the Hinkle and Mitchell’s results with the structural data.
5.3. CuA potential
Another redox center to be noted is CuA. It is generally assumed that its dielectric distance from the P-side is zero because CuA is located outside the membrane region. However, this center is located inside the low-dielectric protein region generally isolated from the P-side solution, therefore our electrostatic calculations yield non-zero potential of CuA. The question arises with respect to the electron transfer from the primary electron donor, RubiPy, to CuA. This very fast transfer should generate an appreciable initial jump of potential that had not been observed in experiment. The nature of this disagreement is not clear. It well may be that the protein structure is deformed in the vicinity of RubiPy docking site, so that water and ions penetrate closer to CuA thereby decreasing the dielectric distance of this center from the P-side. In such a case, our electrostatic problem should be solved with a new boundary condition at the P-side, and this would result in decreasing the calculated potential not only at CuA but also at Fe a and other sites (to a lesser extent). There are no structural data at present to undertake such calculations. Instead, we examined an artificial condition when the potential of CuA was set to be zero. There was no major qualitative difference in the PLS candidates produced by the procedure.
5.4. PLS candidates
The computational approach developed in this paper, together with detailed potentiometric data such as obtained by Belevich et al. [9], opens the door for quantitative analysis aiming at determining specific residues that exchange charges inside the membrane protein and produce changes of the membrane potential observed in the experiment. Even though a specific residue that plays the role of the loading site of the pump (PLS) could not be identified, due to uncertainties both in the experimental data and in the theory, it is still remarkable that the PLS could be localized to a relatively small group of candidates. All these candidates: Trp164, His326, PropD, A/a3, Arg473/474 for PLS are located “above” BNC (that is, closer to the P-side).
Recently, Siegbahn and Blomberg [36] have analyzed the rates of the four kinetic phases measured by Belevich et al. [9]. These authors derived energy levels and barriers for likely intermediate states of the pumping process using the reaction mechanism proposed by Belevich et al. and considered in this paper. They concluded that propionate A of heme a3 is the best candidate for PLS. Our analysis shows that PropA of heme a3 is indeed in the group of possible candidates, but less likely candidate for PLS compared with Trp164, His326, Arg474, PropD of heme a3.
5.5. BNC-first model
Brzezinski with coauthors [37–39] have proposed an alternative mechanism where the first proton goes from Glu278 to BNC rather than to PLS. Their studies were on the P to F transition in R. sphaeroides. We have considered such a possibility for the O to E transition in P. denitrificans discussed in section 4.2 and found that it is not supported by our data. Since the only change is that (1 − VPLS) term in V2 is replaced with (1 − VBNC), one should formally consider BNC to be potentiometrically equivalent to PLS in our model. In this case, however, we could not agree quantitatively with the data of Belevich et al. because all our PLS candidates turned out to be “above” BNC. One should keep in mind of course that all these considerations are based on the assumption of equivalence of different transitions in different enzymes.
6. Conclusions
The relative position of the membrane with respect to the protein is a critical factor affecting the dielectric distances in CcO. The lack of structural information about the membrane is a significant impediment for computational analysis of the potentiometric data.
The dielectric distances from the P-side to Fe a and to BNC are larger than 0.33 suggested by geometry/structure. Our estimate is approximately 0.4 or higher; the exact number is difficult to pinpoint, but our estimate in any case is significantly larger than the usually assumed 0.33. The new estimate explains the ratio of the amplitudes of two kinetic phases observed in a non-pumping CcO mutant.
In the proton-pump model with a single proton-loading site (PLS), the most probable candidates for PLS belong to a group of closely related residues above BNC: Trp164, propionate D of heme a3, His326, Args 473/474; while Trp272 and propionate A of heme a3 also cannot be excluded.
Obtaining more definitive predictions with respect to the identity of PLS might be possible if more detailed experimental data for the first 100 μs of the potential kinetics were available. The data should be obtained with a shorter time-step, say 0.1 μs, for better resolution of the kinetic parameters of the signal.
Despite the inherent uncertainties, it is clear that potentiometry in combination with the computational analysis demonstrated here is a powerful technique for characterization of protonpumps. It would be interesting to perform such experiments and analyses on Complex I of the respiratory chain, the proton pumping mechanism of which remains a complete mystery [40,41].
As a final remark, it is worthwhile to mention that impossibility of strict identification of the proton loading site may also be an indication that there is no single PLS for the proton pump in CcO; rather, several residues within the above-found group may serve as PLS with various degrees of participation.
Supplementary Material
Acknowledgments
This work was supported by grants from NSF (PHY 0646273) and NIH (GM54052) to AAS, and also by the grants from Civil Research and Development Foundation (RUC2-2658-MO-05) and the Russian Foundation for Fundamental Research (08-03-00094-a, 05-03-33127-a) to ESM. The authors are grateful to M. Wikström and M. Verkhovsky for providing their original experimental data file and helpful discussions. We are also grateful to the referee for pointing our attention to Ref. [35] and the related comments.
Abbreviations
- CcO
cytochrome c oxidase
- BNC
binuclear center
- PLS
proton-loading site
- N-side
negative side of the membrane
- P-side
positive side of the membrane
- M.P
membrane position
- SD
standard deviation
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at doi:10.1016/j.bbabio.2008.05.006.
Footnotes
Amino acid residues are numbered according to the Paracoccus denitrificans cytochrome c oxidase sequence.
We notice that the multi-exponential curve-fitting/decomposition of the observed transient membrane potential is numerically unstable, and unless the kinetic components have significantly different rates, the accurate determination of the amplitudes and rates of the components, and indeed the number of such components, has practical limitations.
References
- 1.Dracheva SM, Drachev LA, Konstantinov AA, Semenov AY, Skulachev VP, Arutjunjan AM, Shuvalov VA, Zaberezhnaya SM. Electrogenic steps in the redox reactions catalysed by photosynthetic reaction centre complex of Rhodopseudomonas viridis. Eur J Biochem. 1988;171:253–264. doi: 10.1111/j.1432-1033.1988.tb13784.x. [DOI] [PubMed] [Google Scholar]
- 2.Zaslavsky D, Kaulen AD, Smirnova IA, Vygodina T, Konstantinov AA. Flash-induced membrane potential generation by cytochrome c oxidase. FEBS Lett. 1993;336:389–393. doi: 10.1016/0014-5793(93)80843-j. [DOI] [PubMed] [Google Scholar]
- 3.Konstantinov AA, Siletsky S, Mitchell D, Kaulen A, Gennis RB. The role of the two proton input channels in cytochrome c oxidase from Rhodobacter sphaeroides probed by the effects of site-directed mutations on time-resolved electrogenic intraprotein proton transfer. Proc Natl Acad Sci U S A. 1997;94:9085–9090. doi: 10.1073/pnas.94.17.9085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ruitenberg M, Kannt A, Bamberg E, Ludwig B, Michel H, Fendler K. Single-electron reduction of the oxidized state is coupled to proton uptake via the K pathway in Paracoccus denitrificans cytochrome c oxidase. Proc Natl Acad Sci U S A. 2000;97:4632–4636. doi: 10.1073/pnas.080079097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ruitenberg M, Kannt A, Bamberg E, Fendler K, Michel H. Reduction of cytochrome c oxidase by a second electron leads to proton translocation. Nature. 2002;417:99–102. doi: 10.1038/417099a. [DOI] [PubMed] [Google Scholar]
- 6.Bloch D, Belevich I, Jasaitis A, Ribacka C, Puustinen A, Verkhovsky MI, Wikström M. The catalytic cycle of cytochrome c oxidase is not the sum of its two halves. Proc Natl Acad Sci U S A. 2004;101:529–533. doi: 10.1073/pnas.0306036101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Siletsky SA, Pawate AS, Weiss K, Gennis RB, Konstantinov AA. Transmembrane charge separation during the ferryl-oxo →oxidized transition in a nonpumping mutant of cytochrome c oxidase. J Biol Chem. 2004;279:52558–52565. doi: 10.1074/jbc.M407549200. [DOI] [PubMed] [Google Scholar]
- 8.Siletsky SA, Han D, Brand S, Morgan JE, Fabian M, Geren L, Millett F, Durham B, Konstantinov AA, Gennis RB. Single-electron photoreduction of the PM intermediate of cytochrome c oxidase. Biochim Biophys Acta. 2006;1757:1122–1132. doi: 10.1016/j.bbabio.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 9.Belevich I, Bloch DA, Belevich N, Wikström M, Verkhovsky MI. Exploring the proton pump mechanism of cytochrome c oxidase in real time. Proc Natl Acad Sci U S A. 2007;104:2685–2690. doi: 10.1073/pnas.0608794104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chamorovsky SK, Cherepanov DA, Chamorovsky CS, Semenov AY. Correlation of electron transfer rate in photosynthetic reaction centers with intraprotein dielectric properties. Biochim Biophys Acta. 2007;1767:441–448. doi: 10.1016/j.bbabio.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 11.Roux B. Influence of the membrane potential on the free energy of an intrinsic protein. Biophys J. 1997;73:2980–2989. doi: 10.1016/S0006-3495(97)78327-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grabe M, Lecar H, Jan YN, Jan LY. A quantitative assessment of models for voltage-dependent gating of ion channels. Proc Natl Acad Sci U S A. 2004;101:17640–17645. doi: 10.1073/pnas.0408116101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jackson JD. Classical Electrodynamics. Wiley; New York: 1999. [Google Scholar]
- 14.Hassani S. A modern Introduction to its Foundations. Springer; New York: 1999. Mathematical Physics. [Google Scholar]
- 15.Medvedev DM, Medvedev ES, Kotelnikov AI, Stuchebrukhov AA. Analysis of the kinetics of the membrane potential generated by cytochrome c oxidase upon single electron injection. Biochim Biophys Acta. 2005;1710:47–56. doi: 10.1016/j.bbabio.2005.08.008. [DOI] [PubMed] [Google Scholar]
- 16.Jasaitis A, Verkhovsky MI, Morgan JE, Verkhovskaya ML, Wikström M. Assignment and charge translocation stoichiometries of the major electrogenic phases in the region of cytochrome c oxidase with dioxygen. Biochemistry. 1999;38:2697–2706. doi: 10.1021/bi982275l. [DOI] [PubMed] [Google Scholar]
- 17.Baker NA, Sept D, Joseph S, Holst MJ, McCammon JA. Electrostatics of nanosystems: Application to microtubules and the ribosome. Proc Natl Acad Sci U S A. 2001;98:10037–10041. doi: 10.1073/pnas.181342398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holst M, Saied F. Multigrid solution to the Poisson–Boltzmann equation. J Com Chem. 1993;14:105–113. [Google Scholar]
- 19.Holst MJ, Saied F. Numerical solution of the nonlinear Poisson–Boltzmann equation – Developing more robust and efficient methods. J Com Chem. 1995;16:337–364. [Google Scholar]
- 20.Harrenga A, Michel H. The cytochrome c oxidase from Paracoccus denitrificans does not change the metal center ligation upon reduction. J Biol Chem. 1999;274:33296–33299. doi: 10.1074/jbc.274.47.33296. [DOI] [PubMed] [Google Scholar]
- 21.Ribacka C, Verkhovsky MI, Belevich I, Bloch DA, Puustinen A, Wikström M. An elementary reaction step of the proton pump is revealed by mutation of tryptophan-164 to phenylalanine in cytochrome c oxidase from Paracoccus denitrificans. Biochemistry. 2005;44:16502–16512. doi: 10.1021/bi0511336. [DOI] [PubMed] [Google Scholar]
- 22.Popović DM, Stuchebrukhov AA. Electrostatic study of the proton pumping mechanism in cytochrome c oxidase. J Am Chem Soc. 2004;126:1858–1871. doi: 10.1021/ja038267w. [DOI] [PubMed] [Google Scholar]
- 23.Tsukihara T, Shimokata K, Katayama Y, Shimada H, Muramoto K, Aoyama H, Mochizuki M, Shinzawa-Itoh K, Yamashita E, Yao M, Ishimura Y, Yoshikawa S. The low-spin heme of cytochrome c oxidase as the driving element of the proton-pumping process. Proc Natl Acad Sci U S A. 2003;100:15304–15309. doi: 10.1073/pnas.2635097100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modelling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- 25.Roux B, MacKinnon R. The cavity and pore helices in the KcsA K+ channel: Electrostatic stabilization of monovalent cations. Science. 1999;285:100–103. doi: 10.1126/science.285.5424.100. [DOI] [PubMed] [Google Scholar]
- 26.Dorairaj S, Allen TW. On the thermodynamic stability of a charged arginine side chain in a transmembrane helix. Proc Natl Acad Sci U S A. 2007;104:4943–4948. doi: 10.1073/pnas.0610470104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kucerka N, Tristram-Nagle S, Nagle JF. Closer look at structure of fully hydrated fluid phase DPPC bilayers. Biophys J. 2007;90:L83–L85. doi: 10.1529/biophysj.106.086017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Stern HA, Feller SE. Calculation of the dielectric permittivity profile for a nonuniform system: application to a lipid bilayer simulation. J Chem Phys. 2003;118:3401–3412. [Google Scholar]
- 29.Quenneville J, Popović DM, Stuchebrukhov AA. Combined DFT and electrostatics study of the proton pumping mechanism in cytochrome c oxidase. Biochim Biophys Acta. 2006;1757:1035–1046. doi: 10.1016/j.bbabio.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 30.Shinzawa-Itoh K, Aoyama H, Muramoto K, Terada H, Kurauchi T, Tadehara Y, Yamasaki A, Sugimura T, Kurono S, Tsujimoto K, Mizushima T, Yamashita E, Tsukihara T, Yoshikawa S. Structures and physiological roles of 13 integral lipids of bovine heart cytochrome c oxidase. EMBO J. 2007;26:1713–1725. doi: 10.1038/sj.emboj.7601618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Olsson MHM, Sharma PK, Warshel A. Simulating redox coupled proton transfer in cytochrome c oxidase: looking for the proton bottleneck. FEBS Lett. 2005;579:2026–2034. doi: 10.1016/j.febslet.2005.02.051. [DOI] [PubMed] [Google Scholar]
- 32.Rich PR. Towards an understanding of the chemistry of oxygen reduction and proton translocation in the iron–copper respiratory oxidases. Aust J Plant Physiol. 1995;22:479–486. [Google Scholar]
- 33.Popović DM, Stuchebrukhov AA. Proton pumping mechanism and catalytic cycle of cytochrome c oxidase: Coulomb pump model with kinetic gating. FEBS Lett. 2004;566:126–130. doi: 10.1016/j.febslet.2004.04.016. [DOI] [PubMed] [Google Scholar]
- 34.Istratov AA, Vyvenko OF. Exponential analysis in physical phenomena. Rev Sci Instruments. 1999;70:1–25. [Google Scholar]
- 35.Hinkle P, Mitchell P. Effect of membrane potential on equilibrium poise between cytochrome a and cytochrome c in rat liver mitochondria. J Bioenerg. 1970;1:45–60. doi: 10.1007/BF01516088. [DOI] [PubMed] [Google Scholar]
- 36.Siegbahn PEM, Blomberg ML. Energy diagrams and mechanism for proton pumping in cytochrome c oxidase. Biochim Biophys Acta. 2007;1767:1143–1156. doi: 10.1016/j.bbabio.2007.06.009. [DOI] [PubMed] [Google Scholar]
- 37.Brzezinski P, Larsson G. Redox-driven proton pumping by heme-copper oxidases. Biochim Biophys Acta. 2003;1605:1–13. doi: 10.1016/s0005-2728(03)00079-3. [DOI] [PubMed] [Google Scholar]
- 38.Brzezinski P, Ädelroth P. Design principles of proton-pumping haem-copper oxidases. Curr Opin Struct Biol. 2006;16:465–472. doi: 10.1016/j.sbi.2006.06.012. [DOI] [PubMed] [Google Scholar]
- 39.Branden G, Pawate AS, Gennis RB, Brzezinski P. Controlled uncoupling and recoupling of proton pumping in cytochrome c oxidase. Proc Natl Acad Sci U S A. 2006;103:317–322. doi: 10.1073/pnas.0507734103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sazanov LA, Hinchliffe P. Structure of the hydrophylic domain of respiratory complex I. Science. 2006;311:1430–1436. doi: 10.1126/science.1123809. [DOI] [PubMed] [Google Scholar]
- 41.Brandt U. Energy converting NADH: Quinone oxidoreductase (Complex I) Annu Rev Biochem. 2005;75:69–92. doi: 10.1146/annurev.biochem.75.103004.142539. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.