

Abstract
Glutathione (γ-glutamyl-cysteinyl-glycine, GSH) is the most abundant intracellular antioxidant thiol and is central to redox defense during oxidative stress. GSH metabolism is tightly regulated and has been implicated in redox signaling and also in protection against environmental oxidant-mediated injury. Changes in the ratio of the reduced and disulfide form (GSH/GSSG) can affect signaling pathways that participate in a broad array of physiological responses from cell proliferation, autophagy and apoptosis to gene expression that involve H2O2 as a second messenger. Oxidative stress due to oxidant/antioxidant imbalance and also due to environmental oxidants is an important component during inflammation and respiratory diseases such as chronic obstructive pulmonary disease, idiopathic pulmonary fibrosis, acute respiratory distress syndrome, and asthma. It is known to activate multiple stress kinase pathways and redox sensitive transcription factors such as Nrf2, NF-κB and AP-1, which differentially regulate the genes for pro-inflammatory cytokines as well as the protective antioxidant genes. Understanding the regulatory mechanisms for the induction of antioxidants, such as GSH, versus pro-inflammatory mediators at sites of oxidant-directed injuries may allow for the development of novel therapies which will allow pharmacological manipulation GSH synthesis during inflammation and oxidative injury. This article features the current knowledge about the role of GSH in redox signaling, GSH biosynthesis and particularly the regulation of transcription factor Nrf2 by GSH and downstream signaling during oxidative stress and inflammation in various pulmonary diseases. We also discussed the current therapeutic clinical trials using GSH and other thiol compounds, such as N-acetyl-L-cysteine, fudosteine, carbocysteine, erdosteine in environment-induced airways disease.
Keywords: Reactive Oxygen Species, Glutathione, Nrf2, inflammation, lung, COPD
1. Introduction
1.1. Oxidative stress and lung toxicity
Reactive oxygen species (ROS) and reactive nitrogen species (RNS) play important roles in regulation of cell survival. Normally, moderate levels of ROS/RNS may function as signals to promote cell proliferation and survival, whereas a sudden and prolonged surge of ROS/RNS can induce cell death. Under normal physiologic conditions, the balance between generation and elimination of ROS/RNS maintains the functional integrity of redox-sensitive signaling proteins. The redox homeostasis of a cell ensures that endogenous and exogenous stimuli are modulated by the redox homeostasis of a cell. However, altered redox homeostasis leads to oxidative stress, which in turn may lead to aberrant cell death and contribute to disease development (Trachootham et al., 2008).
The lung, owing to its extensive surface area and blood supply, is the only organ in the entire human architecture which has the greatest exposure to atmospheric oxygen and other environmental toxicants. Hence, the lung is susceptible to oxidative injury by virtue of myriads of reactive forms of oxygen species. ROS and RNS are highly unstable due to unpaired electrons that are capable of initiating oxidation (reviewed in Valko et al., 2007). In situ lung injury due to ROS/RNS is linked to oxidation of proteins, DNA, and lipids. Physiologically, ROS/RNS inflict their effects by remodeling the extracellular matrix and blood vessels; stimulate mucus secretion and alveolar repair responses. At the biochemical level, ROS/RNS inactivate antiproteases, induce apoptosis, regulate cell proliferation and modulate the immune-inflammatory system in the lungs and other tissues (Rutkowski et al., 2007). At the molecular level, increased ROS/RNS levels have been implicated in initiating inflammatory responses in the lungs through the activation of transcription factors such as nuclear factor-kappaB (NF-κB) and activator protein-1 (AP-1), signal transduction, chromatin remodeling and gene expression of pro-inflammatory mediators (Rutkowski et al., 2007). This article will discuss the current knowledge about the role of glutathione (GSH) in redox signaling, GSH biosynthesis and particularly the regulation of transcription factor, nuclear redox factor2 (Nrf2) by GSH and downstream signaling during oxidative stress and inflammation in various pulmonary diseases. It will also discuss the current therapeutic clinical trials using GSH and other thiol compounds, such as N-acetyl-L-cysteine (NAC), fudosteine, carbocysteine, erdosteine in airways disease.
2. Oxidant sources in the lungs
2.1. Cell-derived endogenous ROS/Oxidants
Lung is vulnerable to oxidant damage because of its location, anatomy and function (Crystal, 1991). Lung epithelium is constantly exposed to oxidants generated internally as a part of normal metabolism, as well as to oxidants in the ambient air, including ozone, nitrogen dioxide, car exhaust, cigarette smoke etc. A free radical is any species capable of independent existence that contains one or more unpaired electrons (Halliwell and Gutteridge, 1999). The most important ROS of physiological significance are the superoxide anion (O2•−), hydroxyl radical (OH•), nitric oxide (NO), and hydrogen peroxide (H2O2). H2O2 is generated through nonenzymatic or enzymatic dismutation of superoxide. However, the most reactive and harmful ROS is OH•, which can be formed from H2O2 and superoxide but also via the reaction of superoxide with NO to produce peroxynitrite (ONOO−), which decomposes to form nitrogen dioxide (NO2) and OH•. The main cellular sources of ROS in the lung include not only neutrophils, eosinophils and alveolar macrophages (Kinnula et al., 1995), but also alveolar epithelial cells, bronchial epithelial cells and endothelial cells (Kinnula et al., 1992a, 1992b).
2.2. Inhaled Oxidants
The generation of ROS in the lungs is enhanced after exposure to numerous exogenous environmental chemical and physical agents, which include mineral dusts, ozone, nitrogen oxides, sulphur dioxide, ultraviolet and ionizing radiation (Janssen et al., 1993), and tobacco smoke (Church and Pryor, 1985). A direct consequence of cigarette smoking and inhalation of airborne pollutants is lung damage and elevated inflammatory responses in the lungs, and are implicated in the pathogenesis and exacerbations of chronic obstructive pulmonary disease (COPD). In the gas phase, the cigarette smoke comprises of high concentrations of oxidants/free radicals (>1015 molecules/puff) (Church and Pryor, 1985), short-lived oxidants such as O2•− and NO. Nitric oxide and O2•− immediately react to form highly reactive ONOO− molecule. The tar phase of cigarette smoke contains organic radicals, such as long-lived semiquinone radicals, which can react with molecular oxygen in a redox dependent manner to form O2•− to form OH• and H2O2 (Nakayama et al., 1989). The aqueous phase of cigarette smoke condensate may undergo redox recycling for a considerable period of time in the epithelial lining fluid (ELF) of smokers (Nakayama et al., 1989). The tar phase is also an effective metal chelator wherein iron is chelated to produce tar-semiquinone + tar-Fe2+, which can generate H2O2 continuously (Nakayama et al., 1989). It is of much importance to note that cigarette smoke has been recently reported to irreversibly modify GSH levels in airway epithelial cells; a basic mechanism which may underlie cigarette smoke mediated inflammatory lung damage (van der Toorn et al., 2007).
3. GSH Biosynthesis and Regulation
GSH is the predominant non-protein thiol in the cells and is a key player in the maintenance of the cellular redox status, defined as the ratio of the concentration of oxidizing equivalents to that of reducing equivalents (Forman and Dickinson, 2003). GSH exists primarily in two redox forms, i.e., reduced GSH and glutathione disulfide (GSSG, the oxidized form), the latter representing a negligible 1/100th of the total GSH pool. The normal GSH content of a cell ranges from 1 mM-10 mM, levels that are imperative for a cell to maintain and a function of the balance between depletion and synthesis. Cells can excrete GSSG or reduce it back to GSH at the expense of NADPH through the action of GSH reductase. However, de novo synthesis of GSH from its amino-acid constituents is essential for the elevation of GSH that occurs as an adaptive response to oxidative stress. GSH synthesis involves two enzymatic steps catalyzed by glutamate cysteine ligase (GCL, formerly called as γ-glutamylcysteine synthetase) and GSH synthetase (Meister and Anderson, 1983). The enzyme GCL controls the rate of GSH synthesis (Huang et al., 1993) because the rate-limiting step for de novo synthesis of GSH is the cellular levels of the amino acid cysteine. In that regard, the plasma membrane ectoenzyme γ-glutamyl transpeptidase (γ-GT), which is the only enzyme that can break the γ-linkage found in GSH and GSH-conjugates, is essential in providing cysteine. It metabolizes the extracellular GSH and preferentially forms γ-glutamylcysteine, which is taken up by cells and subsequently reduced to form γ-glutamylcysteine bypassing its production by GCL.
GCL is composed of a heterodimer containing a 73 kDa heavy catalytic subunit (GCLC) and a 30 kDa light modifying subunit (GCLM) (Huang et al., 1993). Although the heavy subunit contains all of the catalytic activity, the association of the heavy subunit with the regulatory light subunit can modulate GCL activity. The ratio of the two subunits for physiological function has long been assumed to be 1:1; however, in tissues the ratio varies significantly and usually GCLC:GCLM is significantly greater than 1:1 (Krzywanski et al., 2004). GCL is regulated by GSH through feedback inhibition. Changes in GCL activity can result from regulation at multiple levels affecting only the catalytic or modifier subunit or both (Lu, 1999). Both human GCLC and GCLM promoters have been cloned (Erickson et al., 2002; Galloway et al., 1997). GCL has been shown to be polymorphic and is directly associated with loss in lung function and smoking history (Siedlinski et al., 2008). However, Chappell et al., have found that polymorphism of GCL is not associated with COPD (Chappell et al., 2008). Very recently, it has been reported that patients with cystic fibrosis (CF) with a milder CFTR genotype, exhibit a strong association between functional polymorphisms of the GCLC gene and CF severity (McKone et al., 2006). This study was mainly prompted by an earlier observation that patients with CF have low levels of GSH in the epithelial lining fluid (ELF) (Roum et al., 1993).
3.1 GSH: Oxidative stress and Redox Regulation in Lung Cells
The presence of sulfhydryl group in GSH makes it function as an antioxidant, protecting against free radicals and other oxidants. Increasing intracellular GSH can decrease the release of cytokines and chemokines from lung cells by decreasing NF-κB activation (Antonicelli et al., 2002; Aoki et al., 1996). In addition, GSH is an important regulator of cell proliferation, apoptosis, and gene transcription (Rahman and MacNee 2000a; Luppi et al., 2005) and can be easily assayed in the laboratory (Rahman et al., 2006). Reduced GSH is distributed relatively evenly throughout the cell, with the exception of the lumen of the rough endoplasmic reticulum (ER), where very little is detected. Both endogenous as well as exogenous oxidants require hours to significantly affect GSH levels in the majority of cells in a given population (Ault and Lawrence 2003). Interestingly, cells within a homogeneous cell line population lose GSH at different rates. The last reserves of cellular GSH are however, found within the mitochondria (Ault and Lawrence 2003). GSH and GSSG form one of the most important redox couple in a cell. GSH is not restricted to a particular cell organelle, rather there is a dynamic distribution going on depending on the cell’s redox status (Beck et al., 2001). GSH pathways/bioactivities may greatly differ from cell compartment to cell compartment (mitochondria, ER, cytoplasm and nucleus) and cell to cell (cancer cell, epithelial cells, immune system cells) (Table 1), and airway surface liquid compartments (airway surfaces, alveolar surfaces) and hence the cytosolic redox ratios of GSH/GSSG will be a reflective of any of these specific sub-compartments.
Table 1.
Levels of intra-cellular GSH in different cell types
| Cell type | Basal GSH level (nmol/mg protein) |
|---|---|
| Human alveolar epithelial cells, A549 | 150 |
| Human primary airway epithelial cells | 60 |
| Human hepatocarcinoma, HepG2 | 71 |
| Bovine arterial endothelial cells | 20 |
| Rat lung epithelial, L2 | 18 |
| Human umbilical vein endothelial cells | 4 |
| Human monocytes/macrophages (MonoMac Cells) | 10 |
GSH is vital in the lung, defending the airspace epithelium from damage in response to oxidants and inflammation and GSH redox status is critical for the transcriptional regulation of many pro-inflammatory genes (Figure 1). This is illustrated by findings in various pulmonary diseases where decreases in the levels of GSH in the lung lining fluid have been shown to occur in idiopathic pulmonary fibrosis (IPF), acute respiratory distress syndrome (ARDS), CF, lung allograft patients and human immunodeficiency virus (HIV) positive patients (Rahman and MacNee, 1999 and 2000a) (Table 2). In contrast, an increase in total GSH concentration, which also includes the oxidized form GSSG, has been reported in the bronchial and alveolar fluid in patients with mild asthma (Rahman and MacNee, 2000b). Furthermore, GSH has recently been reported to attenuate IL-13 induced asthma in mice (Lowry et al., 2008). Thus, low levels of GSH in the lung lining fluid of patients with inflammatory respiratory diseases may render them more susceptible to the deleterious effects of subsequent exposure to inhaled toxicants and may also perpetuate the inflammatory response. It has now become evident that small changes in the cellular redox status may alter signaling pathways.
Fig. 1.

Oxidative alteration in the intracellular redox ratio of GSH/GSH levels and redox regulation of pro-inflammatory and anti-inflammatory diseases. Oxidative alteration in the intracellular redox ratio of GSH/GSSG levels by environmental oxidants can modulate NF-κB and AP-1 activation leading to transcription of pro-inflammatory genes. The expression of pro-inflammatory mediators would lead to chronic inflammation which is involved in the pathogenesis of various pulmonary diseases, such as COPD, IPF, ARDS, and asthma. On the other hand during oxidative stress the ARE present in the human GCLC and GCLM promoters is trans-activated by Nrf2 due to alteration in GSH and GSSG ratio leading to synthesis of GSH. The imbalance of antioxidant and pro-inflammatory genes can lead to chronic inflammation. Various thiol compounds, such as N-acetyl-L-cysteine (NAC), N-acystelyn (NAL), N-isobutyrylcysteine (NIC) and erdosteine can supply cysteine for GSH biosynthesis.
Table 2.
Epithelial lining fluid GSH concentration in inflammatory lung diseases
| Concentration (μM) |
||
|---|---|---|
| Control | Patients | |
| Controls (non-smokers) | 339 ± 112 | - |
| Smokers | 544 ± 97.6 | - |
| Idiopathic pulmonary fibrosis | 429 ± 34 | 97.0 ± 18 |
| Acute respiratory distress syndrome | 651 ± 103.1 | 31.5 ± 8.4 |
| Lung allograft | 302.6 ± 40.8 | 94.0 ± 9.7 |
| Cystic fibrosis | 257 ± 21 | 78.0 ± 13 |
| HIV-seropositive | 245 ± 12 | 170 ± 23 |
| Asthma (μM/mg protein) | 23.3 ± 3 | 36.5 ± 9.4 |
| COPD (stable) (nmol/mg protein) | 4 | 10 |
| COPD (during exacerbations) (nmol/mg protein) | 4 | 2 |
The GSSG/GSH ratio can serve as a good indicator of the cellular redox state (Park et al., 1998). A very recent report has suggested that changing GSH redox balance, increasing GSH level, and the GSH/GSSG ratio by γ-glutamycysteinyl ester (γ-GCE), ameliorate bronchial asthma by altering the Th1/Th2 cell imbalance through IL-12 production from antigen presenting cell (APC) and suppressing chemokine production and eosinophil migration itself (Koike et. al., 2007). Although the ER may be the major site of GSSG accumulation, the formation of GSSG from GSH due to oxidation in the cytosol triggers dynamic redistribution of GSH and GSSG between their respective organelles and the cytosol (Banhegyi et al., 1999, Banhegyi et al., 2003). Hence, cytosolic GSH concentration is important for biological cellular redox state. It is noteworthy that high levels of GSSG do not necessarily reflect that ER is storage or accumulation point for GSSG but as per recent reports by itself is an important generator of oxidative stress. Therefore more GSH is oxidized to GSSG in the ER (Chakravarti et al., 2006). Besides, GSH is known to be an important redox buffer within the ER and is required to balance the ER generated oxidative stress (Jessop and Bulleid 2004).
Several enzymes/proteins involved in the redox system of the cell and their genes such as MnSOD, GCLC, GSH peroxidase (GPx), thioredoxin (Trx) reductase, glutaredoxin1 and metallothionein are induced by modulation of cellular GSH/GSSG levels in response to various oxidative stresses, including hyperoxia and inflammatory mediators such as tumor necrosis factor-α (TNF-α) and lipopolysaccharide in lung cells (Das, 2001; Peltoniemi et al., 2006). It has been shown that GSH depletion due to GCL inhibition by buthionine sulphoximine (BSO) sensitizes both alveolar A549 and bronchial epithelial 16-HBE cells to the injurious effects of hyperoxia and H2O2, resulting in an increased membrane permeability and activation of NF-κB (Rahman et al., 2001). In contrast, pre-treatment of these cell lines with hyperoxia prior to H2O2 exposure protects against the cytotoxic effects of H2O2 as well as preventing NF-κB activation. These protective effects were due to an adaptive increase in GSH in response to pre-treatment with hyperoxia. Therefore, modulation of intracellular GSH can determine the course of tolerance to subsequent oxidant exposure. A recent study has indicated that oxidant induced suppression of GSH synthesis in human bronchial cells may be reversed by erythromycin (He et al., 2008). However, more such studies are required to validate the results so as to have a therapeutic impact.
Normally, GSSG represents less than 1% of total GSH pool. When H2O2 or ONOO− is transiently elevated, an elevation in GSSG, also transient, can occur, providing a possible mechanism for signaling by means of thiol-disulfide exchange. In this scenario, signaling is indirectly dependent upon ROS generation. Nonetheless, as this mechanism requires a change in GSSG that is usually only observed during oxidative stress, such signaling is more likely an oxidative stress response rather than physiologic redox signaling. Interesting to note is a recent report wherein it was shown that the sputum levels of GSSG and nitrosothiols such are elevated in COPD subjects the increase was associated with neutrophilic inflammation (Beeh et al., 2004). Therefore, increased GSSG is not only an indicator for oxidative stress but its presence in the sputum may serve as a marker of oxidative stress in lung diseases.
There have been recent interests in the role of thiol containing enzymes in redox regulation of signaling pathways which is comprehensively reviewed by Bindoli et al. (Bindoli et al., 2008). Peroxiredoxins are a class of enzymes that can reduce H2O2 using thiols (Rhee et al., 1999) in a reaction similar to that catalyzed by the selenium in GPx, some using Trx as a co-substrate and at least one using GSH (Chen et al., 2000). Interestingly, they contain reactive cysteine residues in their unprotonated form, i.e. thiolate (S−) and the catalytic reaction includes several steps, one of which being the formation of a protein-sulfenate intermediate (PSO−), which then react with a thiol that can be the second reactive thiol present in most 2-Cys peroxiredoxins (Wood et al., 2003a) or GSH (Chen et al., 2000). Peroxiredoxins might play an important role in controlling redox signaling pathways (Wood et al., 2003b). Transgenic mice overexpressing peroxiredoxin6 have been demonstrated to show increased resistance to lung injury during hyperoxia (Wang et al., 2006). The levels of another thiol, glutaredoxin, have been shown to be modulated in the lungs and sputum of smokers and in COPD subjects and in mouse models of allergic airway diseases (Peltoniemi et al., 2006; Reynaert et al., 2007). The role of various thiols and the mechanism of their redox modulation leading to parenchymal lung diseases have been reviewed in details by Kinnula et al. (Kinnula et al., 2007). Protein disulfide bonds rarely form in the cytosol because of the high concentrations of GSH. By contrast, a relatively higher concentration of GSSG exists in the ER lumen (Hwang et al., 1992). Such an oxidizing condition within the ER lumen allows the formation of native disulfide bonds in the ER through a complex process involving not only disulfide-bond formation, but also the isomerization of non-native disulfide bonds., i.e. folding of proteins (Chakravarti et al., 2006). Very recently Bindoli et al. have suggested that the status of thiol redox may be attributed to the oxidant status of the cell (Bindoli et al., 2008). The thiol redox state is modulated by Trx and GSH systems that are in close redox link with H2O2 via peroxiredoxins and GPx, respectively. Particularly, the redox state of Trx integrates the extent of cell oxidation to a rather specific physiologic response, which may be different in different cellular compartment. For example, in the mitochondria the redox state of Trx influences the membrane-permeability conditions, in the cytosol controls the mitogen activated protein kinase (MAPK) signaling pathways, and within the nucleus regulates the binding of transcription factors to DNA.
4. Regulation of GSH biosynthesis by pro-inflammatory mediators and growth factors
The synthesis of GSH is upregulated during oxidative stress and inflammation. Thus, an increase in GCL expression would be expected under oxidative stress and the first demonstration of that was with the redox cycling and GSH conjugating quinine and menadione (Shi et al., 1994). Exposure of alveolar epithelial cells in vitro to oxidants, oxidant-generating systems, and lipid peroxidation products, such as H2O2, hyperoxia, ozone, menadione, and 4-hydroxy-2-nonenal, all lead to short-term falls in intracellular GSH associated with increased GSSG levels, followed by increases in GSH levels or upregulation of GCLC mRNA in alveolar epithelial cells, endothelial and other cells in vitro, and in vivo in rats (Paget et al., 1998; Parmentier et al., 2000; Pietarinen-Runtti et al., 1998; Usatyuk et al., 2006). A recent report indicates that chronic alcoholism may also lead to alteration of systemic and pulmonary GSH redox system (Yeh et al., 2007). The GCLM is also concomitantly induced in response to oxidants and phenolic antioxidants in rat lung epithelial L2 cells, suggesting that concomitant induction of both subunits may be a potential mechanism to enhance cellular GSH synthesis, and so develop cellular tolerance to oxidative stress (Tian et al., 1997). While the mRNAs for both subunits increase in response to oxidative stress, there may be a disproportional increase in the proteins (Krzywanski et al., 2004). Thus, the short-term effects of various oxidants and oxidant-generating systems appear to upregulate the gene for GSH synthesis, possibly providing a protective/adaptive mechanism against subsequent oxidative stress.
Identification and characterization of the types of diverse stimuli that act as potent inducers of GCL should aid in the development of effective pharmacological strategies for antioxidant treatment involving GSH regulation in inflammatory lung diseases. To this effect, several studies have been directed towards understanding and elucidating the molecular mechanisms of GSH synthesis and regulation in type II alveolar epithelial cells in response to various environmental, oxidants, antioxidants and inflammatory stimuli. Several investigators have reported that the promoter (5′-flanking) region of human GCLC gene is regulated by a putative c-Jun homodimeric complex-AP-1 sequence (Tanaka et al., 1998; Tomonari et al., 1997). This sequence is located at the proximal region of the GCLC TATA box in various cell lines, including human alveolar epithelial cells (Tanaka et al., 1998, Tomonari et al., 1997). Mulcahy and co-workers (Monva and Mulcahy, 1998; Mulcahy et al., 1997), however, have reported a distal antioxidant response element (ARE) containing an embedded phorbol myristate acetate (PMA)-responsive element (TRE/AP-1) and an electrophile responsive element (EpRE or its functional equivalent, ARE), which play a key role in the regulation of the GCLC and GCLM, respectively, in response to a planar aromatic xenobiotic, the phenolic antioxidant β-naphthoflavone specifically in a liver cell line (HepG2 cells) (Erickson et al., 2002). They further suggested a differential induction of mafF, mafG and mafK expression by electrophile-response-element activator in the regulation of GCLC regulation in a variety of cell lines (Moran et al., 2002). It has been shown that H2O2-dependent activation of GCLC-ARE4 reporter occurs via the MAPK pathways without oxidation of cellular GSH or Trx-1 suggesting that redox GSH status of the cells is not required for regulation of GCLC or ARE (Harper et al., 2001). Mulcahy and colleagues also showed that the internal AP-1 site is important for the constitutive expression of the GCLM gene (Monva and Mulcahy 1998). However, Galloway and co-workers (Galloway et al., 1997) were unable to demonstrate a role for ARE in the induction of GCLM by oxidants such as tert-butyl hydroquinone in HepG2 cells. They suggested that an AP-1 site was the critical element for the basal regulation of this subunit. Therefore, it is likely that the expression of the GCL subunit genes is regulated by different regulatory signals in response to diverse stimuli in specific cells.
Exposure to phenolic antioxidants such as dietary 2(3)-tert-butylated-4-hydroxyanisole and butylated hydroxytoluene as well as the synthetic indolic antioxidant, 5,10-dihydroindeno(1,2-b) indole and pyrrolidine dithiocarbamate, a sulfhydryl-modifying antioxidant compound, upregulate GCLC and GCLM in human endothelial cells and other cell lines (Galloway et al., 1997; Tu and Anders, 1998). These effects of phenolic antioxidants are associated with AP-1/ARE transactivation (Meyer et al., 1993; Pinkus et al., 1996). Taken together, antioxidants protect cells from oxidants by either scavenging these molecules directly or by regulating intracellular GSH levels through the induction of GCL. It should however, be kept in mind that recent studies have shown that many compounds characterized originally as antioxidants have been also shown to exert direct effects upon several signal transduction enzymes independent of their antioxidant function (Dickinson et al., 2003; Biswas et al., 2005; Kode et al., 2008). For example vitamin E has significant effects upon signaling independent of its lipid peroxidation inhibitory role (Azzi et al., 1998).
Transforming growth factor-β1 (TGF-β1) is a multifunctional growth factor that modulates cellular proliferation and induces differentiation and synthesis of extracellular matrix proteins including collagens and fibronectin in many types of lung cells. De Boer have shown increased expression of TGF-β1 in bronchiolar and alveolar epithelium in idiopathic IPF and COPD patients, and higher levels in BAL of atopic asthmatics as compared to healthy subjects (de Boer et al., 1998). TGF-β1 also down-regulates GCLC mRNA and GSH synthesis in in vitro human alveolar epithelial cells and pulmonary artery endothelial cells (White et al., 1992). We, and other workers have shown that GCLC mRNA expression is under the control of the AP-1-like transcription factor (Rahman et al., 1996a), and that TGF-β1 may decrease GCLC gene expression via an ATF4/Nrf2/ARE-1-dependent mechanism (Jardine et al., 2002; Bakin et al., 2005). It has been shown that downregulation of GCLC in response to TGF-β1 in lung epithelial cells was mediated via an AP-1 heterodimer consisting of c-Jun and Fra-1 (Jardine et al., 2002) whereas another study has reported that ATF3, a negative regulator of Nrf2-ARE binding, upregulated by TGF-β1 downregulated Nrf2 by sequestering p300 protein away from Nrf2 (Bakin et al., 2005). Thus, higher levels of TGF-β1 may down-regulate GSH synthesis in lungs of patients with inflammatory diseases such as IPF and COPD. It has been shown that GSH levels in bronchoalveolar larvage fluid (BALF) is decreased during exacerbations of COPD though GSH levels were increased in BALF of smokers and stable COPD patients compared to non-smokers (Drost et al., 2005). Recent studies have clearly highlighted the downregulation of Nrf2 (GSH levels) in pulmonary macrophages and in lungs of patients with COPD via loss of GCL and Nrf2 positive regulator DJ-1 and post-translational modifications of Keap1-Bach1 equilibrium (Harju et al., 2002; Malhotra et al., 2008; Goven et al., 2008; Suzuki et al., 2008). Moreover, decreased GSH-levels may also have direct functional consequences leading to inflammation. In vitro studies showed that GSH (in the concentration range normally found in ELF) suppressed fibroblast proliferation (Cantin et al., 1990). In addition, depletion of GSH in response to TGF-β1 appears to be a key requirement for subsequent collagen I mRNA expression in murine embryo fibroblasts (Liu et al., 2004). This induction was attenuated by pre-treatment with NAC, GSH, or GSH ester. The relevance of GSH regulation and subsequent tolerance/susceptibility in lung epithelial cells in response to pro-/anti-inflammatory mediators and/or oxidants under chronic inflammation in vivo is presently not clearly understood.
4.1 Redox regulation of ARE (Nrf and Maf proteins)
Transcription factor Nrf2 is an essential component for the ARE-mediated induction of phase II detoxifying and GCL genes in response to electrophiles and phenolic antioxidants in HepG2 cells (Wild et al., 1999). It is now known that Nrf proteins bind to the ARE consensus sequence, which shows striking similarity to a binding motif referred to as the Maf recognition element, also known as the erythroid transcription factor (NF-E2) binding sequence. NF-E2 or Maf recognition elements are specifically recognized by either homodimers of small Maf family members (MafK, MafG, MafF) or by heterodimeric proteins composed of Cap’n’Collar (CNC) subfamily of basic leucine zipper (bZip) transcription factors and small Maf partners which mediate expression through AP-1/EpRE sequences. The NF-E2/AP-1 element and the ARE, which has been identified in several phase II genes, possesses striking similarity.
A protein Kelch-like ECH-associated protein1 (Keap1) which plays a role in the ARE-mediated signaling, has been identified as a negative regulator of Nrf2 that also acts as a possible intracellular sensor for thiol-active xenobiotics in HepG2 cells (Itoh et al., 1999). Keap1 binds and retains Nrf2 in an inactive form in the cytoplasm, a form in which Nrf2 is unable to function as a transcription factor. During transition to oxidizing conditions (represented by oxidation of GSH by ROS to GSSG), Nrf2 dissociates from Keap1 allowing it to translocate to the nucleus where it heterodimerizes with small Maf proteins and activates ARE-driven transcription (Dinkova-Kostova et al., 2002). The actual mechanism of dissociation of Nrf2 from Keap1 is currently not clear, but it is thought to involve thiol modifications or phosphorylation of Keap1, Nrf2, or both or via kinase signaling mechanism. Indeed, there is evidence in a cell-free system that cysteine thiols of Keap1 are reactive to inducers of phase 2 genes (Dinkova-Kostova et al., 2002). Four of the most reactive cysteines resided in the region required for binding to Nrf2. Exposure of the Keap1/Nrf2 complex to the inducers resulted in the dissociation of the complex. Thus, the sulfhydryl groups of Keap1 are sensors regulating the induction of phase 2 enzymes (Wakabayashi et al., 2004). Therefore, under the conditions of initial depletion of intracellular GSH, Trx and/or production of ROS may lead to the dissociation of the complex and activation of Nrf2 (Kim et al., 2003). Furthermore, it has been shown that scaffolding of Keap1 to the actin cytoskeleton controls the function of Nrf2, and under oxidative stress and electrophilic stress disruption of the actin cytoskeleton promotes nuclear entry of an Nrf2 reporter protein (Kang et al., 2004).
It has been also demonstrated that mice with site-directed mutation of the Nrf2 gene (Nrf2−/−) were more susceptible to the injurious effects of hyperoxia as noted by marked increase in pulmonary hyper permeability, macrophage inflammation, and epithelial injury compared to the wild type mice (Hayes and McLellan, 1999). A significant reduction in the mRNA expression of ARE-responsive lung antioxidant and phase 2 enzymes, some of which included heme-oxygenase-1, GPx2, and NAD(P)H: quinone oxidoreductase, was also observed in Nrf2−/− mice compared to normal mice. GPx2, the major cigarette smoke-inducible isoform has now been confirmed to be regulated by Nrf2 (Singh et al., 2006). Thus Nrf2 appears to protect against pulmonary hyperoxia injury in mice, presumably by upregulating the transcription of lung antioxidant defense enzymes. However, it remains to be explained as to the role of Keap1 and Nrf2, if any, in the induction of phase II genes in particular GCL in lung cells in response to oxidative stress. Keap1 also appears to regulate the degradation of Nrf2 via a proteasome-dependent pathway and that mouse macrophages deficient in Keap1, Nrf2 accumulates in the nucleus (Zhang and Hannink, 2003). Electrophilic compounds have also been shown to cause Nrf2 nuclear translocation, concomitant with protein stabilization. Furthermore, electrophilic lipids, like prostaglandins, isoprostanes and 4-hydroxy-2-nonenal also cause the dissociation of Nrf2 from Keap1, resulting in the activation of the ARE.
As described previously, signals from ROS or electrophilic insults target the Nrf2-Keap1 complex, dissociating Nrf2 from Keap1, leading to translocation of Nrf2 to the nuclei and transactivate the target genes. Keap1 is now assumed to be a substrate-specific adaptor of Cul3-based E3 ubiquitin ligase (Kobayashi and Yamamoto, 2005). Direct participation of Keap1 in the ubiquitination and degradation of Nrf2 is now thought to be plausible. The Nrf2-Keap1 system is present not only in mammals but also in fish suggesting that its roles in cellular defense are conserved throughout evolution among vertebrates (Kobayashi and Yamamoto, 2005). As discussed in the following section, Nrf2 has an important role in regulation of the GCLC and GCLM genes. Recently, it has been shown that GSH levels also determine the expression of Nrf2. In an elegant experiment by Lee et al. (Lee et. al., 2008), it was shown that murine embryonic fibroblasts (MEFs) can survive in the presence of GSH inhibitor buthionine-(S,R)-sulfoximine (BSO), even though most intracellular GSH was depleted. As a cellular adaptive mechanism, BSO treatment effectively activated the Nrf2 pathway, which led to up-regulation of antioxidant enzymes in these cells through the extracellular signal-regulated kinase cascade. Nrf2-deficient MEFs lost the inducibility of antioxidant genes, which resulted in higher levels of ROS accumulation, caspase-3 activation, and cell death when compared to wild-type cells. On the other hand, Nrf2-deficient cells could be more sensitized to doxorubicin-induced cell death by BSO pre-incubation, while wild-type cells were not. In addition, BSO-mediated cell death was facilitated by administering Nrf2 siRNA to chemoresistant human ovarian cancer cells. These results indicated that Nrf2 is the primary factor inducing the cell survival system under GSH depletion.
The aryl hydrocarbon receptor (AHR) and NF-E2 p45-related factor (Nrf2) are two distinct transcription factors involved in the regulation of drug-metabolizing enzymes. Spate of evidences from several studies implies that AHR and Nrf2 have direct links, but the underlying molecular mechanism still remains to be fully explored. It was recently reported that Nrf2 gene transcription is directly modulated by AHR activation (Miao et. al., 2005). DNA sequence analyzes of the mouse Nrf2 promoter revealed one xenobiotic response element (XRE)-like element (XREL1) located at −712 and two additional XRE-like elements located at +755 (XREL2) and +850 (XREL3). Functional analysis using luciferase assay showed that XREL1, XREL2, and XREL3 are all inducible by 2,3,7,8-tetrachlorodibenzo-p-dioxin treatment, with XREL2 being the most potent.
More recently, it was reported that the Nrf2 expression could also be regulated in a reoxygenation-specific manner (Leonard et al., 2006). It was also shown that there was a nuclear accumulation of Nrf2 in response to reoxygenation during ischemic reperfusion injury. Further more, ROS during reoxygenation using NAC resulted in inhibition of Nrf-2 activation. (Leonard et. al., 2006). Thus, although it appears that redox state of a cell is a major regulator of Nrf2 expression; several other factors as described above also modulate the expression of Nrf2. It is possible that new biochemical factors may be identified in future, which may also play a role in the regulation of expression of Nrf2 gene.
4.2. Nrf2 in GSH biosynthesis in response to inhaled oxidants
Nrf1 and Nrf2, which are important in the transcriptional regulation of human and mouse GCL subunit expressions and activitiy and in regulating cellular GSH levels. The ARE present in the human GCLC and GCLM promoters is trans-activated by Nrf2 in response to α-naphthoflavone, pyrrolidine dithiocarbamate, and tert-butylhydroquinone (TBH) and other exogenous agents (Erikson et al., 2002). Although Nrf1 knockout mice have been reported to die in utero, the hepatocytes from the fetus and the embryonal fibroblasts from these animals have lower GSH levels and are more susceptible to oxidative stress than wild-type (WT) mice (Chen et al., 2003; Kwong et al., 1999). Lower GSH levels have been observed in mice deficient in Nrf2 and are found to be more susceptible to acetaminophen-induced liver injury compared to their WT counterparts (Chan et al., 2001). Whereas, Nrf1 and Nrf2 knockout mice exhibit lower GCLC expression (Chan et al., 2001, Chen et al., 2003), over-expression of the same has been shown to induce the human GCLC promoter activity (Wild et al., 1999). Interestingly, TBH also induces expression of rat GCLC and enhances activity of the rat GCLC promoter even in absence of ARE in the promoter region of GCLC (Yang et al., 2002; Yang et al., 2001). In such a situation it appears that TBH mediated induction of GCLC is essentially mediated by AP-1 (Yang et al., 2002).
Nrf2 mediated induction of human GCLM and GCLC promoters has been reported to be a result of trans-activation via Jun or Maf proteins (Erickson et al., 2002,). The role of Nrf2 in GCL expression has been further established using Nrf2 knockout mice, which demonstrated increased susceptibility to oxidant/acetaminophen-induced lung/liver injury, a phenomenon that was attributed to decreased GCL expression and GSH levels (Chan et al., 2001; Reddy et al., 2007). Over-expression of Nrf2 increased human GCLC and GCLM promoter activity (Jeyapaul and Jaiswal, 2000; Wild et al., 1999) and restored GCL subunit expression and GSH levels in Nrf2 knockout fibroblasts (Chan and Kwong 2000). A similar effect has been also reported in Nrf1 knockout rat fetal hepatocytes, where lower GCLC expression was observed (Chen et al., 2003). Nrf1 over-expression was also found to induce the human GCLC promoter activity via ARE (Jeyapaul and Jaiswal, 2000), an effect that is also reported for the mouse GCLC and GCLM promoters (Lee et al., 2003).
Human and rat GCL subunits might have similar regulatory mechanisms (Lu, 1999). It is of interest to note that although the 1.8-kb 5′ flanking region of the rat GCLC does not contain any consensus ARE element, the reporter activity however, driven by a recombinant rat GCLC-luciferase construct is induced by TBH treatment (Yang et al., 2002). A time-dependent induction in the expression of both genes was recorded in response to TBH. Although TBH treatment induced rat GCLC promoter activity in a wide variety of cell types, however, the induction levels were different in Nrf2 knockout and Nrf1 knockout cell types (Yang et al., 2005). It was previously shown that the rat GCLC promoter is induced by treatment with acetaldehyde and TBH (Yang et al., 2001 and 2002). Consensus NF-κB and AP-1 binding sites are present in the rat GCLC −595/+2 promoter fragment (Yang et al., 2001) and the nuclear binding of the two factors was found to be positive after treatment of cells with acetaldehyde (Yang et al., 2001).
In F1 fibroblast cells (Nrf1 knockout) and F2 fibroblast cells (Nrf2 knockout) the AP-1 family members, c-Fos and c-Jun levels were found to be decreased by upto 70 to 85%, JunB levels did not show any change. While JunD levels were found to be unchanged in F1 cells, slightly decreased levels were reported in F2 cells. The Fos related antigen (Fra), Fra-2 levels were slightly increased in F1 cells, and Fra-1 and Jun activating binding protein-1 (JAB-1) levels were reported to be markedly induced, especially in F2 cells (Yang et al., 2005). Among the NF-κB family members, p50 and p65 were shown to be reduced by upto 60 to 70%, RelB levels were slightly decreased and c-Rel levels were reported to be induced. The molecular mechanism underlying the alterations in the protein levels of Fra-1, p50, and p65, c-Jun, c-Fos is possibly at the transcriptional level, as mRNA levels of these genes were found to be decreased (increased in the case of Fra-1) in F1 and F2 cells when compared to WT cells.
The above alterations significantly decreased the TBH dependent nuclear binding of AP-1 and NF-κB. However, transfection of F2 cells with Nrf2 expression vector interestingly increased the base line levels of p50, p65, c-Jun, GCLC and c-Fos, decreased expression of Fra-1, and restored the cell response to TBH. Thus it appears that Nrf2 is intimately related to the expression of the aforementioned genes and may be an important regulator of GCLC and GCLM promoter activation and synthesis of GSH (Yang et al., 2005). In a recent report the role of Nrf2 in biosynthesis of GSH has been confirmed wherein it was shown that resveratrol, a red wine polyphenol induces GSH synthesis via activation of Nrf2 in human lung epithelial cells (Biswas et al., 2005; Kode et al., 2008).
5. Therapies/clinical trials with GSH analogs
Extracellular augmentation of GSH has been tried through intravenous administration of GSH, oral ingestion of GSH, and aerosol inhalation of nebulized GSH in an attempt to reduce inflammation in various lung diseases (Rahman et al., 2006; Rahman and MacNee, 1999). Various GSH analogues and substitutes are in vogue for therapeutic application (Figure 2) in various respiratory diseases which are descried below:
Fig. 2.
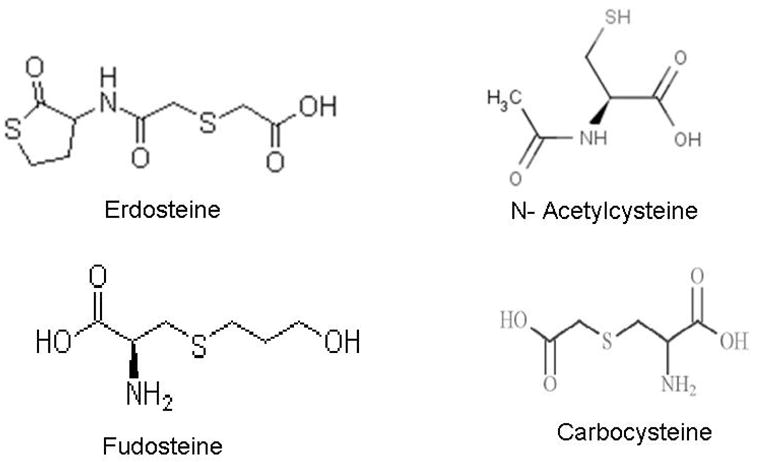
Chemical structures of key thiol compounds in clinical trials.
5.1. N-acetyl-L-cysteine (NAC)
The free thiol group of NAC is capable of interacting with the electrophilic group of free radicals thereby forming NAC thiol, with NAC disulphide or NAC conjugates as a major end product (Cotgreave, 1997). The availability of cysteine for GSH is a fundamental factor in the regulation of GSH production. Radioactivity associated with administration of 14C-NAC distributed to most tissues examined within 1 hour of administration with peak radioactivity levels occurring within 1–4 hours and for a majority of the tissues examined, radioactivity remained elevated for up to 12 hours or more (Arfsten et al., 2007). The results of this study demonstrated the possibility that NAC could provide some benefit in preventing or reducing toxicity related to exposure to chemical irritants in some tissues by increasing tissue NAC and/or cysteine levels, GSH concentrations, and GST activity. However, the finding of this study that GxP activity is elevated, albeit transiently, following repeat administration of NAC suggests that repeated administration of NAC may induce oxidative stress in some tissues and further studies are needed to confirm this finding (Arfsten et al., 2007).
NAC may also reduce cystine to cysteine, which is an important mechanism for intracellular GSH elevation in vivo in lungs (Burgunder et al., 1989). It not only reduces disulphide bonds (a property of a good reducing agent), but also has the potential to interact directly with oxidants. NAC is also used as a mucolytic agent, to reduce mucus viscosity and to improve mucociliary clearance. A number of systematic reviews have evaluated the effects of treatment with mucolytic agents in patients with COPD (Grandjean et al, 2000; Dekhuijzen, 2004). Mucolytic treatment was associated with a significant reduction of 0.79 exacerbations per patient per year compared to placebo, a 29% decrease (Table 3) (Grandjean et al, 2000; Dekhuijzen, 2004; Decramer et al., 2001; Decramer et al., 2005; Poole and Black, 2003). Although, how mucolytic agents work is still unknown, they may reduce exacerbations by altering mucus production, by breakdown of sulphydryl group, or through antibacterial or immunostimulatory effects (Poole and Black, 2001 and 2003). Pharmacological administration of NAC has been used in an attempt to enhance lung GSH in patients with COPD with varying success (Bridgemen et al., 1994; Rasmusse and Glennow 1988). Van Schooten et al. (Van Schooten et al., 2002) reported in a randomized, double-blind, placebo-controlled phase II trial that a 6-month oral dose of 600 mg b.i.d. reduced various plasma and BALF oxidative biomarkers in smokers. A recent report has suggested that NAC attenuates lung ischemia-reperfusion injury after lung transplantation (Inci et al., 2007). In addition, on the mechanistic front, NAC has been shown to attenuate allergic lung disease by regulating the activation of NF-κB and hypoxia-inducible factor-1 (HIF-1) (Lee et al., 2007) and also attenuates acute lung injury caused due to phorbol myristate acetate in isolated rat lungs (Chuang et al., 2007).
Table 3.
Clinical Trials conducted for the efficacy of thiols in smokers and COPD subjects
| SN | Name of Trial | Antioxidant used | Aim of Study | Disease | Outcome |
|---|---|---|---|---|---|
| 1 | BRONCUS | NAC | Effect on FEV1 | COPD | No effect. A reduction in lung over inflation in patients with severe COPD without inhaled glucocorticoids |
| 2 | Systematic Cochrane review of 23 randomized controlled trials | NAC (2 months of oral therapy) | Effect of NAC and antibiotics on number of days of disability | COPD | No effect on lung function. 29% decrease in exacerbations. Good reduction in days of disabibility (0.85 days/patient/month |
| 3 | Systematic Cochrane review of randomized controlled trials of 11 out of 39 reviewed trials | NAC | Used a validated score for evaluation of quality of each study | COPD | 9 trials showed prevention of exacerbations and 5 showed improvement in symptoms compared to 34.6% patients receiving placebos |
| 4 | A meta analysis of published trials | NAC (600 mg bid for 12 months) | To assess the possible prophylactic benefit of prolonged treatment Effect on H2O2 and TBARS in EBC |
COPD COPD |
23% decrease in acute exacerbation No change in TBARS, reduced H2O2 |
| 5 | EQUALIFE studies | Vectrine-erdosteine, thiol compound, 300mg bid for months | Exacerbation rate, hospitalization, lung function and quality of life in 124 patients | COPD | Decreased exacerbations and fewer days in hospital. No loss of lung function and improvement in health-related quality of life |
| 6 | PEACE study | Carbocysteine (carbocisteine) | Effect on rate of exacerbations | COPD | Long-term (one year) use of carbocysteine (1500 mg/day) produced reduction in numbers of exacerbations in patients with COPD |
BRONCUS = Bronchitis Randomized on N-acetyl-L-cysteine Cost Utility Study, FEV1 = Forced expiratory volume in 1 second, TBARS = Thiobarbituric acid reactive substances, NAC = N-acetyl-L-cysteine, EBC = Exhaled Breath Condensate.
Similarly, it has been shown that treatment with NAC (600 mg once daily for 12 months) also reduce the concentration of H2O2 in exhaled breath condensate compared with placebo in stable COPD patients (Kasielski and Nowak, 2001). Another recent clinical trial also proved that oral administration of NAC 600 mg b.i.d. for 2 months rapidly reduces the H2O2 in airways of stable COPD patients (De Benedetto et al., 2005). This study was mainly aimed at determining the utility of H2O2 as a tool for assessment and monitoring of oxidative stress in COPD. The mechanism for this effect of NAC on H2O2 is not known but presumably NAC acts as a sink for ROS, which are increased in these patients (Rahman, 2005). However, a multicentre study using NAC delivered by metered dose inhalers in patients with chronic cough failed to show a positive effect on wellbeing, sensation of dyspnoea, cough or lung function (Dueholm et al., 1992). Recently, NAC has been shown to downregulate inflammatory responses in CF patients (Tirouvanziam et al., 2006)
Whilst there is some evidence that the administration of NAC provides benefit for some COPD patients, it is not clear whether this could be used as maintenance therapy (Decramer et al., 2001). A phase III multicentre Bronchitis Randomized on NAC Cost- Utility Study (BRONCUS) has recently been completed, with the aim of addressing this question and determining whether the effectiveness of NAC as an “antioxidant” results in an alteration in the rate of decline in FEV1, exacerbation rate and quality of life (QoL) in patients with moderate-to-severe COPD (Gerrits et al., 2003) (Table 3). The results of this trial showed no effect on decline in FEV1, but a reduction in lung overinflation in patients with severe COPD and in exacerbation rate in patients who were not treated with inhaled glucocorticoids (Decramer et al., 2005). The variability in all the current studies using NAC 600 mg p.o. q.d. i.e. orally once a day may simply reflect the fact that the dose is not high enough. Thus, further studies are required at higher doses (1,200 or 1,800 mg/day) or using other thiol agents with greater bioavailability in order to observe any clinical benefit on lung function, reduced exacerbation rate and improved health status. IPF is associated with reduced pulmonary GSH and oxidative stress (Beeh et al., 2002; Rahman et al., 1999a). Ongoing clinical trials with NAC in these patients suggest that this thiol compound is well tolerated and may increase pulmonary levels of GSH.
5.1.1. Pharmacology and Pharmacokinetics of NAC
Following an oral dose of 600 mg, NAC is rapidly absorbed with a peak of 4.6 μM after 60 min (Tsikas et al., 1998), the plasma half-life being 2.5 hours. Following oral administration, reduced NAC has a terminal half-life of 6.25h (Holdiness, 1991). No NAC is detectable 10–12 hours after administration (De Caro et al., 1989). It has been estimated that the oral bioavailability of the intact NAC molecule was about 10% (Borgstrom and Kagedal, 1990). NAC when orally administered is metabolized into another compounds since accompanying increase in non-protein and protein sulphydryl groups were found in plasma (Cotgreave et al., 1987; De Caro et al., 1989). It is important to note that increasing the dose also increased NAC bioavailability and time for maximal plasma concentration (Borgstrom and Kagedal, 1990) and higher concentration in plasma may be achieved after intravenous administration (Crouch and Rusho, 2005). The volume of distribution ranges from 0.33 to 0.47 L/kg and protein binding is significant, reaching approximately 50% 4 hours post-administration (Holdiness, 1991). Pharmacokinetic information is not available regarding the penetrability of NAC across the blood-brain barrier or placenta, or into breast milk. Whereas, renal clearance has been reported as 0.190 to 0.211 L/h/kg, up to approximately 70% of the total body clearance is non-renal. Post-absorption, NAC is rapidly metabolized to cysteine, which has reducing and antioxidant properties due to the presence of the thiol group and also is a direct precursor of the GSH. Because of its ability to reduce disulfide bounds it is widely used to reduce viscosity and elasticity of the mucus (Aruoma et al., 1989). In order to exert some effects in the lungs, concentration of NAC or its derivatives have to be sufficiently elevated in bronchial epithelium or in ELF. Nevertheless, no NAC was found in BALF after 2-weeks of NAC intake (200 mg t.i.d) by healthy volunteers (Cotgreave et al., 1987). The details of pharmacology and pharmacokinetics of NAC in vivo and its implications in patients with COPD is nicely reviewed by Sadowska et al. (Sadowska et al., 2007).
5.2. N-acystelyn
N-acystelyn (NAL), a lysine salt of NAC, has a neutral pH in solution, whereas NAC is acidic but retains the mucolytic and antioxidant (reducing) actions of NAC. NAL can be aerosolised into the lung without causing significant side effects (Gillissen et al., 1997); Antonicelli et al. (Antonicelli et al., 2002) compared the effect of NAL and NAC in vitro and found that both drugs enhance intracellular GSH in alveolar epithelial cells and inhibited H2O2 and O2 N- released from human blood-derived neutrophils from smokers with COPD. NAL also inhibited ROS generation induced by serum-opsonized zymosan by human polymorphonuclear neutrophils. This in vitro inhibitory response was comparable to the effects of NAC (Ekberg-Jansson et al., 1999). Antonicelli et al. (Antonicelli et al., 2002) have shown that NAL inhibited oxidant-mediated IL-8 release in alveolar epithelial A549 cells suggesting an anti-inflammatory effect of NAL. Therefore, NAL may represent an interesting alternative approach to augment the antioxidant screen and thereby inhibiting inflammatory responses in the lungs and can be administered by inhalation.
5.3. N-isobutyrylcysteine
Since NAC becomes hydrolyzed in biological systems, the measured bioavailability of the drug is low. Thus, it was speculated that a drug might be synthesized that possessed greater bioavailability than NAC, and could be used as a more effective treatment for COPD. N-isobutyrylcysteine (NIC) is a NAC-like thiol compound that does not undergo effective first pass hydrolysis and hence has a higher oral bioavailability than NAC (Ekberg-Jansson et al., 1999). The oral bioavailability can be as high as 80%, dependent on food intake. However, when evaluated as a therapy for exacerbations of chronic bronchitis, NIC performed no better than placebo, and not as well as NAC (Gillissen et al., 1997). Recently, a study of NIC also failed to reduce exacerbation rates in patients with COPD (Ekberg-Jansson et al., 1999).
5.4. Erdosteine
Erdosteine is a new thiol compound that also acts as an antioxidant, but in addition has mucoactive properties and reduces bacterial adhesiveness (Moretti and Marchioni, 2007). In the “EQUALIFE” randomized placebo-controlled clinical study, erdosteine was dosed orally 300 mg b.i.d. for 8 months (Moretti et al., 2004). Patients receiving erdosteine had significantly fewer exacerbations and spent less days in hospital than the placebo group (Moretti et al., 2004). Moreover, patients receiving erdosteine showed no reduction in lung function over this period and a significant improvement in health-related QoL (Moretti et al., 2004). It is not clear whether clinical benefit is due to its antioxidant or mucolytic actions since the findings are consistent with the Cochrane review meta-analysis on mucolytic reagents in chronic bronchitis (Stey et al., 2000). It is possible that erdosteine may reduce bacterial colonization through a direct effect on adhesion (Moretti et al., 2004). A clinical trial on the combination of steroids and erdosteine in patients with COPD is needed.
5.5. Fudosteine
Fudosteine, [(−)-(R)-2-amino-3-(3-hydroxypropylthio)] propionic acid, has been used as mucoactive agent with indications for chronic respiratory diseases such as bronchial asthma, chronic bronchitis, pulmonary emphysema, COPD, bronchiectasis, pulmonary tuberculosis, pneumoconiosis, atypical mycobacterial disease, and diffuse panbronchiolitis (Komatsu et al., 2005). Fudosteine are a cysteine donating compounds that increase the cysteine levels of the cells and have greater bioavailability than NAC. These thiol compounds are well tolerated and have been shown to increase mitochondrial levels of GSH in alveolar type II cells (Guidot and Brown, 2000). GSH esters, particularly GSH monoethyl esters can increase the GSH levels of these cells by cleavage of an ester bond (an ethyl group esterified to glycine). GSH esters have been shown to increase GSH levels in the lungs of rats; however, this compound can be cytotoxic and variation in the uptake levels of GSH has been shown in various cellular models (Butterworth et al., 1993). Fudosteine inhibits MUC5AC mucin hypersecretion by reducing the expression of the MUC5AC gene (Rhee et al., 2008). Although EGFR is a common molecule for the activation of MUC5AC gene expression, the inhibitory effect of fudosteine is not related to EGFR. With respect to the MUC5AC mucin secretion pathways, fudosteine inhibited the phosphorylation of ERK1/2 and p38 MAPK in LPS-treated rats and of ERK1/2 in TNF-α-treated NCI-H292 cells (Rhee et al., 2008). These findings suggest that fudosteine may be useful in controlling stress-related mucus secretion states in patients with asthma, bronchiectasis or COPD.
5.6. Carbocysteine
S-carboxymethylcysteine (carbocysteine or S-CMC) is a muco-active drug with in vitro free radical scavenging and anti-inflammatory properties. Carbocysteine is available as an oral preparation both as S-CMC and its lysine salt (S-CMC-lys). The lysine group is cleaved on gastric absorption to form the active drug S-CMC. Carbocysteine is well absorbed when taken orally and peak serum concentrations are achieved at 1–1.7 hours and the plasma half life is 1.33 hours. It achieves good translocation into lung tissue and bronchial secretions (Braga et al., 1982). Approximately 30–60% of the drug is excreted unchanged in urine. There is evidence from animal models that carbocysteine increases chloride transport across the airway epithelium and this may also contribute to its muco-regulatory action (Colombo et al., 1994). Evidence from animal models of respiratory epithelial repair demonstrates that carbocysteine reverses neutral endopeptidase levels and cough sensitivity (but not methacholine-induced bronchoconstriction) in rodents with antigen-damaged tracheal epithelium in a dose-related manner (Katayama et al., 2001). Animal studies have demonstrated the anti-inflammatory action of carbocysteine in models of pulmonary inflammation involving several different cytokine profiles. Oral pre-treatment with S-CMC-lys appears to attenuate neutrophil recruitment in acute IL-1β-induced airway inflammation and neutrophil, macrophage and eosinophil migration to the pleural space in carrageenan-induced pleurisy (Asti et al., 1995). Administration of carbocysteine to rats with sulfur dioxide-induced airway inflammation attenuates the secretion of abnormal mucous glycoproteins and reduces inflammatory cells, free radical and elastase activity in their BALF (Ishibashi et al., 2001). Carbocysteine has been clinically used to treat patients with COPD. A recent PEACE study has revealed that the COPD patients treated with carbocysteine experienced decreased numbers of exacerbations per year (Zheng et al., 2008). Non-significant interactions were found between the preventive effects and COPD severity, smoking, as well as concomitant use of inhaled corticosteroids. Furthermore, carbocysteine (carbocisteine) was well-tolerated.
5.7. GSH peroxidase mimetic
Small molecules with enzymatic activity similar to GPx have been developed, such as the seleno-organic compound ebselen. Selenium is an important element in the GPx catalysis of the reaction between GSH and ROS. Consequently, ebselen increases the efficiency of GSH as an antioxidant, and thus may be used as a therapy against oxidative stress and inflammation. Recent studies have shown that ebselen inhibits airway inflammation (neutrophil recruitment and chemokine expression) in response to LPS in various animal models (Haddad et al., 2002; Zhang et al., 2002). It would be interesting to see whether similar results can be obtained by ebselen in inhibiting the airway inflammation of cigarette smokers.
6. GSH and lung diseases
The lung being directly exposed to the air is susceptible to attack from airborne materials, cigarette smoke, environmental oxidants and other pollutants and toxins (Kidd, 1985). GSH and GSH- associated enzymes present in the lower respiratory tract are believed to act as a first line of defense against such attacks by external agents (Deleve and Kaplowitz, 1990; Pacht et al., 1991). Sustained oxidative challenge leads to depletion of lung GSH along with other antioxidants.
GSH deficiencies have now been documented in number of lung diseases, which include ARDS, COPD, asthma, CF, IPF and neonatal lung disease (Lomaestro and Malone, 1995). ARDS patients having sepsis have been found to be deficient in GSH in the ELF compared to healthy subjects (Bunnell and Pacht, 1993) and a large percentage of GSH is in its oxidized, GSSG form. This is indicative of an oxidative stress in the lower respiratory tract. Treatment with NAC to such patients significantly decreased the time spent in the hospital (Suter et al., 1994). Asthmatics also demonstrate increased ROS generation during airway inflammation. Patients with mild form of asthma have a capacity to adaptive increases in their lung alveolar GSH levels (Smith et al., 1993). IPF patients however, the GSH levels are only about 255 of the normal levels, which is the cause of the pathophysiology underlying the development and progression of this disease (Deleve and Kaplowitz, 1990).
Infants born pre-maturely at 25 weeks of gestational age have been found to have decreased levels of GSH in the lungs compared to those who are born at 40 weeks (Grigg et al, 1993). Infants born at 35-weeks also showed lower levels of lung GSH and particularly susceptible to chronic lung diseases later in their lives. Thus many lung diseases are related to the status of GSH in the lungs and lung compartments.
7. Future Perspectives
Therapies with GSH and its analogues as discussed above have been widely practiced and studied. Several clinical trials have also been undertaken (Table 3). Most of these studies have revealed that such a mode of treatment may not necessarily have positive outcome. Several studies have reported undesirable effects suggesting that direct GSH therapy may not be an appropriate way of increasing GSH levels in lung ELF and cells in COPD. The bioavailability of GSH, pH, osmolality in the inflammatory micro-environment, and the resultant formation of toxic products (GSSG and GSH-adducts) are further challenges for direct GSH administration. Alternative formulations may address bioavailability, such as liposomal delivery, but at present it seems that direct administration of GSH will not be successful in treating COPD. Increasing the activity of GCL would also be expected to increase cellular GSH levels, and induction of GCL by pharmacological agents also holds great promise in protection against chronic inflammation and oxidant-mediated injury in COPD. Given the critical role of the GSH pathway in the detoxification of many drugs and xenobiotics, the reported differences in basal tissue distribution among mouse, rat and canine has far-reaching implications in comparing responses of these species in safety testing of GSH and or its analogues as therapeutic agents (Mattes et al., 2006).
Repeated administration of NAC may induce oxidative stress in some tissues and further studies are needed to confirm this finding (Arfsten et al., 2007). NAC may also reduce cystine to cysteine, which is an important mechanism for intracellular GSH elevation in vivo in lungs (Burgunder et al., 1989). It not only reduces disulphide bonds (a property of a good reducing agent), but also has the potential to interact directly with oxidants. Since NAC becomes hydrolyzed in biological systems, the measured bioavailability of the drug is low. This aspect also requires attention for further research. Similarly, it is not clear whether clinical benefit of erdosteine, fudosteine and carbocysteine is due to its antioxidant or mucolytic actions which are consistent with the Cochrane review meta-analysis on mucolytic reagents in chronic bronchitis (Stey et al., 2000). Overall, it may be pertinent to suggest that therapeutic research involving GSH or its analogues or thiol compounds must be carefully designed keeping in mind the species barrier, bioavailability, half life of the analogues, and generation of toxic byproducts by the analogues and interference with metabolic and signaling pathways in a cell or tissue. Since patients may have different level of antioxidants in them and since the oxidative states may differ considerably depending on nutritional, behavioral and occupational states, proper understanding of the above issues may lead to development of better thiol/GSH-dependent therapeutic strategies for pulmonary diseases.
Acknowledgments
This work was supported by the National Institutes of Health (NIH) R01-HL085613 and National Institute of Environmental Health Sciences Center (NIEHS) Grant ES-01247.
This work was supported by the National Institutes of Health (NIH)-NHLBI R01-HL085613 and National Institute of Environmental Health Sciences Center (NIEHS) Grant ES-01247.
Abbreviations
- AP-1
Activator Protein-1
- ARDS
Acute respiratory distress syndrome
- ARE
Antioxidant response element
- BALF
Bronchoalveolar Larvage Fluid
- CF
Cystic Fibrosis
- COPD
Chronic Obstructive Pulmonary Disease
- EGF
Epidermal growth factor
- ELF
Epithelial Lining Fluid
- EpRE
Electrophilic Response Element
- ER
Endoplasmic reticulum
- ERK
extracellular regulated protein kinase
- γ-GT
γ-glutamyl transpeptidase
- GCL
Glutamate Cysteine Ligase
- GCLC
Glutamate-Cysteine Ligase, Catalytic subunit
- GCLM
Glutamate-Cysteine Ligase, Modifying subunit
- GPx
Glutathione peroxidase
- GSH
Glutathione
- GSSG
Glutathione Disulfide
- H2O2
Hydrogen peroxide
- IL-8
Interleukin-8
- IL-13
Interleukin-13
- IPF
Idiopathic pulmonary fibrosis
- MAPK
Mitogen activated protein kinase
- NAC
N-acetyl-L-cysteine
- NAL
N-acystelyn
- NF-κB
Nuclear Factor-kappaB
- NO2
Nitrogen dioxide
- NO•
Nitric Oxide
- Nrf
cap ‘n’ collar-basic leucine zipper (CNC-bZIP) family protein or nuclear redox factor
- •OH
Hydroxyl radical
- ONOO-
Peroxinitrite
- ROS
Reactive Oxygen Species
- RNS
Reactive Nitrogen Species
- TGF- β1
Transforming Growth Factor-β1
- TNF-α
Tumor Necrosis Factor-alpha
- Trx
Thioredoxin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Antonicelli F, Parmentier M, Drost EM, Hirani N, Rahman I, Donaldson K, MacNee W. Nacystelyn inhibits hydrogen peroxide mediated interleukin-8 expression in human alveolar epithelial cells. Free Rad Biol Med. 2002;32:492–502. doi: 10.1016/s0891-5849(01)00820-6. [DOI] [PubMed] [Google Scholar]
- Aoki T, Suzuki Y, Suzuki K, Miyata A, Oyamada Y, Takasugi T, Mori M, Fujita H, Yamaguchi K. Modulation of ICAM-1 expression by extra-cellular glutathione in hyperoxia-exposed human pulmonary artery endothelial cells. Am J Respir Cell Mol Biol. 1996;15:319–327. doi: 10.1165/ajrcmb.15.3.8810635. [DOI] [PubMed] [Google Scholar]
- Arfsten DP, Johnson EW, Wilfong ER, Jung AE, Bobb AJ. Distribution of radio-labeled N-Acetyl-L-Cysteine in Sprague-Dawley rats and its effect on glutathione metabolism following single and repeat dosing by oral gavage. Cutan Ocul Toxicol. 2007;26:113–134. doi: 10.1080/15569520701212233. [DOI] [PubMed] [Google Scholar]
- Aruoma OI, Halliwell B, Hoey BM, Butler J. The antioxidant action of N-acetylcysteine: its reaction with hydrogen peroxide, hydroxyl radical, superoxide, and hypochlorous acid. Free Radic Biol Med. 1989;6:593–597. doi: 10.1016/0891-5849(89)90066-x. [DOI] [PubMed] [Google Scholar]
- Asti C, Melillo G, Caselli GF, Daffonchio L, Hernandez A, Clavenna G, Omini C. Effectiveness of carbocysteine lysine salt monohydrate on models of airway inflammation and hyperresponsiveness, Pharmacol. Res. 1995;31:387–392. doi: 10.1016/1043-6618(95)80094-8. [DOI] [PubMed] [Google Scholar]
- Ault JG, Lawrence DA. Glutathione distribution in normal and oxidatively stressed cells. Exp Cell Res. 2003;285:19–14. doi: 10.1016/s0014-4827(03)00012-0. [DOI] [PubMed] [Google Scholar]
- Azzi A, Aratri E, Boscoboinik D, Clement S, Ozer NK, Ricciarelli R, Spycher S. Molecular basis of α-tocopherol control of smooth muscle cell proliferation. Biofactors. 1998;7:3–14. doi: 10.1002/biof.5520070102. [DOI] [PubMed] [Google Scholar]
- Bakin AV, Stourman NV, Sekhar KR, Rinehart C, Yan X, Meredith MJ, Arteaga CL, Freeman ML. Smad3-ATF3 signaling mediates TGF-beta suppression of genes encoding phase II detoxifying proteins. Free Radic Biol Med. 2005;38:375–387. doi: 10.1016/j.freeradbiomed.2004.10.033. [DOI] [PubMed] [Google Scholar]
- Banhegyi G, Csala M, Nagy G, Sorrentino V, Fulceri R, Benedetti A. Evidence for the transport of glutathione through ryanodine receptor channel type 1. Biochem J. 2003;376:807–812. doi: 10.1042/BJ20031419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banhegyi G, Lusini L, Puskas F, Rossi R, Fulceri R, Braun L, Mile V, di Simplicio P, Mandl J, Benedetti A. Preferential transport of glutathione versus glutathione disulfide in rat liver microsomal vesicles. J Biol Chem. 1999;274:12213–12216. doi: 10.1074/jbc.274.18.12213. [DOI] [PubMed] [Google Scholar]
- Beck MJ, McLellan C, Lightle RL, Philbert MA, Harris C. Spatial glutathione and cysteine distribution and chemical modulation in the early organogenesis-stage Rat Conceptus in utero. Toxicol Sci. 2001;62:92–102. doi: 10.1093/toxsci/62.1.92. [DOI] [PubMed] [Google Scholar]
- Beeh KM, Beier J, Haas IC, Kornmann O, Micke P, Buhl R. Glutathione deficiency of the lower respiratory tract in patients with idiopathic pulmonary fibrosis. Eur Respir J. 2002;19:1119–1123. doi: 10.1183/09031936.02.00262402. [DOI] [PubMed] [Google Scholar]
- Beeh KM, Beier J, Koppenhoefer N, Buhl R. Increased glutathione disulfide and nitrosothiols in sputum of patients with stable COPD. Chest. 2004;126:1116–1122. doi: 10.1378/chest.126.4.1116. [DOI] [PubMed] [Google Scholar]
- Bindoli A, Fukuto JM, Forman HJ. Thiol Chemistry in Peroxidase Catalysis and Redox Signaling. Antioxidants & Redox Signaling. 2008;10:1549–15643. doi: 10.1089/ars.2008.2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biswas SK, McClure D, Jimenez LA, Megson IL, Rahman I. Curcumin induces glutathione biosynthesis and inhibits NF-kappaB activation and interleukin-8 release in alveolar epithelial cells: mechanism of free radical scavenging activity. Antioxid Redox Signal. 2005;7:32–41. doi: 10.1089/ars.2005.7.32. [DOI] [PubMed] [Google Scholar]
- Borgstrom L, Kagedal B. Dose dependent pharmacokinetics of N-acetylcysteine after oral dosing to man. Biopharm Drug Dispos. 1990;11:131–136. doi: 10.1002/bdd.2510110205. [DOI] [PubMed] [Google Scholar]
- Braga PC, Borsa M, De Anglis L, Bossi R, Allegra L, Scaglione F, Scarpazza G. Pharmacokinetic behaviour of S-carboxymethyl-cysteine-lys in patients with chronic bronchitis. Clin Ther. 1982;4:480–488. [PubMed] [Google Scholar]
- Bridgeman MM, Marsden M, Selby C, Morrison D, MacNee W. Effect of N-acetyl cysteine on the concentrations of thiols in plasma bronchoalveolar lavage fluid and lining tissue. Thorax. 1994;49:670–675. doi: 10.1136/thx.49.7.670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunnell E, Pacht ER. Oxidized glutathione is increased in alveolar fluid of patients with ARDS. Am Rev Resp Dis. 1993;148:1174–1178. doi: 10.1164/ajrccm/148.5.1174. [DOI] [PubMed] [Google Scholar]
- Burgunder JM, Varriale A, Lauterburg BH. Effect of N-acetylcysteine on plasma cysteine and glutathione following paracetamol administration. Eur J Clin Pharmacol. 1989;36:127–131. doi: 10.1007/BF00609183. [DOI] [PubMed] [Google Scholar]
- Butterworth M, Upshall DG, Hobbs M, Cohen GM. Elevation of cysteine and replenishment of glutathione in rat lung slices by cysteine isopropylester and other cysteine precursors. Biochem Pharmacol. 1993;45:1769–1774. doi: 10.1016/0006-2952(93)90432-v. [DOI] [PubMed] [Google Scholar]
- Cantin AM, Larivee P, Begin R. Extracellular glutathione suppresses human lung fibroblast proliferation. Am J Respir Cell Mol Biol. 1990;3:79–85. doi: 10.1165/ajrcmb/3.1.79. [DOI] [PubMed] [Google Scholar]
- Chakravarthi S, Jessop CE, Bulleid NJ. The role of glutathione in disulphide bond formation and endoplasmic-reticulum-generated oxidative stress. EMBO Rep. 2006;7:271–275. doi: 10.1038/sj.embor.7400645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan JY, Kwong M. Impaired expression of glutathione synthetic enzyme genes in mice with targeted deletion of the Nrf2 basic-leucine zipper protein. Biochim Biophys Acta. 2000;1517:19–26. doi: 10.1016/s0167-4781(00)00238-4. [DOI] [PubMed] [Google Scholar]
- Chan K, Han XD, Kan YW. An important function of Nrf2 in combating oxidative stress: detoxification of acetaminophen. Proc Natl Acad Sci USA. 2001;98:4611–4616. doi: 10.1073/pnas.081082098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chappell S, Daly L, Mogan K, Guetta-Baranes T, Roca J, Rabonivich R, Lotya J, Millar AB, Donnelly SC, Keatings V, MacNee W, Stolk J, Hiemstra PS, Miniati M, Monti S, O’Connor CM, Kalsheker N. Genetic varients of microsomal epoxide hydrolase and glutamate-cysteine ligase in COPD. Eur Respir J. 2008 doi: 10.1183/09031936.00065308. In Press. [DOI] [PubMed] [Google Scholar]
- Chen JW, Dodia C, Feinstein SI, Jain MK, Fisher AB. 1-Cys peroxiredoxin, a bifunctional enzyme with glutathione peroxidase and phospholipase A2 activities. J Biol Chem. 2000;275:28421–28427. doi: 10.1074/jbc.M005073200. [DOI] [PubMed] [Google Scholar]
- Chen L, Kwong M, Lu R, Ginzinger D, Lee C, Leung L, Chan JY. Nrf1 is critical for redox balance and survival of liver cells during development. Mol Cell Biol. 2003;23:4673–4686. doi: 10.1128/MCB.23.13.4673-4686.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang IC, Liu DD, Kao SJ, Chen HI. N-acetylcysteine attenuates the acute lung injury caused by phorbol myristate acetate in isolated rat lungs. Pulm Pharmacol Ther. 2007;20:726–733. doi: 10.1016/j.pupt.2006.08.010. [DOI] [PubMed] [Google Scholar]
- Church DF, Prior WA. Free-radical chemistry of cigarette smoke and its toxicological implications. Environ Health Perspect. 1985;64:111–126. doi: 10.1289/ehp.8564111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colombo B, Turconi P, Daffonchino L, Fedele G, Omini C, Cremaschi D. Stimulation of Cl- secretion by the mucoactive drug S-carboxymethylcysteine lysine–salt in the isolated rabbit trachea. Eur Resp J. 1994;7:1622–1628. doi: 10.1183/09031936.94.07091622. [DOI] [PubMed] [Google Scholar]
- Cotgreave IA. N-acetylcysteine: pharmacological considerations and experimental and clinical applications. Adv Pharmacol. 1997;38:205–227. [PubMed] [Google Scholar]
- Cotgreave IA, Berggren M, Jones TW, Dawson J, Moldeus P. Gastrointestinal metabolism of N-acetylcysteine in the rat, including an assay for sulfite in biological systems. Biopharm Drug Dispos. 1987;8:377–386. doi: 10.1002/bdd.2510080408. [DOI] [PubMed] [Google Scholar]
- Crouch BI, Rusho WJ. Intravenous Administration of N-Acetylcysteine. Ann Emerg Med. 2005;46:207–208. doi: 10.1016/j.annemergmed.2005.02.027. [DOI] [PubMed] [Google Scholar]
- Crystal RG. Oxidants and respiratory tract epithelial injury: pathogenesis and strategies for therapeutic intervention. Am J Med. 1991;91:39S–44S. doi: 10.1016/0002-9343(91)90282-3. [DOI] [PubMed] [Google Scholar]
- Das KC. c-Jun NH2-terminal kinase-mediated redox dependent degradation of I-κB: role of Trx in NF-κB activation. J Biol Chem. 2001;276:4662–4670. doi: 10.1074/jbc.M006206200. [DOI] [PubMed] [Google Scholar]
- De Benedetto F, Aceto A, Dragani B, Spacone A, Formisano S, Pela R, Donner CF, Sanguinetti CM. Long-term oral N-acetylcysteine reduces exhaled hydrogen peroxide in stable COPD. Pulm Pharmacol Ther 2005. 2005;18:41–47. doi: 10.1016/j.pupt.2004.09.030. [DOI] [PubMed] [Google Scholar]
- de Boer WI, van Schadewijk A, Sont JK, Sharma HS, Stolk J, Hiemstra PJ, van Krieken JH. Transforming growth factor β1 and recruitment of macrophages and mast cells in airways in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 1998;158:1–7. doi: 10.1164/ajrccm.158.6.9803053. [DOI] [PubMed] [Google Scholar]
- De Caro L, Ghizzi A, Costa R, Longo A, Ventresca GP, Lodola E. Pharmacokinetics and bioavailability of oral acetylcysteine in healthy volunteers. Arzneimittelforschung. 1989;39:382–386. [PubMed] [Google Scholar]
- Decramer M, Dekhuijzen PN, Troosters T, van Herwaarden C, Rutten-van Mölken M, van Schayck CP, Olivieri D, Lankhorst I, Ardia A. The Bronchitis Randomized On NAC Cost-Utility Study (BRONCUS): hypothesis and design. BRONCUS-trial Committee. Eur Respir J. 2001;17:329–336. doi: 10.1183/09031936.01.17303290. [DOI] [PubMed] [Google Scholar]
- Decramer M, Rutten-van Molken M, Dekhuijzen PN, Troosters T, van Herwaarden C, Pellegrino R, van Schayck CP, Olivieri D, Del Donno M, De Backer W, Lankhorst I, Ardia A. Effects of N-acetylcysteine on outcomes in chronic obstructive pulmonary disease (Bronchitis Randomized on NAC Cost-Utility Study, BRONCUS): a randomized placebo-controlled trial. Lancet. 2005;365:1552–1560. doi: 10.1016/S0140-6736(05)66456-2. [DOI] [PubMed] [Google Scholar]
- Dekhuijzen PN. Antioxidant properties of N-acetylcysteine: their relevance in relation to chronic obstructive pulmonary disease. Eur Respir J. 2004;23:629–636. doi: 10.1183/09031936.04.00016804. [DOI] [PubMed] [Google Scholar]
- Deleve LD, Kaplowitz N. Importance of regulation of hepatic glutathione. Semin Liver Dis. 1990;10:251–266. doi: 10.1055/s-2008-1040481. [DOI] [PubMed] [Google Scholar]
- Dickinson DA, Iles KE, Zhang H, Blank V, Forman HJ. Curcumin alters EpRE and AP-1 binding complexes and elevates glutamate-cysteine ligase gene expression. FASEB J. 2003;17:473–475. doi: 10.1096/fj.02-0566fje. [DOI] [PubMed] [Google Scholar]
- Dinkova-Kostova AT, Holtzclaw WD, Cole RN, Itoh K, Wakabayashi N, Katoh Y, Yamamoto M, Talalay P. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci USA. 2002;99:11908–11913. doi: 10.1073/pnas.172398899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drost EM, Skwarski KM, Sauleda J, Soler N, Roca J, Agusti A, MacNee W. Oxiative stress and airway inflammation in severe exacerbations of COPD. Thorax. 2005;60:293–300. doi: 10.1136/thx.2004.027946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dueholm M, Nielson C, Thorshauge H, Evald T, Hansen NC, Madsen HD, Maltbaek N. N-acetylcysteine by metred dose inhaler in the treatment of chronic bronchitis: a multi-centre study. Respir Med. 1992;86:89–92. doi: 10.1016/s0954-6111(06)80220-9. [DOI] [PubMed] [Google Scholar]
- Ekberg-Jansson A, Larson M, MacNee W, Tunek A, Wahlgren L, Wouters EF, Larsson S. N-isobutyrylcysteine, a donor of systemic thiols, does not reduce the exacerbation rate in chronic bronchitis. Eur Respir J. 1999;13:829–834. doi: 10.1034/j.1399-3003.1999.13d22.x. [DOI] [PubMed] [Google Scholar]
- Erickson A, Nevarea Z, Gipp JJ, Mulcahy RT. Identification of a variant antioxidant response element in the promoter of the human glutamate-cysteine ligase modifier subunit gene. J Biol Chem. 2002;277:30730–30737. doi: 10.1074/jbc.M205225200. [DOI] [PubMed] [Google Scholar]
- Forman HJ, Dickinson DA. Oxidative signaling and glutathione synthesis. Biofactors. 2003;17:1–12. doi: 10.1002/biof.5520170101. [DOI] [PubMed] [Google Scholar]
- Galloway DC, Blake DG, Shepherd AG, McLellan LI. Regulation of human κ-glutamylcysteine synthetase: co-ordinate induction of the catalytic and regulatory subunits in HepG2 cells. Biochem J. 1997;328:99–104. doi: 10.1042/bj3280099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerrits CM, Herings RM, Leufkens HG, Lammers JW. N-acetylcysteine reduces the risk of re-hospitalization among patients with chronic obstructive pulmonary disease. Eur Respir J. 2003;21:795–798. doi: 10.1183/09031936.03.00063402. [DOI] [PubMed] [Google Scholar]
- Gillissen A, Jaworska M, Orth M, Coffiner M, Maes P, App EM, Cantin AM, Schultze-Werninghaus G. Nacystelyn a novel lysine salt of N-acetylcysteine, to augment cellular antioxidant defence in vitro. Respir Med. 1997;91:159–168. doi: 10.1016/s0954-6111(97)90052-4. [DOI] [PubMed] [Google Scholar]
- Goven D, Boutten A, Lecon-Malas V, Marchal-Somme J, Amara N, Crestani B, Fournier M, Leseche G, Soler P, Boczkowski J, Bonay M. Altered Nrf2/Keap1-Bach1 equilibrium in pulmonary emphysema. Thorax. 2008 doi: 10.1136/thx.2007.091181. In press. [DOI] [PubMed] [Google Scholar]
- Grandjean EM, Berthet P, Ruffmann R, Leuenberger P. Efficacy and oral long-term N-acetylcysteine in chronic bronchopulmonary disease: a meta-analysis of published double-blind, placebo-controlled clinical trials. Clin Ther. 2000;22:209–221. doi: 10.1016/S0149-2918(00)88479-9. [DOI] [PubMed] [Google Scholar]
- Grigg J, Barber A, Silverman M. Bronchoalveolar lavage glutathione in intubated premature infants. Arch Dis Child. 1993;69:49–51. doi: 10.1136/adc.69.1_spec_no.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guidot DM, Brown LA. Mitochondrial glutathione replacement restores surfactant synthesis and secretion in alveolar epithelial cells of ethanol-fed rats. Alcohol Clin Exp Res. 2000;24:1070–1076. [PubMed] [Google Scholar]
- Haddad el-B, McCluskie K, Birrell MA, Dabrowski D, Pecoraro M, Underwood S, Chen B, De Sanctis GT, Webber SE, Foster ML, Belvisi MG. Differential effects of ebselen on neutrophil recruitment, chemokine, and inflammatory mediator expression in a rat model of lipopolysaccharide-induced pulmonary inflammation. J Immunol. 2002;169:974–982. doi: 10.4049/jimmunol.169.2.974. [DOI] [PubMed] [Google Scholar]
- Halliwell B, Gutteridge JMC. Free radicals in biology and medicine. 3. Clarendon Press Oxford; 1999. [Google Scholar]
- Harju T, Kaarteenaho-Wiik R, Soini Y, Sormunen R, Kinnula VL. Diminished immunoreactivity of gamma-glutamylcysteine synthetase in the airways of smokers’ lung. Am J Respir Crit Care Med. 2002;166:754–9. doi: 10.1164/rccm.2112014. [DOI] [PubMed] [Google Scholar]
- Harper R, Wu K, Chang MM, Yoneda K, Pan R, Reddy SPM, Wu R. Activation of nuclear factor-kB transcriptional activity in airway epithelial cells by thioredoxin but not by N-acetyl-L-cysteine and glutathione. Am J Respir Cell Mol Biol. 2001;25:178–185. doi: 10.1165/ajrcmb.25.2.4471. [DOI] [PubMed] [Google Scholar]
- Hayes JD, McLellan LI. Glutathione and glutathione-dependent enzymes represent a co-ordinately regulated defense against oxidative stress. Free Radic Res. 1999;31:273–300. doi: 10.1080/10715769900300851. [DOI] [PubMed] [Google Scholar]
- He Z, Li B, Yu L, Liu Q, Zhong N, Ran P. Suppression of oxidant-induced glutathione synthesis by erythromycin in human bronchial epithelial cells. Respiration. 2008;75:202–209. doi: 10.1159/000111569. [DOI] [PubMed] [Google Scholar]
- Holdiness MR. Clinical pharmacokinetics of N-acetylcysteine. Clin Pharmacokinet. 1991;20:123–134. doi: 10.2165/00003088-199120020-00004. [DOI] [PubMed] [Google Scholar]
- Huang CS, Chang LS, Anderson ME, Meister A. Catalytic and regulatory properties of the heavy subunit of rat kidney γ-glutamylcysteine synthetase. J Biol Chem. 1993;268:19675–19680. [PubMed] [Google Scholar]
- Hwang C, Sinskey AJ, Lodish HF. Oxidized redox state of glutathione in the endoplasmic reticulum. Science. 1992;257:1496–1502. doi: 10.1126/science.1523409. [DOI] [PubMed] [Google Scholar]
- Inci I, Zhai W, Arni S, Hillinger S, Vogt P, Weder W. N-acetylcysteine attenuates lung ischemia-reperfusion injury after lung transplantation. Ann Thorac Surg. 2007;84:240–246. doi: 10.1016/j.athoracsur.2007.03.082. [DOI] [PubMed] [Google Scholar]
- Ishibashi Y, Okamura T, Masumoto Y, Tachiiri T, Momo K. Effects of carbocysteine on airway inflammation and related events in SO2 – exposed rats. Nihon Kokyuki Gakkai Zasshi. 2001;39:17–23. doi: 10.1007/BF02712616. [DOI] [PubMed] [Google Scholar]
- Itoh K, Wakabayash N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev. 1999;13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen YM, Van Houten B, Borm PJ, Mossman BT. Cell and tissue responses to oxidative damage. Lab Invest. 1993;69:261–274. [PubMed] [Google Scholar]
- Jardine H, MacNee W, Donaldson K, Rahman I. Molecular Mechanism of TGF-β1-induced Glutathione Depletion in Alveolar Epithelial Cells Involvement of AP-1/ARE and Fra-1. J Biol Chem. 2002;277:21158–21166. doi: 10.1074/jbc.M112145200. [DOI] [PubMed] [Google Scholar]
- Jessop CE, Bulleid NJ. Glutathione directly reduce an oxidoreductase in the endoplasmic reticulum of mammalian cells. J Biol Chem. 2004;279:55341–55347. doi: 10.1074/jbc.M411409200. [DOI] [PubMed] [Google Scholar]
- Jeyapaul J, Jaiswal AK. Nrf2 and c-Jun regulation of antioxidant response element (ARE)-mediated expression and induction of gamma-glutamylcysteine synthetase heavy subunit gene. Biochem Pharmacol. 2000;59:1433–1439. doi: 10.1016/s0006-2952(00)00256-2. [DOI] [PubMed] [Google Scholar]
- Kang MI, Kobayashi A, Wakabayashi N, Kim SG, Yamamoto M. Scaffolding of Keap1 to the actin cytoskeleton controls the function of Nrf2 as key regulator of cytoprotective phase 2 genes. Proc Natl Acad Sci USA. 2004;101:2046–51. doi: 10.1073/pnas.0308347100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasielski M, Nowak D. Long-term administration of N-acetylcysteine decreases hydrogen peroxide exhalation in subjects with chronic obstructive pulmonary disease. Respir Med. 2001;95:448–456. doi: 10.1053/rmed.2001.1066. [DOI] [PubMed] [Google Scholar]
- Katayama N, Fujimura M, Ueda A, Kita T, Abo M, Tachibana H, Myou S, Kurashima K. Effects of carbocysteine on antigen-induced increases in cough sensitivity and bronchial responsiveness in guinea pigs. J Pharmacol Exp Ther. 2001;297:975–980. [PubMed] [Google Scholar]
- Kidd P. The free radical oxidant toxins of polluted air. In: Levine SA, Kidd PM, editors. Antioxidant adaptation-Its role in free radical pathology. San Leandro, CA: Biocurrents; 1985. pp. 69–103. [Google Scholar]
- Kim YC, Yamaguchi Y, Kondo N, Masutani H, Yodoi J. Thioredoxin-dependent redox regulation of the antioxidant responsive element (ARE) in electrophile response. Oncogene. 2003;22:1860–1865. doi: 10.1038/sj.onc.1206369. [DOI] [PubMed] [Google Scholar]
- Kinnula VL, Chang L, Everitt JI, Crapo JD. Oxidants and antioxidants in alveolar epithelial type II cells: in situ, freshly isolated, and cultured cells. Am J Physiol. 1992a;262:L69–L77. doi: 10.1152/ajplung.1992.262.1.L69. [DOI] [PubMed] [Google Scholar]
- Kinnula VL, Chang L, Ho YS, Crapo JD. Hydrogen peroxide release from alveolar macrophages and alveolar type II cells during adaptation to hyperoxia in vivo. Exp Lung Res. 1992b;18:655–673. doi: 10.3109/01902149209031700. [DOI] [PubMed] [Google Scholar]
- Kinnula VL, Crapo JD, Raivio KO. Generation and disposal of reactive oxygen metabolites in the lung. Lab Invest. 1995;73:3–19. [PubMed] [Google Scholar]
- Kinnula VL, Vuorinen K, Ilumets H, Rytilä P, Myllärniemi M. Thiol proteins, redox modulation and parenchymal lung disease. Curr Med Chem. 2007;14:213–222. doi: 10.2174/092986707779313345. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Yamamoto M. Molecular Mechanisms Activating the Nrf2-Keap1 Pathway of Antioxidant Gene Regulation. Antioxid Redox Signal. 2005;7:385–394. doi: 10.1089/ars.2005.7.385. [DOI] [PubMed] [Google Scholar]
- Kode A, Rajendrasozhan S, Caito S, Yang SR, Megson IL, Rahman I. Resveratrol induces glutathione synthesis by activation of Nrf2 and protects against cigarette smoke-mediated oxidative stress in human lung epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2008;294:L478–L488. doi: 10.1152/ajplung.00361.2007. [DOI] [PubMed] [Google Scholar]
- Koike Y, Hisada T, Utsugi M, Ishizuka T, Shimizu Y, Ono A, Murata Y, Hamuro J, Mori M, Dobashi K. Glutathione redox regulates airway hyperresponsiveness and airway inflammation in mice. Am J Respir Cell Mol Biol. 2007;37:322–329. doi: 10.1165/rcmb.2006-0423OC. [DOI] [PubMed] [Google Scholar]
- Komatsu H, Yamaguchi S, Komorita N, Goto K, Takagi S, Ochi H, Okumoto T. Inhibition of endotoxin- and antigen-induced airway inflammation by fudosteine, a mucoactive agent. Pulm Pharmacol Ther. 2005;18:121–127. doi: 10.1016/j.pupt.2004.11.002. [DOI] [PubMed] [Google Scholar]
- Krzywanski DM, Dickinson DA, Iles KE, Wigley AF, Franklin CC, Liu RM, Kavanagh TJ, Forman HJ. Variable regulation of glutamate cysteine ligase subunit proteins affects glutathione biosynthesis in response to oxidative stress. Arch Biochem Biophys. 2004;423:116–125. doi: 10.1016/j.abb.2003.11.004. [DOI] [PubMed] [Google Scholar]
- Kwong M, Kan YW, Chan JY. The CNC basic leucine zipper factor, Nrf1, is essential for cell survival in response to oxidative stressinducing agents. J Biol Chem. 1999;274:37491–37498. doi: 10.1074/jbc.274.52.37491. [DOI] [PubMed] [Google Scholar]
- Lee HR, Cho JM, Shin DH, Yong CS, Choi HG, Wakabayashi N, Kwak MK. Adaptive response to GSH depletion and resistance to l -buthionine-( S,R )-sulfoximine: involvement of Nrf2 activation. Molecular and Cellular Biochemistry. 2008:1573–4919. doi: 10.1007/s11010-008-9853-y. (Online) [DOI] [PubMed] [Google Scholar]
- Lee JM, Calkins MJ, Chan K, Kan YW, Johnson JA. Identification of the NF-E2-related factor-2-dependent genes conferring protection against oxidative stress in primary cortical astrocytes using oligonucleotide microarray analysis. J Biol Chem. 2003;278:12029–12038. doi: 10.1074/jbc.M211558200. [DOI] [PubMed] [Google Scholar]
- Lee KS, Kim SR, Park HS, Park SJ, Min KH, Lee KY, Choe YH, Hong SH, Han HJ, Lee YR, Kim JS, Atlas D, Lee YC. A novel thiol compound, N-acetylcysteine amide, attenuates allergic airway disease by regulating activation of NF-kappaB and hypoxia-inducible factor-1alpha. Exp Mol Med. 2007;39:756–768. doi: 10.1038/emm.2007.82. [DOI] [PubMed] [Google Scholar]
- Leonard MO, Kieran NE, Howell K, Burne MJ, Varadarajan R, Dhakshinamoorthy S, Porter AG, O’Farrelly C, Rabb H, Taylor CT. Reoxygenation-specific activation of the antioxidant transcription factor Nrf2 mediates cytoprotective gene expression in ischemia-reperfusion injury. FASEB J. 2006;20:2624–2626. doi: 10.1096/fj.06-5097fje. [DOI] [PubMed] [Google Scholar]
- Liu RM, Liu Y, Forman HJ, Olman M, Tarpey MM. Glutathione regulates transforming growth factor beta stimulated collagen production in fibroblasts. Am J Physiol Lung Cell Mol Physiol. 2004;286:L121–L128. doi: 10.1152/ajplung.00231.2003. [DOI] [PubMed] [Google Scholar]
- Lomaestro BM, Malone M. Glutathione in health and disease: pharmacotherapeutic issues. Ann Pharmacother. 1995;29:1263–1273. doi: 10.1177/106002809502901213. [DOI] [PubMed] [Google Scholar]
- Lowry MH, McAllister BP, Jean JC, Brown LA, Hughey RP, Cruikshank WW, Amar S, Lucey EC, Braun K, Johnson P, Wight TN, Joyce-Brady M. Lung lining fluid glutathione attenuates IL-13 induced asthma. Am J Respir Cell Mol Biol. 2008;38:509–516. doi: 10.1165/rcmb.2007-0128OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu SC. Regulation of hepatic glutathione synthesis: current concepts and controversies. FASEB J. 1999;13:1169–1183. [PubMed] [Google Scholar]
- Luppi F, Aarbiou J, van Wetering S, Rahman I, de Boer WI, Rabe KF, Hiemstra PS. Effects of cigarette smoke condensate on proliferation and wound closure of bronchial epithelial cells in vitro: role of glutathione. Resp Res. 2005;6:140–148. doi: 10.1186/1465-9921-6-140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra D, Thimmulappa R, Navas-Acien A, Sandford A, Elliott M, Singh A, Chen L, Zhuang X, Hogg J, Pare P, Tuder RM, Biswal S. Decline in Nrf2 regulated antioxidants in COPD lungs due toloss of its positive regulator DJ-1. Am J Respir Crit Care Med. 2008 doi: 10.1164/rccm.200803-380OC. In press. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Mattes WB, Daniels KK, Summan M, Xu ZA, Mendrick DL. Tissue and species distribution of the glutathione pathway transcriptome. Xenobiotica. 2006;36:1081–1121. doi: 10.1080/00498250600861793. [DOI] [PubMed] [Google Scholar]
- McKone EF, Shao J, Frangolias DD, Keener CL, Shephard CA, Farin FM, Tonelli MR, Pare PD, Sandford AJ, Aitken ML, Kavanagh TJ. Variants in the glutamate-cysteine-ligase gene are associated with cystic fibrosis lung disease. Am J Respir Crit Care Med. 2006;174:415–419. doi: 10.1164/rccm.200508-1281OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meister A, Anderson ME. Glutathione. Ann Rev Biochem. 1983;52:711–760. doi: 10.1146/annurev.bi.52.070183.003431. [DOI] [PubMed] [Google Scholar]
- Meyer M, Schreck R, Baeuerle PA. Hydrogen peroxide and antioxidants have opposite effects on activation of NF-kB and AP-1 in intact cells: AP-1 as secondary antioxidant-responsive factor. EMBO J. 1993;12:2005–2015. doi: 10.1002/j.1460-2075.1993.tb05850.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miao W, Hu L, Scrivens PJ, Batist G. Transcriptional regulation of NF-E2 p45-related factor (NRF2) expression by the aryl hydrocarbon receptor-xenobiotic response element signaling pathway: direct cross-talk between phase I and II drug-metabolizing enzymes. J Biol Chem. 2005;280:20340–20348. doi: 10.1074/jbc.M412081200. [DOI] [PubMed] [Google Scholar]
- Monova H, Mulcahy RT. An electrophile response element (EpRE) regulates β-naphthoflavone induction of the human γ-glutamylcysteine synthetase regulatory subunit gene: Constitutive expression is mediated by an adjacent AP-1 site. J Biol Chem. 1998;273:14683–14689. doi: 10.1074/jbc.273.24.14683. [DOI] [PubMed] [Google Scholar]
- Moran JA, Dahl EL, Mulcahy RT. Differential induction of mafF, mafG and mafK expression by electrophile-response-element activators. Biochem J. 2002;361:371–377. doi: 10.1042/0264-6021:3610371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretti M, Bottrighi P, Dallari R, Da Porto R, Dolcetti A, Grandi P, Garuti G, Guffanti E, Roversi P, De Gugliemo M, Potena A. The effect of longterm treatment with erdosteine on chronic obstructive pulmonary disease: the EQUALIFE Study. Drugs Exp Clin Res. 2004;30:143–152. [PubMed] [Google Scholar]
- Moretti M, Marchioni CF. An overview of drdosterine antioxidant activity in experimental research. Pharmacol Res. 2007;55:249–254. doi: 10.1016/j.phrs.2006.12.006. [DOI] [PubMed] [Google Scholar]
- Mulcahy RT, Wartman MA, Bailey HH, Gipp JJ. Constitutive and β-napthoflavonone-induced expression of the human γ-glutamylcysteine synthetase heavy subunit gene is regulated by a distal antioxidant response element/TRE sequence. J Biol Chem. 1997;272:7445–7454. doi: 10.1074/jbc.272.11.7445. [DOI] [PubMed] [Google Scholar]
- Nakayama T, Church DF, Pryor WA. Quantitative analysis of the hydrogen peroxide formed in aqueous cigarette tar extracts. Free Radical Biol Med. 1989;7:9–15. doi: 10.1016/0891-5849(89)90094-4. [DOI] [PubMed] [Google Scholar]
- Pacht ER, Timerman AP, Lykens MG, Merola AJ. Deficiency of alveolar fluid glutathione of patients with sepsis and ARDS. Chest. 1991;100:1397–1403. doi: 10.1378/chest.100.5.1397. [DOI] [PubMed] [Google Scholar]
- Paget MS, Kang JG, Roe JH, Buttner MJ. σR, an RNA Polymerase sigma factor that modulates expression of the thioredoxin system in response to oxidative stress in Streptomyces coelicolor A3(2) EMBO J. 1998;17:5776–5782. doi: 10.1093/emboj/17.19.5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park EM, Park YM, Gwak YS. Oxidative damage in tissues of rats exposed to cigarette smoke. Free Radic Biol Med. 1998;25:79–86. doi: 10.1016/s0891-5849(98)00041-0. [DOI] [PubMed] [Google Scholar]
- Parmentier M, Hirani N, Rahman I, Donaldson K, Antonicelli F. Regulation of LPS-mediated IL-1beta release by N-acetyl-L-cysteine in THP-1 cells. Eur Respir J. 2000;16:933–939. doi: 10.1183/09031936.00.16593300. [DOI] [PubMed] [Google Scholar]
- Peltoniemi MJ, Rytilä PH, Harju TH, Soini YM, Salmenkivi KM, Ruddock LW, Kinnula VL. Modulation of glutaredoxin in the lung and sputum of cigarette smokers and chronic obstructive pulmonary disease. Respir Res. 2006;7:133–141. doi: 10.1186/1465-9921-7-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pietarinen-Runtti P, Raivio KO, Saksela M, Asikainen TM, Kinnula VL. Antioxidant enzyme regulation and resistance to oxidants of human bronchial epithelial cells cultured under hypoxic conditions. Am Respir Cell Mol Biol. 1998;19:286–292. doi: 10.1165/ajrcmb.19.2.2836. [DOI] [PubMed] [Google Scholar]
- Pinkus R, Weiner LM, Daniel V. Role of oxidants and antioxidants in the induction of AP-1, NF-κB and glutathione S-transferase gene expression. J Biol Chem. 1996;271:13422–13429. doi: 10.1074/jbc.271.23.13422. [DOI] [PubMed] [Google Scholar]
- Poole PJ, Black PN. Oral mucolytic drugs for exacerbations of chronic obstructive pulmonary disease: systematic review. BMJ. 2001;322:1271–1274. doi: 10.1136/bmj.322.7297.1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole PJ, Black PN. Preventing exacerbations of chronic bronchitis and COPD: therapeutic potential of mucolytic agents. Am J Respir Med. 2003;2:367–370. doi: 10.1007/BF03256664. [DOI] [PubMed] [Google Scholar]
- Rahman I. The role of oxidative stress in the pathogenesis of COPD: implications for therapy. Treat Respir Med. 2005;4:175–200. doi: 10.2165/00151829-200504030-00003. [DOI] [PubMed] [Google Scholar]
- Rahman I, Bel A, Mulier B, Lawson MF, Harrison DJ, MacNee W, Smith CA. Transcriptional regulation of γ-glutamylcysteine synthetase-heavy subunit by oxidants in human alveolar epithelial cells. Biochem Biophys Res Commun. 1996a;229:832–837. doi: 10.1006/bbrc.1996.1888. [DOI] [PubMed] [Google Scholar]
- Rahman I, Kode A, Biswas SK. Assay for quantitative determination of glutathione and glutathione disulfide levels using enzymatic recycling method. Nat Protoc. 2006;1:3159–3165. doi: 10.1038/nprot.2006.378. [DOI] [PubMed] [Google Scholar]
- Rahman I, MacNee W. Role of oxidants/antioxidants in smoking-induced lung diseases. Free Radic Biol Med. 1996b;21:669–681. doi: 10.1016/0891-5849(96)00155-4. [DOI] [PubMed] [Google Scholar]
- Rahman I, MacNee W. Regulation of redox glutathione levels and gene transcription in lung inflammation: therapeutic approaches. Free Radical Bio Med. 2000a;28:1405–1420. doi: 10.1016/s0891-5849(00)00215-x. [DOI] [PubMed] [Google Scholar]
- Rahman I, MacNee W. Oxidative stress and regulation of glutathione in lung inflammation. Eur Respir J. 2000b;16:534–554. doi: 10.1034/j.1399-3003.2000.016003534.x. [DOI] [PubMed] [Google Scholar]
- Rahman I, Mulier B, Gilmour PS, Watchorn T, Donaldson K, Jeffery PK, MacNee W. Oxidant-mediated lung epithelial cell tolerance: the role of intracellular glutathione and nuclear factor-κB. Biochem Pharmacol. 2001;62:787–794. doi: 10.1016/s0006-2952(01)00702-x. [DOI] [PubMed] [Google Scholar]
- Rahman I, Skwarska E, Henry M, Davis M, O’Connor CM, FitzGerald MX, Greening A, MacNee W. Systemic and pulmonary oxidative stress in idiopathic pulmonary fibrosis. Free Radic Biol Med. 1999;27:60–68. doi: 10.1016/s0891-5849(99)00035-0. [DOI] [PubMed] [Google Scholar]
- Rasmusse JB, Glennow C. Reduction in days of illness after long-term treatment with N-acetylcysteine controlled-release tablets in patients with chronic bronchitis. Eur J Respir Dis. 1988;1:351–355. [PubMed] [Google Scholar]
- Reddy NM, Kleeberger SR, Cho HY, Yamamoto M, Kensler TW, Biswal S, Reddy SP. Deficiency in Nrf2-GSH signaling impairs type II cell growth and enhances sensitivity to oxidants. Am J Respir Cell Mol Bio. 2007;37:3–8. doi: 10.1165/rcmb.2007-0004RC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynaert NL, Wouters EF, Janssen-Heininger YM. Modulation of glutaredoxin-1 expression in a mouse model of allergic airway disease. Am J Respir Cell Mol Biol. 2007;36:147–151. doi: 10.1165/rcmb.2006-0259RC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee CK, Kang CM, You MB, Yoon HK, Kim YK, Kim KH, Moon HS, Park SH, Song JS. Effect of fudosteine on mucin production. Eur Respir J. 2008 doi: 10.1183/09031936.00018508. In Press. [DOI] [PubMed] [Google Scholar]
- Rhee SG, Kang SW, Netto LE, Seo MS, Stadtman ER. A family of novel peroxidases, peroxiredoxins. Biofactors. 1999;10:207–209. doi: 10.1002/biof.5520100218. [DOI] [PubMed] [Google Scholar]
- Roum JH, Buhl R, McElvaney NG, Borok Z, Crystal RG. Systemic deficiency of glutathione in cystic fibrosis. J Appl Physiol. 1993;75:2419–2424. doi: 10.1152/jappl.1993.75.6.2419. [DOI] [PubMed] [Google Scholar]
- Rutkowski R, Pancewicz SA, Rutkowski K, Rutkowska J. Reactive oxygen and nitrogen species in inflammatory process. Pol Merkur Lekarski. 2007;23:131–136. [PubMed] [Google Scholar]
- Sadowska AM, Manuel-y-Keenoy B, De Backer WA. Antioxidant and anti-inflammatory efficacy of NAC in the treatment of COPD: Discordant in vitro and in vivo dose-effects: A review. Pulm Pharmac Ther. 2007;20:9–22. doi: 10.1016/j.pupt.2005.12.007. [DOI] [PubMed] [Google Scholar]
- Shi MM, Kugelman A, Iwamoto T, Tian L, Forman HJ. Quinone-induced oxidative stress elevates glutathione and induces γ-glutamylcysteine synthetase activity in rat lung epithelial L2 cells. J Biol Chem. 1994;269:26512–26517. [PubMed] [Google Scholar]
- Siedlinski M, Postma DS, van Diemen CC, Blokstra A, Smit HA, Boezen HM. Lung function loss, smoking, vitamin C intake, and polymorphisms of the glutamate-cysteine ligase genes. Am J Respir Crit Care Med. 2008;178:13–19. doi: 10.1164/rccm.200711-1749OC. [DOI] [PubMed] [Google Scholar]
- Singh A, Rangasamy T, Thimmulappa RK, Lee H, Osburn WO, Brigelius-Flohé R, Kensler TW, Yamamoto M, Biswal S. Glutathione peroxidase 2, the major cigarette smoke-inducible isoform of GPX in lungs, is regulated by Nrf2. Am J Respir Cell Mol Biol. 2006;35(6):639–50. doi: 10.1165/rcmb.2005-0325OC. Erratum in: Am. J. Respir. Cell Mol. Biol. 2007. 36, 642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JJ, Houston M, Anderson J. Increased levels of glutathione in bronchoalveolar lavage from patients with asthma. Am Rev Resp Dis. 1993;147:1461–1464. doi: 10.1164/ajrccm/147.6_Pt_1.1461. [DOI] [PubMed] [Google Scholar]
- Stey C, Steurer J, Bachmann S, Medici TC, Tramer MR. The effect of oral N-acetylcysteine in chronic bronchitis: a quantitative systematic review. Eur Respir J. 2000;16:253–262. doi: 10.1034/j.1399-3003.2000.16b12.x. [DOI] [PubMed] [Google Scholar]
- Suter PM, Domenighetti G, Schaller MD, Laverrière MC, Ritz R, Perret C. NAC enhances recovery from acute lung injury in man. Chest. 1994;105:190–194. doi: 10.1378/chest.105.1.190. [DOI] [PubMed] [Google Scholar]
- Suzuki M, Betsuyaku T, Ito Y, Nagai K, Nasuhara Y, Kaga K, Kondo S, Nishimura M. Downregulated NF-E2-related Factor 2 in pulmonary macrophages of aged smokers and COPD patients. Am J Respir Cell Mol Biol. 2008 doi: 10.1165/rcmb.2007-0424OC. In press. [DOI] [PubMed] [Google Scholar]
- Tanaka T, Uchiumi T, Kohno K, Tomonari A, Nishio K, Saijo N, Kondo T, Kuwano M. Glutathione homeostasis in human hepatic cells: Overexpression of γ-glutamylcysteine synthetase gene in cell lines resistant to buthionine sulfoximine: an inhibitor of glutathione synthesis. Biochem Biophys Res Commun. 1998;246:398–403. doi: 10.1006/bbrc.1998.8631. [DOI] [PubMed] [Google Scholar]
- Tian L, Shi MM, Forman HJ. Increased transcription of the regulatory subunit of γ-glutamylcysteine synthetase in rat lung epithelial L2 cells exposed to oxidative stress or glutathione depletion. Arch Biochem Biophys. 1997;342:126–133. doi: 10.1006/abbi.1997.9997. [DOI] [PubMed] [Google Scholar]
- Tirouvanziam R, Conrad CK, Bottiglieri T, Herzenberg LA, Moss RB, Herzenberg LA. High-dose oral N-acetylcysteine, a glutathione prodrug, modulates inflammation in cystic fibrosis. Proc Natl Acad Sci USA. 2006;103:4628–4633. doi: 10.1073/pnas.0511304103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomonari A, Nishio K, Kurokawa H, Arioka H, Ishida T, Fukumoto H, Fukuoka K, Nomoto T, Iwamoto Y, Heike Y, Itakura M, Saijo N. Identification of cis-acting DNA elements of the human γ-glutamylcysteine synthetase heavy subunit gene. Biochem Biophys Res Commun. 1997;232:522–527. doi: 10.1006/bbrc.1997.6319. [DOI] [PubMed] [Google Scholar]
- Trachootham D, Lu W, Ogasawara MA, Nilsa RD, Huang P. Redox Regulation of Cell Survival. Antioxid Redox Sign. 2008;10:1343–1374. doi: 10.1089/ars.2007.1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsikas D, Sandmann J, Ikic M, Fauler J, Stichtenoth DO, Frölich JC. Analysis of cysteine and N-acetylcysteine in human plasma by high-performance liquid chromatography at the basal state and after oral administration of N-acetylcysteine. J Chromatogr B Biomed Sci Appl. 1998;708:55–60. doi: 10.1016/s0378-4347(97)00670-1. [DOI] [PubMed] [Google Scholar]
- Tu Z, Anders MW. Up-regulation of glutamate-cysteine ligase gene expression by butylated hyoxytoluene is mediated by transcription factor AP-1. Biochem Biophys Res Commun. 1998;244:801–805. doi: 10.1006/bbrc.1998.8345. [DOI] [PubMed] [Google Scholar]
- Usatyuk PV, Parinandi NL, Natarajan V. Redox regulation of 4-hydroxy-2-nonenal-mediated endothelial barrier dysfunction by focal adhesion, adherens, and tight junction proteins. J Biol Chem. 2006;281:35554–35566. doi: 10.1074/jbc.M607305200. [DOI] [PubMed] [Google Scholar]
- Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- van der Toorn M, Smit-de Vries MP, Slebos DJ, de Bruin HG, Abello N, van Oosterhout AJ, Bischoff R, Kauffman HF. Cigarette smoke irreversibly modifies glutathione in airway epithelial cells. Am J Physiol Lung Cell Mol Physiol. 2007;293:L1156–L1162. doi: 10.1152/ajplung.00081.2007. [DOI] [PubMed] [Google Scholar]
- Van Schooten FJ, Besaratinia A, De Flora S, D’Agostini F, Izzotti A, Camoirano A, Balm AJ, Dallinga JW, Bast A, Haenen GR, Van’t Veer L, Baas P, Sakai H, Van Zandwijk N. Effects of oral administration of N-acetyl-L-cysteine: a multibiomarker study in smokers. Cancer Epidemiol Biomarkers Prev. 2002;11:167–175. [PubMed] [Google Scholar]
- Wakabayashi N, Dinkova-Kostova AT, Holtzclaw WD, Kang MI, Kobayashi A. stress by induction of the phase 2 response: fate of cysteines of the Keap1 sensor modified by inducers. Proc Natl Acad Sci USA. 101:2040–2045. doi: 10.1073/pnas.0307301101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Phelan SA, Manevich Y, Feinstein SI, Fisher AB. Transgenic mice overexpressing peroxiredoxin 6 show increased resistance to lung injury in hyperoxia. Am J Respir Cell Mol Biol. 2006;34:481–486. doi: 10.1165/rcmb.2005-0333OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White AC, Das SK, Fanburg BL. Reduction of glutathione is associated with growth restriction and enlargement of bovine pulmonary artery endothelial cells produced by transforming growth factor-β1. Am J Respir Cell Mol Biol. 1992;6:364–368. doi: 10.1165/ajrcmb/6.4.364. [DOI] [PubMed] [Google Scholar]
- Wild AC, Moinova HR, Mulcahy RT. Regulation of γ-glutamylcysteine synthetase subunit gene expression by the transcription factor Nrf2. J Biol Chem. 1999;274:33627–33636. doi: 10.1074/jbc.274.47.33627. [DOI] [PubMed] [Google Scholar]
- Wood ZA, Poole LB, Karplus PA. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science. 2003b;300:650–653. doi: 10.1126/science.1080405. [DOI] [PubMed] [Google Scholar]
- Wood ZA, Schroder E, Robin HJ, Poole LB. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem Sci. 2003a;28:32–40. doi: 10.1016/s0968-0004(02)00003-8. [DOI] [PubMed] [Google Scholar]
- Yang H, Magilnick N, Lee C, Kalmaz D, Ou X, Chan JY, Lu SC. Nrf1 and Nrf2 Regulate Rat Glutamate-Cysteine Ligase Catalytic Subunit Transcription Indirectly via NF-kappaB and AP-1. Mol Cell Biol. 2005;25:5933–5946. doi: 10.1128/MCB.25.14.5933-5946.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Wang J, Huang ZZ, Ou X, Lu SC. Cloning and characterization of the 5′-flanking region of the rat glutamate-cysteine ligase catalytic subunit. Biochem J. 2001;357:447–455. doi: 10.1042/0264-6021:3570447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, Zeng Y, Lee TD, Yang Y, Ou X, Chen L, Haque M, Rippe R, Lu SC. Role of AP-1 in the coordinate induction of rat glutamate-cysteine ligase and glutathione synthetase by tert-butylhydroquinone. J Biol Chem. 2002;277:35232–35239. doi: 10.1074/jbc.M203812200. [DOI] [PubMed] [Google Scholar]
- Yeh MY, Burnham EL, Moss M, Brown LA. Chronic alcoholism alters systemic and pulmonary glutathione redox status. Am J Respir Crit Care Med. 2007;176:270–276. doi: 10.1164/rccm.200611-1722OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang DD, Hannink M. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol. 2003;23:8137–8151. doi: 10.1128/MCB.23.22.8137-8151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Nomura A, Uchida Y, Iijima H, Sakamoto T, Iishii Y, Morishima Y, Mochizuki M, Masuyama K, Hirano K, Sekizawa K. Ebselen suppresses late airway responses and airway inflammation in guinea pigs. Free Radic Biol Med. 2002;32:454–464. doi: 10.1016/s0891-5849(01)00825-5. [DOI] [PubMed] [Google Scholar]
- Zheng JP, Kang J, Huang SG, Chen P, Yao WZ, Yang L, Bai CX, Wang CZ, Wang C, Chen BY, Shi Y, Liu CT, Chen P, Li Q, Wang ZS, Huang YJ, Lui ZY, Chen FP, Yuan JZ, Yuan BT, Qian HP, Zhi RC, Zhong NS. Effect of carbocisteine on acute exacerbation of chronic obstructive pulmonary disease (PEACE Study): a randomized placebo-controlled study. Lancet. 371:2013–2018. doi: 10.1016/S0140-6736(08)60869-7. [DOI] [PubMed] [Google Scholar]