

Abstract
We have evaluated a technology called Transcriptionally Active PCR (TAP) for high throughput identification and prioritization of novel target antigens from genomic sequence data using the Plasmodium parasite, the causative agent of malaria, as a model. First, we adapted the TAP technology for the highly AT-rich Plasmodium genome, using well-characterized P. falciparum and P. yoelii antigens and a small panel of uncharacterized open reading frames from the P. falciparum genome sequence database. We demonstrated that TAP fragments encoding six well-characterized P. falciparum antigens and five well-characterized P. yoelii antigens could be amplified in an equivalent manner from both plasmid DNA and genomic DNA templates, and that uncharacterized open reading frames could also be amplified from genomic DNA template. Second, we showed that the in vitro expression of the TAP fragments was equivalent or superior to that of supercoiled plasmid DNA encoding the same antigen. Third, we evaluated the in vivo immunogenicity of TAP fragments encoding a subset of the model P. falciparum and P. yoelii antigens. We found that antigen-specific antibody and cellular immune responses induced by the TAP fragments in mice were equivalent or superior to those induced by the corresponding plasmid DNA vaccines. Finally, we developed and demonstrated proof-of-principle for an in vitro humoral immunoscreening assay for down-selection of novel target antigens. These data support the potential of a TAP approach for rapid high throughput functional screening and identification of potential candidate vaccine antigens from genomic sequence data.
Keywords: Malaria, PCR, Plasmodium, Antigen, Genome, Screening
1. Introduction
Despite the completion of the genomic sequence of a large number of pathogens, there is not yet a means to efficiently analyze such data to identify which antigens among many thousands are appropriate targets for prophylactic or therapeutic interventions. To individually screen each gene for immunogenic potential by conventional methods would be unfeasible. The development of screening methods is also limited by the ability to validate the appropriate recall immune responses that can be induced against identified targets. Reliance on high throughput technologies is, therefore, critical to taking advantage of the wealth of genomic data for practical use. One solution to these problems is to render amplified PCR products transcriptionally active by adding promoter and terminator sequences to them. Previously described methods using “linear expression elements” (LEEs) [1] that could be transfected into cultured cells or injected into animals to express the coding sequence were not successful when applied to Plasmodium (J. Aguiar, unpublished). More recently, Liang et al. developed a technology called Transcriptionally Active PCR (TAP) that allowed the direct use of linear DNA fragments for in vitro expression, functional studies and in vivo immunization [2]. The entire process consists of only two PCR amplification steps, the first using gene-specific primers to amplify the gene of interest, and the second “nested” step using a mixture of DNA fragments to attach functional promoter and terminator sequences onto this fragment. The promoter element is from the human CMV immediate early gene plus a shortened and modified intron from the same gene (850 bp), and the transcription termination element is from SV40 (200 bp). After these two reactions are completed, the PCR product can be used directly. The technology speeds up the process from gene selection to protein expression by eliminating previously required molecular cloning, bacteria transformation and growth, and plasmid purification manipulations.
In proof-of-principle studies, Liang et al. demonstrated that TAP fragments and supercoiled DNA plasmids encoding the same antigens (e.g. hepatitis B surface antigen) induced comparable levels of in vitro expression and antibodies in mice [2]. TAP fragments encoding the target gene of interest could also be tagged with a well characterized, highly immunoreactive epitope derived from the influenza hemagglutinin (HA) protein (YPYDVPDYA) [3–8]. HA-tagged proteins resulting from transfection into cultured cells could be identified with anti-HA antibodies, facilitating purification of the expressed protein, subcellular localization or immunoprecipitation studies, and enabling rapid in vitro immune screening studies [2].
Herein, we have evaluated the TAP technology for functional screening of genomic sequence data in the context of malaria. Malaria is one of the world’s most important infectious diseases, responsible for 300–500 million cases and 1.5–2.7 million deaths annually [9]. Malaria is an attractive model for the development and validation of approaches to translate genomic information to vaccine development both because of the critical need for effective anti-malarial interventions, and because the Plasmodium parasite is a complex pathogen which requires the induction of multiple immune responses against multiple target antigens. The feasibility of a malaria vaccine is supported by existing examples of immunity both experimentally, in humans or animals immunized with radiation attenuated Plasmodium sporozoites, and in individuals living in holoendemic areas [10, 11]. However, the specific target antigens and epitopes of this protection are poorly characterized. The 23 Mb genome of P. falciparum is predicted to encode more than 5,300 proteins [12], each of which is a potential target of protective immune responses. The current generation of subunit vaccines against malaria is based on a single or few antigens and therefore might elicit too narrow a breadth of response and fail to provide optimal protection on genetically diverse backgrounds. Moreover, immune reactivity against those characterized antigens is relatively weak and seems unable to account for the protective effects observed with whole organism vaccination. Therefore, we are pursuing an alternative approach based on the presumption that mimicking the protection induced by whole organism vaccination may require a vaccine as complex as the whole organism [13, 14]. This approach requires the identification of an unprecedented number of parasite-derived target antigens, in order to reproduce the breadth and multiplicity of the whole organism-induced protective immunity. Antigens identified by high throughput screening techniques such as TAP will likely help to identify good candidate antigens for development of the next generation of subunit vaccines.
The purpose of this study was to validate the TAP technology as a means to identify and characterize potential immunodominant antigenic targets of protective immune responses from the P. falciparum genomic sequence data. Specifically, we attempted to: 1) Define and standardize PCR conditions to effectively amplify the AT rich P. falciparum genes as well as murine P. yoelii genes from both plasmid DNA and genomic templates; 2) Determine optimal conditions for in vitro transfection of a range of cell lines; 3) Use the above conditions to generate TAP constructs that incorporate a HA epitope tag (EpiTAP) as a tool for screening known and unknown P. falciparum genes against sera for recognition by individuals with a history of malaria exposure; and 4) Compare the DNA immunization properties of TAP fragments vs. supercoiled plasmids encoding well characterized P. yoelii and P. falciparum genes from both pre-erythrocytic and erythrocytic stages of the parasite life cycle in terms of immunogenicity and protection against sporozoite challenge.
2. Materials and Methods
2.1. Mice
Four to six week-old female BALB/c (H-2d haplotype), C57BL/6J (H-2b), or A/J (H-2a) mice were purchased from Jackson Laboratories (Bar Harbor, MA).
2.2. Parasites
P. yoelii (17XNL non-lethal strain, clone 1.1) was maintained by alternating passages of the parasites in Anopheles stephensi mosquitoes and CD1 mice. For parasite challenge studies, sporozoites were isolated from the thorax of infected mosquitoes by the discontinuous gradient technique [15] or the Ozaki technique [16].
2.3. Plasmids, DNA, and oligonucleotide primers
Genomic DNA was obtained from P. yoelii strain 17XNL and P. falciparum strain 3D7 using standard methods [17]. Plasmid DNA was obtained from Vical Inc. (San Diego, CA) or PureSyn Inc. (Malvern, PA) with the exception of the green fluorescent protein (GFP) control template which was provided in the TAP Express kit (Gene Therapy Systems, Inc. San Diego, CA). The following plasmids were used as templates for PCR amplifications: VR2507 (PyCSP), VR2533 (PyMSP1), VR2513 (PyHEP17), VR2514 (PySSP2), VR2510 (PfCSP), VR2535 (PfMSP1-42kD; 42kD C-terminal region), VR2525 (PfAMA1), VR2526 (PfEBA175-RII; Region II), VR2523 (PfExp1) and VR2519 (PfSSP2) [18, 19]. The following plasmids were used for in vivo immunization: VR2515 (PyHEP17) [20, 21], VR2516 (PyCSP) [22], VR2525 (PfAMA1) [23] and VR2526 (PfEBA175-RII) [24]. Plasmids for in vitro amplification were based on the VR1012 backbone [25], and for in vivo immunizations on the VR1020 backbone [26], which is essentially the same as VR1012 but also includes a tissue plasminogen activator leader sequence fused to the gene of interest. Unless otherwise specified the full Plasmodium spp. gene sequence was amplified. Plasmids and TAP PCR products were diluted to desired concentrations in sterile PBS for in vivo immunization studies. Oligonucleotide primers were synthesized by Genset Corp (La Jolla, CA) and sequences are listed in Tables 1 and 2.
Table 1.
TAP & EpiTAP Oligos for Known Antigens*
| PfCSP-5′ | 5′-ATGATGAGAAAATTAGCTA |
| PfCSP-3′ | 5′-TCAATTAAGGAACAAGAAGGATAAT |
|
| |
| PfSSP2-5′ | 5′-ATGAATCATCTTGGGAATGT |
| PfSSP2-3′ | 5′-TCATATTTAATTCCACTCGT |
|
| |
| PfEXP1-5′ | 5′-ATGAAAATCTTATCAGTATT |
| PfEXP1-3′ | 5′-TCATTAGTGTTCAGGGCCA |
|
| |
| PfAMA1-5′ | 5′-ATGAGAAAATTATACTGCG |
| PfAMA1-3′ | 5′-TCAATAGTATGGTTTTTCCATCAG |
|
| |
| PfEBA175-RII-5′ | 5′-ATGAAATGTAATATTAGTAT |
| PfEBA175-RII-3′ | 5′-TCACATACTTGAAAAAGCCTC |
|
| |
| PfMSP1-42kD-5′ | 5′-ATGGGAGAAGCAATATCTGTC |
| PfMSP1-42kD-3′ | 5′-TCAAATGAAACTGTATAATA |
|
| |
| PyHEP17-5′ | 5′-ATGAAAATCAATATAGCTTC |
| PyHEP17-3′ | 5′-GTTTTGAACATTTGGGGTG |
|
| |
| PyCSP-5′ | 5′-ATGAAGAAGTGTACCATTTTAG |
| PyCSP-3′ | 5′-TCAATTAAAGAATACTAATACTAAT |
|
| |
| PySSP2-5′ | 5′-ATGAAGCTCTTAGGAAATAG |
| PySSP2-3′ | 5′-TCATTAGTTCCAGTCATT |
|
| |
| PyMSP1-5′ | 5′-ATGAAGGTGATTGGACTTTT |
| PyMSP1-3′ | 5′-TCAATTATCGGGATGTGTC |
Plasmodium spp. sequence only.
See text for incorporated universal TAP or EpiTAP sequences.
Table 2.
EpiTAP Oligos for Unknown Proteins*
| PFC0450w-5′ | 5′-ATGAGCATGTTTCTTAATATAC |
| PFC0450w-3′ | 5′-TCAAATTTTATTTAATTCATATTG |
|
| |
| PFC0700c-5′ | 5′-ATGTCCAAATTCAACATTCTG |
| PFC0700c-3′ | 5′-TCAGCCCAAAGATTCCTCAA |
|
| |
| PFC0210c-5′ | 5′-ATGAGAAAATTAGCTATTTTATC |
| PFC0210c-3′ | 5′-TCAATTAAGGAACAAGAAGGATA |
|
| |
| PF14_0074-5′ | 5′-ATGGAAAACGAGTATGCAAC |
| PF14_0074-3′ | 5′-TCATATATTGCTTAATAGACAAC |
|
| |
| PF14_0751-5′ | 5′-ATGGATGGACCACTGGCC |
| PF14_0751-3′ | 5′-TCACCGTTCTAATTCTTTTAC |
|
| |
| PFL0800c-5′ | 5′-ATGAATGCCTTAAGAAGATTAC |
| PFL0800c-3′ | 5′-TCAATCATCTGATAAACTTTC |
|
| |
| PFI0165c-5′ | 5′-ATGGATTTGATGAATGATGAG |
| PFI0165c-3′ | 5′-TCATTTCACATGTAACGAATTG |
|
| |
| PFA0515w-5′ | 5′-ATGAAATGTACAAGTGTTAATATA |
| PFA0515w-3′ | 5′-TCATTTCATATGATTTTCTATAAA |
|
| |
| PFD0425w-5′ | 5′-ATGGAAGGCTTTGTTGCTTTG |
| PFD0425w-3′ | 5′-TCAATTTTGAACGTAAACACTATGAG |
Plasmodium spp. sequence only.
See text for incorporated universal TAP or EpiTAP sequences.
2.4. TAP & Epi-TAP Products
TAP and Epi-TAP PCR products were generated as instructed by the manufacturer (Gene Therapy Systems) using their TAP and Epi-TAP Express™ kits but modified for Plasmodium. In the first step, primers were designed incorporating either TAP or Epi-TAP universal sequences (Fig. 1). The 5′ primers begin with the universal sequence 5′-CTGCAGGCACCGTCGTCGACTTAACA-3′ or 5′-ACGATGTTCCGGATTACGCTAGCCTCCCAGTT-3′ for TAP and EpiTAP respectively, and continue with a start codon ATG either from the Plasmodium spp. gene of interest or added in-frame as part of the primer if only a certain domain is being amplified in accordance with the manufacturer’s instructions. The ATG is then followed by 15–20 bp of the 5′ sequence of the Plasmodium spp. gene of interest. The 3′ primers for both TAP and EpiTAP begin with the universal sequence 5′-CATCAATGTATCTTATCATGTCTGA-3′ and continue with a stop codon TCA either from the Plasmodium spp. gene of interest or added in-frame as part of the primer if only a certain domain is being amplified in accordance with the manufacturer’s instructions. The TCA is then followed by 15–20 bp’s of the anti-sense terminal sequence of the Plasmodium spp. gene. For a list of Plasmodium spp primer sequences see Tables 1 and 2. PCR amplifications were performed using the Clontech Advantage PCR kit (Palo Alto, CA) or the Expand Long Template PCR System kit (Roche Diagnostics, Indianapolis, IN) and a GeneAmp PCR 2400 System Thermocycler (Perkin Elmer, Wellesley, MA). For the first reaction, 30 pmol of each primer and 5–10 ng of plasmid DNA template or 50–100 ng of genomic DNA template were used, with the following cycling conditions: TAP: 94°C for 1 min, then 32 cycles of three-temperature PCR at 94°C for 30 sec, 60°C for 1 min, 68°C for 2 min, followed by a final extension time at 68°C for 10 min; EpiTAP: 94°C for 3 min, then 32 cycles of three-temperature PCR at 94°C for 30 sec, 60°C for 1 min, 68°C for 3 min, followed by a final extension time at 68°C for 10 min. Products were run on a 1.0% agarose gel to assess quality and quantity. The second TAP and EpiTAP reactions used a 1:20 dilution of the first step product, and proprietary CMV promoter and SV40 terminator fragments as templates. The promoter and terminator fragments overlap with the first step PCR product at, respectively, 5′ and 3′ end of the universal region and, as the result of the second step PCR, the promoter, the gene of interest and the terminator are linked in the correct order to form a transcriptionally active PCR (TAP) fragment. As stated previously, the addition of the CMV promoter and SV40 terminator fragments increase the size of the end product by approximately 1000 bp from the first step. This was performed under the following PCR conditions: TAP: 94°C for 1 min, then 32 cycles of 94°C for 30 sec, 60°C for 2 min, 68°C for 3 min, followed by a final extension at 68°C for 10 min; EpiTAP: 94°C for 1 min, then 32 cycles of 94°C for 30 sec, 60°C for 1.5 min, 68°C for 3 min, followed by a final extension at 68°C for 10 min. Quality and quantity were again assessed by gel electrophoresis, and products were purified using a PCR purification kit (Qiagen, Inc., Valencia, CA) to remove primers and reagents. The PCR products were directly used for in vitro transfection, or ethanol-precipitated and resuspended in saline to a final concentration of 1 mg/ml for in vivo immunizations.
Figure 1. Schematic of TAP/EpiTAP PCR Reaction.
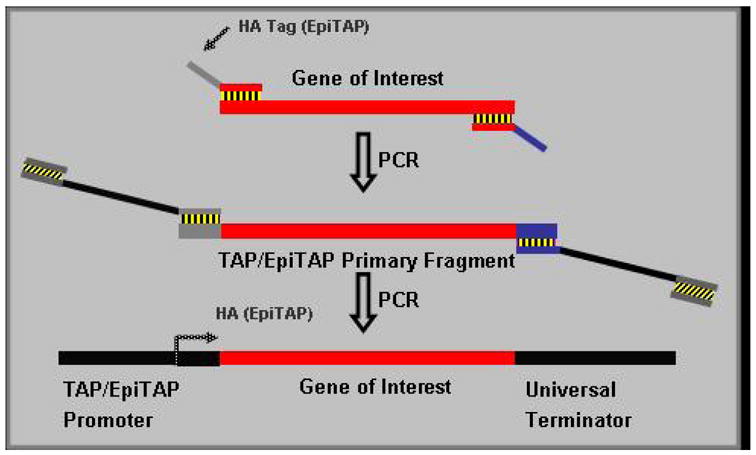
TAP and Epi-TAP PCR products were generated as instructed by the manufacturer (Gene Therapy Systems) using their TAP and Epi-TAP Express™ kits but modified for Plasmodium using a two step PCR reaction. In the first step, primers were designed incorporating a universal sequence for either a TAP or Epi-TAP amplification with the Epi-TAP primer including an HA epitope tag. The 5′ primers begin with one of these universal sequences and continue with a start codon ATG which is followed by 15–20 bp of the 5′ sequence of the Plasmodium spp. gene of interest. The 3′ primers for both TAP and EpiTAP begin with the same universal sequence and continue with a stop codon TCA followed by 15–20 bp’s of the anti-sense terminal sequence of the Plasmodium sp. gene. The second TAP and EpiTAP reactions used a 1:20 dilution of the first step product, and proprietary CMV promoter and SV40 terminator fragments as templates. The promoter and terminator fragments overlap with the first step PCR product at, respectively, 5′ and 3′ end of the universal region and, as the result of the second step PCR, the promoter, the gene of interest and the terminator are linked in the correct order to form a transcriptionally active PCR (TAP) fragment.
2.5. Synthetic Peptides
Peptides representing defined P. yoelii CD8+ and/or CD4+ T cell epitopes (see Table S1, Supplemental Data) were synthesized commercially (Research Genetics, Huntsville, AL; or AnaSpec Inc, San Jose, CA) at > 95% purity for use in cellular immunity assays
2.6. In vitro Transfection
Transient transfection assays were performed using COS-7 (ATCC #CRL-1651), CHO-K1 (ATCC #CRL-9618) and UM-449 melonoma (Vical Inc., San Diego CA) cell lines. Adherent cells were plated on a 96-well plate at ~10,000 cells/well in 100 μl of complete media [RPMI-1640 with 20% Fetal Calf Serum (FCS), 2 mM L-glutamine, 50 U/ml Penicillin and Streptomycin], and left overnight in an atmosphere of 37°C and 5% CO2, to achieve approximately 50–70% confluency by the following day. TAP and EpiTAP transfections were performed using GenePORTER-2 transfection reagent (Gene Therapy Systems) as described by the manufacturer. DNA was incubated with reagents in serum-free OptiMEM media (Gibco/Invitrogen, Carlsbad CA) according to the kit protocol, growth media was aspirated from the cell culture, and 100 μl/well of the DNA/Geneporter mixture was then plated on top of the adherent cells. Cultures were incubated for 4 h at 37°C, after which an equal volume of serum-containing media (RPMI 20% FCS) was added to each well. Plates were incubated for an additional 24–48 h. Transient expression of the transfected gene was confirmed by immunostaining using an antigen-specific monoclonal antibody (mAb) as the primary antibody and FITC-conjugated goat anti-mouse IgG as the secondary antibody (Kirkegaard and Perry, Gaithersburg, MD).
2.7. In vitro humoral immunoscreening assay
HA-EpiTAP fragments encoding target open reading frames (ORFs) were used as capture antigens for ELISA assays. HA-EpiTAP fragments encoding each of the ORFs were individually amplified and transfected into CHO-K1 cells using GenePORTER-2 as described above, in a 96-well format in duplicate sets. After 48 h, in one set of transfections the supernatant was removed, and in the other set the cells were lysed with 200 μl of MilliQ purified water (Millipore, Billerica, MA). The supernatant and lysate were transferred to respective sets of 96-well plates that were prepared as follows: the wells were pre-coated with anti-HA antibody (Covance, mAb, HA.11; Cat# MMS-101P) at 1.0 μg/ml in PBS at 50 μl/well, washed 4 × with 200 μl/ml wash buffer (PBS with 0.05% Tween 20), and blocked with a 5% milk/PBS buffer for 2 h at room temperature followed by another 4 washes. The lysates/supernatants were added at a volume of 50 μl/well, incubated at 4°C overnight to allow for the HA epitope tagged proteins to bind to the capturing anti-HA antibodies, and then washed 4 times. Pooled sera from individuals resident in hyperendemic area of Kenya (hyper-immune sera; [27] or malaria-naïve control sera, diluted 1:100 in the 5% milk/PBS solution, were applied to the capture antigen and incubated for 2 h at room temperature followed by another 4 washes. Antigen-bound antibody was detected with conjugated anti-human antibody by adding 50 μl/well of Peroxidase Labeled Goat Anti-Human IgG (KPL, Gaithersburg, MD) diluted 1:4000 in 5% milk/PBS solution and incubating for 1 h at room temperature. After a final 4 washes, 100 μl/well of ABTS Substrate Solution A and B (KPL) in equal amounts were added. The reaction was allowed to develop for 20 min at room temperature and then stopped with 100 μl/well of ABTS Peroxidase Stop Solution (KPL). Responses were read on a Spectra MAX 190 (Molecular Devices Corp., Sunnyvale CA) and considered positive if the corrected optical density (OD) 405 nm for consistency against malaria-immune sera was at least 0.2 OD units above the mean + 3 standard deviation (SD) of the malaria-naive control sera. During assay development, a fluorescein-labeled anti-HA mouse mAb (Boehringer Mannheim) was used to probe HA-Epi-TAP transfected, fixed and permeabilized cells to verify the effectiveness of the transfection procedure. Six well-characterized P. falciparum antigens (PfCSP, PfSSP2, PfExp1, PfMSP1-42kD, PfAMA1 or PfEBA175-RII) were used to develop the assay, and a panel of six novel proteins (PFC0450w, PFC0700c, PFC0210c, PF14_0074, PF14_0751, PFL0800c) identified from the P. falciparum genomic sequence database [12, 28] were screened using the refined assay.
2.8. In vivo immunizations
In the first series of studies, BALB/c mice (n=5/group) were immunized 3 times at 3 wk intervals intramuscularly in each tibialis anterior muscle, with 50 μg of plasmid DNA or TAP fragments encoding the pre-erythrocytic stage antigen PyHEP17 in a total volume of 100 μl split between the two legs. Alternatively, C57BL/6J mice (n=5/group) were immunized 3 times at 3 wk intervals intradermally at the base of the tail with 50 μg of plasmid DNA or TAP fragments encoding erythrocytic stage antigens PfEBA175-RII or PfAMA1 in a total volume of 50 μl split between 3 sites. In the second series of studies, BALB/c mice (n=4/group) were immunized with 100 μg or 50 μg of PyCSP plasmid or 50 μg PyCSP-TAP, and A/J mice (n=4/group) were immunized with 100 μg, 50 μg, 20 μg or 10 μg of PyHEP17 plasmid or 50 μg, 20 μg or 10 μg of PyHep17-TAP. Non-immunized mice and mice immunized with empty vector plasmid VR1020 (100 μg/mouse) served as controls. Blood was collected 10 days post each immunization for evaluation of antibody responses. Splenocytes were harvested at 14 days post 3rd immunization for evaluation of T cell responses. For evaluation of protection, mice were challenged by tail-vein injection with 50,000 infectious sporozoites at 2 wks after the third immunization and the protective activity determined 42 hours later by previously reported methods using TaqMan™ RT-PCR [29].
2.9. Indirect fluorescent antibody test (IFAT)
For sporozoite IFAT, P. yoelii sporozoites (17XNL strain) or P. falciparum sporozoites (3D7 strain) were isolated from the salivary glands of An. Stephensi mosquitoes, using standard gradient isolation procedures, counted, and diluted to 10 μl aliquots of approximately 1000 sporozoites each in a standard IFAT slide depression chamber (Cel-Line/Erie Scientific Co.). For blood stage IFAT, parasitized erythrocytes collected from parasitemic mice (P. yoelii) or in vitro culture (P. falciparum) were enumerated and diluted to deliver approximately 1000 parasitized erythrocytes per 10 μl. Air-dried slides were stored at −70°C until immediately prior to use. Mouse sera (pooled, n=5/group) were diluted two-fold from 1:20 to 1:40, in 2% BSA/PBS and assayed, together with positive control sera (anti-PyCSP mAb NYS1 for sporozoite IFAT; anti-PyHEP17 mAb NYLS3 and anti-PfEBA175 or anti-PfAMA1 polyclonal rabbit sera for blood stage IFAT) and negative controls (PBS, naïve mouse sera). Aliquots of 10 μl each serum dilution were added per well, and antibody recognition of P. yoelii or P. falciparum sporozoite or blood stage parasites was evaluated as described previously [30]. IFAT results are expressed as endpoint serum dilution at which fluorescence was scored as positive.
2.10. Enzyme-linked immunosorbent assay (ELISA)
Antibodies against recombinant PyCSP protein or a PyHEP17 synthetic peptide were assessed as previously described [31]. Results are reported as OD 0.5 units which is the reciprocal of the serum dilution at which the mean OD reading is 0.5.
2.11. IFN-γ ELIspot
The number of epitope-specific IFN-γ-secreting cells was determined by ex vivo ELIspot. Multiscreen MAHA-S 4510 plates (Millipore, Bedford, MA) were coated with 75 μl of PBS containing 10 μg/ml of rat anti-murine IFN-γ mAb (clone R4-6A2, BD Pharmingen, San Diego, CA) and incubated overnight at room temperature. Plates were washed twice with 200 μl/well complete media (10% FCS in RPMI-1640 with 25 mM Hepes, 2 mM L-glutamine, 50 U/ml Penicillin and Streptomycin), and blocked with 200 μl/well of complete medium for 3–4 h at 37°C in an atmosphere of 5% CO2. After blocking, plates were washed once with complete media before the addition of target and effector cells. Target cells (A20.2J; H-2d) (ATCC clone HB-98) were used at a concentration of 5×106 cells/ml in complete media. Cells were pulsed with or without peptide representing defined T cell epitopes at a concentration of 10 μg/ml for 1 h at 37°C in a 5% CO2 incubator with gentle re-suspension every 15 min, then irradiated with 16,666 Rads using a 137Cs gamma or 60Co irradiator. Target cells were then washed once with complete media, re-suspended at 1.5×106 cells/ml and 20 μg/ml peptide added back (for final concentration of 10 μg/ml). For effectors, freshly harvested mouse splenocytes were washed and re-suspended to a final concentration of either 5×106 or 2.5×106 cells/ml in complete media. Effector splenocytes at 100 μl/well were plated in quadruplicates into plates pre-coated with anti-IFN-γ mAb. Then, 100 μl of the appropriate target cell/peptide combination was added to each well, and plates incubated for 36 h at 37°C and 5% CO2. Plates were then washed 3 times with PBS-T (PBS 0.05% Tween20), and 100 μl/well of biotinylated anti-IFN-γ secondary antibody (XMG1.2, Pharmingen, San Diego, CA) at 2 μg/ml in PBS-T was added to each well. Plates were incubated at room temperature for 3 h. Plates were washed 6 times with PBS-T and 100 μl of Streptavidin-Horse Radish Peroxidase conjugate (KPL, Gaithersburg, MD) at 1:800 dilution in PBS-T was added to each well. After 1 h incubation at room temperature, plates were washed 6 times with PBS-T followed by 3 times with PBS alone, and developed with DAB reagent (KPL) according to manufacturer’s instructions. After 10–15 min, the reaction was stopped by extensive washing with H2O and plates were air-dried and stored in the dark. Spots were counted with an automated KS ELIspot reader (Carl Zeiss Vision, Germany). Results were expressed as IFN-γ spot forming cells per 1×106 splenocytes (SFC/million splenocytes). Responses were considered positive if the response to test peptide (PyCSP) was significantly different (p<0.05) as compared with the response to no peptide and if the stimulation index (S.I. = response with test peptide/response with control peptide) was greater than 2.0.
2.12. Intracellular cytokine staining (ICS) and FACS analysis
A20.2J target cells were pulsed with or without peptide (10 μg/ml) for 1 h at 37°C in 5% CO2 and irradiated, as described above. Splenocytes (5×106 cells/ml) from immunized or naive mice were aliquoted in duplicate in 96-well round-bottom plates at 100 μl/well. Then, 100 μl of each A20.2J target cells/peptide combination (1.5 × 106 cells/ml) was added to the appropriate wells. Brefeldin-A (GolgiPlug™, Pharmingen, San Diego, CA) was added to the culture at a final concentration of 10 μg/ml. Cells were incubated for 16 h at 37°C in 5% CO2. Following culture, plates were spun at 1,200 rpm for 5 min, cells were washed once with cold PBS, the supernatant was discarded, and the cell pellet was resuspended in the remaining liquid by gentle vortexing. Cell surface markers were stained with a combination of 0.3–0.5 μl/well of anti-CD8-APC, anti-CD4-PERCP, or anti-CD62L-FITC Abs (Pharmingen) in a final volume of 100 μl in FACS wash on ice in the dark for 20 min. After the surface staining, cells were washed twice with FACS wash and permeabilized in 100 μl of Cytofix/Cytoperm buffer (Pharmingen) for 20 min on ice in the dark according to manufacturer’s directions. Cells were then washed with 100 μl of Perm/Wash buffer and stained intracellularly with 0.5 μl/well of PE-conjugated mAbs to IFN-γ, IL-2, or TNF-α (Pharmingen) in Perm/Wash buffer in a final volume of 100 μl on ice in the dark for 20 min. Cells were then washed twice with Perm/Wash, once with FACS wash, resuspended in 100 μl of FACS wash, and stored at 4°C prior to analysis. Samples were acquired by four-color fluorescent activated cell sorting using the FACSCaliburTM (Becton Dickinson Immunocytometry Systems, San Jose, CA). For each sample, approximately 100,000 events were acquired. Cytokine expression on gated CD8+ or CD4+ T cells was analyzed using the CellQuest software (Becton Dickinson) and expressed as % of positive cells. Negative control samples, consisting of cells from naïve mice incubated with peptide-pulsed A20.2J cells or splenocytes from immunized mice incubated with A20.2J cells not pulsed with peptide, consistently showed < 0.2% background.
2.13. Cytotoxic T Lymphocyte (CTL) Assay
Standard restimulated CTL assays were carried out as described previously [32]. For effector cells, splenocytes from immunized mice were incubated in the presence of peptide PyCSP280-295 containing the minimal 9-mer CTL epitope (residues 280-288) (2.5 μM), or a pool of PyHEP17 15-mer peptides PyHEP61-75, PyHEP66-80, and PyHEP71-85 (10 μg/ml each) (Table S1). For target cells, MHC-matched (H-2d) P815 mastocytoma cells (ATCC clone TIB 64) were pulsed with the PyCSP280-288 or with the same PyHEP17 peptides used for the stimulation, or with a pool of PyHEP17 9-mer peptides (PyHEP61-69, PyHEP70-78, PyHEP76-84, and PyHEP84-92; Table S1), or without peptide. The percent lysis was determined according to the formula (experimental release − medium control release)/(maximum Triton-X100 release − medium control release) × 100. The percent specific lysis was calculated by subtracting the percent lysis of targets without peptide from the percent lysis of targets with specific peptide. In all experiments, spontaneous release (minimum release/maximum release) was < 30%.
2.14. Lymphocyte Proliferation Assay (LPA)
Splenocytes from immunized mice were cultured in quadruplicate at a concentration of 1.25 to 5×105 cells in 200 μl of complete medium in a flat bottom 96-well tissue culture plate in the presence or absence of synthetic peptides representing defined CD4+ T cell epitopes (10 μg/ml), or with mitogen (Con A at 5 μg/ml) for 5 days. Wells were then pulsed with 1.0 μCi 3H-methyl thymidine (Dupont NEN) overnight, and uptake assessed by liquid scintillation spectroscopy (Beckman LS6800). Results were expressed as a stimulation index (S.I.) = (c.p.m. sample/c.p.m. control without peptide). The response to a peptide was considered positive if the S.I. was greater than 2.0.
2.15. Data Analysis and Statistical Analysis
For each quadruplicate (ELISA and ELIspot assays), outliers were rejected if any single quadruplicate value contributed more than 50% SD of the quadruplicate and if its value was 3-fold greater or less than the average of the remaining 3 values. After removing outliers, the mean OD (ELISA) or SFC (ELIspot) obtained in negative control wells (malaria-naïve sera or PBS) was subtracted from the value of each well. Negative counts were converted to zero. Antibody levels were log-transformed before analysis. Immunological outcomes expressed as means (antibody OD values, number of FACs events, concentration of cytokines as determined by reference to positive standard controls, % specific lysis value, LPA counts per minute) were compared between groups by the Student’s t-test (unpaired) for independent samples. Those outcomes expressed as proportions (prevalence of antibodies, frequency of cytokine responses) were compared between groups using the Chi-square test (uncorrected) or Fisher’s exact test (two-tailed) (if the expected cell value is less than five). Assessment of statistical significance was performed by Student’s unpaired t-test on log transformed data. In all analyses, P < 0.05 was considered statistically significant.
3. Results
3.1. TAP and Epi-TAP amplification of Plasmodium antigens
Six well-characterized P. falciparum proteins (pre-erythrocytic stage PfCSP, PfSSP2, PfExp1, and erythrocytic stage PfAMA1, PfMSP1-42kD, PfEBA175-RII) were evaluated to validate the TAP technology in the context of the highly AT rich P. falciparum parasite and with the end goal of identifying promising antigens for next generation malaria vaccines (Table 3). Four P. yoelii genes (representing the respective orthologues PyCSP, PyHEP17, PySSP2, and PyMSP1) were evaluated as an example of a second Plasmodium parasite species with a different AT ratio, and to also allow for direct assessment of protection in the P. yoelii rodent model. Standard PCR protocols recommended by the manufacturer (Gene Therapy Systems) were not effective for generating TAP and EpiTAP fragments from Plasmodium genes. In order to effectively amplify the highly AT rich P. falciparum genes as well as the P. yoelii genes from both plasmid and genomic DNA templates, therefore, we systematically modified the standard parameters provided by the manufacturer to identify a narrow range of PCR conditions for each reaction step.
Table 3.
Known Plasmodium spp. Antigen TAP PCR Reaction Details
| Plasmodium spp. gene | Size (bp)* | Region Amplified | Yield^ |
|---|---|---|---|
| PyCSP | 1173 | Full length | + |
| PyMSP1 | 1386 | Full length | ++ |
| PyHEP17 | 665 | Full length | +++ |
| PySSP2 | 2478 | Full length | + |
| PfCSP | 1194 | Full length | ++ |
| PfMSP1-42kD | 1191 | 42kD C-terminal region | +++ |
| PfAMA1 | 1869 | Full length | +++ |
| PfEBA175-RII | 1851 | Region II | ++ |
| PfEXP1 | 489 | Full length | + |
| PfSSP2 | 1728 | Full length | +/− |
Size is for malaria antigen alone without promoter/terminator sequences.
For final product; Scale: −, +/−, +, ++, +++
Unless otherwise specified the following conditions were used: First Reaction: 94°C for 1 min, then 32 cycles of three-temperature PCR at 94°C for 30 sec, 60°C for 1 min, 68°C for 2 min, followed by a final extension time at 68°C for 10 min. Second Reaction: 94°C for 1 min, then 32 cycles of 94°C for 30 sec, 60°C for 2 min, 68°C for 3 min, followed by a final extension at 68°C for 10 min.
Annealing and extension temperatures and times were systematically varied and evaluated, including two-temperature conditions for the first TAP reaction (in contrast to the manufacturer’s protocol where annealing and extension steps are performed at the same temperature) and three temperature conditions as per standard PCR protocol (where annealing and extension steps are performed at different temperatures). Annealing temperatures of 60°C minimized smearing seen at lower temperatures. Yield of PCR product was less at higher temperatures. In addition, much longer annealing times than prescribed in the kit protocol were needed to allow for proper amplification. The optimized protocol as described in the Experimental Procedures section could amplify all evaluated Plasmodium candidate vaccine antigens with either plasmid or genomic DNA templates, except for PfLSA1 (Fig. 2A and Fig. 2B), representing a 91% success rate (10/11 known Plasmodium genes amplified successfully). It should be noted that 77% of the PfLSA1 gene is known to be repetitive [33], likely explaining our failure to amplify this gene. The final products contained DNA fragments of the gene of interest flanked by promoter and terminator, which were ~ 1,050 bp larger than primary DNA fragments in agreement with the expected size following addition of the promoter (850 bp) and terminator (200 bp) to the target gene of interest.
Figure 2. A) Transcriptionally active PCR (TAP) first reaction.

94°C × 1 min then, 32 cycles of 94°C × 30 s, 60°C × 1 min and 68°C × 2 min, followed by 68°C × 10 min. Samples were run on a 1% agarose gel. For P. yoelii and P. falciparum gels, 1st and 2nd lanes for each construct represent reactions with plasmid and genomic DNA templates respectively. Constructs in P. yoelii by gel lane, 1&2: PyCSP 3&4: PyMSP1, 5&6: PyHEP17 (size difference due to amplification of exon with genomic template), 7&8: PySSP2, 9: positive control (GFP Template). Constructs in P. falciparum gel by lane, 1&2: PfCSP, 3&4: PfMSP1-42kD (42kD C-terminal region), 5&6: PfAMA1, 7&8: PfEBA175-RII (Region II), 9&10: PfExp1 (size difference due to amplification of exon with genomic template), 11&12: PfSSP2. 17X genomic for P. yoelii and 3D7 genomic for P. falciparum. B) Transcriptionally active PCR (TAP) second reaction: 94°C × 1 min then, 32 cycles of 94°C × 30 s, 60°C × 2 min and 68°C × 3 min, followed by 68°C × 10 min. Samples were run on a 1% agarose gel. For P. yoelii and P. falciparum gels, 1st and 2nd lanes for each construct are derived from dilutions of 1st TAP reactions used for template (plasmid and genomic origin, respectively). Constructs in P. yoelii gel by lane, 1&2: PyCSP, 3&4: PyMSP1, 5&6: PyHEP17 (size difference due to amplification of exon with genomic template), 7&8: PySSP2, 9&10: positive control (GFP Template) & negative control (no template). Constructs in P. falciparum gel by lane, 1&2: PfCSP, 3&4: PfMSP1-42kD, 5&6: PfAMA1, 7&8: PfEBA175-RII, 9&10: PfExp1 (size difference due to amplification of exon with genomic template), 11&12: PfSSP2.
Overall we achieved similar results with EpiTAP fragments, but some minor modifications were necessary to further optimize yields with P. falciparum antigens (Table 4).
Table 4.
Known Plasmodium spp. Antigen Epi-TAP PCR Product Details
| Plasmodium spp. gene | Size (bp)* | Region Amplified | Yield^ | Comments |
|---|---|---|---|---|
| PyCSP | 1173 | Full length | + | |
| PyMSP1 | 1386 | Full length | +++ | |
| PyHEP17 | 665 | Full length | +++ | |
| PySSP2 | 2478 | Full length | + | |
| PfCSP | 1194 | Full length | + | |
| PfMSP1-42kD | 1191 | 42kD C-terminal region | +++ | |
| PfAMA1 | 1869 | Full length | +++ | 1st reaction annealing done at 60°C for 1.5 min. and the final extension time done at 68°C for 3 min. |
| PfEBA175-RII | 1851 | Region II; FVO | +++ | 1st reaction annealing done at 54°C for 1.5 min. and the final extension time done at 68°C for 3 min. |
| PfEXP1 | 489 | Full length | ++ | |
| PfSSP2 | 1728 | Full length | +++ | 1st reaction annealing done at 60°C for 1.5 min. and the final extension time done at 68°C for 3 min. |
Size is for malaria antigen alone without promoter/terminator sequences
For final product; Scale: −, +/−, +, ++, +++
Unless otherwise specified the following conditions were used: First Reaction: 94°C for 3 min, then 32 cycles of three-temperature PCR at 94°C for 30 sec, 60°C for 1 min, 68°C for 3 min, followed by a final extension time at 68°C for 10 min. Second Reaction: 94°C for 1 min, then 32 cycles of 94°C for 30 sec, 60°C for 1.5 min, 68°C for 3 min, followed by a final extension at 68°C for 10 min.
3.2. Adaptation of standard parameters for Epi-TAP to genome-wide screening of P. falciparum
After establishing the success of the TAP/Epi-TAP technology for well-characterized Plasmodium genes, we applied the Epi-TAP technology to amplify unknown antigens derived from the P. falciparum genome. A panel of 9 genes (PFL0800c, PFI0165c, PFA0515w, PFD0425w, PFC0210c, PFC0450w, PF14_0751, PF14_0074, PFC0700c) was selected from the genomic sequence database for TAP amplification. These genes represented a subset of the 27 putative proteins assayed for recognition by human peripheral blood mononuclear cells derived from individuals experimentally immunized with radiation attenuated P. falciparum sporozoites [28]. The initial selection of these antigens was based on data derived from MudPIT of P. falciparum sporozoite preparations [34]. For those genes that were multi-exon, oligos were designed to amplify the largest exon.
Conditions for EpiTAP-1 and EpiTAP-2 amplification were systematically varied in an attempt to identify a single amplification condition that resulted in successful amplification of Epi-TAP products from genomic DNA template for each of these unknowns. However, some modifications were necessary in order to amplify some of the antigens (Table 5). It was also noted that the efficiency of TAP amplification was influenced by the size of the ORF. Larger ORFs (>3000 bp) were problematic, and strategies were employed to optimize their amplification by using a high fidelity PCR kit and modifying reaction conditions as outlined in Table 5. This is not unusual for standard PCR amplification with longer templates [35, 36] so this effect is presumed to be due to the PCR process per se rather than something specific for the TAP approach. In summary, with minor modifications to the PCR conditions, we were able to generate TAP fragments for each of the nine ORFs representing potentially novel P. falciparum proteins.
Table 5.
Unknown Plasmodium falciparum Protein EpiTAP PCR Product Details
| Plasmodium spp. gene | Size (bp)* | Region Amplified | Yield^ | Comments |
|---|---|---|---|---|
| PFC0450w | 327 | Full length | ++ | 1st reaction annealing done at 54°C for 45 sec. |
| PFC0700c | 924 | Full length | ++ | |
| PFC0210c | 1194 | Full length | ++ | 1st reaction annealing done at 60°C for 1.5 min. 2nd reaction ramp up done at 94°C for 3 min. |
| PF14_0074 | 1185 | Full length | ++ | |
| PF14_0751 | 564 | Full length | ++ | 1st reaction annealing done at 54°C for 45 sec. |
| PFL0800c | 624 | Full length | +++ | 1st reaction annealing done at 54°C for 1.5 min. |
| PFI0165c | 7212 | Full length | +++ | 1st Reaction: 92°C for 2 min, then 35 cycles at 92°C for 10 sec, 40°C for 30sec, 60°C for 12min, followed by a final extension time at 60°C for 10 min. |
| PFA0515w | 4413 | Full length | ++ | |
| PFD0425w | 2952 | Single exon | ++ | 1st Reaction: 92°C for 2 min, then 35 cycles at 92°C for 10 sec, 45°C for 30sec, 60°C for 12min, followed by a final extension time at 60°C for 10 min. |
Size is for malaria antigen alone without promoter/terminator sequences
For final product; Scale: −, +/−, +, ++, +++
Unless otherwise specified the following conditions were used: First Reaction: 94°C for 3 min, then 32 cycles of three-temperature PCR at 94°C for 30 sec, 60°C for 1 min, 68°C for 3 min, followed by a final extension time at 68°C for 10 min. Second Reaction: 94°C for 1 min, then 32 cycles of 94°C for 30 sec, 60°C for 1.5 min, 68°C for 3 min, followed by a final extension at 68°C for 10 min.
3.3. In vitro transfection of TAP and Epi-TAP fragments
We determined optimal conditions for in vitro transfection in a range of cell lines, including UM-449, CHO-K1 and COS-7 cells, in 96-well format. Using a systematic approach, both GenePORTER-1 and GenePORTER-2 (Gene Therapy Systems) were evaluated with varying amounts of DNA and reagents within the ranges recommended by the manufacturer. Using ~5 μl of reagent per μg of DNA together with 25 μl of DNA Diluent A per μg of DNA, GenePORTER-2 demonstrated the highest transfection efficiency with ranges up to 50–70%. After comparisons of in vitro transfection activities of supercoiled plasmids and TAP/EpiTAP expression fragments, the CHO-K1 cell line was selected for subsequent assays, based on the percentage and equivalency of transfected cells and the magnitude/intensity of antigen expression (data not presented). These findings were consistent with earlier reports [2]. Despite systematically varying the conditions, however, fragments > 2 Kb proved difficult to successfully transfect, therefore reducing the panel of amplified ORFs that could be use in further cellular assays from nine to the six chosen below.
3.4. In vitro humoral immunoscreening assay for high throughput screening of antibody recognition
We developed an assay in which the Epi-TAP derived malarial proteins expressed in CHO-K1 cell lysates or supernatants bind to a 96-well plate pre-coated with anti-HA mAb, and are then over-laid with malaria-immune sera (or appropriate control sera) to measure antibody binding via ELISA. The major difficulty encountered was high background activity, but this was greatly reduced by: (i) coating plates with 1 μg/ml of Ha.11 capture antibody; (ii) using a 5% milk/PBS blocking solution instead of BSA; (iii) diluting sera and HRP-conjugated secondary antibody in a 2% milk/PBS solution; and (iv) allowing lysates/supernatants to incubate overnight at 4°C. These strategies greatly reduced but did not completely eliminate background reactivity, presumed to be a consequence of the influenza-derived HA tag since most people are exposed to and have immune responses against this antigen. This may perhaps be overcome in future studies by incorporating the use of an alternate epitope tag derived from a pathogen to which most people are not exposed.
Next, we demonstrated proof-of-principle for this assay, by using HA-tagged Epi-TAP fragments of well-characterized malaria antigens PfCSP, PfSSP2, PfExp1, PfMSP1-42kD, PfAMA1 or PfEBA175-RII as capture antigens. We demonstrated that the EpiTAP fragments encoding these antigens were recognized by malaria immune serum as compared with negative control sera (Table 6). We subsequently applied this assay to the 6 uncharacterized ORFs, and were able to reproducibly detect antibodies against these newly described P. falciparum antigens (Table 6). We further demonstrated antigen-dependent, differential recognition amongst cellular versus secreted fraction of transfected cell cultures. Cellular fractions (representing lysates of transfected cells) from 6/6 unknowns and supernatant fractions from 2/6 unknowns were recognized by malaria-immune sera, but not by malaria- naïve control sera (Table 6). Assays were reproducible with positive responses with unknowns being detected in as many as 5/8 assays. Thus, this novel humoral immune screening assay allows for the identification of those antigens recognized by malaria-immune sera but not malaria-naïve sera.
Table 6.
Humoral immunoscreening
| Antigen # | Locus | Cell fraction Positive by IS assay | Spnt fraction Positive by IS assay |
|---|---|---|---|
| CSP | + (1/3) | - | |
| SSP2 | +/− (2/2) | - | |
| EXP1 | + (1/4) | - | |
| MSP1-42kD | +/− (1/2) | - | |
| AMA1 | + (3/6) | - | |
| EBA175-RII | +/− (1/3) | - | |
| 2 | PFL0800c | + (2/3) | - |
| 4 | PFC0210c | + (1/1) | - |
| 11 | PFC0450w | + (1/4) | + (1/4) |
| 12 | PF14_0074 | + (1/1) | - |
| 13 | PFC0700c | + (3/5) | + (1/5) |
| 25 | PF14_0751 | + (5/8) | - |
Cell lysates or supernatants from CHO-K1 transfected with HA-EpiTAP fragments of well-characterized P. falciparum antigens (PfCSP, PfSSP2, PfExp1, PfMSP1-42kD, PfAMA1 or PfEBA175-RII) or novel P. falciparum antigens (antigen 2, 4, 11, 12, 13, or 25; [14]; GenBank locus ID as indicated) were screened by a modified immunoscreening ELISA technique against malaria-immune or malaria-naïve sera, as described in Materials and Methods. Responses were considered positive if the corrected OD405 nm against malaria-immune sera was at least 0.2 OD units above the mean + 3 SD of the malaria-naive control. Numbers in parenthesis represent the number of positive assays/total number of assays, for each antigen.
3.5. Antibody responses induced in vivo by immunization with TAP fragments
We demonstrated that linear TAP fragments encoding P. falciparum and P. yoelii antigens were able to induce parasite-specific antibody responses in mice equivalent to or better than those induced by the corresponding supercoiled plasmid DNA. Using IFAT assays, the TAP fragments encoding liver/blood stage antigen PyHEP17 (in BALB/c mice) or the erythrocytic stage antigens PfAMA1 and PfEBA175-RII (in C57BL/6 mice) induced comparable or superior antibody titers to the corresponding plasmid DNA vaccines (Fig. 3). In relation to sporozoite stage IFAT, the overall levels of antibodies varied depending on the construct. TAP fragments encoding the pre-erythrocytic stage antigen PyCSP elicited lower parasite specific antibodies by IFAT than the corresponding plasmid, but this was not statistically significant (p ≥ 0.1; Fig. 4A). However, when ELISA titers from mice immunized with either TAP fragments or plasmids carrying PyCSP were compared, there were notable differences in response with the plasmid generating 5 to 6-fold higher titers than the TAP fragment (Fig. 4B).
Figure 3. Antibody responses against blood-stage antigens.

BALB/c (PyHEP17) or C57BL/6 mice (PfAMA1 or PfEBA-175) were immunized IM or ID, respectively, with 50 μg plasmid DNA or TAP fragments encoding PyHEP17 or PfAMA1 or PfEBA-175 3 times at 3-week intervals. Sera were collected 2 wk after each immunization. Parasite-specific antibodies were assayed by Indirect Fluorescent Antibody Test (IFAT) against P. yoelii (PyHEP17) or P. falciparum (PfAMA1 or PfEBA175-RII) parasitized erythrocytes using pooled sera (n=5/group).
Figure 4. Antibody responses against sporozoite stage antigens.

BALB/c mice were immunized IM with 100 or 50 μg plasmid DNA or 50 μg of TAP fragments encoding PyCSP or empty VR1012 plasmid (plasmid control) 3 times at 3-week intervals. Sera were collected 2 wk after each immunization. Control sera were collected from unimmunized naïve mice. Sporozoite-specific antibodies were assayed by (A) Indirect Fluorescent Antibody Test (IFAT) against P. yoelii sporozoites using pooled sera (n=4/group) or (B) ELISA against recombinant PyCSP capture antigen, using sera collected post first (white bars with black dots), second (white bars with black stripes; shown for ELISA only) or third (black bars with white dots) immunizations. Histograms represent (A) logarithmically transformed geometric mean IFAT +/− standard deviation or (B) geometric mean ELISA OD 0.5 +/− standard deviation of pooled sera.
3.6. Cellular responses induced in vivo by immunization with TAP fragments
We also demonstrated the capacity of TAP fragments encoding PyHEP17 to induce antigen-specific CD8+ and CD4+ T cell responses in BALB/c mice. Overall, the responses were equivalent to or greater than those induced by immunization with plasmid DNA encoding the same gene. In ELIspot assays, antigen-specific IFN-γ secretion induced by PyHEP17 TAP fragments was more than 2-fold higher than cytokine production induced by PyHEP17 plasmid (Fig. 5A). Similarly, for PyHEP17 CTL responses, the TAP fragments were slightly more immunogenic than the supercoiled plasmids (Fig. 5B). However, PyHEP17-specific IFN-γ or TNF-α responses against PyHEP17 peptide epitopes, as evaluated by ICS, were mixed, with plasmid inducing stronger responses to some peptides and TAP inducing stronger responses to others (Fig. 5C). There was no difference between PyHEP17 TAP and plasmid with regard to CD4+ T cell responses as measured by lymphocyte proliferation (Fig. 5D).
Figure 5. T cell responses against PyHEP17.

BALB/c (PyHEP 17) were immunized IM with 50 μg plasmid DNA or TAP fragments encoding PyHEP17 3 times at 3-week intervals. Mice were sacrificed 2 wk after last immunization and splenocytes harvested. Recall T cell responses were assayed by (A) IFN-γ ELIspot (n=5/group), (B) Cytotoxic T lymphocyte (CTL) assays (n=5/group), (C) Intracellular Cytokine Staining (ICS) assays (n=3/group) or (D) Lymphoproliferation Assay (LPA) (n=5/group) using synthetic peptides representing defined CD8+ and/or CD4+ T cell epitopes on PyHEP17 (Table S1). CTL peptide pools: 15-mers (PyHEP61-75, PyHEP66-80, and PyHEP71-85); 9-mers (PyHEP61-69, PyHEP70-78, PyHEP76-84, and PyHEP84-92). Histograms represent the mean response for each group +/− standard deviation.
For PyCSP, antigen-specific CD8+ T cell IFN-γ responses were approximately 3-fold higher with the plasmid than the corresponding TAP construct (Fig. 6). CD4+ T cell IFN-γ responses as evaluated by ICS appeared equivalent between plasmid and TAP products, but responses were very low (data not presented).
Figure 6. T cell responses against PyCSP.

BALB/c mice were immunized IM with 100 or 50 μg plasmid DNA or 50 μg of TAP fragments encoding PyCSP 3 times at 3-week intervals. Mice were sacrificed 2 wk after last immunization and splenocytes harvested. Recall T cell responses were assayed by Intracellular Cytokine Staining (ICS) (n=4/group) using synthetic peptides representing defined immunodominant or subdominant CD8+ or CD4+ T cell epitopes on PyCSP (Table S1). Histograms represent the mean response for each group +/− standard deviation.
3.7. In vivo protective efficacy of TAP fragments
In limited protection studies using TaqMan™ RT-PCR [29], BALB/c mice immunized with circular plasmid DNA encoding PyCSP were protected against sporozoite challenge, but animals immunized with TAP fragments encoding the same gene were not protected (data not presented). In addition, neither TAP fragments nor supercoiled plasmid encoding PyHEP17 protected A/J or CAF1/J mice against similar challenge (data not presented), although the failure of PyHEP17 plasmid to consistently protect mice has been noted in other studies [21, 29].
4. Discussion
The large amount of genetic information currently being generated in multiple pathogen models brings not only a potential goldmine of information to the laboratory bench but also the enormous challenge of mining this information in a rapid, efficient, and focused manner. Herein, we have evaluated and verified TAP in the context of malaria as a functional genomics tool that allows thousands of antigen genes to be individually amplified in a transcriptionally active form so that their biological function, DNA or protein subunit vaccine immunological potency can be directly assessed in a high throughput manner. This study represents, to the best of our knowledge, the first demonstration of the use of transcriptionally active PCR products or any linear PCR fragments as a useful high throughput tool for screening genomic sequence data to identify potential target antigens for vaccine development. Recently, protein-based approaches have proved promising in this regard [37, 38], We further show that transcriptionally active PCR products or linear PCR fragments can be immunogenic in vivo, and are capable of inducing both antibody and cellular immune responses at levels equivalent to those induced by the corresponding supercoiled plasmid DNA. Thus, a TAP-based approach allows for the relative prioritization amongst target antigens included in a high throughput genomic screening strategy on the basis of their immune reactivity.
Under optimized conditions, TAP and EpiTAP products were successfully amplified using either genomic or plasmid DNA templates. While some constructs amplified more easily than others, this difficulty could be overcome with slight modification of the PCR annealing time and temperatures. As with a previous study, amplification to a ready-to-use product could be performed within a day, highlighting the high throughput capabilities of the TAP system [2]. This contrasts with the standard time and labor intensive method of cloning DNA or PCR fragments encoding the gene-of-interest into a replication competent expression vector, transforming and growing bacteria, and purifying the plasmid. In vitro expression of the TAP and Epi-TAP encoded antigens was demonstrated following transfection of multiple cell lines, including COS-7, CHO-K1 and UM449 cells.
One application of these high throughput in vitro screening assays for monitoring T cell and antibody responses is to survey the cellular and humoral immune responses from malaria exposed individuals. The cellular immunoscreening assay uses, for example, PBMC from humans immunized with radiation-attenuated P. falciparum sporozoites for quantification of IFN-γ using the ELIspot assay or other cellular assays such as ICS. IFN-γ is proposed as the primary marker of cellular immunogenicity since we [39] and others have shown that the protective immunity against pre-erythrocytic stage malaria induced by immunization with irradiated sporozoites is mediated, at least in part, by IFN-γ. The antigen-transfected cells serve as the target cells (APCs) for the cellular immune assay, and are transferred to standard ELIspot plates precoated with anti-IFN-γ mAbs. T-cells from sporozoite immunized or naturally exposed individuals (effector cells) are transferred to each well containing the transfected APCs, cultured for 24–36 h, and subsequently processed as per a conventional ELIspot assay. The humoral immunoscreening uses sera or plasma from individuals naturally or experimentally exposed to malaria. The assumption underlying the use of specimens from individuals exposed to whole parasite (either volunteers experimentally immunized with irradiated sporozoites or naturally exposed to malaria in the field) is that such individuals would develop a vigorous and multifaceted immune response, including antibodies and CTL and helper T lymphocyte components, directed against multiple antigens derived from the whole organism to which they have been exposed. It is presumed that the entire repertoire of parasite-induced T cell specificities is represented in these individuals. Their lymphocytes should recognize the subset of P. falciparum proteins expressed by irradiated sporozoites on the surface of infected hepatocytes (enabling cellular immunoscreening). Also, their antibodies should recognize the subset of P. falciparum antigens expressed on the surface of sporozoites or antigens released into the blood and lymph (enabling humoral screening). These assays will quantify the capacity of each antigen to induce recall humoral and cellular immune responses from individuals immune to malaria.
The outcome of such screening assays would be the identification of novel targets of T cell and antibody responses and prioritization of these antigens for vaccine development based on their immune reactivity. Recognition by recall immune responses demonstrates that the epitopes derived from that antigen are not only generated in vivo in the course of natural infection, but also that a B cell or T cell repertoire exists which is capable of recognizing that particular antigen/epitope. Our criteria for prioritization and down-selection of target antigens for vaccine development is based on the assumption that the magnitude of immune response generated by malaria-immune volunteers to the antigen of interest will reflect the protective capacity of that antigen, although it is recognized that this may not in fact be the case. This may be particularly relevant for malaria where there are no known correlates of protective immunity, although IFN-γ responses against liver-stage antigens and IgG1 and/or IgG3 responses against blood-stage antigens are implicated. It should be also noted that proteins that are antigenic may not necessarily be immunogenic and capable of inducing protective immunity against pathogen challenge. Thus, additional characterization of proteins identified by TAP immunoscreening approaches (using for example, subcellular localization studies, gene knockout studies, rodent ortholog protection studies) will be needed to validate their potential as vaccine candidates. Although in some disease models subcellular localization studies could be carried out using TAP fragments, this is not feasible in the malaria model given the failure of Plasmodium sequences to function in transfected eukarotic cells and the absence of many of the target organelles.
In the current report, we have developed and verified the 96-well format humoral immune screening assay, designed to detect the capacity of the Epi-TAP encoded antigens to be recognized by malaria-immune sera, and demonstrated that both well characterized as well as newly described P. falciparum antigens from the genomic sequence database were recognized by malaria immune sera but not malaria-naïve sera. In other studies, we have established proof-of-principle for the cellular immune screening assay, using supercoiled plasmids (data not presented).
We also evaluated the immunogenic potential of TAP fragments in mice, measuring parasite-specific and antigen-specific antibody responses as well as CD4+ and CD8+ T cell responses, and comparing these responses to those induced by the corresponding supercoiled plasmid DNA. In vivo, TAP proved to act as well or sometimes better than plasmid constructs in eliciting immune responses. We demonstrated antibody production against a variety of malaria antigens, supporting previous findings comparing antibody responses to immunization with TAP and plasmid constructs encoding HepB-SAg [2]. In addition, we also demonstrated that antigen-specific T cell responses can be induced by in vivo immunization with linear TAP fragments, and that the T cell responses induced by TAP fragments are comparable to or exceed those induced by supercoiled plasmids.
Future directions include expanding on the foundation developed here to apply the TAP technology to amplify and characterize a comprehensive set of unknown antigens derived from the P. falciparum genome; specifically, the subset of P. falciparum proteins expressed by irradiated sporozoites in hepatocytes (by cellular immunoscreening), and the subset of P. falciparum proteins expressed on the surface of merozoites, apical organelles, and infected erythrocytes (by humoral immunoscreening) to determine the capacity of each putative antigen to be recognized by malaria-immune T cells or B cells.
5. Conclusion
We have demonstrated the feasibility of TAP as a rapid, high throughput approach that capitalizes on genomic sequence data to enable the identification and characterization of potential antigenic targets of protective immune responses. Antigens identified by such screening methods would represent good candidates for inclusion in next generation vaccines. We have further demonstrated that TAP fragments elicit comparable antibody responses in vivo when compared to plasmid, at least in mice. We have also established that TAP fragments can induce antigen-specific CD8+ and CD4+ T cell responses following in vivo immunization, although this activity is more variable and gene-dependent, in particular for CD8+ T cell responses. In our limited series of studies, we have been unable to demonstrate a capacity of TAP fragments to confer protection against parasite challenge. However, the TAP approach is designed for high throughput in vitro screening studies, and not small-scale in vivo immunization studies. Hence our results support the potential of TAP for application to genome wide screening for target antigen identification for use in development of vaccines and diagnostics to combat naturally occurring and genetically engineered infectious diseases.
Supplementary Material
Acknowledgments
We thank Arnel Belmonte and Martha Sedegah for providing the P. yoelii sporozoites, and Erika Belikova for assistance with the TaqMan studies. This work was supported by funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health under contract N01-AI-95362 and under grant R44 AI047641-02 awarded to Gene Therapy Systems, Inc, and by funds allocated to the Naval Medical Research and Development Center by the US Army Medical Research Material Command (work units 61102A.S13.F.A0009 and 62787A.870.F.A0228). All experiments reported herein were conducted in compliance with the Animal Welfare Act and in accordance with the principles set forth in the “Guide for the Care and Use of Laboratory Animals” (Institute of Laboratory Animal Resources, National Research Council, National Academy Press, 1996). The studies utilizing human specimens reported herein were approved by the Naval Medical Research Center Institutional Review Board in compliance with all applicable Federal regulations governing the protection of human subjects. This work was prepared as part of official U.S. Government duties. Title 17 U.S.C. §105 provides that ‘Copyright protection under this title is not available for any work of the United States Government.’ Title 17 U.S.C. §101 defines a U.S. Government work as a work prepared by a military service member or employee of the U.S. Government as part of that person’s official duties. The views expressed in this article are those of the authors and do not necessarily reflect the official policy or position of the Department of the Navy, Department of Defense, nor the U.S. Government.
List of abbreviations
- APC
antigen presenting cell
- bp
base pair
- CTL
cytotoxic T lymphocyte
- ELISA
enzyme-linked immunosorbent assay
- FCS
fetal calf serum
- FITC
fluorescein isothiocyanate
- GFP
green fluorescent protein
- h
hours
- HA
influenza hemagglutinin
- HTL
helper T lymphocyte
- IFAT
indirect fluorescent antibody test
- LEE
linear expression element
- LPA
lymphoproliferation assay
- ICS
intracellular cytokine staining
- min
minutes
- OD
optical density
- ORF
open reading frame
- PBMC
peripheral blood mononuclear cell
- PBS
phosphate buffered saline
- P. falciparum
Plasmodium falciparum
- P. yoelii
Plasmodium yoelii
- RT-PCR
real time PCR
- SD
standard deviation
- sec
seconds
- SFC
spot forming cell
- SI
stimulation index
- TAP
Transcriptionally Active PCR
- wk
week
Appendix A. Supplementary data
Supplementary data associated with this article can be found, in the online version, at xxx
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Sykes KF, Johnston SA. Linear expression elements: rapid, in vivo, method to screen gene functions. Nat. Biotechnol. 1999;17:355–9. doi: 10.1038/7908. [DOI] [PubMed] [Google Scholar]
- 2.Liang X, Teng A, Braun DM, Felgner J, Wang Y, Baker SI, Chen S, Zelphati O, Felgner PL. Transcriptionally active polymerase chain reaction (TAP): high throughput gene expression using genome sequence data. J Biol Chem. 2002;277:3593–8. doi: 10.1074/jbc.M110652200. [DOI] [PubMed] [Google Scholar]
- 3.Niman HL, Thompson AM, Yu A, Markman M, Willems JJ, Herwig KR, Habib NA, Wood CB, Houghten RA, Lerner RA. Anti-peptide antibodies detect oncogene-related proteins in urine. Proc Natl Acad Sci U S A. 1985;82:7924–8. doi: 10.1073/pnas.82.23.7924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Field J, Nikawa J, Broek D, MacDonald B, Rodgers L, Wilson IA, Lerner RA, Wigler M. Purification of a RAS-responsive adenylyl cyclase complex from Saccharomyces cerevisiae by use of an epitope addition method. Mol Cell Biol. 1988;8:2159–65. doi: 10.1128/mcb.8.5.2159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen YT, Holcomb C, Moore HP. Expression and localization of two low molecular weight GTP-binding proteins, Rab8 and Rab10, by epitope tag. Proc Natl Acad Sci U S A. 1993;90:6508–12. doi: 10.1073/pnas.90.14.6508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bastos R, Lin A, Enarson M, Burke B. Targeting and function in mRNA export of nuclear pore complex protein Nup153. J Cell Biol. 1996;134:1141–56. doi: 10.1083/jcb.134.5.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Laminet AA, Apell G, Conroy L, Kavanaugh WM. Affinity, specificity, and kinetics of the interaction of the SHC phosphotyrosine binding domain with asparagine-X-X-phosphotyrosine motifs of growth factor receptors. J Biol Chem. 1996;271:264–9. doi: 10.1074/jbc.271.1.264. [DOI] [PubMed] [Google Scholar]
- 8.Smith S, Blobel G. Colocalization of vertebrate lamin B and lamin B receptor (LBR) in nuclear envelopes and in LBR-induced membrane stacks of the yeast Saccharomyces cerevisiae. Proc Natl Acad Sci U S A. 1994;91:10124–8. doi: 10.1073/pnas.91.21.10124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Snow RW, Trape JF, Marsh K. The past, present and future of childhood malaria mortality in Africa. Trends Parasitol. 2001;17:593–7. doi: 10.1016/s1471-4922(01)02031-1. [DOI] [PubMed] [Google Scholar]
- 10.Hoffman SL, Goh LM, Luke TC, Schneider I, Le TP, Doolan DL, Sacci J, de la Vega P, Dowler M, Paul C, Gordon DM, Stoute JA, Church LW, Sedegah M, Heppner DG, Ballou WR, Richie TL. Protection of humans against malaria by immunization with radiation-attenuated Plasmodium falciparum sporozoites. J Infect Dis. 2002;185:1155–64. doi: 10.1086/339409. [DOI] [PubMed] [Google Scholar]
- 11.Andersen E, Jones TR, Purnomo, Masbar S, Wiady I, Tirtolusumo S, Bangs MJ, Charoenvit Y, Gunawan S, Hoffman SL. Assessment of age-dependent immunity to malaria in transmigrants. Am J Trop Med Hyg. 1997;56:647–9. doi: 10.4269/ajtmh.1997.56.647. [DOI] [PubMed] [Google Scholar]
- 12.Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, Carlton JM, Pain A, Nelson KE, Bowman S, Paulsen IT, James K, Eisen JA, Rutherford K, Salzberg SL, Craig A, Kyes S, Chan MS, Nene V, Shallom SJ, Suh B, Peterson J, Angiuoli S, Pertea M, Allen J, Selengut J, Haft D, Mather MW, Vaidya AB, Martin DM, Fairlamb AH, Fraunholz MJ, Roos DS, Ralph SA, McFadden GI, Cummings LM, Subramanian GM, Mungall C, Venter JC, Carucci DJ, Hoffman SL, Newbold C, Davis RW, Fraser CM, Barrell B. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002;419:498–511. doi: 10.1038/nature01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoffman SL, Doolan DL. Malaria vaccines-targeting infected hepatocytes. Nat Med. 2000;6:1218–9. doi: 10.1038/81315. [DOI] [PubMed] [Google Scholar]
- 14.Doolan DL, Aguiar JC, Weiss WR, Sette A, Felgner PL, Regis DP, Quinones-Casas P, Yates JR, 3rd, Blair PL, Richie TL, Hoffman SL, Carucci DJ. Utilization of genomic sequence information to develop malaria vaccines. J Exp Biol. 2003;206:3789–802. doi: 10.1242/jeb.00615. [DOI] [PubMed] [Google Scholar]
- 15.Pacheco ND, Strome CP, Mitchell F, Bawden MP, Beaudoin RL. Rapid, large-scale isolation of Plasmodium berghei sporozoites from infected mosquitoes. J Parasitol. 1979;65:414–7. [PubMed] [Google Scholar]
- 16.Ozaki LS, Gwadz RW, Godson GN. Simple centrifugation method for rapid separation of sporozoites from mosquitoes. J Parasitol. 1984;70:831–3. [PubMed] [Google Scholar]
- 17.Sambrook J, Manniatis T, Fritsch E. Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, N.Y: 1989. [Google Scholar]
- 18.Doolan DL, Hoffman SL. DNA-based vaccines against malaria: status and promise of the Multi-Stage Malaria DNA Vaccine Operation. Int J Parasitol. 2001;31:753–62. doi: 10.1016/s0020-7519(01)00184-9. [DOI] [PubMed] [Google Scholar]
- 19.Kumar S, Villinger F, Oakley M, Aguiar JC, Jones TR, Hedstrom RC, Gowda K, Chute J, Stowers A, Kaslow DC, Thomas EK, Tine J, Klinman D, Hoffman SL, Weiss WW. A DNA vaccine encoding the 42 kDa C-terminus of merozoite surface protein 1 of Plasmodium falciparum induces antibody, interferon-gamma and cytotoxic T cell responses in rhesus monkeys: immuno-stimulatory effects of granulocyte macrophage-colony stimulating factor. Immunol Lett. 2002;81:13–24. doi: 10.1016/s0165-2478(01)00316-9. [DOI] [PubMed] [Google Scholar]
- 20.Doolan DL, Hedstrom RC, Rogers WO, Charoenvit Y, Rogers M, de la Vega P, Hoffman SL. Identification and characterization of the protective hepatocyte erythrocyte protein 17 kDa gene of Plasmodium yoelii, homolog of Plasmodium falciparum exported protein 1. J Biol Chem. 1996;271:17861–8. doi: 10.1074/jbc.271.30.17861. [DOI] [PubMed] [Google Scholar]
- 21.Doolan DL, Sedegah M, Hedstrom RC, Hobart P, Charoenvit Y, Hoffman SL. Circumventing genetic restriction of protection against malaria with multigene DNA immunization: CD8+ cell-, interferon gamma-, and nitric oxide-dependent immunity. J Exp Med. 1996;183:1739–46. doi: 10.1084/jem.183.4.1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sedegah M, Jones TR, Kaur M, Hedstrom R, Hobart P, Tine JA, Hoffman SL. Boosting with recombinant vaccinia increases immunogenicity and protective efficacy of malaria DNA vaccine. Proc Natl Acad Sci U S A. 1998;95:7648–53. doi: 10.1073/pnas.95.13.7648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jones TR, Gramzinski RA, Aguiar JC, Sim BK, Narum DL, Fuhrmann SR, Kumar S, Obaldia N, Hoffman SL. Absence of antigenic competition in Aotus monkeys immunized with Plasmodium falciparum DNA vaccines delivered as a mixture. Vaccine. 2002;20:1675–80. doi: 10.1016/s0264-410x(01)00513-8. [DOI] [PubMed] [Google Scholar]
- 24.Sim BK, Narum DL, Liang H, Fuhrmann SR, Obaldia N, 3rd, Gramzinski R, Aguiar J, Haynes JD, Moch JK, Hoffman SL. Induction of biologically active antibodies in mice, rabbits, and monkeys by Plasmodium falciparum EBA-175 region II DNA vaccine. Mol Med. 2001;7:247–54. [PMC free article] [PubMed] [Google Scholar]
- 25.Hartikka J, Sawdey M, Cornefert-Jensen F, Margalith M, Barnhart K, Nolasco M, Vahlsing HL, Meek J, Marquet M, Hobart P, Norman J, Manthorpe M. An improved plasmid DNA expression vector for direct injection into skeletal muscle. Hum Gene Ther. 1996;7:1205–17. doi: 10.1089/hum.1996.7.10-1205. [DOI] [PubMed] [Google Scholar]
- 26.Luke CJ, Carner K, Liang X, Barbour AG. An OspA-based DNA vaccine protects mice against infection with Borrelia burgdorferi. J Infect Dis. 1997;175:91–7. doi: 10.1093/infdis/175.1.91. [DOI] [PubMed] [Google Scholar]
- 27.Doolan DL, Hoffman SL, Southwood S, Wentworth PA, Sidney J, Chesnut RW, Keogh E, Appella E, Nutman TB, Lal AA, Gordon DM, Oloo A, Sette A. Degenerate cytotoxic T cell epitopes from P. falciparum restricted by multiple HLA-A and HLA-B supertype alleles. Immunity. 1997;7:97–112. doi: 10.1016/s1074-7613(00)80513-0. [DOI] [PubMed] [Google Scholar]
- 28.Doolan DL, Southwood S, Freilich DA, Sidney J, Graber NL, Shatney L, Bebris L, Florens L, Dobano C, Witney AA, Appella E, Hoffman SL, Yates JR, 3rd, Carucci DJ, Sette A. Identification of Plasmodium falciparum antigens by antigenic analysis of genomic and proteomic data. Proc Natl Acad Sci U S A. 2003;100:9952–7. doi: 10.1073/pnas.1633254100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Witney AA, Doolan DL, Anthony RM, Weiss WR, Hoffman SL, Carucci DJ. Determining liver stage parasite burden by real time quantitative PCR as a method for evaluating pre-erythrocytic malaria vaccine efficacy. Mol Biochem Parasitol. 2001;118:233–45. doi: 10.1016/s0166-6851(01)00372-3. [DOI] [PubMed] [Google Scholar]
- 30.Charoenvit Y, Mellouk S, Cole C, Bechara R, Leef MF, Sedegah M, Yuan LF, Robey FA, Beaudoin RL, Hoffman SL. Monoclonal, but not polyclonal, antibodies protect against Plasmodium yoelii sporozoites. J Immunol. 1991;146:1020–5. [PubMed] [Google Scholar]
- 31.Charoenvit Y, Leef MF, Yuan LF, Sedegah M, Beaudoin RL. Characterization of Plasmodium yoelii monoclonal antibodies directed against stage-specific sporozoite antigens. Infect Immun. 1987;55:604–8. doi: 10.1128/iai.55.3.604-608.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sedegah M, Finkelman F, Hoffman SL. Interleukin 12 induction of interferon gamma-dependent protection against malaria. Proc Natl Acad Sci U S A. 1994;91:10700–2. doi: 10.1073/pnas.91.22.10700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhu J, Hollingdale MR. Structure of Plasmodium falciparum liver stage antigen-1. Mol Biochem Parasitol. 1991;48:223–6. doi: 10.1016/0166-6851(91)90117-o. [DOI] [PubMed] [Google Scholar]
- 34.Florens L, Washburn MP, Raine JD, Anthony RM, Grainger M, Haynes JD, Moch JK, Muster N, Sacci JB, Tabb DL, Witney AA, Wolters D, Wu Y, Gardner MJ, Holder AA, Sinden RE, Yates JR, Carucci DJ. A proteomic view of the Plasmodium falciparum life cycle. Nature. 2002;419:520–6. doi: 10.1038/nature01107. [DOI] [PubMed] [Google Scholar]
- 35.Cheng S, Fockler C, Barnes WM, Higuchi R. Effective amplification of long targets from cloned inserts and human genomic DNA. Proc Natl Acad Sci U S A. 1994;91:5695–9. doi: 10.1073/pnas.91.12.5695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Barnes WM. PCR amplification of up to 35-kb DNA with high fidelity and high yield from lambda bacteriophage templates. Proc Natl Acad Sci U S A. 1994;91:2216–20. doi: 10.1073/pnas.91.6.2216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Davies DH, McCausland MM, Valdez C, Huynh D, Hernandez JE, Mu Y, Hirst S, Villarreal L, Felgner PL, Crotty S. Vaccinia virus H3L envelope protein is a major target of neutralizing antibodies in humans and elicits protection against lethal challenge in mice. J Virol. 2005;79:11724–33. doi: 10.1128/JVI.79.18.11724-11733.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Davies DH, Liang X, Hernandez JE, Randall A, Hirst S, Mu Y, Romero KM, Nguyen TT, Kalantari-Dehaghi M, Crotty S, Baldi P, Villarreal LP, Felgner PL. Profiling the humoral immune response to infection by using proteome microarrays: high-throughput vaccine and diagnostic antigen discovery. Proc Natl Acad Sci U S A. 2005;102:547–52. doi: 10.1073/pnas.0408782102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Doolan DL, Hoffman SL. The complexity of protective immunity against liver-stage malaria. J Immunol. 2000;165:1453–62. doi: 10.4049/jimmunol.165.3.1453. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.