


Abstract
The DNA repair enzyme human uracil DNA glycosylase (UNG) scans short stretches of genomic DNA and captures rare uracil bases as they transiently emerge from the DNA duplex via spontaneous base pair breathing motions. The process of DNA scanning requires that the enzyme transiently loosen its grip on DNA to allow stochastic movement along the DNA contour, while engaging extrahelical bases requires motions on a more rapid timescale. Here, we use NMR dynamic measurements to show that free UNG has no intrinsic dynamic properties in the millisecond to microsecond and subnanosecond time regimes, and that the act of binding to nontarget DNA reshapes the dynamic landscape to allow productive millisecond motions for scanning and damage recognition. These results suggest that DNA structure and the spontaneous dynamics of base pairs may drive the evolution of a protein sequence that is tuned to respond to this dynamic regime.
Biomolecules can have highly dynamic structures that undergo motions over many different time scales and are often essential to their biological functions. We are interested in the role of DNA and enzyme dynamics in the recognition of damaged bases in DNA. One important example is the recognition and removal of uracil in DNA by the enzyme human uracil DNA glycosylase (UNG), which uses an extrahelical base recognition mechanism (1). UNG is essential for the prevention of C→T transition mutations arising from cytosine deamination (2), cytotoxic U:A pairs arising from incorporation of dUTP in DNA and for increasing Ig gene diversity during the acquired immune response (3). A central event in all of these UNG-mediated processes is the singling out of target U:A or U:G base pairs in a background of approximately 109 T:A or C:G base pairs in the human genome. We have previously used NMR dynamic measurements of free DNA and its complex with UNG to establish that enzymatic discrimination of thymine from uracil is initiated by dynamic opening of T:A and U:A base pairs (4,5), leading to an extrahelical state of T or U that places these bases in a transient exosite binding pocket on the enzyme (Figure 1) (6). This capture mechanism requires UNG to scan along nontarget DNA, transiently pausing to sample base pair opening events that occur with an estimated rate of ∼8 ms−1 under physiological conditions (7). These intrinsic dynamics of DNA may in turn guide the evolution of complementary dynamic fluctuations in UNG that allow the enzyme to scan the DNA chain and recognize transient states of damaged bases in DNA.
Figure 1.

The uracil target search mechanism of human UNG. The search process begins by diffusion to a nontarget site in DNA followed by short-range scanning of the DNA helix. During a short scanning event lasting about 5 ms, UNG can trap thymine or uracil bases (X) in a specific exosite binding pocket as they spontaneously emerge from the DNA base stack with estimated rates of 8 ms−1 at 37°C (7). A molecular sieving mechanism is used to reject the methyl group of thymine and allow uracil (U) to selectively proceed to the enzyme active site. The scanning and target discrimination step is probed by the NMR experiments in this work.
Flexibility in structure is thought to be an important aspect of enzyme regulation and function (8,9). For example, if the free enzyme has a rigid structure, the binding energy of the substrate may be used to drive formation of the active state of the enzyme from an inactive form that exists in the absence of substrate. Alternatively, the free enzyme may be flexible and exist in a dynamic equilibrium between active and inactive states. In such an enzyme, substrate encounter may select for the active conformer from the preexisting population. NMR dynamic studies on several enzymes have found evidence for conformer selection as the free proteins were observed in a skewed equilibrium between active and inactive states in the absence of substrate (10–14). Of course, any real process would likely incorporate elements from both of these rigid formalisms. Here, we report an intriguing example of dynamics in the process of damage recognition by UNG that is distinct from either of the formal models described above. We find that the free enzyme is rigid in the millisecond to microsecond and subnanosecond time regimes, but that binding to nontarget DNA flattens its conformational free energy landscape allowing UNG to sample states that are energetically inaccessible in the absence of DNA. The energetic effects of nontarget DNA binding are likely important factors that enable UNG to scan DNA and capture rapidly emerging extrahelical bases.
METHODS
Preparation of enzyme
BL21 (DE3) Codon Plus cells were transformed with a PET21a plasmid containing the UNG gene under the control of an Isopropyl β-D-1-thiogalactopyranoside (IPTG)-inducible T7 promoter. For the backbone assignment experiments, isotopically enriched UNG was obtained by growing cells in morphaline propanol sulfonic acid (MOPS) minimal media containing 15NH4Cl, 13C-labeled glucose and 99% 2H2O resulting in ∼70% deuterium incorporation at non-exchangeable positions. For enzyme used in the dynamic experiments, cells were grown in MOPS media containing 99% 2H2O using a 99% deuterated glucose as the sole carbon source. Heavy water and all other isotopically enriched chemicals were obtained from Cambridge Isotope Laboratories (CIL), Inc. The protocol for over expression and purification of UNG has been described previously (15).
Oligonucleotides
Fluorescence anisotropy measurements made use of a DNA duplex containing a 5′-FAM labeled strand (5′-FAM-T1C2G3A4T5C6G7A8T9G10-3′) hybridized to its unlabeled complement strand. Both single strands were synthesized in-house using standard solid phase phosphoramidite chemistry. The single-stranded oligonucleotides were purified using a preparative Phenomenex Jupiter column and their concentrations were measured with UV/VIS absorbance using calculated molar extinction coefficients. The FAM-labeled DNA strand was combined with a 1.2-fold excess of its unlabeled complement and hybridization was accomplished by heating to 50°C and slowly cooling to room temperature. A native 15% polyacrylamide gel was used to assess whether hybridization was successful. Unlabeled DNA of the same sequence 5′-C1T2G3G4A5T6C7C8A9G10-3′ was used in the NMR studies and was ordered from Integrated DNA Technologies (IDT), Inc. and purified as described above.
Enzyme–DNA dissociation constant measurements
The binding affinity of UNG for nontarget DNA was determined by following the increase in fluorescence anisotropy of the 5′-FAM-labeled DNA as UNG was added to the solution. Binding experiments were performed at 20°C using several NaCl concentrations in the range 10–100 mM in a buffer containing 10 mM NaH2PO4 (pH 7.0), 1 mM ethylenediaminetetraacetic acid (EDTA) and 1 mM Dithiothreitol (DTT). The anisotropy increase as a function of UNG concentration was fitted to Equation (1) where C = ([Pt] + [Lt] + Kd), and the
| 1 |
nonlinear regression software program Prism was used to obtain the dissociation constant (Kd). A standard buffer 10 mM NaH2PO4 (pH 7.0), 75 mM NaCl, 1 mM EDTA and 1 mM DTT were chosen for all NMR experiments as it was found to be optimal for protein solubility and stoichiometric DNA binding.
NMR resonance assignments
The backbone NH, Cα, Cβ and C′ resonance assignments for 13C, 15N and 2H-labeled UNG were made using TROSY detected versions of the HNCACB, HN(CO)CACB, HNCO, HN(CA)CO (16) and HNN (17) 3D correlation experiments. Experiments were preformed using an 800 MHz Varian INOVA system equipped with a cryoprobe. Resultant data were processed using the NMRPipe software package (18) and analyzed using CARA (19). Assignments for UNG bound to nontarget DNA were obtained by tracking the chemical shift perturbations of the backbone amide resonances as DNA was added to a solution of enzyme.
Quantification of dynamics in UNG
Protein dynamic measurements were performed using UNG (0.92 mM) that was doubly labeled with 15N and 2H. For measurements on the nontarget DNA complex, approximately 4-fold stoichiometric excess of DNA (4.0 mM) was present to ensure that the enzyme was ∼99% bound under experimental conditions. Millisecond timescale dynamics were measured using a modified spin-state selective TROSY detected pulse sequence with a 60 ms constant time Carr–Purcell–Meibloom–Gill period (TCPMG) and a 3.8 kHz B1 CPMG pulse strength (20). Observed transverse relaxation rates were calculated by comparison of measured peak intensities to those obtained from a reference experiment without a CPMG delay period using the Equation (2).
| 2 |
Errors in the measured relaxation rates were estimated by measuring variances in peak intensities obtained from acquiring duplicate data points and propagating that error into the measured values for Robs2. The observed relaxation rates at each νcpmg were fitted to Equation (3), which describes dynamic exchange between two states a and b with an exchange constant kex = kforward + kreverse, an amplitude factor (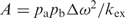 , where papb is the product of the fractional populations of the two exchanging states), and Δω, the difference in resonance frequencies between states a and b.
, where papb is the product of the fractional populations of the two exchanging states), and Δω, the difference in resonance frequencies between states a and b.
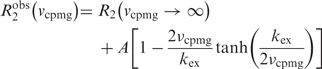 |
3 |
In order to determine the field dependent ‘A’ term from Equation (3), experiments were conducted at 600 and 800 MHz static magnetic field strengths. Peak intensities were measured using SPARKY (21) and the data for each residue at the two magnetic fields were fitted using Prism software. The amplitude factors (A) and kex values for each residue were determined by simultaneous nonlinear regression fitting of the dispersion data at both fields to Equation (3) (Supplementary Table 1). The kex terms from the individually fitted residues were within error of one another which justified a global fitting procedure where a common kex was shared by all dynamic residues.
Normal mode analysis
The structural coordinates for UNG bound to nontarget DNA (pdb code 2OXM) were used as input for the elNemo webserver (http://www.igs.cnrs-mrs.fr/elnemo/) with the coordinates for the bound DNA molecule removed. The five lowest frequency normal modes of this structure were computed using the default parameters by representing the protein backbone as rigid blocks consisting of two amino acids each with an 8 Å interaction cutoff distance. The lowest frequency mode is shown in the Supplementary Video.
RESULTS
NMR assignments and dynamic behavior of free UNG
In order to probe the conformational dynamics of the backbone amides of human UNG, it was first necessary to make residue-specific NMR assignments for the 210 nonproline residues of the free enzyme. A standard series of heteronuclear triple resonance NMR experiments were performed using 15N, 13C and 2H-labeled enzyme (Supplementary Figure 1). Ultimately, 204 of the 210 backbone 15N, Cα, C′ and Cβ resonances of nonproline amino acids of UNG were assigned. Of the six unassigned resonances (none of which were observed in these experiments), three are located in the flexible N-terminus, two are located in the dynamic DNA binding site (Ser247 and Tyr248, see below) and the final residue (Leu207) is in the protein core.
NMR is uniquely capable of detecting chemical dynamics with rate constants spanning many orders of magnitude (22). However, DNA scanning and base pair opening occur on the millisecond timescale leading us to focus on similar timescale motions that might be present in the free or DNA-bound enzyme (1,4,7). This time regime is best probed using relaxation dispersion NMR experiments (23,24), which can quantify rates of exchange of magnetic nuclei between different chemical environments [Equation (3)]. The exchange contribution to the observed transverse relaxation rate [Robs2, Equation (3)] is measured as a function of an applied external field (νcpmg), and may be obtained using Equation (3) provided conditions of fast exchange hold. If νcpmg is much smaller than the rate of exchange between states, this field will have little effect on the measured relaxation rate. Conversely, if νcpmg is of a much greater frequency than the exchange rate between states, the exchange component to transverse relaxation [Rex, the second term in Equation (3)] will be attenuated, and Robs2 will approach the intrinsic rate [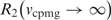 ].
].
The relaxation dispersion profiles for the vast majority of the backbone amides of free 15N, 2H-labeled UNG showed a flat νcpmg field dependence indicating that very few residues possessed millisecond timescale dynamics [selected residues of free UNG (blue) are shown in Figure 2a]. The exceptions were Ser247, Tyr248 and Ser169, which hydrogen bond to the DNA backbone in the complex, and the resonance of Asn204 which is a key catalytic residue that hydrogen bonds to H3 and O4 of the fully extrahelical uracil base, but does not interact with DNA in the nontarget complex (6). We also investigated whether free UNG possessed rapid dynamics in the nanosecond time regime by performing a heteronuclear 1H–15N Nuclear Overhauser Effect (NOE) experiment (22). The measured heteronuclear 1H–15N NOE's were large and relatively constant over the protein structure providing no evidence for significant dynamic motions in this rapid time regime (Supplementary Figure 2).
Figure 2.

Dynamics of free UNG and its complex with nontarget DNA. (a) Global fits of the relaxation dispersion profiles of the amide nitrogens of the UNG–DNA complex (green). Selected residues of the UNG–DNA complex were used to globally optimize a single exchange rate constant [kex, Equation (3)]. All residues are located in close proximity to the DNA binding pocket of UNG. With the exception of Ser169, horizontal lines are drawn through the data for free UNG (blue) to emphasize the νcpmg field independent behavior for resonances of the free enzyme. (b) Dynamic regions that interact with the DNA strand containing the extrahelical thymine in the exosite (PDB 2OXM). The locations of the backbone amides used in the global fitting in (a) are colored red in this view. The extrahelical thymine is shown in ball and stick representation and the other DNA strand is omitted for clarity. (c) Global view of the dynamics and chemical shift perturbations induced by nontarget DNA binding. All backbone amides in UNG that show significant exchange in the DNA-bound state are colored red. The width of the backbone of UNG is drawn proportional to the amide chemical shift perturbation brought about by nontarget DNA binding (Figure 3c). The magnitudes of the chemical shift changes between free and bound UNG are not well correlated with the exchange contribution to the line widths in the bound state indicating that exchange involves two bound states. The bound DNA is shown in cyan looking down the helical axis.
Nontarget DNA binding induces millisecond-timescale dynamics in UNG
To investigate the scanning and target search steps (Figure 1), we then interrogated the dynamics of UNG while it was bound to a 10-mer nontarget DNA duplex. Using fluorescence anisotropy methods, the equilibrium dissociation constant for a fluorescein end-labeled version of the 10-mer duplex was first determined to ensure that UNG would be fully saturated with DNA in the NMR dynamic experiments (KD = 29 ± 0.4 µM) (Figure 3a). This KD value is low enough to ensure that 99% of the UNG molecules are DNA-bound at the millimolar enzyme and DNA concentrations used in the NMR experiments (see below). To obtain assignments for the nontarget complex, the 10-mer duplex was titrated into an NMR sample of UNG and 1H–15N HSQC spectra were acquired at each titration point. The complex was in fast exchange with the free enzyme and DNA on the chemical shift timescale, allowing the resonance assignments for the complex to be obtained by following the chemical shift changes over the course of the titration. Ambiguous assignments in the complex were resolved using a 15N-edited 3D HMQC-NOESY-HSQC experiment. Backbone amides whose chemical shifts were directly perturbed by DNA binding exclusively mapped to the DNA binding surface as indicated by comparison with the crystallographic structure of the nontarget complex (Figure 3b, c) (6). Thus, the solution complex recapitulates the crystallographic binding mode, facilitating direct comparisons.
Figure 3.

Nontarget DNA binding to human UNG. (a) Fluorescence anisotropy measurements were used to determine the dissociation constant of UNG for a 5′-fluorescein-labeled 10-mer duplex DNA of the sequence 5′ TCGATCGATG 3′. (b) The crystal structure of human UNG bound to nontarget DNA (yellow) showing the central thymidine in the transient exosite specific for T and U (Figure 1) (6). (c) The same view of UNG as panel (b) but the heteronuclear (1H–15N) weighted chemical shift perturbations of its backbone amides upon addition of nontarget DNA are color coded onto the surface. The DNA molecule is removed for clarity.
We then performed relaxation dispersion measurements on the UNG–DNA complex at two static field strengths (600 and 800 MHz). In contrast to the free enzyme, a select group of residues in the complex displayed field-dependent dispersion profiles and elevated exchange contributions to transverse relaxation (Figure 2a, green data points) (Supplementary Figure 3). These interesting residues are found in five elements of the primary sequence that we have designated as catalytic strand, phosphate pincers I and II, minor groove finger and second sphere (Figure 2a–c). The catalytic group comprises residues 144–148 and contains Gln144, Asp145 and His148 all of which have been implicated in base flipping or catalysis (25). Phosphate pincer regions I and II comprise residues 246–249 and residue Ser169, respectively. The pincer regions contact the phosphate backbone of the DNA strand containing the extrahelical base in both the early exosite complex and the final catalytic complex with uracil. These interactions are important in providing the binding energy that drives the uracil base from the exosite to the active site (Figure 1). The finger region is comprised of residues 270–274 and projects into the DNA minor groove, filling the hole left behind by the flipped base. Although residues Leu272 and Val274 in this region can be fitted to relaxation dispersion curves (Figure 2a), residues 270, 271 and 273 cannot, due to extremely weak signals arising from unfavorable exchange parameters. Finally, the second sphere residues 153–159 form part of an extended loop that runs anti-parallel to and contacts the strand containing the catalytic residues Gln144, Asp145 and His148.
Of the dozen amides that could be cleanly fitted to Equation (3), similar values of kex were extracted even though these groups were dispersed in the primary sequence and in regions of different secondary structure (Supplementary Table 1). This result suggested that a single global motion around the DNA binding site, with a rate constant kex, was sufficient to model the dispersion data. Indeed, global fitting of the entire set of curves yielded a single exchange rate kex = 900 ± 200 s−1 (green curves, Figure 2a). The observed effects cannot arise from exchange of the enzyme between a free and bound state because (i) the association rate constant of human UNG for DNA (k = 7 × 107 M−1s−1, see Supplementary Figure 4) (7,26,27) makes the observed pseudo-first-order exchange rate between free and bound enzyme (kex = kon[DNA] + koff) >280 000 s−1 under the conditions of the NMR dynamic studies ([DNA] = 4 mM). This large exchange rate is over 300-fold greater than the measured exchange rates, and in general, is too rapid for measurements by NMR relaxation dispersion experiments; (ii) the measured line widths of the exchanged broadened resonances were independent of DNA concentration; and (iii) the magnitudes of the chemical shift changes between free and bound UNG are not well correlated with the exchange contribution to the line widths in the bound state (Figure 2c).
It should be noted that the dispersion profiles for the majority of amino acids shown in Figure 2a have elevated R2obs values even at the highest νcpmg compared to the nondynamic residues of free and bound UNG (Supplementary Figure 2). This indicates the presence of additional contributions to the NMR line widths for these amide resonances that could not be refocused by the external applied field [vcpmg, Equation (3)]. We were unable to refocus these exchange contributions to the line widths using an off-resonance 15N R1ρ relaxation experiment capable of sampling frequencies as fast as 5000 s−1 (data not shown) (28). The origin of this additional contribution is unknown, but must involve motions that are slower than the rotational correlation time of UNG. As with the free enzyme, no fast nanosecond-timescale dynamics were observed in the complex, as evidenced by the large and invariant heteronuclear amide NOE's (Supplementary Figure 2).
DISCUSSION
This study provides the first characterization of a DNA repair enzyme dynamically inspecting undamaged DNA. The data clearly reveal millisecond time scale motions of UNG that are induced upon binding to nontarget DNA. Since the induced dynamic behavior is localized to residues known to be involved in nontarget DNA binding and target searching (6), the motions are likely to be functionally relevant to the scanning and target search steps of DNA repair as depicted in Figure 1. Although NMR is a powerful method for detecting dynamic motions of biomolecules, the measurements by themselves do not reveal the structural changes that are being sampled. Thus, other experimental observations must be utilized to link the observed dynamics to structure and the process of searching and recognizing DNA damage.
The regions of UNG that show induced dynamics upon binding to nontarget DNA reflect a subset of regions that undergo a clamping movement upon binding to both nontarget and target DNA (6,29). The relative displacements in the backbone amide positions of UNG at early and late points along the reaction coordinate are shown in Figure 4a. The early displacements, which are relatively large, involve transitioning from the free enzyme to the exosite complex with nontarget DNA (blue line, Figure 4a). The late displacements, which are very small, reflect structural changes that occur upon transitioning from the exosite complex to the specific uracil complex (green line, Figure 4a). Thus, the enzyme has largely assumed its catalytically active closed conformation at the exosite complex even though the flipped base is only one-sixth of the way along the 160° rotation leading to the active site. The inference is that the observed dynamic fluctuations of the DNA bound enzyme reflects exchange between a predominantly closed conformation as observed in the exosite crystal structure and another bound state that is intermediate between the free state and the exosite complex. Such fluctuations on the millisecond time scale are consistent with the estimated millisecond life time of UNG at individual base pairs as it stochastically slides along the DNA contour (7). Oscillations between a looser binding state and a closed state that allows sampling of base pair opening events provide a plausible mechanism for scanning and pausing along the DNA contour to inspect the duplex for uracil bases (1,7).
Figure 4.

Backbone atom displacements of UNG upon DNA binding and lowest frequency NMA of the enzyme. (a) Comparison between amide nitrogen displacement in the lowest frequency normal mode of the exosite DNA complex (black dashed line) and the observed amide displacements between free UNG (pdb code 2OXM) and the exosite complex (pdb code 1AKZ) (blue line). (b) The two extrema of the lowest frequency normal mode of UNG indicate an open to closed conformational transition. A video of the atom displacements in this normal mode trajectory is available as Supplementary Video 1.
If an open to closed conformational transition is responsible for the observed dynamic behavior of the DNA-bound enzyme, then it would be expected that the equilibrium scaffold of UNG would possess low-energy motions that allow this type of reversible clamping motion. To investigate this possibility, we performed a normal mode analysis (NMA) of UNG using the crystallographic coordinates from the exosite complex (pdb code 2OXM) (30). The lowest frequency mode obtained from this analysis resulted in atom displacements that recapitulate those observed in moving from the free enzyme to the exosite complex (dashed line, Figure 4a). The two extrema from this lowest frequency mode are shown in Figure 4b with the exosite DNA duplex included for reference. A video of the complete trajectory is available as Supplementary Video 1 online. The mechanism of short-range DNA scanning that is suggested by this analysis involves oscillation of the enzyme between an open state that allows stochastic, thermally driven translocation along the DNA strand and a closed state where the pincer and finger regions interact more intimately with the DNA major and minor grooves (Figure 4b). The closed state seen in the exosite complex is especially well poised for the rapid capture of thymine and uracil bases that spontaneously emerge from the DNA base stack (6,7).
A surprising observation is that UNG is not dynamic until it binds to nontarget DNA. This property distinguishes UNG from several previously studied enzyme systems where pre-existing and catalytically competent dynamic motions were detected in the free enzyme (10–14). The induced flexibility of the UNG backbone upon DNA binding is distinct from several transcription factors that are highly dynamic in the free state and assume a rigid conformation only upon binding to their cognate sequences (31,32). Given the high concentration of nonspecific DNA binding sites in the cell nucleus and UNG's micromolar affinity for nontarget DNA (Figure 3a), the enzyme would be expected to be bound to DNA at all times in vivo. This environment would select for dynamic properties of the enzyme–DNA complex that optimize efficient repair rather than properties of the free enzyme which may be under different selection pressures.
A functional model for the role of nontarget DNA binding in the function of UNG is summarized in Figure 5. The absence of significant dynamics in the free enzyme indicates that free UNG inhabits a narrow conformational energy well and is unable to sample conformations that resemble the bound state. Thus, the free energy of DNA binding is used to populate unstable states that are inaccessible in the absence of DNA, and it is only upon binding to nontarget DNA that dynamic modes relevant to the search process become activated. The bound states of UNG are most reasonably assigned to a weakly interacting open conformation that is competent for sliding along the DNA contour and resembles the free enzyme. The second closed state resembles the early exosite structure and is poised to detect extrahelical bases. The closed state may also possess additional dynamic motions in the sub-millisecond regime that allow efficient capture of transiently emerging bases. These findings reveal how the free energy of DNA binding can be used to modulate the conformational and dynamic landscape of an enzyme allowing it to scan the DNA contour and respond to intrinsic base pair dynamics.
Figure 5.

The free energy of nontarget DNA binding is used to alter the dynamic landscape of UNG. Free UNG (pdb code 1AKZ) populates a single open conformation within a steep energy well. The free energy of DNA binding is used to destabilize regions of the UNG structure and lower the activation barrier for UNG to sample an open and closed state in the search complex (see text). The free energy differences between the free enzyme and the complex are drawn arbitrarily.
SUPPLEMENTARY DATA
Supplementary Data is available at NAR Online.
FUNDING
National Institutes of Health (grant GM56834-13 to J.T.S.). Funding to open access charge: GM56834-13.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
The content of the publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does the mention of trade names, commercial products, or organizations imply endorsement by the US Government.
REFERENCES
- 1.Stivers JT. Extrahelical damaged base recognition by DNA glycosylase enzymes. Chemistry. 2008;14:786–793. doi: 10.1002/chem.200701501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hagen L, Pena-Diaz J, Kavli B, Otterlei M, Slupphaug G, Krokan HE. Genomic uracil and human disease. Exp. Cell Res. 2006;312:2666–2672. doi: 10.1016/j.yexcr.2006.06.015. [DOI] [PubMed] [Google Scholar]
- 3.Imai K, Slupphaug G, Lee WI, Revy P, Nonoyama S, Catalan N, Yel L, Forveille M, Kavli B, Krokan HE, et al. Human uracil-DNA glycosylase deficiency associated with profoundly impaired immunoglobulin class-switch recombination. Nat. Immunol. 2003;4:1023–1028. doi: 10.1038/ni974. [DOI] [PubMed] [Google Scholar]
- 4.Cao C, Jiang YL, Stivers JT, Song F. Dynamic opening of DNA during the enzymatic search for a damaged base. Nat. Struct. Mol. Biol. 2004;11:1230–1236. doi: 10.1038/nsmb864. [DOI] [PubMed] [Google Scholar]
- 5.Cao C, Jiang YL, Krosky DJ, Stivers JT. The catalytic power of uracil DNA glycosylase in the opening of thymine base pairs. J. Am. Chem. Soc. 2006;128:13034–13035. doi: 10.1021/ja062978n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Parker JB, Bianchet MA, Krosky DJ, Friedman JI, Amzel LM, Stivers JT. Enzymatic capture of an extrahelical thymine in the search for uracil in DNA. Nature. 2007;449:433–438. doi: 10.1038/nature06131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Porecha RH, Stivers JT. Uracil DNA glycosylase uses DNA hopping and short-range sliding to trap extrahelical uracils. Proc. Natl Acad. Sci. USA. 2008;105:10791–10796. doi: 10.1073/pnas.0801612105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hammes-Schiffer S, Benkovic SJ. Relating protein motion to catalysis. Annu. Rev. Biochem. 2006;75:519–541. doi: 10.1146/annurev.biochem.75.103004.142800. [DOI] [PubMed] [Google Scholar]
- 9.Jencks WP. Catalysis in Chemistry and Enzymology. New York: Dover; 1987. [Google Scholar]
- 10.Eisenmesser EZ, Millet O, Labeikovsky W, Korzhnev DM, Wolf-Watz M, Bosco DA, Skalicky JJ, Kay LE, Kern D. Intrinsic dynamics of an enzyme underlies catalysis. Nature. 2005;438:117–121. doi: 10.1038/nature04105. [DOI] [PubMed] [Google Scholar]
- 11.Volkman BF, Lipson D, Wemmer DE, Kern D. Two-state allosteric behavior in a single-domain signaling protein. Science. 2001;291:2429–2433. doi: 10.1126/science.291.5512.2429. [DOI] [PubMed] [Google Scholar]
- 12.Ishima R, Freedberg DI, Wang YX, Louis JM, Torchia DA. Flap opening and dimer-interface flexibility in the free and inhibitor-bound HIV protease, and their implications for function. Struct. Fold. Des. 1999;7:1047–1055. doi: 10.1016/s0969-2126(99)80172-5. [DOI] [PubMed] [Google Scholar]
- 13.Cole R, Loria JP. Evidence for flexibility in the function of ribonuclease A. Biochemistry. 2002;41:6072–6081. doi: 10.1021/bi025655m. [DOI] [PubMed] [Google Scholar]
- 14.Rozovsky S, Jogl G, Tong L, McDermott AE. Solution-state NMR investigations of triosephosphate isomerase active site loop motion: ligand release in relation to active site loop dynamics. J. Mol. Biol. 2001;310:271–280. doi: 10.1006/jmbi.2001.4673. [DOI] [PubMed] [Google Scholar]
- 15.Mol CD, Arvai AS, Slupphaug G, Kavli B, Alseth I, Krokan HE, Tainer JA. Crystal structure and mutational analysis of human uracil-DNA glycosylase: structural basis for specificity and catalysis [see comments] Cell. 1995;80:869–878. doi: 10.1016/0092-8674(95)90290-2. [DOI] [PubMed] [Google Scholar]
- 16.Salzmann M, Pervushin K, Wider G, Senn H, Wuthrich K. [13C]-constant-time [15N,1H]-TROSY-HNCA for sequential assignments of large proteins. J. Biomol. NMR. 1999;14:85–88. doi: 10.1023/a:1008346931993. [DOI] [PubMed] [Google Scholar]
- 17.Panchal SC, Bhavesh NS, Hosur RV. Improved 3D triple resonance experiments, HNN and HN(C)N, for HN and 15N sequential correlations in (13C, 15N) labeled proteins: application to unfolded proteins. J. Biomol. NMR. 2001;20:135–147. doi: 10.1023/a:1011239023422. [DOI] [PubMed] [Google Scholar]
- 18.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 19.Keller R. The computer aided resonance assignment tutorial. Goldau, Switzerland: Cantina Verlag; 2004. ISBN 3-85600-112-3. [Google Scholar]
- 20.Loria JP, Rance M, Palmer AG. A TROSY CPMG sequence for characterizing chemical exchange in large proteins. J. Biomol. NMR. 1999;15:151–155. doi: 10.1023/a:1008355631073. [DOI] [PubMed] [Google Scholar]
- 21.Goddard TD, Kneller DG. San Francisco: University of California; SPARKY 3. [Google Scholar]
- 22.Palmer AG., 3rd NMR characterization of the dynamics of biomacromolecules. Chem. Rev. 2004;104:3623–3640. doi: 10.1021/cr030413t. [DOI] [PubMed] [Google Scholar]
- 23.Palmer AG, 3rd, Kroenke CD, Loria JP. Nuclear magnetic resonance methods for quantifying microsecond-to-millisecond motions in biological macromolecules. Methods Enzymol. 2001;339:204–238. doi: 10.1016/s0076-6879(01)39315-1. [DOI] [PubMed] [Google Scholar]
- 24.Loria JP, Berlow RB, Watt ED. Characterization of enzyme motions by solution NMR relaxation dispersion. Acc. Chem. Res. 2008;41:214–221. doi: 10.1021/ar700132n. [DOI] [PubMed] [Google Scholar]
- 25.Stivers JT, Jiang YL. A mechanistic perspective on the chemistry of DNA repair glycosylases. Chem. Rev. 2003;103:2729–2759. doi: 10.1021/cr010219b. [DOI] [PubMed] [Google Scholar]
- 26.Krosky DJ, Song F, Stivers JT. The origins of high-affinity enzyme binding to an extrahelical DNA base. Biochemistry. 2005;44:5949–5959. doi: 10.1021/bi050084u. [DOI] [PubMed] [Google Scholar]
- 27.Stivers JT, Pankiewicz KW, Watanabe KA. Kinetic mechanism of damage site recognition and uracil flipping by escherichia coli uracil DNA glycosylase. Biochemistry. 1999;38:952–963. doi: 10.1021/bi9818669. [DOI] [PubMed] [Google Scholar]
- 28.Massi F, Johnson E, Wang C, Rance M, Palmer AG., 3rd NMR R1 rho rotating-frame relaxation with weak radio frequency fields. J. Am. Chem. Soc. 2004;126:2247–2256. doi: 10.1021/ja038721w. [DOI] [PubMed] [Google Scholar]
- 29.Parikh SS, Walcher G, Jones GD, Slupphaug G, Krokan HE, Blackburn GM, Tainer JA. Uracil-DNA glycosylase-DNA substrate and product structures: conformational strain promotes catalytic efficiency by coupled stereoelectronic effects. Proc. Natl Acad. Sci. USA. 2000;97:5083–5088. doi: 10.1073/pnas.97.10.5083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Suhre K, Sanejouand YH. ElNemo: a normal mode web server for protein movement analysis and the generation of templates for molecular replacement. Nucleic Acids Res. 2004;32:W610–W614. doi: 10.1093/nar/gkh368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cordier F, Hartmann B, Rogowski M, Affolter M, Grzesiek S. DNA recognition by the brinker repressor–an extreme case of coupling between binding and folding. J. Mol. Biol. 2006;361:659–672. doi: 10.1016/j.jmb.2006.06.045. [DOI] [PubMed] [Google Scholar]
- 32.Vise PD, Baral B, Latos AJ, Daughdrill GW. NMR chemical shift and relaxation measurements provide evidence for the coupled folding and binding of the p53 transactivation domain. Nucleic Acids Res. 2005;33:2061–2077. doi: 10.1093/nar/gki336. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.