


Abstract
Herein, we describe a case study into the population dynamics of in vitro selection, using RNA-cleaving DNAzymes as a model system. We sought to understand how the composition of the population can change over time in response to different levels of selection pressure, and how well these changes are correlated with selection of the target phenotype. The model population is composed of 857 DNAzyme clones representing 215 discrete sequence classes, which had previously been identified from two parallel selection experiments, conducted under an increasingly stringent, or permissive and constant selection time pressure. In this report, we determined the principal phenotypic properties (i.e. kobs, maximum cleavage yield and PCR efficiency) from a sample of 58 clones representing 46 different major and minor sequence classes from various rounds of each selection experiment. Interestingly, a positive correlation between the catalytic rate constant and the corresponding frequency and temporal position of a given DNAzyme was not consistently observed; however, the strength of the correlation was qualitatively higher under conditions of more stringent selection time pressure. These results suggest that the selective sampling paradigm on which in vitro selection is based, may underestimate the true functional capacity of any given random-sequence library.
INTRODUCTION
DNAzymes and deoxyribozymes are synonymous terms used to describe single-stranded DNA molecules that can function as enzymes by catalyzing specific chemical transformations. Since the report of the first DNAzyme in 1994 (1), many additional examples have been identified. To date, DNAzymes can catalyze more than a dozen different types of reactions and can provide rate enhancements as high 1010-fold over the corresponding uncatalyzed reaction (2). A continuing challenge in this nascent field is to map the fundamental limits of DNAzyme-mediated catalysis (3), in terms of both their functional versatility and functional proficiency as enzymes. However, there is an underlying question as to whether their true catalytic potential is limited by the lack of functional groups in DNA (relative to RNA and proteins), or simply by the method through which they are identified. So far, DNAzymes have not been found in nature, but have been exclusively isolated through a process known as ‘in vitro selection’.
In vitro selection is a selective amplification technique that can identify DNAzymes and other functional nucleic acids like ribozymes and aptamers (4), from chemically synthesized libraries of random sequences. There is a practical limit to the number of different sequences that can be probed in any particular experiment, which is typically well below the total theoretical number of permutations (i.e. sequence space) associated with a random sequence of a given length. For instance, it would require ∼3 × 107 kg of DNA to create a complete sequence library that includes all 450 (or ∼1030) different sequences of an oligonucleotide with 50 random nucleotide positions (5). Thus, an in vitro selection experiment can practically search only ∼1014 different sequences. From this example, it should be apparent that sequence space cannot be explored exhaustively, which underscores the importance of exploring sequence space more effectively. There are a number of experimental variables that must be considered in the design and implementation of in vitro selection experiments, and understanding how these variables can affect the outcome may facilitate the identification of new DNAzymes and other functional nucleic acids. Toward this end, a number of studies have provided insight into the significance of various factors including the length (6–9), nucleotide composition (10) and structural complexity of random-sequence libraries (11,12), as well as the type of divalent metal cofactors (13), selection time (14) and excess sequence elements (6,15).
The population dynamics of in vitro selection is another factor that should be considered, but is often taken for granted because it is not an independent variable that can be directly controlled. Nevertheless, it is important to understand how the population composition can change over time in response to different levels of selection pressure, because of the selective sampling paradigm on which in vitro selection is based. In vitro selection does not systematically screen every individual sequence for activity. Instead, only a very small fraction of the initial library (i.e. <10−12 of ∼1014 different sequences), typically from a single point in time (i.e. the terminal round), is actually tested for activity. The cost, time and labor-benefit of selective sampling is certainly justified when the population dynamics follow the expected pattern, characterized by a consistent increase in the frequency (i.e. copy number) of sequences with the target phenotype over successive rounds of selection. However, deviations from this presumed trajectory will reduce the effectiveness of this strategy, which is a possibility that has not been adequately addressed.
A few studies have used mathematical models to simulate how the composition of aptamer populations change in response to certain variables during in vitro selection (16–18), but the general validity of these theoretical studies may be limited by simplifying assumptions. Moreover, they cannot reproduce the complexity and stochastic effects that are inherent to real experimental model systems. For this reason, we previously initiated a case study into the population dynamics of in vitro selection (19), using a real population of RNA-cleaving DNAzymes as a model system. Beginning from the same population, two parallel selection experiments were conducted under an increasingly stringent or constant and permissive level of selection time pressure, and the change in population composition was documented by sequencing a total of 857 clones isolated from 17 different rounds of selection. This comprehensive sequencing effort not only allowed bulk changes in the composite population to be monitored, but also changes in the specific frequency of any one of the 215 discrete sequence classes that were ultimately identified. Interestingly, and contrary to expectations, we observed dynamic fluctuations in the structure of the population, including the presence of many transient sequence classes that peaked in frequency at different rounds of selection. The frequency of only one sequence class appeared to follow a consistently increasing trajectory.
In the current study, we sought to gain further insight into the population dynamics of this model system. In particular, we wanted to understand how well DNAzyme fitness was correlated with the selection of the target phenotype. Herein, fitness is defined simply as a measure of survival and reproductive success under the imposed selection conditions. Therefore, the fitness of a given DNAzyme should be directly correlated to, and manifested by, its frequency and temporal position along the generational timeline of an in vitro selection experiment. The catalytic rate constant for RNA cleavage (i.e. kobs) was the target phenotype of the original selection experiments, and represents our primary interest in this report. However, we examined a total of three different phenotypic characteristics including the kobs, maximum cleavage yield (Ymax) and PCR amplification efficiency, because each is suspected to play a role in the overall fitness of a DNAzyme. These phenotypic properties were measured for a diverse sample of DNAzyme clones representing both major and minor sequence classes from many different generations of each selection experiment.
MATERIALS AND METHODS
Oligonucleotides and reagents
Oligonucleotides were prepared by automated DNA synthesis using cyanoethylphosphoramidite chemistry (Keck Biotechnology Resource Laboratory, Yale University; Mobix Central Facility, McMaster University). DNA and RNA oligonucleotides were purified by 10% denaturing (7 M urea) polyacrylamide gel electrophoresis (PAGE) and their concentrations were determined by spectroscopic methods. Nucleoside 5′-triphosphates and [γ-32P]ATP were purchased from Amersham Pharmacia. T4 DNA ligase, T4 polynucleotide kinase (PNK), calf intestine alkaline phosphatase and T7 RNA polymerase were purchased from MBI Fermentas. All chemical reagents were purchased from Sigma. The 50-nt RNA substrate (S) was produced by in vitro transcription using T7 RNA polymerase and an appropriate double-stranded DNA template generated by PCR as described previously (14). The sequences of relevant oligonucleotides used in the library and for PCR are provided in Supplementary Figure S1. The sequences of all characterized DNAzyme clones are provided in Supplementary Figures S2 and S3.
In vitro selection and sequencing of selected DNA populations
Details of the in vitro selection scheme and subsequent cloning and sequence analysis of selected DNA populations have been described elsewhere (14,19).
Construction of fitness landscapes
The landscapes were constructed by grouping individual clones into common sequence classes as described previously (19), and then normalizing the absolute number of clones in a given class by the total number of clones sequenced in a particular generation to give the normalized frequency of each class in each generation. In an effort to emphasize the pattern of dominant sequence classes and minimize the visual clutter created by transient sequence classes composed of relatively few clones, we originally imposed an arbitrary cumulative threshold value of 0.1 as a criterion for inclusion into the landscape (19). In other words, if the sum of the normalized frequency values calculated across all generations of a given sequence class was ≥0.1 (i.e. ∼5 or more clones), the sequence class was included in the fitness landscape. This requirement was imposed after the normalized frequency values were calculated for all sequence classes. In this study, the aforementioned landscapes were modified to include several examples of minor sequence classes so that the phenotypic properties of both major and minor classes could subsequently be compared. It should be noted that the total sequence diversity exhibited in experiment A (i.e. 113 classes) and B (i.e. 172 classes) was simply too large to clearly illustrate in fitness landscapes; therefore, the exclusion of many minor sequence classes was considered necessary.
Construction of substrate-DNAzyme cis constructs
The 50-nt RNA substrate was first ligated to the upstream 14-nt DNA sequence (A1 = 5′ gga aac tag aca ga) in a separate large-scale ligation reaction. Typically, 2500 pmol of RNA substrate was combined with 2750 pmol ligation splint (5′ tct ctc tcc tct gtc tag) and 3000 pmol of A1, heated for 30 s at 90°C and allowed to cool at room temperature for ∼10 min. After cooling, 10 μl of 10 × ligase buffer (supplied by Fermentas) and 10 μl of T4 DNA ligase (5 Weiss units/μl) were added to initiate the ligation reaction. The reaction mixture (100 μl, total volume) was incubated at room temperature for 4 h or overnight. The ligated A1-RNA substrate was recovered by standard ethanol precipitation and purified by 10% denaturing PAGE. The A1-RNA substrate construct was 5′-32P labeled with PNK, extracted twice with phenol/chloroform/isoamyl alcohol (25:24:1), and ethanol precipitated before being used in separate small-scale ligation reactions with specific deoxyribozymes. Thirty picomoles of A1-substrate was combined with 90 pmol of 5′-phosphorylated deoxyribozyme, 36 pmol of ligation splint (5′ gcg tac gtg tcg aac ctg att cg) and 45 pmol of substrate cleavage blocker (5′ ttt acg taa cgc acc cat ctc tct cct ctg tct ag), heated at 90°C for 30 s and allowed to cool at room temperature for ∼5–10 min. The substrate cleavage blocker is a synthetic DNA oligonucleotide complementary to a section of the RNA substrate surrounding the cleavage site, and was used to minimize deoxyribozyme-mediated cleavage during ligation. After cooling, 1.25 μl of 10 × ligase buffer (supplied by Fermentas), 4 μl of T4 DNA ligase (5 Weiss units/μl) and DEPC-treated H2O were added to a final volume of 25 μl. The reaction was incubated at 37°C for 1 h, then ethanol precipitated and purified by 10% denaturing PAGE.
Kinetic analyses of DNAzymes
The rate constants for DNAzyme-catalyzed RNA cleavage were determined in an intramolecular self-cleavage format, which by default, is a single turnover reaction. This format is used during in vitro selection (as opposed to an intermolecular trans reaction), and therefore is most appropriate for the current study. Each 5′-32P labeled substrate-DNAzyme cis construct was heated at 90°C for 30 s, and allowed to cool at room temperature for ∼10 min. An equal volume of 2× reaction buffer (200 mM KCl, 800 mM NaCl, 15 mM MgCl2, 15 mM MnCl2, 100 mM HEPES pH 7.0 at 23°C) was then added to initiate the reaction. The reaction was terminated after a designated period of time by the addition of quenching buffer containing 60 mM EDTA, 7 M urea and loading dye solution. The cleavage products from a reaction time course (consisting of ∼13–16 different time points) were separated by denaturing 10% PAGE, and quantitated using a PhosphorImager and ImageQuant software. A graph of fraction cleaved versus time (t) was plotted for each timecourse, and the experimental data fit to either a single exponential equation Y = Ymax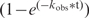 , or a double exponential equation
, or a double exponential equation 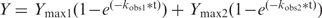 , using nonlinear regression analysis in GraphPad Prism 4, from which the observed rate constant (kobs) and maximum cleavage yield (Ymax) were determined. All cleavage reactions were observed to follow single-exponential kinetics, unless otherwise specifically noted. For clarity, the kinetic parameters from only the first phase (i.e. kobs1 and Ymax1) of biphasic reactions were plotted in Figure 4B and C. Kinetic parameters were determined from at least two independent experiments that typically differed by <30% for kobs values and <5% for Ymax values. The kobs values presented in Figure 3 represent initial rate constants that were determined previously by linear regression, as described elsewhere (19).
, using nonlinear regression analysis in GraphPad Prism 4, from which the observed rate constant (kobs) and maximum cleavage yield (Ymax) were determined. All cleavage reactions were observed to follow single-exponential kinetics, unless otherwise specifically noted. For clarity, the kinetic parameters from only the first phase (i.e. kobs1 and Ymax1) of biphasic reactions were plotted in Figure 4B and C. Kinetic parameters were determined from at least two independent experiments that typically differed by <30% for kobs values and <5% for Ymax values. The kobs values presented in Figure 3 represent initial rate constants that were determined previously by linear regression, as described elsewhere (19).
Figure 4.

The fitness and phenotypic landscapes of selection experiment A. (A) The fitness landscape adapted from ref. 19, shows changes in the frequency of specific DNAzyme sequence classes over multiple generations of in vitro selection. The selection time used for each generation is indicated. The number of clones observed in each sequence class was normalized to the total number of clones sequenced per generation. Sequence classes were arbitrarily arranged in an order that illustrates the evolutionary succession between competing classes, but is not intended to reflect any sequence homology. (B) Catalytic rate constants are shown for a representative sample of 21 DNAzyme clones isolated from different sequence classes and different generations of the selection experiment. The spatial arrangement and order of sequence classes from the fitness landscape in part A, have been preserved in this graph to facilitate comparison. Rate constants ranged in value from 0.14 min−1 (E2) to 2.52 min−1 (E18–G21), with no inactive clones. Sequence classes E23 and E77 exhibited biphasic kinetics for which only the first phase (kobs1) has been graphed: E23 kobs1 = 0.61 min−1, kobs2 = 0.024 min−1; E77 kobs1 = 0.36 min−1, kobs2 = 0.025 min−1. (C) Maximum cleavage yields (Ymax) corresponding to the same DNAzyme clones reported in (B) are shown. Only data from the first phase (Ymax1) of the reaction catalyzed by the clones in sequence classes E23 and E77 are graphed: E23 Ymax1 = 26%, Ymax2 = 34%; E77 Ymax1 = 35%, Ymax2 = 32%.
Figure 3.

Genotypic and phenotypic progression of the composite population, adapted from ref. 19. The diversity ratio and catalytic rate constant (kobs) were determined over multiple generations, to trace changes in both the genotypic and phenotypic character of the composite population. The diversity ratio is a measure of the sequence diversity within the population, and is defined as the number of different sequence classes divided by the number of sequenced clones. In experiment A, the selection pressure was increased by decreasing the selection time from 5 h to 5 s. In experiment B, the selection time was maintained at 5 h in every generation.
Determination of PCR amplification efficiency
PAGE-purified synthetic DNA oligonucleotides corresponding to specific DNAzyme clones were used as the template for PCR1 reactions. The product of each PCR1 reaction was used as the input template for the corresponding PCR2 reaction. A master reaction mixture composed of all components necessary for PCR, except the DNAzyme template, was prepared and then separated into 25-μl aliquots. Each aliquot contained 0.2 mM of each type of dNTP, 2.5 μl of 10× PCR buffer (supplied by Biotools), 0.5 μM each of forward and reverse primers, 1.25 U Tth DNA polymerase (Biotools) and 0.25 μl of 50× SYBR green (Molecular Probes). PCR reactions were conducted in a 96-well plate format using the Stratagene Mx3000P QPCR System. PCR amplification was monitored in real time using SYBR green as a fluorescence reporter. Each PCR cycle consisted of 94°C for 30 s, 50°C for 45 s and 72°C for 40 s.
The PCR amplification efficiency for a given DNAzyme sequence was determined using the following steps: (i) The cycle threshold (Ct) was measured for a set of four reactions representing 10-fold serial dilutions of the DNAzyme template (the cycle threshold refers to the cycle at which the level of fluorescence crosses a predefined threshold above background). (ii) A graph of Ct versus the logarithm of the dilution factor was plotted, from which the slope was used to calculate the percent amplification efficiency (E) using the equation E = 10−(1/slope) – 1 (20). Each PCR efficiency value was determined from duplicate trials.
RESULTS
A model system based on RNA-cleaving DNAzymes
The model system described herein was originally developed through two previous studies (14,19). A recap of the relevant experimental details is presented below, in order to provide sufficient context to understand the results of new experiments.
We previously devised an in vitro selection strategy to isolate RNA-cleaving DNAzymes from a library of chimeric molecules, each containing 80 random-sequence deoxyribonucleotides and 50 fixed-sequence ribonucleotides (14). Details of the selection scheme are provided in Figure 1, which has been adapted from ref. 14. A single selection experiment was initially conducted for seven rounds under a constant, and relatively permissive, 5-h selection time. Detectable cleavage products were not observed for the first four rounds of selection; however, three different catalytic subpopulations appeared in generation 5 (G5), each corresponding to a unique cleavage site along the 50-nt RNA substrate domain. In G7, one of the three subpopulations of DNAzymes was chosen to serve as the common starting pool for two subsequent selection experiments conducted in parallel. This subpopulation exhibited robust cleavage of the RNA substrate at a specific 5′-GG dinucleotide junction, and a preliminary assessment of the population composition revealed extensive genetic variation (i.e. 43 unique sequence classes were identified from 48 sequenced clones). We therefore reasoned that this system could provide an interesting case study to investigate the dynamics of population change over multiple rounds of in vitro selection (19). The two experiments, denoted as A and B, differed in the stringency of the selection time, which served as the main form of selection pressure (Figure 2). The same selection buffer was used throughout both experiments, and consisted of 100 mM KCl, 400 mM NaCl, 7.5 mM MgCl2, 7.5 mM MnCl2 and 50 mM HEPES pH 7.0 at 23°C. In experiment A, the selection time was progressively reduced from 5 h (G7) to 30 min (G8), to 5 min (G9–G11), to 30 s (G12–G14) and finally to 5 s (G15–G24). In experiment B, a constant selection time of 5 h was used during each selection round (G7–G30). The relatively large number of selection rounds conducted in each experiment was intended to broaden the evolutionary timescale, in order to increase the sensitivity of the model system for detecting slow or latent evolutionary trajectories.
Figure 1.

In vitro selection strategy. (A) Library design. Each molecule in the library contains 80 random-sequence nucleotides (N80) as the putative DNAzyme domain, and a substrate domain composed of 50 fixed-sequence ribonucleotides (shown in red). The random domain is flanked by a 9-nt forward primer binding site (FPBS), and a 15-nt reverse-primer-binding site (RPBS). RPBS1 was used from generations 0 to 7. In generation 8, the population was split into two parallel selection experiments and different reverse primer binding sites (RPBS2) were used to minimize cross contamination during PCR. The RNA substrate is preceded by a 14-nt fixed DNA sequence intended to facilitate the separation of cleavage products during PAGE. (B) In vitro selection cycle. Each round of selection consists of steps 1–9. In step 1, the 32P-radiolabeled DNAzyme domain is 5′-phosphorylated, and then ligated to the substrate domain in step 2. The ligation product is purified by denaturing polyacrylamide gel electrophoresis (PAGE) in step 3, and incubated with the selection buffer (containing the divalent metal cofactors Mn2+ and Mg2+) to promote metal-dependent DNAzyme-mediated cleavage of the attached RNA substrate in step 4. The cleavage reaction is allowed to proceed for a designated period and stopped with the addition of the metal-chelating agent, EDTA. Although cleavage can potentially occur anywhere along the 50 ribonucleotides comprising the substrate domain, the population of DNAzymes described herein cleaved at one specific site (denoted by the arrow). The 3′-cleavage fragment(s) containing the DNAzyme domain is subsequently purified by PAGE in step 5. Two consecutive polymerase chain reactions (PCR) are used in steps 6 and 7 to amplify the cleavage fragments. The forward and reverse primers used in PCR 1 are denoted as P1 and P2, respectively. P1 contains extra nucleotides at the 5′ end (denoted in blue) that introduce a new forward priming site for the second PCR. PCR 2 uses the same reverse primer as PCR 1, but a different forward primer denoted as P3. The P3 primer contains a 3′-terminal ribonucleotide. In step 8, the double-stranded PCR product is treated with NaOH to cleave the embedded ribonucleotide, which regenerates the single-stranded DNAzyme domain. The DNAzyme domain is isolated by denaturing PAGE (step 9), and used to initiate the next round of selection.
Figure 2.
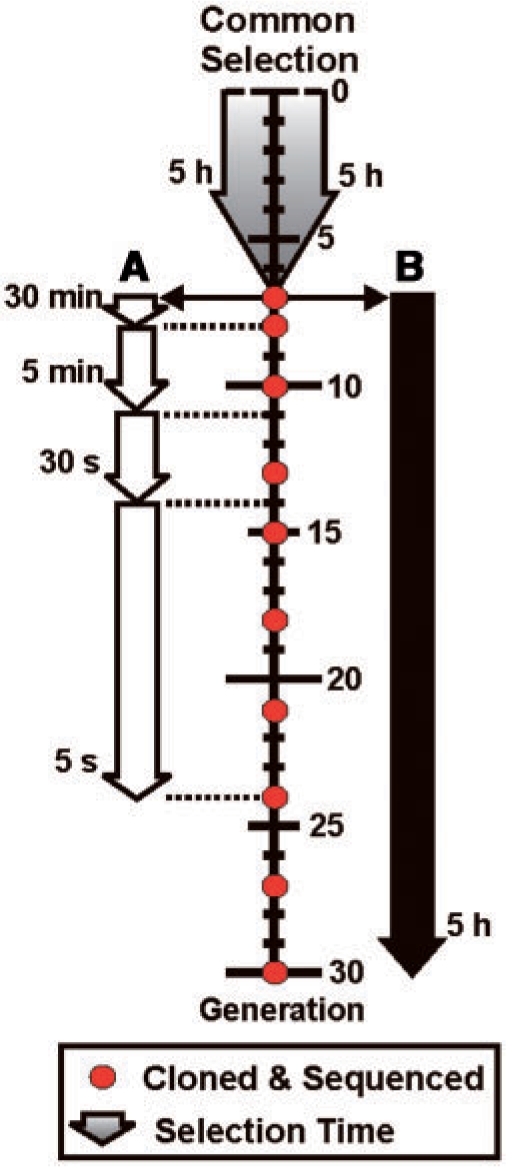
Summary of the selection time used during each round of selection. A single selection experiment was conducted from generation 0 to 7 using a selection time of 5 h. The population at generation 7 served as the common starting pool for two parallel selection experiments conducted under different levels of selection time pressure. In experiment A, the selection time was progressively reduced over a total of 24 rounds. In experiment B, the selection time was maintained at a constant value over a total of 30 rounds. The sequences of ∼50 random clones were determined at each generation denoted by a red circle, in both experiments A and B.
To minimize PCR cross-contamination between the two parallel selection experiments, different primer-binding sites were appended to the 3′ end of each population of DNAzymes in G8. Approximately 50 clones from each of generations 7, 8, 10, 13, 15, 18, 21, 24, 27 and 30 were sequenced for both experiments A (up to G24 only) and B. A total of 215 different sequence classes were identified from 857 sequenced clones (19). In general, individual clones fell into discrete sequence classes as reflected by an average sequence identity of >90% within classes, versus an average sequence identity of only ∼30% between classes. Furthermore, the maximum sequence identity shared between any two classes did not exceed 56%. A complete summary of the distribution and frequency of clones within each sequence class and selection experiment is described elsewhere (19).
Changes in the catalytic rate constant and sequence diversity of the composite population were traced over the course of each selection experiment (Figure 3, adapted from ref. 19). In experiment A, a relatively rapid reduction in the sequence diversity occurred until G15, after which point the sequence diversity ratio (calculated by dividing the number of discrete sequence classes by the number of sequenced clones) fluctuated around an average value of ∼0.2 (corresponding to ∼10 sequence classes). The composite population in experiment A also experienced an ∼17-fold increase in the catalytic rate constant between G7 (kobs = 0.036 min−1) and G15 (kobs = 0.62 min−1), but did not increase significantly beyond G15 when the selection time was held constant. A very different scenario was observed in experiment B, which was subjected to a constant 5-h selection time during every round of selection. Interestingly, a steady decrease in the sequence diversity occurred despite the fact that the catalytic rate constant did not increase between G7 and G30.
Experiment A: mapping the catalytic rate constant and maximum cleavage yield onto the fitness landscape
The distribution of fitness values in a population of DNAzymes can be visualized as a three-dimensional landscape to study evolutionary changes over time, in analogy to the original concept of fitness landscapes proposed by Sewell Wright (21). In the context of this study, fitness landscapes were used to show the change in frequency of specific DNAzyme sequence classes over successive generations of selective amplification. Figure 4A shows the fitness landscape corresponding to 16 different sequence classes, which account for ∼14% of the total number of classes, and ∼66% of the total number of clones identified in experiment A (19). This landscape has been modified from the original version (19), which contained only major sequence classes based on normalized frequency, and as defined by an arbitrary cumulative threshold value of 0.1 (see ‘Materials and Methods’ section). Figure 4A now contains several minor classes that appear transiently and in low copy number, which were added so that better insight could be gained from subsequent comparative analyses of fitness and phenotype between classes. The remaining sequence classes were excluded from the landscape simply to minimize visual clutter, and their absence is not expected to change any conclusions reported herein. However, this restricted sample set should be taken into consideration when interpreting the fitness landscape. For instance, while it may not be readily apparent from the landscape shown in Figure 4A, the sequence diversity has converged significantly, as previously indicated in Figure 3.
In general, the fitness landscape is characterized by a relatively continuous transition in dominant sequence classes; that is, sequence classes that dominate near the beginning of the selection experiment are superseded by other classes in subsequent rounds of selection. Presumably, the increase in selection pressure (i.e. decrease in selection time) helps to elicit changes in the temporal distribution of sequences by eliminating DNAzymes with lower catalytic activity. Only one sequence class, E18, appears to rise steadily to dominance over repeated rounds of selection. It should be noted that the staggered appearance of the sequence classes across generational time is probably due in part to the sample size used to construct the landscapes (i.e. ∼50 clones sequenced per generation), particularly during the early generations when the sequence diversity was relatively high.
By itself, the fitness landscape in Figure 4A does not show any phenotypic information. To determine how well the frequency and temporal position of a given sequence class correlates with the target phenotype, we constructed a phenotypic landscape (in analogy to the preceding fitness landscape) based on the catalytic rate constants of individual DNAzyme clones. Figure 4B shows the kobs values of 21 representative clones chosen from among each sequence class, and in some cases from more than one generation within a given sequence class. It should be noted that all rate constants were determined in a ‘cis’ format, in which the substrate domain is covalently attached to the DNAzyme domain, as is the case during in vitro selection. The measured rate constants range in value from 0.14 min−1 (E2) to 2.52 min−1 (E18-G21), and no clones were inactive. A general correlation is observed between the catalytic rate constant and the frequency/position of a given sequence class along the landscape. For instance, sequence classes that were eliminated during earlier selection rounds tend to have lower rate constants than classes that survived until later rounds (under increased selection time pressure). However, there were also some notable deviations from this trend. For instance, sequence classes E42 and E14 exhibit high rate constants of 2.1 and 1.8 min−1, respectively, but nevertheless decreased in frequency after generation 15. This observation is contrary to expectations. Sequence class E75 actually exhibits lower rate constants (i.e. 0.87 min−1 and 0.31 min−1 for different clones from G15 and G21, respectively) than E42 and E14, but represents a greater fraction of the population in later generations (i.e. from G18 onward). Another interesting pair of sequence classes are E88 and E25, which both appear very late in the selection experiment, despite having competitive rate constants of 1.6 and 1.8 min−1, respectively. E88 appears for the first time in G24 as a very minor sequence class, and E25 appears only transiently in G15 before making a resurgence in generations 21 and 24.
Another phenotypic characteristic that could potentially influence the frequency and temporal position of different DNAzyme sequence classes is the maximum cleavage yield (Ymax). It is not unusual for DNAzyme catalyzed reactions to exhibit incomplete substrate conversion, which can be due to misfolding and/or other factors discussed elsewhere (22). Figure 4C shows the corresponding Ymax values for each of the clones reported in Figure 4B. The maximum cleavage yields range from 26% (E23) to 80% (E18); however, it should be noted that the clones from sequence classes E23 and E77 exhibited biphasic kinetics, which are characterized by a rapid initial phase of substrate cleavage followed by a slower second phase. For clarity, only the value of the first phase (Ymax1) is shown in Figure 4C (and similarly for the corresponding kobs1 values presented in Figure 4B). The average Ymax value derived from representative clones of the seven consecutive sequence classes from E18 to E14 is ∼69%, which is ∼9% higher than the average value for the nine consecutive sequence classes from E74 to E121 (calculated using the sum of Ymax1 and Ymax2 for the biphasic reactions exhibited by E23 and E77); this observation is consistent with the dominance of the former group of sequences during later rounds in the experiment.
Experiment B: mapping the catalytic rate constant and maximum cleavage yield onto the fitness landscape
Figure 5A shows the fitness landscape corresponding to 38 different sequence classes, which account for ∼22% of the total number of classes, and ∼63% of the total number of clones identified in experiment B (19). This landscape has been modified from the original version described previously (19), by incorporating several examples of minor sequence classes to augment the existing collection of major classes (as defined previously for experiment A). Similar to the results from experiment A, the population in experiment B is subject to dynamic changes, as reflected by the continuous transition in dominating sequence classes. Moreover, almost no sequence classes appear to rise steadily in frequency. However, in contrast to the relatively narrow and peaked distribution of classes observed in the fitness landscape of experiment A (Figure 4A), the shape of the landscape from experiment B is characterized by a broader and more flat distribution of classes (Figure 5A).
Figure 5.

The fitness and phenotypic landscapes of selection experiment B. (A) The fitness landscape adapted from ref. 19, shows changes in the frequency of specific DNAzyme sequence classes over multiple generations of in vitro selection. The number of clones observed in each sequence class was normalized to the total number of clones sequenced per generation. Sequence classes were arbitrarily arranged in an order that illustrates the evolutionary succession between competing classes, but is not intended to reflect any sequence homology. Blank ‘spacer’ intervals were inserted into the graph to provide an unobstructed view of certain classes. Sequence class E8/9R is labeled as such, because it is the suspected recombination product of class E8 and E9 (19). (B) Catalytic rate constants are shown for a representative sample of 34 DNAzyme clones isolated from different sequence classes and different generations of the selection experiment. The spatial arrangement and order of sequence classes from the fitness landscape in (A), have been preserved in this graph to facilitate comparison. Rate constants ranged in value from 0.02 min−1 (E2–G21) to 1.44 min−1 (E18), with no inactive clones. (C) Maximum cleavage yields (Ymax) corresponding to the same DNAzyme clones reported in (B) are shown.
The corresponding phenotypic landscapes in Figure 5B and C show the kinetic parameters (kobs and Ymax, respectively) for 34 individual clones representing various sequence classes. The measured rate constants range in value from 0.02 min−1 (E2-G21) to 1.44 min−1 (E18), and no inactive clones were observed. These phenotypic landscapes reveal some interesting trends. A number of sequence classes that were eliminated by G15, actually exhibit higher rate constants than the classes that supplanted them in subsequent generations. For instance, E18, which exhibited the highest rate constant among the tested clones (i.e. kobs = 1.4 min−1), was only observed transiently in generations 8, 10 and 15. In general, there does not appear to be any consistent positive correlation between the target phenotype and the frequency/position of DNAzymes along the fitness landscape. Higher cleavage yields might therefore be expected to compensate for the lower rate constants, in order to produce the observed distribution of sequence classes. The magnitude of the Ymax values plotted in Figure 5C are relatively comparable across the landscape, indicating that differential cleavage yields were not a primary determinant of the selection outcome. In fact, the average Ymax value for the 15 consecutive classes from E38 to E198 is actually ∼10% higher than the average value for the 15 consecutive classes from E3 to E53, which dominated later in the selection experiment.
Intra-class phenotypic variations
On average, clones were assigned to a common sequence class when they shared >90% nucleotide identity; nevertheless, we wondered if small sequence variations would cause any significant phenotypic differences between clones from the same class. To address this issue, we chose a sample of sequence classes (i.e. E18, E25, E75, E14, E8, E2 and E4), and determined the rate constants for different clones within each class (Figures 4B and 5B). The following pairs of clones are from the same generation within each given class, so the kobs value of only one clone per pair could be explicitly shown in Figure 4B: E18G15–C1 (1.0 min−1) and E18G15–C6 (1.0 min−1); E42G15–C16 (2.1 min−1) and E42G15–C24 (1.1 min−1); E74G15–C30 (0.48 min−1) and E74G15–C33 (1.3 min−1). The observed variation in catalytic activity between clones from the same sequence class ranged from neutral (e.g. E8) to as high as ∼5-fold (e.g. E14). Therefore, intra-class phenotypic variations could potentially contribute to the dynamic fluctuations observed in the frequency and temporal position of some sequence classes. These sequence variations were likely acquired as spontaneous mutations during PCR, which can have maladaptive, neutral or adaptive consequences for the fitness of a DNAzyme. For instance, the E14 class identified in experiment A, appears to be a case where an adaptive mutation may have extended its evolutionary trajectory along the fitness landscape (Figure 4A). In G10, class E14 exhibited a rate constant of only 0.34 min−1, but this value increased to 1.7 and 1.8 min−1 in the following generations (Figure 4B). If not for the acquisition of an adaptive mutation(s), E14 would likely have been eliminated during earlier rounds of selection along with other slower DNAzyme classes such as E105.
In a different study, we demonstrated that the catalytic activity of E14 is mediated by a small DNAzyme motif known as ‘8-17’, which is embedded near the 5′ end of the random-sequence domain (15). The 8-17 DNAzyme contains a well-defined catalytic core measuring only 14–15 nt in length, and forms a three-way helical junction structure when it binds to the substrate through Watson–Crick base-pairing on either side of the cleavage site (Figure 6A). Sequence elements peripheral to this motif, and the nucleotides immediately adjacent to it (which are required for substrate binding), were shown to be largely unnecessary for activity as determined by deletion analyses (15). Therefore, it is reasonable to attribute variations in the activity of different E14 clones to specific mutations in the 8-17 catalytic core. Furthermore, 8-17 has been the subject of systematic mutational analyses reported elsewhere (23,24), so the functional consequences of different mutations can be readily interpreted. Figure 6B traces the change in the 8-17 motif of E14 over successive generations. In generations 7 and 8, E14 is composed of a single type of 8-17 motif. However, in G10, the E14 class is composed of 67% of the original ‘wild-type’ sequence, and 33% of a new sequence containing a C12T substitution. This C12T mutation is associated with a higher rate constant (23,24), consistent with the measured values shown in Figure 6B. In generation 13, the C12T mutant accounts for 89% of the E14 class, and appears to go to fixation by G15, representing 100% of the class thereafter. Interestingly, E14 was also identified as a transient class that appeared as a single clone in G10, G13, G15 and G18 in experiment B. The adaptive C12T mutation was also observed in G10 and G15 of experiment B, but only in combination with other mutations that are expected to be maladaptive.
Figure 6.

Catalytic activity of E14 is mediated by an 8-17 motif. (A) Schematic diagram of the library design showing the relative location of an embedded 8–17 motif, and presumed folding into its secondary structure. The 8–17 motif engages the substrate sequence through Watson–Crick base-pairing (vertical lines) on either side of a specific GG dinucleotide cleavage site, to form a three-way helical junction. The 8–17 is composed of 15 nt, characterized by a 3-bp stem terminating in a tri-loop, and a 5-nt single-stranded bulge region. Only the four underlined nucleotides are highly conserved. The specific 8–17 sequence observed in E14 is shown. (B) Sequence analysis showing acquisition and subsequent fixation of an adaptive mutation at position 12 (boxed) in the 8–17 motif of E14, over successive generations (G) of in vitro selection. The percentage of the population represented by a given 8–17 sequence is indicated for each generation, along with the actual number of clones. Catalytic rate constants are provided for three representative clones from the indicated generations.
Different selection pressure promotes divergent evolutionary trajectories
E14 is actually just one of 29 sequence classes that were identified in both experiment A and B, of which six of the most prominent (based on copy number) are presented in Figures 4 and 5 (i.e. E18, E42, E14, E23, E2 and E25). Experiments A and B started with the same G7 pool of sequences, so the relatively low level of recurrence (29/215 ≈ 13%) observed between these experiments underscores how easily the outcome of selection can change in response to different selection pressures. Figure 7 illustrates the remarkably different evolutionary trajectories taken by the same sequence class (E18) in experiment A versus B. In experiment A, the frequency of E18 progressively increases until it reaches a maximum of ∼73% of the total population in G21 (before declining slightly to 55% in G24). In contrast, E18 appears only transiently (i.e. ∼2% of the population) in generations 8, 10 and 15 of experiment B. We determined the rate constants of specific clones from both experiments to eliminate the possibility that intra-class phenotypic variations might account for the observed difference in trajectories; however, all the tested clones exhibited comparable rate constants >1 min−1 (Figure 7). It should also be noted that 103 of the 106 clones comprising the E18 class contain identical 8-17 motifs, including all three clones from experiment B.
Figure 7.

Divergent evolutionary trajectories of sequence class E18. The catalytic rate constant and maximum cleavage yield are shown for select clones isolated from the designated generations. In experiment A, the selection time was progressively decreased from 5 h to 5 s. In experiment B, the selection time was maintained at 5 h.
PCR amplification efficiency
We sought to determine if there was any correlation between the frequency/temporal distribution of sequence classes along the fitness landscape, and the efficiency with which they are amplified during PCR. We speculated that significant variations in amplification efficiency could obscure the expected relationship between the catalytic rate constant and corresponding frequency/temporal position of a given sequence class. For instance, a relatively high amplification efficiency might compensate for a low catalytic activity, and relatively low amplification efficiency might hamper the benefits of high catalytic activity. The frequency of specific DNAzymes in the fitness landscape would therefore reflect the total contribution made by each of these factors (and Ymax).
A representative sample of sequences were chosen from experiments A and B, and the amplification efficiency was determined for both of the PCR1 and PCR2 amplification steps involved in the selection scheme (Figure 1B). A total of 480 PCR reactions were conducted for this purpose (Figure 8). It should be noted that the forward primer differed between PCR 1 and PCR 2, while the reverse primer differed between experiments A and B. The amplification efficiency of 13 sequences chosen from experiment A ranged in value from 69% to 80%, with an average value of 75% for both PCR 1 and PCR 2. The amplification efficiency of 17 sequences from experiment B ranged in value from 66% to 92%, with an average value of 82% for PCR 1 and 78% for PCR 2. According to the results shown in Figure 8, there does not appear to be any systematic correlation between PCR amplification efficiency and the frequency/position of individual DNAzymes in the fitness landscapes.
Figure 8.

PCR amplification efficiency. Each value represents the average of two duplicate experiments, and error bars correspond to the range. The efficiency of both PCR 1 (yellow bars) and PCR 2 (blue bars) are shown. The sequence classes are arranged in the same relative order (from left to right) as they appear in the fitness landscapes of Figures 4 and 5. The forward primer differs between PCR 1 and PCR 2, while the reverse primer differs between Experiments A and B. A representative sample of sequence classes are shown for experiments A and B.
DISCUSSION
The selective sampling paradigm of in vitro selection
With the isolation of many hundreds of different deoxyribozymes, ribozymes and aptamers over the last 18 years, it has become increasing apparent that random-sequence space contains many types of functional nucleic acids beyond what can be found in nature. Significant progress has been made toward mapping the boundaries of nucleic acid function, although the true functional limits may be difficult to define because of the sheer scale of global sequence space. Nevertheless, we can make inferences about the global distribution, and functional potential of nucleic acids, by investigating subsets of sequence space through in vitro selection. Although the smaller number of sequences probed in an in vitro selection experiment is more manageable, the question of how best to access functional molecules is still very relevant, since the distribution of target sequences is not known a priori. Furthermore, in vitro selection relies on selective sampling, rather than the systematic screening of each sequence, to identify molecules with the target phenotype. The challenge, therefore, is to implement a selection strategy that can reliably separate and detect the rare functional sequences, from the majority of nonfunctional sequences present in the initial random-sequence library.
With in vitro selection, the general expectation is that sequences with the target phenotype should increase in frequency with repeated cycles of selective amplification. Typically, the experiment is terminated after a plateau in the activity of the composite population is reached (or as otherwise determined by the specific reasoning of the experimenter), and the population from the terminal round is then subjected to molecular cloning and sequencing to identify specific sequences that can be studied in isolation. If the terminal pool consists of multiple sequences, they may be grouped into major and minor classes according to their copy number, and the major classes are usually assumed to represent the best candidates for detailed functional analysis. In this way, only a very small sample of the genetic diversity in the original library is subject to any real scrutiny. It follows that the effectiveness of this selective sampling process will depend on the strength of the relationship between the target phenotype, and the frequency and temporal position of sequences in the selection experiment.
Understanding population dynamics in a model system of RNA-cleaving DNAzymes
The beginning and end of most in vitro selection experiments are generally well characterized, but the path in between has seldom been examined in detail, if at all. This scenario essentially precludes an analysis of the population dynamics, and the ability to detect any important changes in the composition of the population. Several notable exceptions do exist, all of which involve model systems based on pools of partially randomized variants of the Tetrahymena self-splicing ribozyme, undergoing in vitro evolution for improved and/or altered function (25–30).
In previous work, we established a different type of model system to study population dynamics, based on a pool of unrelated RNA-cleaving DNAzyme sequences undergoing in vitro selection. Changes in the composite population and the frequency of individual sequence classes were traced over time, and in response to either increasingly stringent (experiment A), or constant and permissive (experiment B) selection time pressure. Dynamic fluctuations in the population composition were observed over time, and simple computer simulations were used as a preliminary effort to investigate the influence of the three phenotypic characteristics expected to have the greatest impact on the survival and reproduction of DNAzymes; the catalytic rate constant (i.e. the target phenotype), maximum cleavage yield and PCR amplification efficiency. The utility of these computer simulations was limited by several factors including a simple algorithm, a small sample set (consisting of only eight sequence classes), and the assignment of hypothetical and arbitrary values to each of the phenotypic properties. In the current study, we sought to extend our understanding of the population dynamics in this model system. Toward this end, we determined the actual phenotypic values for a sample of 58 representative DNAzyme clones distributed between and within, major and minor sequence classes, and various rounds of selection.
In general, a positive correlation was observed between the target phenotype and the fitness of a given sequence (as reflected by the frequency and temporal position along the generational timeline), but the strength of this correlation depended directly on the stringency of the applied selection pressure. In experiment A, which was subjected to relatively stringent time pressure, a larger fraction of the population that was found in later rounds of the experiment in higher frequencies, exhibited higher catalytic rate constants than those that disappeared in earlier rounds. In contrast, under the relatively permissive time pressure used in experiment B, the sequences found in later rounds of the experiment often exhibited lower activity than those observed in earlier rounds. Nevertheless, it should be noted that while fitness was more strongly correlated to the target phenotype in experiment A, the population in this selection pathway was still susceptible to unexpected changes such as the decline of relatively fast DNAzymes (e.g. E42 and E14), and the sudden appearance of ‘new’ DNAzymes (e.g. E88) late in the selection.
It has become clear from our investigation that some DNAzymes, which we refer to as ‘latent’, can only be accessed under special circumstances. First, the identification of latent DNAzymes may require a thorough analysis of the population diversity. This is particularly important when the target phenotype is not strongly correlated with the frequency and temporal position of a DNAzyme. For instance, we identified several relatively fast DNAzymes in earlier rounds of selection that were not present in the terminal round population. A similar scenario was observed in the original selection experiment from which the well known 8-17 and 10-23 DNAzymes were isolated (31); the 8-17 motif was part of a subpopulation of sequences that dominated in rounds 6–8, but was ultimately superseded by a different subpopulation in rounds 9 and 10, which contained the 10-23 motif. A thorough survey of the activity between different classes can also be important, because relatively fast DNAzymes were not only identified in the major sequence classes, but also among some of the minor classes. Furthermore, phenotypic variations may exist within classes, which can lead to an underestimation of catalytic activity, depending on the representative clone(s) chosen for analysis.
Second, the manifestation of some latent DNAzymes may depend on the acquisition of one or more adaptive mutations, which can occur by chance during PCR. For instance, E14 likely acquired an adaptive mutation that served to extend its evolutionary trajectory, and therefore likelihood of detection (Figure 6). Because only a small fraction of the total sequence space is typically included in the initial random-sequence library, some of the functional molecules will invariably be represented by suboptimal sequences. However, the optimal sequence variant may be located only a short mutational distance away, and could potentially be accessed by random mutagenesis. It should be noted that the acquisition of such adaptive mutations will occur serendipitously, and can only be indirectly manipulated by interference with the PCR mutation rate (32).
The identification of latent DNAzymes can also be facilitated by the application of stringent selection pressure, which appears to strengthen a positive correlation between the target phenotype and frequency/temporal position of a DNAzyme. This conclusion is supported by the fact that E18 was reliably identified in experiment A (under stringent time pressure), but appeared only transiently in experiment B (under permissive time pressure), despite having one of the highest measured catalytic rate constants in this study. In general, the low level of recurrence observed between the outcome of experiments A and B is predicted to be largely due to the difference in selection pressure. However, it should be noted that there are several general factors that can influence the probability of recurrence in any replicate in vitro selection/evolution experiments, as discussed in detail elsewhere (33).
Limitations of the study
The unexpected fluctuations observed in the evolutionary trajectories of some DNAzyme sequence classes, cannot be adequately explained even when all three phenotypic properties are taken into consideration. Therefore, we can deduce that additional factors are obscuring the expected relationship between fitness and phenotype. It is beyond the objective and scope of this study to try to elucidate all of these factors, and their precise contributions to each of the many evolutionary trajectories observed herein. Such efforts would likely be futile, given the complexity of the problem. However, we can speculate about several factors that could potentially cause fluctuations in the population composition.
A combination of subsampling and genetic drift is one such factor that may help to explain the population fluctuations. Some form of subsampling occurs between each step in the in vitro selection cycle. For instance, only a fraction of the total product from PCR1 is used to seed PCR2, and only a fraction of the DNA pool is recovered after each PAGE purification step. The subsampling that occurs with each of these laboratory manipulations can potentially have a ‘bottleneck’ effect that decreases the effective population size (33), which in turn may cause a decrease in the sequence diversity and hasten genetic drift.
Another intriguing possibility is that some form of stabilizing or negative selection could be acting on the population. In general, stabilizing selection acts to remove phenotypic extremes when an intermediate phenotype represents an optimum for fitness (34). In our model system, DNAzymes were provided with both 7.5 mM Mg2+ and 7.5 mM Mn2+ as cofactors to facilitate the cleavage reaction during the main selection step of each round. However, DNAzymes that can function in a lower concentration of cofactors (i.e. only 5 mM Mg2+ is provided in the substrate-DNAzyme ligation reaction), would be susceptible to negative selection during the ligation step of each round. We tested this hypothesis by conducting simple cleavage assays to verify DNAzyme activity under the ligation buffer conditions (data not shown). Most of the DNAzymes exhibited some level of cleavage activity (typically <10-fold lower than the activity exhibited in the original selection buffer), indicating that negative selection effects could have factored into the observed frequencies of different DNAzymes.
In this study, we used a relatively large model system that consisted of more than 800 sequenced DNAzyme clones; nevertheless, we cannot exclude the possibility that some of the observed fluctuations in DNAzyme frequency may be artifacts of the sampling size (i.e. ∼50 clones/round). However, the sample size used herein meets or exceeds the typical size used in most in vitro selection experiments, and should provide relevant insight into the potential issues these experiments may encounter.
Although we have investigated the population dynamics of in vitro selection through a specific case study involving RNA-cleaving DNAzymes, our findings should be of general interest regardless of the target phenotype and model system. We suspect that most selection experiments can be susceptible to the same type of unpredictable population changes observed herein, because the selective amplification and sampling process of in vitro selection is fundamentally conserved, even if the specific details may differ from one selection experiment to another. The potential generality of our conclusions has been evidenced in the outcome of another in vitro selection experiment recently conducted by our laboratory (35). In this other experiment, we sought to isolate RNA-cleaving DNAzymes that could efficiently cleave pyrimidine–pyrimidine junctions. In addition to a different library design featuring a chimeric substrate and 20-nt random region, we reduced the total number of experimental steps involved in each round of selection to minimize the potential effects of subsampling. The number of PCR reactions per round was also reduced to minimize potential PCR bias, and the ligation step was removed to alleviate the negative selection effect described earlier. Despite these alterations (and in the presence of increasingly stringent time pressure), we still observed examples where the frequency and temporal position of a given sequence was not directly correlated with the target phenotype. Due to the scarcity of in vitro selection studies that have examined the population dynamics in sufficient detail, it remains to be determined just how pervasive this effect may be.
CONCLUSIONS
By scrutinizing not just the outcome of in vitro selection, but the path taken to get there, we have discerned significant deviations from the expected pattern of evolutionary change in a model population of RNA-cleaving DNAzymes. Our results demonstrate that the presence of the target phenotype in the initial random-sequence library is necessary, but not sufficient, to guarantee detection through in vitro selection. The particular design and implementation of a selection strategy can dramatically alter the outcome, which underscores the inherent difficulty in exploring sequence space effectively, and mapping the functional limits of DNAzymes accurately. Two important measures can be taken to improve the likelihood of identifying ‘latent’ functional sequences, which can only be accessed under certain conditions. First, in the context of catalysis, the application of stringent selection pressure (i.e. reaction time) strengthens a positive correlation between the frequency/temporal position and the target phenotype (i.e. magnitude of the catalytic rate constant). Second, because this positive correlation can potentially be obscured by unpredictable factors, there may be a significant benefit to assessing the population composition at multiple stages in the selection experiment, and assessing the activity of representative clones from both major and minor sequence classes.
SUPPLEMENTARY DATA
Supplementary Dataare available at NAR Online.
FUNDING
A Discovery grant from the Natural Sciences and Engineering Research Council of Canada; Y.L. is a Canada Research Chair; K.S. holds a Natural Sciences and Engineering Research Council Doctoral Canada Graduate Scholarship. Funding for open access charge: the Canada Research Chairs Program.
Conflict of interest statement. None declared.
Supplementary Material
REFERENCES
- 1.Breaker RR, Joyce GF. A DNA enzyme that cleaves RNA. Chem. Biol. 1994;1:223–229. doi: 10.1016/1074-5521(94)90014-0. [DOI] [PubMed] [Google Scholar]
- 2.Baum DA, Silverman SK. Deoxyribozymes: useful DNA catalysts in vitro and in vivo. Cell Mol. Life Sci. 2008;65:2156–2174. doi: 10.1007/s00018-008-8029-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schlosser K, Li Y. DNAzyme-mediated catalysis with only guanosine and cytidine nucleotides. Nucleic Acids Res. 2009;37:413–420. doi: 10.1093/nar/gkn930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Silverman SK. Artificial functional nucleic acids: aptamers, ribozymes and deoxyribozymes identified by in vitro selection. In: Lu Y, Li Y, editors. Functional Nucleic Acids for Analytical Applications. New York: Springer; 2009. pp. 47–108. [Google Scholar]
- 5.Achenbach JC, Chiuman W, Cruz RP, Li Y. DNAzymes: from creation in vitro to application in vivo. Curr. Pharm. Biotechnol. 2004;5:321–336. doi: 10.2174/1389201043376751. [DOI] [PubMed] [Google Scholar]
- 6.Sabeti PC, Unrau PJ, Bartel DP. Accessing rare activities from random RNA sequences: the importance of the length of molecules in the starting pool. Chem. Biol. 1997;4:767–774. doi: 10.1016/s1074-5521(97)90315-x. [DOI] [PubMed] [Google Scholar]
- 7.Coleman TM, Huang F. Optimal random libraries for the isolation of catalytic RNA. RNA Biol. 2005;2:129–136. doi: 10.4161/rna.2.4.2285. [DOI] [PubMed] [Google Scholar]
- 8.Legiewicz M, Lozupone C, Knight R, Yarus M. Size, constant sequences, and optimal selection. RNA. 2005;11:1701–1709. doi: 10.1261/rna.2161305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Knight R, Yarus M. Finding specific RNA motifs: function in a zeptomole world? RNA. 2003;9:218–230. doi: 10.1261/rna.2138803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Knight R, De Sterck H, Markel R, Smit S, Oshmyansky A, Yarus M. Abundance of correctly folded RNA motifs in sequence space, calculated on computational grids. Nucleic Acids Res. 2005;33:5924–5935. doi: 10.1093/nar/gki886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gevertz J, Gan HH, Schlick T. In vitro RNA random pools are not structurally diverse: a computational analysis. RNA. 2005;11:853–863. doi: 10.1261/rna.7271405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carothers JM, Oestreich SC, Davis JH, Szostak JW. Informational complexity and functional activity of RNA structures. J. Am. Chem. Soc. 2004;126:5130–5137. doi: 10.1021/ja031504a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang W, Billen LP, Li Y. Sequence diversity, metal specificity, and catalytic proficiency of metal-dependent phosphorylating DNA enzymes. Chem. Biol. 2002;9:507–517. doi: 10.1016/s1074-5521(02)00127-8. [DOI] [PubMed] [Google Scholar]
- 14.Schlosser K, Li Y. Tracing sequence diversity change of RNA-cleaving deoxyribozymes under increasing selection pressure during in vitro selection. Biochemistry. 2004;43:9695–9707. doi: 10.1021/bi049757j. [DOI] [PubMed] [Google Scholar]
- 15.Schlosser K, Lam JC, Li Y. Characterization of long RNA-cleaving deoxyribozymes with short catalytic cores: the effect of excess sequence elements on the outcome of in vitro selection. Nucleic Acids Res. 2006;34:2445–2454. doi: 10.1093/nar/gkl276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Irvine D, Tuerk C, Gold L. SELEXION. Systematic evolution of ligands by exponential enrichment with integrated optimization by non-linear analysis. J. Mol. Biol. 1991;222:739–761. doi: 10.1016/0022-2836(91)90509-5. [DOI] [PubMed] [Google Scholar]
- 17.Sun F, Galas D, Waterman MS. A mathematical analysis of in vitro molecular selection-amplification. J. Mol. Biol. 1996;258:650–660. doi: 10.1006/jmbi.1996.0276. [DOI] [PubMed] [Google Scholar]
- 18.Vant-Hull B, Payano-Baez A, Davis RH, Gold L. The mathematics of SELEX against complex targets. J. Mol. Biol. 1998;278:579–597. doi: 10.1006/jmbi.1998.1727. [DOI] [PubMed] [Google Scholar]
- 19.Schlosser K, Li Y. Diverse evolutionary trajectories characterize a community of RNA-cleaving deoxyribozymes: a case study into the population dynamics of in vitro selection. J. Mol. Evol. 2005;61:192–206. doi: 10.1007/s00239-004-0346-7. [DOI] [PubMed] [Google Scholar]
- 20.Meijerink J, Mandigers C, van de Locht L, Tonnissen E, Goodsaid F, Raemaekers J. A novel method to compensate for different amplification efficiencies between patient DNA samples in quantitative real-time PCR. J. Mol. Diagn. 2001;3:55–61. doi: 10.1016/S1525-1578(10)60652-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wright S. The roles of mutation, inbreeding, cross-breeding, and selection in evolution. In: Jones DF, editor. Proceedings of the 6th International Congress of Genetics. Vol. 1. New York: Ithaca; 1932. pp. 356–366. [Google Scholar]
- 22.Carrigan MA, Ricardo A, Ang DN, Benner SA. Quantitative analysis of a RNA-cleaving DNA catalyst obtained via in vitro selection. Biochemistry. 2004;43:11446–11459. doi: 10.1021/bi049898l. [DOI] [PubMed] [Google Scholar]
- 23.Schlosser K, Gu J, Sule L, Li Y. Sequence–function relationships provide new insight into the cleavage site selectivity of the 8-17 RNA-cleaving deoxyribozyme. Nucleic Acids Res. 2008;36:1472–1481. doi: 10.1093/nar/gkm1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Peracchi A, Bonaccio M, Clerici M. A mutational analysis of the 8-17 deoxyribozyme core. J. Mol. Biol. 2005;352:783–794. doi: 10.1016/j.jmb.2005.07.059. [DOI] [PubMed] [Google Scholar]
- 25.Beaudry AA, Joyce GF. Directed evolution of an RNA enzyme. Science. 1992;257:635–641. doi: 10.1126/science.1496376. [DOI] [PubMed] [Google Scholar]
- 26.Lehman N, Joyce GF. Evolution in vitro of an RNA enzyme with altered metal dependence. Nature. 1993;361:182–185. doi: 10.1038/361182a0. [DOI] [PubMed] [Google Scholar]
- 27.Lehman N, Joyce GF. Evolution in vitro: analysis of a lineage of ribozymes. Curr. Biol. 1993;3:723–734. doi: 10.1016/0960-9822(93)90019-k. [DOI] [PubMed] [Google Scholar]
- 28.Tsang J, Joyce GF. Evolutionary optimization of the catalytic properties of a DNA-cleaving ribozyme. Biochemistry. 1994;33:5966–5973. doi: 10.1021/bi00185a038. [DOI] [PubMed] [Google Scholar]
- 29.Tsang J, Joyce GF. Specialization of the DNA-cleaving activity of a group I ribozyme through in vitro evolution. J. Mol. Biol. 1996;262:31–42. doi: 10.1006/jmbi.1996.0496. [DOI] [PubMed] [Google Scholar]
- 30.Lehman N, Donne MD, West M, Dewey TG. The genotypic landscape during in vitro evolution of a catalytic RNA: implications for phenotypic buffering. J. Mol. Evol. 2000;50:481–490. doi: 10.1007/s002390010051. [DOI] [PubMed] [Google Scholar]
- 31.Santoro SW, Joyce GF. A general purpose RNA-cleaving DNA enzyme. Proc. Natl Acad. Sci. USA. 1997;94:4262–4266. doi: 10.1073/pnas.94.9.4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cadwell RC, Joyce GF. Mutagenic PCR. PCR Methods Appl. 1994;3:S136–S140. doi: 10.1101/gr.3.6.s136. [DOI] [PubMed] [Google Scholar]
- 33.Lehman N. Assessing the likelihood of recurrence during RNA evolution in vitro. Artif. Life. 2004;10:1–22. doi: 10.1162/106454604322875887. [DOI] [PubMed] [Google Scholar]
- 34.Diaz Arenas C, Lehman N. Darwin's concepts in a test tube: parallels between organismal and in vitro evolution. Int. J. Biochem. Cell Biol. 2009;41:266–273. doi: 10.1016/j.biocel.2008.08.034. [DOI] [PubMed] [Google Scholar]
- 35.Schlosser K, Gu J, Lam JC, Li Y. In vitro selection of small RNA-cleaving deoxyribozymes that cleave pyrimidine-pyrimidine junctions. Nucleic Acids Res. 2008;36:4768–4777. doi: 10.1093/nar/gkn396. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.