


Abstract
The progress of replication forks is often threatened in vivo, both by DNA damage and by proteins bound to the template. Blocked forks must somehow be restarted, and the original blockage cleared, in order to complete genome duplication, implying that blocked fork processing may be critical for genome stability. One possible pathway that might allow processing and restart of blocked forks, replication fork reversal, involves the unwinding of blocked forks to form four-stranded structures resembling Holliday junctions. This concept has gained increasing popularity recently based on the ability of such processing to explain many genetic observations, the detection of unwound fork structures in vivo and the identification of enzymes that have the capacity to catalyse fork regression in vitro. Here, we discuss the contexts in which fork regression might occur, the factors that may promote such a reaction and the possible roles of replication fork unwinding in normal DNA metabolism.
INTRODUCTION
The idea that replication forks break down and act as a significant source of genome instability has gained widespread acceptance over the last decade. This popularity has not arisen because of a realization that lesions within the template DNA can block replication—such inhibition was demonstrated many decades ago (1,2). Rather, the potential links between genome instability and replication fork breakdown have gained prominence because of in vivo and in vitro studies that suggest movement of replication forks is inhibited far more frequently than previously suspected (3). Central to many models of how impeded replication forks are processed is the idea that a blocked fork can be unwound to generate a four-stranded DNA structure resembling a Holliday junction formed during recombination, first proposed in 1976 (Figure 1) (4,5). This so-called ‘fork regression’ has been proposed by many groups to aid the repair of damaged forks although the details of how such regressed forks (also referred to as ‘chicken foot’ structures (6)) might be processed differ between models. Indeed, at first glance, it is difficult to see how unwinding of a blocked replication fork could enhance the efficiency of repair of such a fork. The original block would be unaffected by this unwinding (Figure 1) and so would presumably be able to block the progression of a reassembled replisome. However, the strand switching involved in regression might facilitate excision repair of single-stranded DNA lesions by relocation of such damage into regions of duplex DNA, a process that might also facilitate lesion bypass rather than repair (see below, Figure 5). More generally, access of repair enzymes to DNA lesions could be hampered by the presence of a blocked replication fork, and fork unwinding might relieve this inhibition (see below, Figures 6–8).
Figure 1.

Blockage of replication fork movement and the possible unwinding of leading and lagging daughter strands to form a four-stranded DNA structure. 3′ ends of DNA strands are indicated by arrowheads.
Figure 5.

Fork regression followed by reversal. (A) A lesion in the leading strand template could result in the formation of a blocked fork with a gap on the leading strand. (B) Fork regression would reposition the 3′ end of the blocked leading strand so that it would be paired with the nascent lagging strand, whilst the DNA lesion would be relocated back into the reformed parental duplex. (C and D) Bypass of the lesion could be effected by extension of the leading strand using the lagging strand as a template followed by reversal of fork regression. (E and F) Repositioning of the lesion back into the parental duplex could also facilitate repair rather than bypass. Extension of the leading strand using the nascent lagging strand and reversal of regression would reconstitute a fork structure on to which the replication apparatus could be reloaded.
Figure 6.
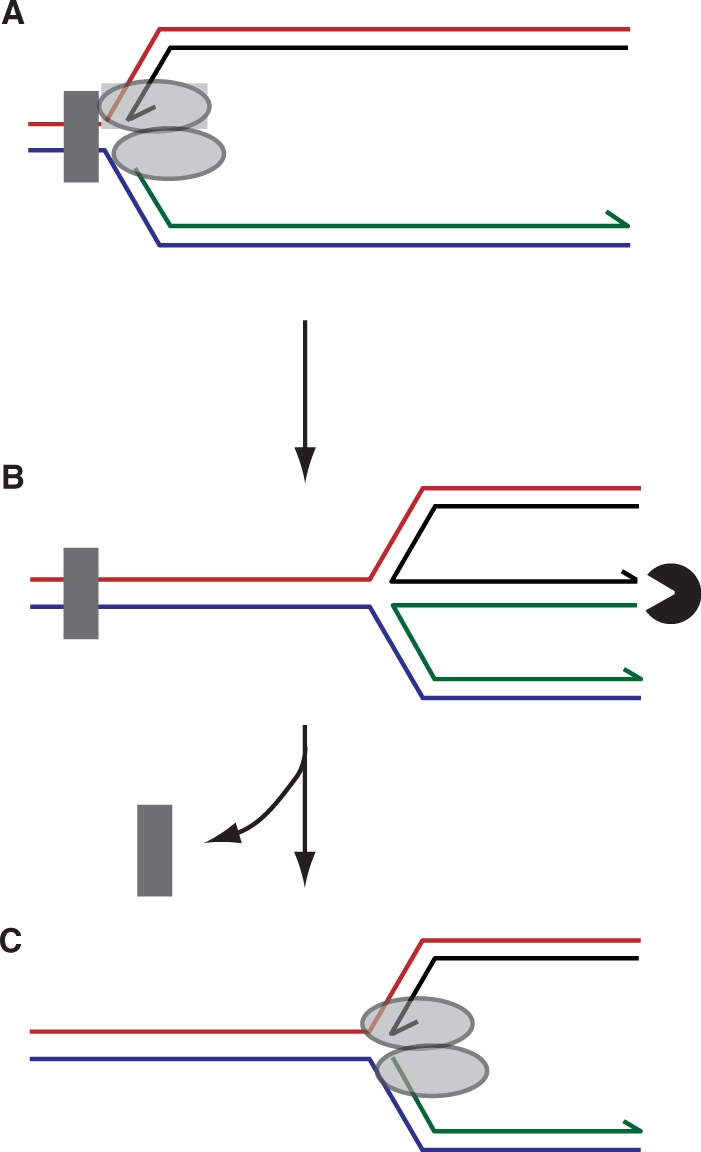
Restoration of a fork structure after regression by degradation of the extruded duplex arm. Blockage of a replisome (A) followed by fork regression and exonuclease-mediated degradation of the dsDNA end (B) would restore a fork structure onto which the replisome could be reassembled (C). Concomitantly, repositioning of the blocking lesion away from the fork may facilitate access of repair enzymes to the lesion. Note that, in this model, a block is depicted in which both leading and lagging strand synthesis is inhibited resulting in formation of a blunt dsDNA end by regression. A leading strand template-specific lesion might result in an extruded duplex arm with an extended ssDNA overhang (Figure 5B) rather than a blunt dsDNA end. Given the DNA structure specificities exhibited by exonucleases, different extruded DNA ends would require exonucleases with appropriate specificities.
Figure 7.
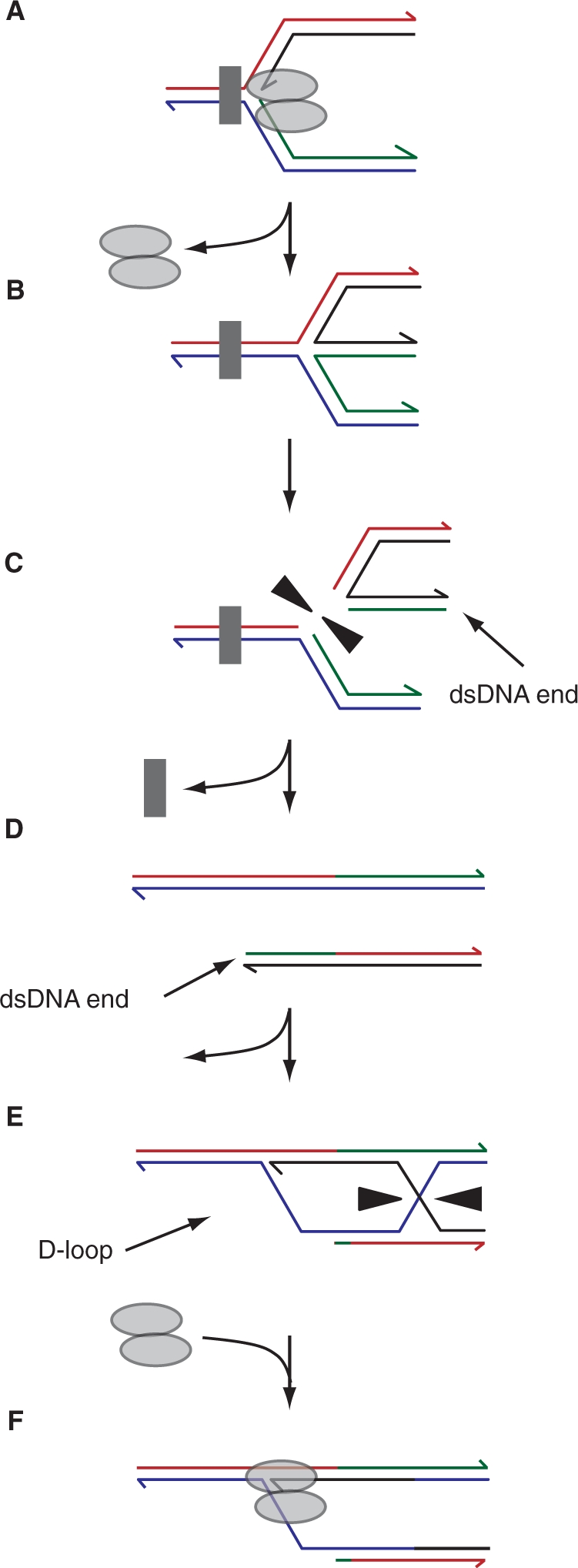
Regression and Holliday junction cleavage. Blockage of a fork followed by regression and cleavage of the four-stranded structure (A–C) would generate a dsDNA end (D). Processing of this end to promote loading of strand exchange proteins would result in D-loop formation with the intact sister duplex (E). Loading of the replication machinery onto this D-loop and resolution of the connected Holliday junction would restore an intact replication fork (F). Assuming the original block could be cleared, possibly aided by repositioning of the block away from the fork to promote access by repair enzymes (C), then replication fork progression could resume.
Figure 8.

Regression followed by recombination of the extruded dsDNA end. Any exonucleolytic processing of the dsDNA end generated by fork regression might result in generation of a 3′ ssDNA tail rather than complete degradation (A–C). Recombination between this ssDNA and the homologous sequence in the reformed parental duplex would result in formation of a D-loop linked to two Holliday junctions (D). Cleavage of the two linked Holliday junctions by resolvases (as shown in E) or dissolution by a RecQ-type helicase in conjunction with a topoisomerase, coupled with reassembly of the replisome at the D-loop would restore a replication fork (F).
This review aims to summarize the evidence that replication fork unwinding does occur, the situations in which it might occur and with what frequency. The possible consequences of fork regression and subsequent processing of these regressed forks on genome duplication and stability will also be explored. As will be seen, there are many unanswered questions within this field, in part because much of the data concerning fork regression is indirect in nature. We have aimed therefore for a balanced overview of this area, which might give the impression of equivocation. However, we believe that the many uncertainties regarding fork regression demand such an approach until more direct evidence becomes available.
WHEN MIGHT FORK REGRESSION OCCUR?
The assumption behind most models invoking replication fork reversal is that fork unwinding occurs only upon disengagement of the replication machinery from the forked DNA. Consideration of the disposition of components of the replication machinery at the replication fork supports this assumption. Firstly, disruption of the primer-template junction on the leading strand upon fork regression would inhibit interaction of the leading strand polymerase with DNA. Secondly, fork regression would also displace the replicative helicase away from the ends of the newly synthesized DNA strands.
If fork unwinding occurs after, or concomitant with, disengagement of the replication machinery from the fork, when might such disengagement occur? The high processivities of replication suggest that the probabilities of active replisomes disengaging from the chromosome spontaneously are very low. Disengagement of the replication machinery might therefore occur only upon blockage of replication. Many types of DNA lesion within single strands of template DNA are known to inhibit replicative DNA polymerases (7,8) whilst interstrand cross-links inhibit processes involving DNA strand separation (9). Such DNA damage can arise due to both endogenous and exogenous agents (10). Moreover, proteins bound to the template DNA, in particular transcribing RNA polymerases, present barriers to fork progression both in vivo and in vitro (11,12) as do covalent protein–DNA complexes (13). Protein–DNA barriers to replication also include pre-programmed blocks such as the Escherichia coli Tus-ter system and the eukaryotic replication fork barrier (RFB) located within rDNA repeats (14–16).
If replication fork regression occurs only upon blockage of a fork and disengagement of the replication enzymes from the fork, then the frequency of fork regression will be determined, at least in part, by the frequency of blockage and the stability of replisome association with a blocked fork. These two critical parameters will be dealt with below.
FREQUENCY OF REPLICATION BLOCKAGE
Bacteria
Analysis of replication blockage often involves the use of DNA-damaging agents such as UV light or depletion of deoxyribonucleotide pools by treatment with hydroxyurea, all of which increase the probability of replication blockage. However, estimating the frequency with which forks come to a halt in vivo in the absence of exogenous agents has proven difficult, largely because of the multiple overlapping mechanisms that exist in cells in order to repair blocked forks. For instance, in E. coli replication might be promoted past a lesion in the leading strand template by a specialized translesion DNA polymerase (17), by processing of forks by the strand exchange protein RecA (18) or by PriC-directed replication fork reloading downstream of the lesion (19). Thus, the phenotypic consequences of inactivation of a single fork repair pathway are often uninformative as regards the probability of replication blockage.
One potential method of avoiding the complications of overlapping pathways is to use mutants deficient specifically in the ability to reload the replication apparatus after blockage. A study of E. coli cells with a temperature-sensitive mutation in DnaC, thought to be required only for the loading of the replicative helicase onto the chromosome, revealed that ∼18% of cells failed to complete chromosome duplication during a single round of replication under non-permissive conditions (20). These data suggest that replication fork blockage is a relatively rare event, at least in E. coli. However, whether the mutant DnaC used in this study is completely defective at the non-permissive temperature is unknown and so cells might have retained at least some ability to rebuild blocked replication forks. Indeed, other studies have suggested a higher frequency of fork blockage in E. coli. Reloading of the replication apparatus back onto the chromosome away from the normal origin of replication, oriC, requires either PriA or PriC in addition to DnaC (21). Strains with deletions in either priA or priC are viable, whereas strains deleted for both genes are inviable, implying that most cells do require the ability to reload replisomes away from oriC even in the absence of elevated levels of DNA damage (21). The lethality of combinations of mutations in recombination and repair genes [see, for example, (22,23)], also hints at a high frequency of replication fork problems.
Eukaryotes
Estimates of the frequency of replication fork blockage in eukaryotes are even less precise. Little is known concerning replication fork reloading mechanisms in eukaryotes (see below) and so specific disabling of any putative replisome reloading mechanism(s), as described above for E. coli, is currently not possible. Mutations in recombination enzymes that may promote fork restart, such as the strand exchange protein Rad51, do lead to loss of viability (24). However, recombination enzymes are involved in many aspects of DNA metabolism, not just the processing of blocked replication forks. Thus the pleiotropic effects of recombination defects make it difficult to correlate viability defects with specific defects in resumption of replication. However, as in E. coli, growth defects exhibited by combinations of mutations in genes encoding recombination and/or repair enzymes do indicate that fork progression is often interrupted [see, for example, (25,26)].
BLOCKED FORK STABILITIES
Bacteria
If fork unwinding is likely to occur only upon fork blockage and replisome disengagement, how long do replisomes remain associated with DNA after blockage? The functional half-life of blocked E. coli replisomes reconstituted in vitro is 4–6 min regardless of whether forks are halted by accumulation of positive torsional strain (27) or by collision with protein–DNA complexes (28). Whether this loss of functionality in vitro is associated with complete dissociation of the replisome remains unknown but these data do imply that at least some components of a blocked replisome have limited stability. However, blocked fork stabilities of minutes in vitro contrast with the observed association of E. coli replication enzymes at the site of a site-specific replication block for several hours in vivo, as measured by live-cell imaging (29,30). Such forks, even after being blocked for 2 h, retained the ability to resume replication rapidly upon removal of the block (29).
Comparison of blocked fork stabilities in vitro and in vivo might suggest the presence of stabilizing factors present within E. coli cells but absent in reconstituted replication systems in vitro. However, the apparent prolonged association of replication enzymes at a blockage site in vivo might also reflect continual dissociation and then reloading of the replication machinery via PriA or PriC (21). It remains possible therefore that disengagement of the replisome, and thus the potential for unwinding of the fork, occurs within a few minutes of fork blockage in E. coli.
Eukaryotes
Saccharomyces cerevisiae replisomes blocked by nucleoprotein complexes or by depletion of deoxynucleotide pools remain associated with blocked forks for up to 60 min in vivo, as measured by chromatin immunoprecipitation and PCR (31,32). These data support extended fork stabilities in vivo. However, whilst fork-reloading systems similar to those found in E. coli remain to be identified in eukaryotes, the ability to initiate replication at recombination intermediates (33) implies such systems do exist. Indeed, restart of at least partially disassembled replication forks has been documented in Xenopus (34). Thus monitoring of blocked fork stabilities in eukaryotic cells by chromatin immunoprecipitation might be complicated by continual replisome dissociation and reloading, as in E. coli. However, although there is little evidence that stabilization factors exist in bacteria, there is evidence of active stabilization of blocked forks in eukaryotes. In yeast, Mrc1 and Tof1 travel with replication forks (35,36) and may play a structural role within the replisome to ensure concerted unwinding and DNA polymerization (37,38) even in the absence of replication blocking agents (39). The replication checkpoint also promotes retention of function of eukaryotic forks blocked by hydroxyurea treatment or DNA alkylation, facilitating continued replication upon removal of the block (40,41). Although the molecular basis of this stabilization by Mec1 and Rad53 kinases in S. cerevisiae is unclear, there is evidence that Mec1 promotes stabilization of DNA polymerases at blocked forks whilst Rad53 promotes retention of the replicative helicase at such forks (32,42). Such fork stabilization mechanisms imply that any regression of forks might be delayed in comparison with blocked forks in bacteria.
This possible reduced opportunity for fork regression in eukaryotes might reflect the multiple origins of replication present in eukaryotic chromosomes. Thus, the problem of a blocked fork in a eukaryotic chromosome might be solved by the arrival of a converging fork from an adjacent origin (43). Rapid initiation of blocked fork processing in eukaryotes might therefore be unnecessary, and possibly even dangerous, given the potential for genome rearrangements upon engagement of recombination mechanisms (44). However, the apparently greater stability of blocked forks in eukaryotes as opposed to bacteria does not exclude the possibility that active processing of blocked forks, including fork regression, is a requirement for the maintenance of genome stability and/or cell viability given that a single unreplicated region within a genome could prove lethal. Frequency of use might not therefore reflect importance.
THE STRUCTURE OF BLOCKED REPLICATION FORKS
Different blocks to DNA replication are likely to result in the formation of different blocked fork structures. The semi-discontinuous nature of DNA synthesis at replication forks suggests that single-strand-specific blocks on the lagging strand template can be bypassed by re-priming lagging strand synthesis on the 3′ side of the lesion, leaving a ssDNA gap in the lagging strand duplex to be repaired later by recombination (Figure 2A) (45–48). Single-strand-specific lesions on the leading strand template may result in a different outcome. The leading strand polymerase might be inhibited but continued movement of the replicative helicase would continue to generate ssDNA, allowing lagging strand synthesis to continue some way beyond the 3′ end of the blocked leading strand resulting in a blocked fork with a leading strand gap, possibly of many hundreds of bases (Figure 2Bi) (49–52). However, recent work using E. coli proteins has suggested that re-priming of leading strand synthesis can occur at a fork in vitro (Figure 2Bii) (19), providing a mechanistic explanation for the repeated detection of what appears to be discontinuous leading strand synthesis in the presence of UV light-induced pyrimidine dimers or defective DNA ligase (53–56). Leading strand lesions might therefore be bypassed by re-priming in the same manner as lagging strand lesions. Such models imply that exposure of cells to DNA-damaging agents that affect primarily single strands of DNA should not have a major impact on replication fork progression, at least in bacteria which do not possess the replication checkpoints found in eukaryotes. However, E. coli cells exposed to UV light experience substantial delays in replication, recovery depends on lesion removal and also on the ability to reload replisomes back onto the chromosome, all of which suggests that re-priming of leading strand synthesis downstream of lesions might not be a frequent event (57–60). The extent to which leading strand synthesis is re-primed downstream of a lesion is therefore unclear.
Figure 2.

Possible structures of forks halted by single-strand-specific blocks such as UV-light-induced pyrimidine dimers located on either the lagging strand template (A) or the leading strand template (B).
In contrast to single-strand-specific blocks, double-strand-specific blocks, such as interstrand cross-links or nucleoprotein complexes, are likely to inhibit both the polymerases and the helicase. Analysis of S. cerevisiae replication forks halted by the preprogrammed nucleoprotein block in the rDNA cluster revealed the presence of only three bases of ssDNA on the leading strand template (61) whilst reconstituted E. coli replisomes blocked by the Tus-ter nucleoprotein complex possessed a ssDNA region on the lagging strand template of 50–70 bases (62). Regardless of the precise disposition of the leading and lagging strands at such blocks, both these studies imply little if any gap in the leading strand and a gap of at most a few tens of bases on the lagging strand.
EVIDENCE THAT FORK REGRESSION DOES OCCUR
Initial observations
The idea of fork regression was first suggested in 1974 (63) and received experimental support in 1976 (4,5). The formation of heavy/heavy DNA using bromodeoxyuridine pulse-labelling of replicating human cells treated with an alkylating agent or with UV light implied that pairing of the nascent DNA strands could occur during DNA replication (4,5). Strauss and co-workers (4) also detected four-armed structures in partially replicated DNA using electron microscopy. These data could be explained by extrusion of a duplex from a replication fork, resulting in the formation of double-stranded DNA in which both strands were newly synthesized (Figure 1). Formation of duplexes, containing only nascent DNA, was detected in several subsequent studies (64–67) but some of this work suggested that branch migration of replication forks was an artefact of DNA extraction or of labelling (64,65). Regardless of whether fork regression could occur in vivo, these studies did highlight the possibility that replication forks had the potential to unwind and form four-stranded DNA structures that resembled Holliday junctions.
Genetic evidence
The concept of fork regression received little attention during the 1980s, possibly due to the lack of evidence that such a reaction had a physiological role. However, fork regression began to be employed to explain various genetic observations in the 1990s. Hyperrecombination was detected in the replication termination region of the E. coli chromosome (68), a region in which protein–DNA complexes (Tus-ter) act as pre-programmed polar blocks to replication (15). Regression of a blocked fork was proposed to initiate these recombination events, with the extruded fourth arm suggested to be a substrate for recombination (68). Other work demonstrated that defects in the E. coli replicative helicase DnaB or the putative accessory replicative helicase Rep resulted in the accumulation of double-stranded DNA breaks (69), which could be explained by regression and subsequent processing of blocked replication forks (70). Fork regression has also been invoked to explain genome instability associated with fork blockage in the S. cerevisiae rDNA region (71,72) and processing of forks blocked by transcribing RNA polymerases (73) and UV light-induced DNA damage (18) in E. coli.
This rebirth of the concept of fork regression led to the application of fork regression models to explain, at least in part, many aspects of cell viability, DNA repair and genome stability in relation to the interplay between replication and recombination in E. coli (3,43,74). Such models were also adopted to explain observations made in eukaryotic systems (44,75).
Thus, the ability of models invoking fork regression to explain at least some mutant phenotypes supports the concept of fork regression, albeit indirectly. However, such models are often contradictory which may reflect the difficulty in extrapolating what might happen in wild-type cells from observations made using mutants. For instance, cells deficient in an enzyme that processes a specific intermediate of recombination might accumulate this intermediate. An increased concentration of this intermediate might then allow processing by a second enzyme which results in a series of reactions that normally does not occur in wild-type cells. Contradictory models might also reflect the use of different methods to block replication. The nature of the initial block will have a profound effect on the structure of the blocked fork (see above) and thus how the blocked fork might be processed, as highlighted by a recent comparison of processing of forks in E. coli blocked either by inactivation of the replicative helicase or by UV light-induced DNA damage (76).
These interpretation problems aside, genetic analyses are a powerful tool in studying fork blockage, regression and processing. Unlike more direct detection methods, genetic assays are exquisitely sensitive to the presence of a blocked and unrepaired replication fork. Just one persistent blocked fork can have major impacts on cell viability, providing a sensitive assay for analysis of blocked fork processing (29,77–80).
Electrophoretic evidence
Electrophoretic techniques provide more direct monitoring of the structures of blocked replication forks. Neutral-neutral 2-D gel electrophoresis has been particularly informative in this regard since separation of DNA is based on both size and shape (81). Thus, branched DNA structures corresponding to replication bubbles, replication forks and four-armed Holliday junction structures can all be resolved (82). This approach has been used to detect DNA structures that might have arisen by fork regression since such Holliday junction-like structures (hereafter referred to as Holliday junctions) appear to migrate as a cone/spike above the ‘Y’ arc within such gels that is sensitive to treatment with a Holliday junction-specific endonuclease (71). Although recent work has highlighted apparent discrepancies between predicted migration patterns of regressed forks and the cone-shaped signal in such gels (83), a subsequent study suggested that exonucleolytic degradation of the regressed fourth arm might explain such discrepancies (84) (Figure 6).
These cone-shaped signals have been detected in a variety of systems including the highly repetitive rDNA array in S. cerevisiae which contains a pre-programmed block to replication (71), hydroxyurea-treated S. cerevisiae cells (41), UV-treated E. coli (18) and a bacteriophage T4 origin of replication (84). However, whether 2-D gels provide an accurate measure of the frequency with which fork regression occurs in vivo is unknown, given that any regressed fork signals in 2-D gels reflect both the rates of formation and dissolution of such structures. Holliday junction structures can spontaneously branch migrate (85) and so any regressed forks could migrate to reform a fork structure or to form two linear duplexes, potentially reducing the intensity of any signal from such structures. Rapid processing of regressed forks in vivo could also mask the actual frequency with which fork regression occurs. The extent to which any fork unwinds to form a fourth duplex arm will also constrain the ability of electrophoretic techniques to detect regressed forks. How many base pairs at a fork might be extruded to form a fourth duplex during regression is unknown and may also differ depending on the nature of the blockage and the mechanism driving fork regression. It is conceivable that extrusion of only a few tens of base pairs might still lead to enzymatic processing of the resultant Holliday junction, but render this regressed fork structure unresolvable from a normal fork in 2-D gels.
Electron microscopy
A second direct detection method is electron microscopy, the technique first used to detect regressed fork structures (4). EM does not suffer from the problem of assigning electrophoretic signals to specific DNA structures since DNA structures are visualized directly. ssDNA and dsDNA can also be distinguished by EM, whereas little work has been done using 2-D gels to analyse structures containing extensive ssDNA regions (82). EM studies have shown that in S. cerevisiae wild-type cells treated with hydroxyurea, blocked forks accumulated only small regions of ssDNA and regressed fork structures were rarely seen (86). In contrast, in cells deficient in the DNA replication checkpoint, extensive regions of ssDNA were seen at blocked forks and many structures resembling regressed forks were observed (86). These data suggest that fork regression might be a rare occurrence upon replication blockage in a wild-type eukaryotic cell, and that fork regression might be a pathological rather than a normal physiological event in eukaryotes (41). However, whilst EM has several advantages over 2-D gels, both techniques are limited by their ‘snapshot’ nature—levels of detectable regressed forks might be influenced significantly by rapid processing and also by limited rather than extensive extrusion of a fourth arm. Thus, direct techniques such as EM and 2-D gels are more appropriate for comparing relative levels of fork regression rather than absolute frequencies of regression.
In summary, fork regression has been detected using direct techniques but the frequency of regression appears low as judged by these methods. This low apparent frequency might be explained by regression being a pathological event (41,86) but it might also reflect the short-lived nature of regressed forks rather than their frequency of occurrence. Indeed, genetic analyses suggest that unwinding of blocked forks, and subsequent processing of these Holliday junctions, could play important roles in the completion of genome duplication and the maintenance of genome stability. Thus, whilst the frequency of fork regression is in question, a low frequency and/or rapid processing does not necessarily preclude an important role for regression in genome duplication.
Discussion up to this point has assumed that fork regression is a favourable reaction, at least under some conditions. Studies demonstrating that DNA topology and specific enzymes can promote regression support this view, providing support for the concept of fork regression.
WHAT DRIVES FORK REGRESSION?
DNA supercoiling
Separation of the parental strands at a replication fork causes overwinding of the parental duplex resulting in an increase in positive superhelicity ahead of the fork, some of which may equilibrate across the fork to form precatenanes (87,88). This torsional strain must be relieved by topoisomerases to allow continued strand separation but DNA ahead of a moving replisome is likely to be overwound to some extent (87). However, analysis of partially replicated plasmid DNA in vitro, in which DNA-intercalating agents were used to generate overwinding of the parental DNA and so mimic the overwinding found near forks in vivo, did not generate the expected numbers of supercoils or precatenanes as judged by gel electrophoretic methods (6,89). Scanning force microscopy and EM of such DNA structures revealed that fork regression had occurred, resulting in rewinding of the parental DNA strands in front of the fork and relief of the mechanical strain caused by binding of intercalating agents (6,89). This ability of positive torsional strain within template DNA to drive fork regression was supported by the use of Holliday junction-specific endonucleases to cleave replication forks reconstituted in vitro and blocked by the accumulation of positive supercoiling (90). In contrast, fork regression was inhibited in negatively supercoiled DNA (90).
These data imply that positive supercoiling may promote fork regression in vivo. Relief of torsional strain by fork regression would come with little thermodynamic cost associated with disruption of base pairs since any base pair disruption during fork regression would be matched by the formation of base pairs between the newly-formed nascent-nascent duplex and the reformed parental duplex (6). One caveat to these studies is that the forked DNA structures were embedded within plasmids and it is unknown whether the small topological domain size of plasmids provide good models for the larger, more fluid domains found in vivo (91,92). Furthermore, the probability of regression occurring via relief of positive torsional strain would depend on the rate of dissociation of the replisome from the blocked fork (assuming replisome dissociation must precede regression) versus the rate at which topoisomerases act in the vicinity of blocked forks to provide alternative relief. Blocked fork stabilities of a few minutes (27,28) might therefore provide sufficient time for topoisomerases to remove positive supercoils ahead of the fork, reducing the likelihood of any regression being driven by positive torsional strain. Even when removal of such excess positive windings was inhibited by use of a partial loss-of-function DNA gyrase mutation in E. coli (the main topoisomerase involved in the removal of positive superhelicity ahead of forks) genetic analysis suggested that although replication fork breakdown was frequent, regression of these forks was infrequent (93). Whether positive supercoiling ahead of blocked forks does result in fork regression in vivo is therefore uncertain.
Helicases/translocases
-
RecG: This is a branched DNA-specific helicase found in E. coli, the absence of which confers moderate defects in DNA repair and recombination (94). RecG is also the first enzyme shown to unwind DNA forks to form Holliday junctions in vitro (73). This monomeric enzyme promotes regression of forked DNA in vitro by simultaneous translocation along the leading and lagging strand templates resulting in coupled unwinding of both daughter strands (Figure 3) (73,90,95). Moreover, RecG can catalyse regression of negatively supercoiled in vitro replication intermediates even though such supercoiling inhibits spontaneous fork regression (96,97). Analysis of small DNA substrates with heterologous arms indicated that RecG preferentially bound to and unwound forks with a lagging strand but no leading strand positioned at the branch point (90) implying that RecG might target forks halted by a leading strand template lesion (Figure 2Bi). However, using forked DNA structures with homologous daughter duplex arms, reflecting the homology found in such structures in vivo, RecG promoted efficient regression of forks with varied dispositions of leading and lagging strands at the branch point (73,98,99). RecG might therefore be able to catalyse fork regression regardless of the disposition of leading and lagging strands at the branch point.
Structural analysis of Thermatoga maritima RecG bound to a forked DNA substrate revealed that the two helicase domains interact with the parental duplex of the fork indicating that the helicase motor of RecG translocates along dsDNA (100). The third domain makes critical contacts with the branch point of the fork (Figure 3A) (100–102). Within this third domain each of the parental strands are located within grooves separated by a so-called ‘wedge’ motif, with each groove able to accommodate ssDNA but not dsDNA (100). These data are consistent with translocation of the helicase domains of RecG along the parental duplex catalysing disruption of hydrogen bonding within the two daughter duplexes, resulting in re-annealing of the parental DNA strands and consequent annealing of the leading and lagging strands (100,101,103).
RecG-catalysed fork regression has been used to explain the DNA repair defects found in cells lacking RecG (73,104) whilst the co-localization of Bacillus subtilis RecG with replication forks in vivo (105) and the interaction of E. coli RecG with the C-terminus of SSB in vitro (106) support a role for RecG in processing replication forks. However, RecG can unwind a range of branched nucleic acid substrates including Holliday junctions, three-strand junctions, D-loops and R-loops (107–110) reactions which are all consistent with the known contacts between RecG and forked DNA (90,100). Whilst any helicase that catalyses fork regression might also be expected to catalyse branch migration of the resultant Holliday junction, unwinding of other branched DNA substrates makes it difficult to establish unequivocally that RecG promotes fork regression in vivo. Indeed, a broad DNA substrate specificity in vitro is a recurring theme amongst enzymes with the ability to catalyse fork regression. However, whilst it is tempting to search for a single in vivo function for any particular enzyme, multiple functions are also possible and might reflect the multiplicity of DNA structures that arise during DNA replication, repair and recombination. Ascribing in vivo function by correlating phenotypes of mutants with in vitro activities of the corresponding enzyme is perhaps the most challenging aspect of analysing helicases involved in genome stability.
-
RuvAB: This is a helicase complex found in E. coli which, in conjunction with the endonuclease RuvC, branch migrates and cleaves Holliday junctions during the late stages of homologous recombination (111,112). This specificity for Holliday junctions is reflected in the structure of the RuvAB complex in which a RuvA tetramer binds to one face of a Holliday junction, whilst two hexamers of RuvB each encircle duplex DNA arms emerging from opposing sides of the RuvA tetramer (Figure 4A) (112). Translocation of the RuvB hexamers along the duplex DNA results in branch migration of the DNA substrate, with the DNA ‘spooling’ across the face of the RuvA tetramer (113–115). Thus, although RuvAB can catalyse unwinding of a range of branched DNA substrates (113,116), the structure of the RuvA tetramer, the interaction of RuvAB with the RuvC Holliday-junction-specific endonuclease and the roles of RuvA, B and C in DNA repair and recombination indicate RuvAB(C) acts primarily on Holliday junctions (112,114).
However, RuvAB has been proposed to catalyse fork regression in vivo in strains bearing defects in replisome components or in the putative accessory replicative helicase Rep (117–119). In vitro, RuvAB can unwind model DNA forks in a direction consistent with regression (Figure 4B) but this directionality is only displayed on DNA substrates in which loading of RuvB is artificially restricted to the parental DNA duplex (116). Moreover, such restriction severely inhibits RuvAB helicase activity (116). In the absence of such a restriction, RuvAB does not appear to promote efficient fork regression in vitro (73), but instead catalyses the opposite reaction resulting in unwinding rather than re-annealing of the parental duplex (Figure 4C) (116,118). These apparent discrepancies might be reconciled if RuvAB, in certain mutant strains, is needed to promote extensive extrusion and/or stabilization of the fourth duplex arm from a fork that has already undergone limited regression, catalysed either by RecG or by positive supercoiling. The very limited processivity of RecG during regression of model fork structures in vitro (120) is compatible with RecG-catalysed initial fork regression followed by RuvAB-catalysed branch migration of the resultant Holliday junction.
-
RecQ family helicases: These are a highly conserved family of enzymes with key roles in the maintenance of genome stability (121). Unicellular organisms tend to contain only a single RecQ family member whereas multi-cellular organisms possess multiple RecQ-type enzymes (75). The single RecQ helicase found in E. coli, the founding member of this helicase family, does not promote regression of model forks in vitro (98,99). However, RecQ-type helicases are thought to perform a variety of functions in vivo which may be reflected in the variety of different domains found within members of this family in addition to the highly conserved helicase domains (122). Humans lacking the Bloom's syndrome helicase (BLM), a member of the RecQ family, display increased genome instability (123) and, whilst the molecular basis of these phenotypes has yet to be fully established, BLM has been shown to catalyse the regression of model fork structures in vitro (98,99). Regression of blocked forks by BLM might provide one mechanism of maintaining genome stability, possibly via template switching to allow replication past ssDNA lesions without running the risk of interchromosomal recombination (4,98,99). A second human RecQ helicase, WRN, has also been shown to promote fork regression in vitro (99,124). Werner syndrome patients lacking WRN also display elevated levels of genome instability and suffer many phenotypes associated with ageing (125). WRN also possesses a 3′ to 5′ exonuclease activity, not seen in other human RecQ helicases, which has been implicated in degradation of the leading strand in model fork substrates (124). Notably, forks containing a gap in the leading strand at the branch point are preferred substrates for WRN-catalysed regression, suggesting WRN exonuclease and helicase activities might co-operate during any fork regression (124).
As with RecG, BLM and WRN have the ability to unwind a range of branched DNA structures (126), thus complicating any correlation between phenotypes of cells lacking each helicase and in vitro helicase activities. Indeed, BLM can resolve a recombination intermediate containing a double Holliday junction in concert with human topoisomerase IIIα (127) and can also disrupt Rad51-ssDNA filaments (128), activities, which could also account for the maintenance of genome stability by BLM. The ability of BLM and WRN to unwind a fork in the direction required for regression can also be inhibited by RPA, a human ssDNA-binding protein, if there is a significant stretch of ssDNA present at the branch point (129). Fork regression activity of BLM and WRN might therefore be restricted to specific blocked fork structures.
A third human RecQ family helicase, RecQ5β, can also unwind model forks with a leading strand gap to promote re-annealing of the parental strands and annealing of the daughter strands (129). Human RecQ5β co-localizes with sites of DNA replication in vivo and interacts physically with PCNA whilst mice and chicken cells lacking RecQ5 have high levels of genome instability, all of which are consistent with a possible role of fork regression in processing blocked forks (129–131). However, in contrast to RecG, BLM and WRN, RecQ5β does not unwind the daughter strands within model forks in a concerted fashion (129). Thus, formation of a Holliday junction from a fork by RecQ5β would presumably occur via two independent unwinding events. Whether such a reaction would result in efficient formation of a Holliday junction from a fork remains unknown but could be tested by the use of large homologous fork structures such as those employed in studies of RecG and BLM (73,98).
UvsW: This helicase plays multiple roles in replication and repair of bacteriophage T4 DNA and can also complement many of the phenotypes of E. coli recG strains (132). The DNA substrate specificity of UvsW is also similar to RecG, with forks and Holliday junctions being preferred substrates suggesting that, like RecG, UvsW may catalyse regression of forks (133,134). Indeed, UvsW is required for accumulation of regressed forks at a T4 origin of replication in vivo, as detected by gel electrophoresis, and catalyses fork regression in vitro, providing compelling evidence that UvsW-driven fork regression is a physiologically important reaction in T4 (135). However, although UvsW might be functionally similar to RecG, it has no structural homology to RecG outside of the two helicase domains but instead resembles eukaryotic Rad54 (136). Although Rad54, an Snf2 family translocase, plays a central role in eukaryotic homologous recombination and can unwind various branched DNA substrates (137), there is currently no evidence that this enzyme is involved in fork regression.
-
Rad5 and HARP: Saccharomyces cerevisiae Rad5 is required for the bypass of UV light-induced DNA damage via a mechanism that does not involve recombination, properties consistent with a template switching mechanism of DNA damage tolerance (138). In vitro, Rad5 can promote regression of model fork substrates and branch migration of the resultant Holliday junction, consistent with damage tolerance via template switching (139). Rad5-catalysed regression occurs via concerted unwinding of the daughter strands at the fork (139) but the enzyme has no detectable helicase activity on non-branched DNA substrates (140) in contrast to RecG, BLM and WRN. This feature of Rad5 may be related to it being a member of the Snf2 family of helicases/translocases, many of which translocate along but do not unwind DNA (141). Snf2 enzymes and RecG both define families within the Superfamily 2 of helicases/translocases (142) and so it is tempting to speculate that the helicase/translocase domains of Rad5, like RecG (100), move along the parental duplex of a fork in an ATP-dependent manner resulting in disruption of daughter duplexes and fork regression.
The recent identification of a human Snf2 family member, HARP, with the ability to re-anneal ssDNA bubbles bound by RPA via ATP-driven translocation (143) is also reminiscent of the dsDNA-specific translocation by RecG that can be coupled to fork regression (90,100). It has been argued that this HARP annealing activity is distinct from fork regression since HARP does not exhibit helicase activity on partial duplex substrates (143). However, Rad5 is also an ATP-driven translocase that does not possess the ability to unwind partial duplexes but can still regress forks in vitro (139). The possibility remains therefore that HARP might catalyse fork regression.
FANCM: This is a component of the Fanconi anaemia (FA) core complex in humans (144), disruption of which results in many chromosome instability phenotypes, both spontaneous and damage-induced (145). FANCM is a Superfamily 2 helicase that, although it cannot unwind partial duplex substrates (144), can unwind both forks and Holliday junctions and can also catalyse regression of model forks in vitro (146,147). Given that FA cells are especially sensitive to DNA cross-linking agents, FANCM-catalysed regression has been suggested to counter the movement of a replisome towards an interstrand cross-link, thus maintaining access of repair enzymes to the lesion (147). The Schizosaccharomyces pombe FANCM homologue Fml1 can also promote regression of a large model fork, and cells lacking Fml1 are also sensitive to DNA cross-linking agents, demonstrating conservation of in vitro and in vivo function of this motor (148). Moreover, the stripped-down S. pombe system has been employed to demonstrate that Fml1 promotes Rad51 function at blocked forks in vivo leading to a model of Fml1-catalysed regression and template switching followed by Rad51-catalysed recombination of the extruded fourth duplex arm (148) (see below). However, whether such a model might appertain to human FANCM is unclear.
Hjm/Hel308: This is a Superfamily 2 helicase from the archaeon Sulfolobus tokodaii that might also promote regression of forks bearing both leading and lagging strands at the branch point (149). However, this reaction appears to be complex since Hjm/Hel308A can promote annealing of complementary DNA strands, and also apparently translocate in both the 3′-5′ direction and the 5′-3′ direction along ssDNA (149). More details concerning this regression reaction are needed.
Figure 3.

(A) X-ray crystal structure of Thermatoga maritima RecG bound to a forked DNA having a lagging strand but no leading strand (100). Helicase domains 1 and 2 are shown in blue whilst domain 3 is in grey. (B) Model of RecG catalysis at forked DNA structures. Dashed arrows indicate relative movements of duplex arms.
Figure 4.

Action of RuvAB on Holliday junctions and forks. (A) Action of RuvAB on Holliday junction structures. Translocation of the two RuvB hexamers along opposing duplex arms results in movement of duplexes as indicated by dashed arrows. Strand separation results from spooling of the DNA strands across the ‘acidic pins’ found on the surface of the RuvA tetramer (176). (B) Catalysis of fork regression by RuvAB would necessitate loading of a single RuvB hexamer onto the parental duplex. (C) Binding of two RuvB hexamers onto opposing duplex arms of a fork would result in unwinding of the junction in the direction opposite to that required for regression.
Strand exchange proteins
Strand exchange proteins play central roles in DNA repair and recombination in all organisms. The strand exchange protein in E. coli, RecA, functions by binding to ssDNA in a cooperative manner to form a dynamic nucleoprotein filament (150). This filament then interacts with dsDNA and, upon association with homologous duplex DNA, can catalyse strand exchange. Such exchange is thought to play key roles in the post-replication repair of ssDNA gaps thought to be generated by re-priming of DNA synthesis downstream of single-strand-specific DNA lesions (45,47). In addition to catalysing exchange of strands between ssDNA and homologous duplex DNA, polymerization of RecA in the 5′ to 3′ direction along ssDNA can also extend into adjacent duplex DNA and drive strand exchange between two homologous duplexes, so-called four strand exchange (151–153).
RecA has been implicated in catalysing fork regression in mutants bearing a temperature-sensitive allele of the replicative helicase gene at the restrictive temperature (154). In vivo labelling studies coupled with 2-D electrophoretic analyses also demonstrated that, in cells exposed to UV light, regressed forks form in an RecA-dependent manner (18,59,155). Moreover, RecA can catalyse regression in vitro of model forks bearing a 2000-nt gap in the leading strand (120,156), reflecting the possible structure of a fork blocked by a lesion in the leading strand template (see Figure 2Bi).
This ability to catalyse regression in vitro appears to be a general property of strand exchange proteins, with T4 bacteriophage UvsX and human Rad51 both being able to promote regression of model forks with a leading strand gap (157,158). The requirement for ssDNA to allow initial binding and nucleation of strand exchange proteins (150,159) implies that blocked forks possessing limited ssDNA would not be a target for these enzymes. Such fork structure specificity is supported by genetic analyses in E. coli strains with mutations in a range of helicases, with the ability of RecA to catalyse fork regression being proposed to depend on the nature of the helicase mutation (154,160).
POSSIBLE FUNCTIONS OF FORK REGRESSION IN NORMAL DNA METABOLISM
Many models of replication fork repair have incorporated fork regression as a critical step on the road to resuscitation of a blocked fork. However, these models are often complex and also contradictory, presenting a confusing picture of how fork regression might aid replication. This confusion is due to: (i) the problem of assigning function to an enzyme in a wild-type organism based on the phenotype of a mutant lacking this enzyme; (ii) difficulties in assigning in vivo function to an enzyme when the enzyme can act on multiple DNA substrates in vitro; (iii) problems associated with detection of regressed forks in vivo; and (iv) the use of different types of block in different studies to analyse fork processing.
If the discrepancies between different models are laid aside, how might fork regression aid genome duplication in the face of blocks to replication? In considering this question, it is critical to consider the nature of replicative blocks. Replication past DNA lesions requires either removal or bypass of the block (161). Such removal or bypass could involve reorientation of the original DNA template (see below) whilst bypass could also be effected by recruitment of a specialized translesion polymerase (7,8). In contrast, the majority of protein–DNA complexes present lower barriers to fork movement such that reloading of the replisome might allow replication to proceed through the original protein–DNA block. Indeed, stochastic blockage by most nucleoprotein complexes is likely the key difference between such ‘accidental’ blocks and pre-programmed protein–DNA blocks such as E. coli Tus-ter that present efficient replicative blocks.
Generalized models of how fork regression might aid genome duplication will now be discussed.
Fork regression and reversal
The most basic model invoking regression involves unwinding of the fork to form a Holliday junction and then reversal of this unwinding process to reform the original fork (4). Why might such a reaction occur? In the case of a DNA lesion within the leading strand template, lagging strand synthesis may be able to continue some way beyond this lesion resulting in the damage being within a region of ssDNA (Figure 2Bi). Thus, no intact complementary strand would be available for excision repair. Fork reversal might promote replication past DNA lesions by provision of an undamaged complementary strand via template switching, the original proposed function of fork regression in which the blocked leading strand is extended using the lagging strand as a template (Figure 5A–D) (4). Reversal of the original regression reaction followed by reloading of the replisome onto the restored fork would result in bypass of the original lesion (Figure 5D). Repair rather than bypass of the original blocking lesion could also be promoted since regression would reposition the lesion opposite the uncorrupted lagging strand template (Figure 5B, E and F). Regression and reversal might therefore facilitate bypass and/or repair of leading strand template lesions. Evidence is accumulating that post-replicative bypass mechanisms are important in eukaryotes (162) and the involvement of Rad5 (see above) in such mechanisms provides indirect evidence for lesion bypass by regression and template switching (139).
Regression followed by reversal may also promote genome duplication in other ways. Movement of the fork backwards via regression could provide a general mechanism of facilitating access of repair enzymes to sites of replicative blocks, assuming that access to lesions is restricted within the context of a blocked replisome. For example, transcribing RNA polymerases stalled by DNA damage may be potent replicative blocks and fork regression, regardless of subsequent processing steps, might facilitate access of repair/displacement systems to such blocks (73).
Regression followed by degradation of the extruded duplex arm
An alternative means of restoring a fork after regression would be to degrade the duplex arm extruded from the fork (70) (Figure 6). This process might promote repair of a DNA lesion by facilitating access of repair enzymes to the DNA damage, as mentioned above. However, unlike reversal, degradation of the extruded duplex would prevent use of the lagging strand for template switching. Given that most ‘accidental’ nucleoprotein blocks to replication are likely to have a low probability of blocking an individual replisome, reinitiation of replication upstream of the block (Figure 6C) might also facilitate movement of the replisome through such a block on the second attempt (80).
Given the multiplicity of exonucleases present in all organisms it might be expected that fork restoration would more likely occur via degradation of the extruded duplex rather than reversal of fork unwinding. E. coli RecBCD is a rapid, processive helicase complex with a potent nuclease activity that can initiate degradation of both strands at a blunt duplex DNA end (163) and has been implicated in degradation of such dsDNA ends at regressed forks in specific mutant backgrounds (70,164,165) (Figure 6B). Whilst no enzyme complex equivalent to RecBCD has been identified in eukaryotes, multiple helicases and nucleases have recently been implicated in processing of DNA ends in eukaryotes in a manner functionally equivalent to RecBCD (166–168). Moreover, S. cerevisiae Exo1 endonuclease may act at reversed forks, although whether degradation occurs via a duplex extruded by fork regression is unclear (169).
A second E. coli exonuclease, RecJ, may also degrade strands at blocked forks. RecA-catalysed reversal of forks blocked by UV lesions (see above) might reposition the lesion back into the parental duplex, facilitating excision repair (18,76). Targeting of the nascent lagging strand by RecQ helicase and RecJ 5′-3′ exonuclease might then restore a fork structure onto which the replication apparatus could be reloaded, allowing resumption of replication (76). However, this model does not invoke re-annealing of the nascent leading and lagging strands. Instead, the leading strand is retained in the RecA nucleoprotein filament whilst the lagging strand is degraded.
Regression followed by cleavage of the Holliday junction
All organisms appear to possess endonucleases that specifically bind to and cleave Holliday junctions symmetrically, resulting in the generation of DNA duplexes (170,171). Such cleavage reactions are critical in resolving Holliday junctions formed during recombination. In the case of a regressed fork, cleavage of the four-stranded structure would generate one intact duplex (after sealing of the nick by DNA ligase) and a second duplex with a dsDNA end (70) (Figure 7A–C). Such cleavage would therefore destroy rather than remodel the blocked fork structure, on the face of it a reckless act. However, dsDNA ends can act as substrates for recombination, with strand exchange between the dsDNA end and an intact sister duplex resulting in D-loop formation and assembly of the replication apparatus onto this D-loop (21,33). At a regressed fork cleaved by a Holliday junction resolvase the end result of such recombination would be re-creation of a replication fork upstream of the original block (Figure 7D–F), the same outcome as expected of regression followed by degradation of the extruded duplex (Figure 6C).
Such a reaction might, as described above, promote access of repair enzymes to the block (Figure 7C). However, with regressed fork cleavage followed by recombination, there would be a risk of inaccurate recombination occurring. Indeed, in E. coli the Holliday junction resolvase RuvC might cleave regressed replication forks only in the absence of RecBCD (70,164,165). The implication is that degradation of the extruded duplex arm (by RecBCD) is a more efficient reaction than cleavage of regressed forks. However, work analysing the interplay between replication and transcription in E. coli implies that direct resetting of a blocked fork by RecBCD is unlikely to provide an efficient means of restarting replication and, by implication, that cleavage of regressed forks by RuvC does (73). These apparent contradictions might simply reflect the different mutant strains analysed in different studies and may provide a good example of how context is critical in understanding blocked fork processing.
Regression followed by recombination of the extruded duplex
Implicit in the reversal followed by degradation model (Figure 6) is the assumption that degradation would halt once the extruded arm was completely degraded. However, this processing could also result in generation of a 3′ ssDNA tail and subsequent homologous recombination (68). For example, in E. coli the highly processive RecBCD helicase/exonuclease (172) might continue to unwind and degrade the nascent leading and lagging strands via the blunt duplex DNA end formed by regression, in effect continuing the regression process. In such a situation, RecBCD might continue until it encounters χ, an octameric DNA sequence that modulates RecBCD activity such that degradation of the strand with a 3′ end is inhibited (173). The resultant 3′ ssDNA tail would then become a target for recombination with strand exchange most likely occurring with the homologous sequence found within the reformed parental duplex. The end result would be reassembly of the replication machinery at a D-loop, generating a replication fork with two Holliday junctions upstream of the fork (70) (Figure 8). Resolution of this double Holliday junction structure would therefore recreate a replication fork.
As with regression followed by Holliday junction cleavage, recombination of the extruded duplex would be accompanied by the risk of inaccurate homologous recombination between non-identical sequences. However, this risk may be reduced by the physical association of the recombining dsDNA end with the target donor sequence (the reformed parental duplex downstream of the regressed fork). In contrast, there would be no direct linkage between donor and target after cleavage of a regressed fork (Figure 7D).
Most enzymatic steps involved in this putative processing have been characterized in detail within other contexts, usually double-strand break repair (33,150,163). Recent work has also suggested a possible alternative mechanism of dealing with a double Holliday junction structure that does not involve symmetrical cleavage of both junctions by endonucleases. The human RecQ-type helicase BLM in combination with human topoisomerase IIIα can catalyse dissolution of such a structure in vitro (127) which may avoid problems associated with directionality of cleavage of Holliday junctions.
The only direct evidence that regression followed by recombination might occur in vivo relates to a bacteriophage T4 origin of replication. During the early stages of infection replication occurs at ori(34) via the formation of an R-loop that initiates DNA synthesis in one direction (174). Initiation of DNA synthesis from the fork structure at the opposite end of the origin bubble requires regression of this fork by UvsW (135)(see above) and subsequent processing by a Holliday junction resolvase (EndoVII) and also an ATPase/exonuclease complex (gp46/47) that generates ssDNA from dsDNA ends to facilitate loading of a strand exchange protein (84). Whilst the Holliday junction resolvase could conceivably operate before the exonuclease, as shown in Figure 7, the T4 origin is not a recombination hotspot (175) which supports an intramolecular (Figure 8) rather than an intermolecular recombination (Figure 7) step (84).
SUMMARY
A large body of evidence is accumulating that suggests replication fork regression does occur and may help to promote genome duplication in wild-type organisms even in the absence of exogenous DNA damage. In particular, the identification of an increasing number of enzymes that can catalyse regression in vitro (Table 1) implies that many organisms possess the potential to unwind blocked replication forks. However, most of the evidence in support of a physiological role for fork regression is indirect. The obvious technical difficulties in direct detection of fork regression in vivo might explain the indirect nature of most of this evidence but it remains possible that regression occurs only infrequently under normal physiological conditions. Ultimately, determining whether fork regression does play a role in genome duplication will require two problems to be addressed. Firstly, many models imply that fork regression can promote repair or bypass of replicative blocks. Direct evidence for this repair or bypass via fork regression is needed. Secondly, an unequivocal link between the ability of an enzyme to catalyse fork regression in vitro with its function in vivo is also required. Until both of these issues are addressed, models of blocked fork processing that invoke regression will remain speculative.
Table 1.
Summary of factors that may promote fork regression
| Factor | Organism | Properties |
|---|---|---|
| DNA supercoiling | All | Non-enzymatic |
| RecG | E. coli | Superfamily 2 helicase, highly conserved in bacteria but no obvious (nuclear-encoded) eukaryotic homologues |
| RuvAB | E. coli | Inefficient initiation of fork regression but could promote branch migration of Holliday junctions formed by regression |
| RecQ homologues | H. sapiens | Highly conserved group of Superfamily 2 helicases. Not all RecQ-type helicases possess regression activity |
| UvsW | T4 bacteriophage | Superfamily 2 helicase, bears functional (but not structural) similarity to RecG |
| Rad5 | S. cerevisiae | Superfamily 2 DNA translocase but no detectable helicase activity |
| FANCM/Fml1 | H. sapiens/S. pombe | Superfamily 2 helicases. FANCM also contains an endonuclease domain |
| Hjm/Hel308 | S. tokodai | Superfamily 2 helicase |
| Strand exchange proteins | E. coli/T4 bacteriophage/H. sapiens | Ubiquitous. Initial binding requires ssDNA, implying regression by these enzymes may be fork structure-specific |
With the exception of RuvAB all helicases/translocases currently suspected of catalysing regression are Superfamily 2 motors. Whether this reflects specific properties of Superfamily 2 motors that are needed for efficient fork regression is unknown.
FUNDING
The Biotechnology and Biological Sciences Research Council (BB/C008316/1, BB/E020690/1 to P.M.) and the Medical Research Council (G0501626 to P.M.). Funding for open access charge: Biotechnology and Biological Sciences Research Council and Medical Research Council.
Conflict of interest statement. None declared.
ACKNOWLEDGEMENTS
The authors would like to thank Len Wu, Piero Bianco and Bob Lloyd for comments on the manuscript, Ian Hickson and Steve West for discussions and Geoff Briggs for supplying the image used in Figure 3A.
REFERENCES
- 1.Setlow RB, Swenson PA, Carrier WL. Thymine dimers and inhibition of DNA synthesis by ultraviolet irradiation of cells. Science. 1963;142:1464–1466. doi: 10.1126/science.142.3598.1464. [DOI] [PubMed] [Google Scholar]
- 2.Hanawalt PC. The U.V. sensitivity of bacteria: its relation to the DNA replication cycle. Photochem. Photobiol. 1966;5:1–12. [PubMed] [Google Scholar]
- 3.Cox MM, Goodman MF, Kreuzer KN, Sherratt DJ, Sandler SJ, Marians KJ. The importance of repairing stalled replication forks. Nature. 2000;404:37–41. doi: 10.1038/35003501. [DOI] [PubMed] [Google Scholar]
- 4.Higgins NP, Kato K, Strauss B. A model for replication repair in mammalian cells. J. Mol. Biol. 1976;101:417–425. doi: 10.1016/0022-2836(76)90156-x. [DOI] [PubMed] [Google Scholar]
- 5.Fujiwara Y, Tatsumi M. Replicative bypass repair of ultraviolet damage to DNA of mammalian cells: caffeine sensitive and caffeine resistant mechanisms. Mutat. Res. 1976;37:91–110. doi: 10.1016/0027-5107(76)90058-0. [DOI] [PubMed] [Google Scholar]
- 6.Postow L, Ullsperger C, Keller RW, Bustamante C, Vologodskii AV, Cozzarelli NR. Positive torsional strain causes the formation of a four-way junction at replication forks. J. Biol. Chem. 2001;276:2790–2796. doi: 10.1074/jbc.M006736200. [DOI] [PubMed] [Google Scholar]
- 7.Prakash S, Johnson RE, Prakash L. Eukaryotic translesion synthesis DNA polymerases: specificity of structure and function. Annu. Rev. Biochem. 2005;74:317–353. doi: 10.1146/annurev.biochem.74.082803.133250. [DOI] [PubMed] [Google Scholar]
- 8.Nohmi T. Environmental stress and lesion-bypass DNA polymerases. Annu. Rev. Microbiol. 2006;60:231–253. doi: 10.1146/annurev.micro.60.080805.142238. [DOI] [PubMed] [Google Scholar]
- 9.Dronkert ML, Kanaar R. Repair of DNA interstrand cross-links. Mutat. Res. 2001;486:217–247. doi: 10.1016/s0921-8777(01)00092-1. [DOI] [PubMed] [Google Scholar]
- 10.Lindahl T. The Croonian Lecture, 1996: endogenous damage to DNA. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1996;351:1529–1538. doi: 10.1098/rstb.1996.0139. [DOI] [PubMed] [Google Scholar]
- 11.Aguilera A. The connection between transcription and genomic instability. EMBO J. 2002;21:195–201. doi: 10.1093/emboj/21.3.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rudolph CJ, Dhillon P, Moore T, Lloyd RG. Avoiding and resolving conflicts between DNA replication and transcription. DNA Repair (Amst) 2007;6:981–993. doi: 10.1016/j.dnarep.2007.02.017. [DOI] [PubMed] [Google Scholar]
- 13.Nakano T, Morishita S, Katafuchi A, Matsubara M, Horikawa Y, Terato H, Salem AMH, Izumi S, Pack SP, Makino K, et al. Nucleotide excision repair and homologous recombination systems commit differentially to the repair of DNA-Protein crosslinks. Mol. Cell. 2007;28:147–158. doi: 10.1016/j.molcel.2007.07.029. [DOI] [PubMed] [Google Scholar]
- 14.Rothstein R, Michel B, Gangloff S. Replication fork pausing and recombination or “gimme a break”. Genes Dev. 2000;14:1–10. [PubMed] [Google Scholar]
- 15.Neylon C, Kralicek AV, Hill TM, Dixon NE. Replication termination in Escherichia coli: structure and antihelicase activity of the Tus-ter complex. Microbiol. Mol. Biol. Rev. 2005;69:501–526. doi: 10.1128/MMBR.69.3.501-526.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tsang E, Carr AM. Replication fork arrest, recombination and the maintenance of ribosomal DNA stability. DNA Repair (Amst) 2008;7:1613–1623. doi: 10.1016/j.dnarep.2008.06.010. [DOI] [PubMed] [Google Scholar]
- 17.Tang M, Pham P, Shen X, Taylor JS, O'Donnell M, Woodgate R, Goodman MF. Roles of E. coli DNA polymerases IV and V in lesion-targeted and untargeted SOS mutagenesis. Nature. 2000;404:1014–1018. doi: 10.1038/35010020. [DOI] [PubMed] [Google Scholar]
- 18.Courcelle J, Donaldson JR, Chow KH, Courcelle CT. DNA damage-induced replication fork regression and processing in Escherichia coli. Science. 2003;299:1064–1067. doi: 10.1126/science.1081328. [DOI] [PubMed] [Google Scholar]
- 19.Heller RC, Marians KJ. Replication fork reactivation downstream of a blocked nascent leading strand. Nature. 2006;439:557–562. doi: 10.1038/nature04329. [DOI] [PubMed] [Google Scholar]
- 20.Maisnier-Patin S, Nordstrom K, Dasgupta S. Replication arrests during a single round of replication of the Escherichia coli chromosome in the absence of DnaC activity. Mol. Microbiol. 2001;42:1371–1382. doi: 10.1046/j.1365-2958.2001.02718.x. [DOI] [PubMed] [Google Scholar]
- 21.Heller RC, Marians KJ. Replisome assembly and the direct restart of stalled replication forks. Nat. Rev. Mol. Cell Biol. 2006;7:932–943. doi: 10.1038/nrm2058. [DOI] [PubMed] [Google Scholar]
- 22.Uzest M, Ehrlich SD, Michel B. Lethality of rep recB and rep recC double mutants of Escherichia coli. Mol. Microbiol. 1995;17:1177–1188. doi: 10.1111/j.1365-2958.1995.mmi_17061177.x. [DOI] [PubMed] [Google Scholar]
- 23.Magner DB, Blankschien MD, Lee JA, Pennington JM, Lupski JR, Rosenberg SM. RecQ promotes toxic recombination in cells lacking recombination intermediate-removal proteins. Mol. Cell. 2007;26:273–286. doi: 10.1016/j.molcel.2007.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsuzuki T, Fujii Y, Sakumi K, Tominaga Y, Nakao K, Sekiguchi M, Matsushiro A, Yoshimura Y, Morita T. Targeted disruption of the Rad51 gene leads to lethality in embryonic mice. Proc. Natl Acad. Sci. USA. 1996;93:6236–6240. doi: 10.1073/pnas.93.13.6236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Torres JZ, Schnakenberg SL, Zakian VA. Saccharomyces cerevisiae Rrm3p DNA helicase promotes genome integrity by preventing replication fork stalling: viability of rrm3 cells requires the intra-S-phase checkpoint and fork restart activities. Mol. Cell Biol. 2004;24:3198–3212. doi: 10.1128/MCB.24.8.3198-3212.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gangloff S, Soustelle C, Fabre F. Homologous recombination is responsible for cell death in the absence of the Sgs1 and Srs2 helicases. Nat. Genet. 2000;25:192–194. doi: 10.1038/76055. [DOI] [PubMed] [Google Scholar]
- 27.Marians KJ, Hiasa H, Kim DR, McHenry CS. Role of the core DNA polymerase III subunits at the replication fork. a is the only subunit required for processive replication. J. Biol. Chem. 1998;273:2452–2457. doi: 10.1074/jbc.273.4.2452. [DOI] [PubMed] [Google Scholar]
- 28.McGlynn P, Guy CP. Replication forks blocked by protein-DNA complexes have limited stability in vitro. J. Mol. Biol. 2008;381:249–255. doi: 10.1016/j.jmb.2008.05.053. [DOI] [PubMed] [Google Scholar]
- 29.Possoz C, Filipe SR, Grainge I, Sherratt DJ. Tracking of controlled Escherichia coli replication fork stalling and restart at repressor-bound DNA in vivo. EMBO J. 2006;25:2596–2604. doi: 10.1038/sj.emboj.7601155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reyes-Lamothe R, Possoz C, Danilova O, Sherratt DJ. Independent positioning and action of Escherichia coli replisomes in live cells. Cell. 2008;133:90–102. doi: 10.1016/j.cell.2008.01.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Calzada A, Hodgson B, Kanemaki M, Bueno A, Labib K. Molecular anatomy and regulation of a stable replisome at a paused eukaryotic DNA replication fork. Genes Dev. 2005;19:1905–1919. doi: 10.1101/gad.337205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cobb JA, Schleker T, Rojas V, Bjergbaek L, Tercero JA, Gasser SM. Replisome instability, fork collapse, and gross chromosomal rearrangements arise synergistically from Mec1 kinase and RecQ helicase mutations. Genes Dev. 2005;19:3055–3069. doi: 10.1101/gad.361805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.San Filippo J, Sung P, Klein H. Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 2008;77:229–257. doi: 10.1146/annurev.biochem.77.061306.125255. [DOI] [PubMed] [Google Scholar]
- 34.Trenz K, Smith E, Smith S, Costanzo V. ATM and ATR promote Mre11 dependent restart of collapsed replication forks and prevent accumulation of DNA breaks. EMBO J. 2006;25:1764–1774. doi: 10.1038/sj.emboj.7601045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Osborn AJ, Elledge SJ. Mrc1 is a replication fork component whose phosphorylation in response to DNA replication stress activates Rad53. Genes Dev. 2003;17:1755–1767. doi: 10.1101/gad.1098303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Katou Y, Kanoh Y, Bando M, Noguchi H, Tanaka H, Ashikari T, Sugimoto K, Shirahige K. S-phase checkpoint proteins Tof1 and Mrc1 form a stable replication-pausing complex. Nature. 2003;424:1078–1083. doi: 10.1038/nature01900. [DOI] [PubMed] [Google Scholar]
- 37.Nitani N, Nakamura K, Nakagawa C, Masukata H, Nakagawa T. Regulation of DNA replication machinery by Mrc1 in fission yeast. Genetics. 2006;174:155–165. doi: 10.1534/genetics.106.060053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lou H, Komata M, Katou Y, Guan Z, Reis CC, Budd M, Shirahige K, Campbell JL. Mrc1 and DNA polymerase e function together in linking DNA replication and the S phase checkpoint. Mol. Cell. 2008;32:106–117. doi: 10.1016/j.molcel.2008.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Szyjka SJ, Viggiani CJ, Aparicio OM. Mrc1 is required for normal progression of replication forks throughout chromatin in S. cerevisiae. Mol. Cell. 2005;19:691–697. doi: 10.1016/j.molcel.2005.06.037. [DOI] [PubMed] [Google Scholar]
- 40.Tercero JA, Diffley JF. Regulation of DNA replication fork progression through damaged DNA by the Mec1/Rad53 checkpoint. Nature. 2001;412:553–557. doi: 10.1038/35087607. [DOI] [PubMed] [Google Scholar]
- 41.Lopes M, Cotta-Ramusino C, Pellicioli A, Liberi G, Plevani P, Muzi-Falconi M, Newlon CS, Foiani M. The DNA replication checkpoint response stabilizes stalled replication forks. Nature. 2001;412:557–561. doi: 10.1038/35087613. [DOI] [PubMed] [Google Scholar]
- 42.Bjergbaek L, Cobb JA, Tsai-Pflugfelder M, Gasser SM. Mechanistically distinct roles for Sgs1p in checkpoint activation and replication fork maintenance. EMBO J. 2005;24:405–47. doi: 10.1038/sj.emboj.7600511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McGlynn P, Lloyd RG. Recombinational repair and restart of damaged replication forks. Nat. Rev. Mol. Cell Biol. 2002;3:859–870. doi: 10.1038/nrm951. [DOI] [PubMed] [Google Scholar]
- 44.Aguilera A, Gomez-Gonzalez B. Genome instability: a mechanistic view of its causes and consequences. Nat. Rev. Genet. 2008;9:204–217. doi: 10.1038/nrg2268. [DOI] [PubMed] [Google Scholar]
- 45.Rupp WD, Wilde CE, III, Reno DL, Howard-Flanders P. Exchanges between DNA strands in ultraviolet-irradiated Escherichia coli. J. Mol. Biol. 1971;61:25–44. doi: 10.1016/0022-2836(71)90204-x. [DOI] [PubMed] [Google Scholar]
- 46.Meneghini R, Hanawalt P. T4-endonuclease V-sensitive sites in DNA from ultraviolet-irradiated human cells. Biochim. Biophys. Acta. 1976;425:428–437. doi: 10.1016/0005-2787(76)90007-1. [DOI] [PubMed] [Google Scholar]
- 47.West SC, Cassuto E, Howard-Flanders P. Mechanism of E. coli RecA protein directed strand exchanges in post-replication repair of DNA. Nature. 1981;294:659–662. doi: 10.1038/294659a0. [DOI] [PubMed] [Google Scholar]
- 48.McInerney P, O'Donnell M. Functional uncoupling of twin polymerases: mechanism of polymerase dissociation from a lagging-strand block. J. Biol. Chem. 2004;279:21543–21551. doi: 10.1074/jbc.M401649200. [DOI] [PubMed] [Google Scholar]
- 49.Svoboda DL, Vos JM. Differential replication of a single, UV-induced lesion in the leading or lagging strand by a human cell extract: fork uncoupling or gap formation. Proc. Natl Acad. Sci. USA. 1995;92:11975–11979. doi: 10.1073/pnas.92.26.11975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cordeiro-Stone M, Makhov AM, Zaritskaya LS, Griffith JD. Analysis of DNA replication forks encountering a pyrimidine dimer in the template to the leading strand. J. Mol. Biol. 1999;289:1207–1218. doi: 10.1006/jmbi.1999.2847. [DOI] [PubMed] [Google Scholar]
- 51.Pagès V, Fuchs RP. Uncoupling of leading- and lagging-strand DNA replication during lesion bypass in vivo. Science. 2003;300:1300–1303. doi: 10.1126/science.1083964. [DOI] [PubMed] [Google Scholar]
- 52.McInerney P, O'Donnell M. Replisome fate upon encountering a leading strand block and clearance from DNA by recombination proteins. J. Biol. Chem. 2007;282:25903–25916. doi: 10.1074/jbc.M703777200. [DOI] [PubMed] [Google Scholar]
- 53.Rupp WD, Howard-Flanders P. Discontinuities in the DNA synthesized in an excision-defective strain of Escherichia coli following ultraviolet irradiation. J. Mol. Biol. 1968;31:291–304. doi: 10.1016/0022-2836(68)90445-2. [DOI] [PubMed] [Google Scholar]
- 54.Gottesman MM, Hicks ML, Gellert M. Genetics and function of DNA ligase in Escherichia coli. J. Mol. Biol. 1973;77:531–547. doi: 10.1016/0022-2836(73)90221-0. [DOI] [PubMed] [Google Scholar]
- 55.Johnston LH, Nasmyth KA. Saccharomyces cerevisiae cell cycle mutant cdc9 is defective in DNA ligase. Nature. 1978;274:891–893. doi: 10.1038/274891a0. [DOI] [PubMed] [Google Scholar]
- 56.Lopes M, Foiani M, Sogo JM. Multiple mechanisms control chromosome integrity after replication fork uncoupling and restart at irreparable UV lesions. Mol. Cell. 2006;21:15–27. doi: 10.1016/j.molcel.2005.11.015. [DOI] [PubMed] [Google Scholar]
- 57.Khidhir MA, Casaregola S, Holland IB. Mechanism of transient inhibition of DNA synthesis in ultraviolet- irradiated E. coli: inhibition is independent of recA whilst recovery requires RecA protein itself and an additional, inducible SOS function. Mol. Gen. Genet. 1985;199:133–140. doi: 10.1007/BF00327522. [DOI] [PubMed] [Google Scholar]
- 58.Courcelle CT, Belle JJ, Courcelle J. Nucleotide excision repair or polymerase V-mediated lesion bypass can act to restore UV-arrested replication forks in Escherichia coli. J Bacteriol. 2005;187:6953–6961. doi: 10.1128/JB.187.20.6953-6961.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Courcelle CT, Chow KH, Casey A, Courcelle J. Nascent DNA processing by RecJ favors lesion repair over translesion synthesis at arrested replication forks in Escherichia coli. Proc. Natl Acad. Sci. USA. 2006;103:9154–9159. doi: 10.1073/pnas.0600785103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rudolph CJ, Upton AL, Lloyd RG. Replication fork stalling and cell cycle arrest in UV-irradiated Escherichia coli. Genes Dev. 2007;21:668–681. doi: 10.1101/gad.417607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gruber M, Wellinger RE, Sogo JM. Architecture of the replication fork stalled at the 3′ end of yeast ribosomal genes. Mol. Cell Biol. 2000;20:5777–5787. doi: 10.1128/mcb.20.15.5777-5787.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hill TM, Marians KJ. Escherichia coli Tus protein acts to arrest the progression of DNA replication forks in vitro. Proc. Natl Acad. Sci. USA. 1990;87:2481–2485. doi: 10.1073/pnas.87.7.2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hotchkiss RD. Models of genetic recombination. Annu. Rev. Microbiol. 1974;28:445–468. doi: 10.1146/annurev.mi.28.100174.002305. [DOI] [PubMed] [Google Scholar]
- 64.Tatsumi K, Strauss B. Production of DNA bifilarly substituted with bromodeoxyuridine in the first round of synthesis: branch migration during isolation of cellular DNA. Nucleic Acids Res. 1978;5:331–347. doi: 10.1093/nar/5.2.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wanka F, Brouns RM, Aelen JM, Eygensteyn A, Eygensteyn J. The origin of nascent single-stranded DNA extracted from mammalian cells. Nucleic Acids Res. 1977;4:2083–2097. doi: 10.1093/nar/4.6.2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nilsen T, Baglioni C. Unusual base-pairing of newly synthesized DNA in HeLa cells. J. Mol. Biol. 1979;133:319–338. doi: 10.1016/0022-2836(79)90396-6. [DOI] [PubMed] [Google Scholar]
- 67.Zannis-Hadjopoulos M, Persico M, Martin RG. The remarkable instability of replication loops provides a general method for the isolation of origins of DNA replication. Cell. 1981;27:155–163. doi: 10.1016/0092-8674(81)90369-x. [DOI] [PubMed] [Google Scholar]
- 68.Louarn JM, Louarn J, Francois V, Patte J. Analysis and possible role of hyperrecombination in the termination region of the Escherichia coli chromosome. J. Bacteriol. 1991;173:5097–5104. doi: 10.1128/jb.173.16.5097-5104.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Michel B, Ehrlich SD, Uzest M. DNA double-strand breaks caused by replication arrest. EMBO J. 1997;16:430–438. doi: 10.1093/emboj/16.2.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Seigneur M, Bidnenko V, Ehrlich SD, Michel B. RuvAB acts at arrested replication forks. Cell. 1998;95:419–430. doi: 10.1016/s0092-8674(00)81772-9. [DOI] [PubMed] [Google Scholar]
- 71.Zou H, Rothstein R. Holliday junctions accumulate in replication mutants via a RecA homolog- independent mechanism. Cell. 1997;90:87–96. doi: 10.1016/s0092-8674(00)80316-5. [DOI] [PubMed] [Google Scholar]
- 72.Defossez PA, Prusty R, Kaeberlein M, Lin SJ, Ferrigno P, Silver PA, Keil RL, Guarente L. Elimination of replication block protein Fob1 extends the life span of yeast mother cells. Mol. Cell. 1999;3:447–455. doi: 10.1016/s1097-2765(00)80472-4. [DOI] [PubMed] [Google Scholar]
- 73.McGlynn P, Lloyd RG. Modulation of RNA polymerase by (p)ppGpp reveals a RecG-dependent mechanism for replication fork progression. Cell. 2000;101:35–45. doi: 10.1016/S0092-8674(00)80621-2. [DOI] [PubMed] [Google Scholar]
- 74.Michel B, Boubakri H, Baharoglu Z, LeMasson M, Lestini R. Recombination proteins and rescue of arrested replication forks. DNA Repair. 2007;6:967–980. doi: 10.1016/j.dnarep.2007.02.016. [DOI] [PubMed] [Google Scholar]
- 75.Wu L, Hickson ID. DNA helicases required for homologous recombination and repair of damaged replication forks. Annu. Rev. Genet. 2006;40:279–306. doi: 10.1146/annurev.genet.40.110405.090636. [DOI] [PubMed] [Google Scholar]
- 76.Belle JJ, Casey A, Courcelle CT, Courcelle J. Inactivation of the DnaB helicase leads to the collapse and degradation of the replication fork: a comparison to UV-induced arrest. J. Bacteriol. 2007;189:5452–5462. doi: 10.1128/JB.00408-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Horiuchi T, Fujimura Y. Recombinational rescue of the stalled DNA replication fork: a model based on analysis of an Escherichia coli strain with a chromosome region difficult to replicate. J. Bacteriol. 1995;177:783–791. doi: 10.1128/jb.177.3.783-791.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bidnenko V, Ehrlich SD, Michel B. Replication fork collapse at replication terminator sequences. EMBO J. 2002;21:3898–307. doi: 10.1093/emboj/cdf369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Lambert S, Watson A, Sheedy DM, Martin B, Carr AM. Gross chromosomal rearrangements and elevated recombination at an inducible site-specific replication fork barrier. Cell. 2005;121:689–702. doi: 10.1016/j.cell.2005.03.022. [DOI] [PubMed] [Google Scholar]
- 80.Payne BT, van Knippenberg IC, Bell H, Filipe SR, Sherratt DJ, McGlynn P. Replication fork blockage by transcription factor-DNA complexes in Escherichia coli. Nucleic Acids Res. 2006;34:5194–5202. doi: 10.1093/nar/gkl682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Friedman KL, Brewer BJ. Analysis of replication intermediates by two-dimensional agarose gel electrophoresis. Methods Enzymol. 1995;262:613–627. doi: 10.1016/0076-6879(95)62048-6. [DOI] [PubMed] [Google Scholar]
- 82.Pohlhaus JR, Kreuzer KN. Formation and processing of stalled replication forks–utility of two-dimensional agarose gels. Methods Enzymol. 2006;409:477–493. doi: 10.1016/S0076-6879(05)09028-2. [DOI] [PubMed] [Google Scholar]
- 83.Fierro-Fernandez M, Hernandez P, Krimer DB, Schvartzman JB. Replication fork reversal occurs spontaneously after digestion but is constrained in supercoiled domains. J. Biol. Chem. 2007;282:18190–18196. doi: 10.1074/jbc.M701559200. [DOI] [PubMed] [Google Scholar]
- 84.Long DT, Kreuzer KN. Regression supports two mechanisms of fork processing in phage T4. Proc. Natl Acad. Sci. USA. 2008;105:6852–6857. doi: 10.1073/pnas.0711999105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Panyutin IG, Hsieh P. The kinetics of spontaneous DNA branch migration. Proc. Natl Acad. Sci. USA. 1994;91:2021–2025. doi: 10.1073/pnas.91.6.2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Sogo JM, Lopes M, Foiani M. Fork reversal and ssDNA accumulation at stalled replication forks owing to checkpoint defects. Science. 2002;297:599–602. doi: 10.1126/science.1074023. [DOI] [PubMed] [Google Scholar]
- 87.Postow L, Crisona NJ, Peter BJ, Hardy CD, Cozzarelli NR. Topological challenges to DNA replication: Conformations at the fork. Proc. Natl Acad. Sci. USA. 2001;98:8219–8226. doi: 10.1073/pnas.111006998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Espeli O, Marians KJ. Untangling intracellular DNA topology. Mol. Microbiol. 2004;52:925–931. doi: 10.1111/j.1365-2958.2004.04047.x. [DOI] [PubMed] [Google Scholar]
- 89.Olavarrieta L, Martinez-Robles ML, Sogo JM, Stasiak A, Hernandez P, Krimer DB, Schvartzman JB. Supercoiling, knotting and replication fork reversal in partially replicated plasmids. Nucleic Acids Res. 2002;30:656–666. doi: 10.1093/nar/30.3.656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.McGlynn P, Lloyd RG. Rescue of stalled replication forks by RecG: simultaneous translocation on the leading and lagging strand templates supports an active DNA unwinding model of fork reversal and Holliday junction formation. Proc. Natl Acad. Sci. USA. 2001;98:8227–8234. doi: 10.1073/pnas.111008698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Postow L, Hardy CD, Arsuaga J, Cozzarelli NR. Topological domain structure of the Escherichia coli chromosome. Genes Dev. 2004;18:1766–1779. doi: 10.1101/gad.1207504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Deng S, Stein RA, Higgins NP. Transcription-induced barriers to supercoil diffusion in the Salmonella typhimurium chromosome. Proc. Natl Acad. Sci. USA. 2004;101:3398–3403. doi: 10.1073/pnas.0307550101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Grompone G, Ehrlich SD, Michel B. Replication restart in gyrB Escherichia coli mutants. Mol. Microbiol. 2003;48:845–854. doi: 10.1046/j.1365-2958.2003.03480.x. [DOI] [PubMed] [Google Scholar]
- 94.McGlynn P, Lloyd RG. Genome stability and the processing of damaged replication forks by RecG. Trends Genet. 2002;18:413–419. doi: 10.1016/s0168-9525(02)02720-8. [DOI] [PubMed] [Google Scholar]
- 95.McGlynn P, Mahdi AA, Lloyd RG. Characterisation of the catalytically active form of RecG helicase. Nucleic Acids Res. 2000;28:2324–2332. doi: 10.1093/nar/28.12.2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.McGlynn P, Lloyd RG, Marians KJ. Formation of Holliday junctions by regression of nascent DNA in intermediates containing stalled replication forks: RecG stimulates regression even when the DNA is negatively supercoiled. Proc. Natl Acad. Sci. USA. 2001;98:8235–8240. doi: 10.1073/pnas.121007798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Slocum SL, Buss JA, Kimura Y, Bianco PR. Characterization of the ATPase activity of the Escherichia coli RecG protein reveals that the preferred cofactor is negatively supercoiled DNA. J. Mol. Biol. 2007;367:647–664. doi: 10.1016/j.jmb.2007.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ralf C, Hickson ID, Wu L. The Bloom's syndrome helicase can promote the regression of a model replication fork. J. Biol. Chem. 2006;281:22839–22846. doi: 10.1074/jbc.M604268200. [DOI] [PubMed] [Google Scholar]
- 99.Machwe A, Xiao L, Groden J, Orren DK. The Werner and Bloom syndrome proteins catalyze regression of a model replication fork. Biochemistry. 2006;45:13939–13946. doi: 10.1021/bi0615487. [DOI] [PubMed] [Google Scholar]
- 100.Singleton MR, Scaife S, Wigley DB. Structural analysis of DNA replication fork reversal by RecG. Cell. 2001;107:79–89. doi: 10.1016/s0092-8674(01)00501-3. [DOI] [PubMed] [Google Scholar]
- 101.Briggs GS, Mahdi AA, Wen Q, Lloyd RG. DNA binding by the substrate specificity (wedge) domain of RecG helicase suggests a role in processivity. J. Biol. Chem. 2005;280:13921–13927. doi: 10.1074/jbc.M412054200. [DOI] [PubMed] [Google Scholar]
- 102.Mahdi AA, McGlynn P, Levett SD, Lloyd RG. DNA binding and helicase domains of the Escherichia coli recombination protein RecG. Nucleic Acids Res. 1997;25:3875–3880. doi: 10.1093/nar/25.19.3875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mahdi AA, Briggs GS, Sharples GJ, Wen Q, Lloyd RG. A model for dsDNA translocation revealed by a structural motif common to RecG and Mfd proteins. EMBO J. 2003;22:724–734. doi: 10.1093/emboj/cdg043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gregg AV, McGlynn P, Jaktaji RP, Lloyd RG. Direct rescue of stalled DNA replication forks via the combined action of PriA and RecG helicase activities. Mol. Cell. 2002;9:241–251. doi: 10.1016/s1097-2765(02)00455-0. [DOI] [PubMed] [Google Scholar]
- 105.Lecointe F, Serena C, Velten M, Costes A, McGovern S, Meile JC, Errington J, Ehrlich SD, Noirot P, Polard P. Anticipating chromosomal replication fork arrest: SSB targets repair DNA helicases to active forks. EMBO J. 2007;26:4239–4251. doi: 10.1038/sj.emboj.7601848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Buss JA, Kimura Y, Bianco PR. RecG interacts directly with SSB: implications for stalled replication fork regression. Nucleic Acids Res. 2008;36:7029–7042. doi: 10.1093/nar/gkn795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Vincent SD, Mahdi AA, Lloyd RG. The RecG branch migration protein of Escherichia coli dissociates R-loops. J. Mol. Biol. 1996;264:713–721. doi: 10.1006/jmbi.1996.0671. [DOI] [PubMed] [Google Scholar]
- 108.Fukuoh A, Iwasaki H, Ishioka K, Shinagawa H. ATP-dependent resolution of R-loops at the ColE1 replication origin by Escherichia coli RecG protein, a Holliday junction-specific helicase. EMBO J. 1997;16:203–209. doi: 10.1093/emboj/16.1.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.McGlynn P, Al-Deib AA, Liu J, Marians KJ, Lloyd RG. The DNA replication protein PriA and the recombination protein RecG bind D-loops. J. Mol. Biol. 1997;270:212–221. doi: 10.1006/jmbi.1997.1120. [DOI] [PubMed] [Google Scholar]
- 110.Whitby MC, Lloyd RG. Targeting Holliday junctions by the RecG branch migration protein of Escherichia coli. J. Biol. Chem. 1998;273:19729–19739. doi: 10.1074/jbc.273.31.19729. [DOI] [PubMed] [Google Scholar]
- 111.Eggleston AK, Mitchell AH, West SC. In vitro reconstitution of the late steps of genetic recombination in E. coli. Cell. 1997;89:607–17. doi: 10.1016/s0092-8674(00)80242-1. [DOI] [PubMed] [Google Scholar]
- 112.West SC. Processing of recombination intermediates by the RuvABC proteins. Ann. Rev. Genet. 1997;31:213–244. doi: 10.1146/annurev.genet.31.1.213. [DOI] [PubMed] [Google Scholar]
- 113.Hiom K, Tsaneva IR, West SC. The directionality of RuvAB-mediated branch migration: in vitro studies with three-armed junctions. Genes Cells. 1996;1:443–451. doi: 10.1046/j.1365-2443.1996.d01-253.x. [DOI] [PubMed] [Google Scholar]
- 114.Hargreaves D, Rice DW, Sedelnikova SE, Artymiuk PJ, Lloyd RG, Rafferty JB. Crystal structure of E.coli RuvA with bound DNA Holliday junction at 6 Å resolution. Nat. Struct. Biol. 1998;5:441–446. doi: 10.1038/nsb0698-441. [DOI] [PubMed] [Google Scholar]
- 115.Yamada K, Miyata T, Tsuchiya D, Oyama T, Fujiwara Y, Ohnishi T, Iwasaki H, Shinagawa H, Ariyoshi M, Mayanagi K, et al. Crystal structure of the RuvA-RuvB complex: a structural basis for the Holliday junction migrating motor machinery. Mol. Cell. 2002;10:671–681. doi: 10.1016/s1097-2765(02)00641-x. [DOI] [PubMed] [Google Scholar]
- 116.McGlynn P, Lloyd RG. Action of RuvAB at replication fork structures. J. Biol. Chem. 2001;276:41938–41944. doi: 10.1074/jbc.M107945200. [DOI] [PubMed] [Google Scholar]
- 117.Baharoglu Z, Petranovic M, Flores MJ, Michel B. RuvAB is essential for replication forks reversal in certain replication mutants. EMBO J. 2006;25:596–604. doi: 10.1038/sj.emboj.7600941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Baharoglu Z, Bradley AS, Le Masson M, Tsaneva I, Michel B. ruvA Mutants that resolve Holliday junctions but do not reverse replication forks. PLoS Genet. 2008;4:e1000012. doi: 10.1371/journal.pgen.1000012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Le Masson M, Baharoglu Z, Michel B. ruvA and ruvB mutants specifically impaired for replication fork reversal. Mol. Microbiol. 2008;70:537–548. doi: 10.1111/j.1365-2958.2008.06431.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Robu ME, Inman RB, Cox MM. Situational repair of replication forks – roles of RecG and RecA proteins. J. Biol. Chem. 2004;279:10973–10981. doi: 10.1074/jbc.M312184200. [DOI] [PubMed] [Google Scholar]
- 121.Bachrati CZ, Hickson ID. RecQ helicases: guardian angels of the DNA replication fork. Chromosoma. 2008;117:219–233. doi: 10.1007/s00412-007-0142-4. [DOI] [PubMed] [Google Scholar]
- 122.Hickson ID. RecQ helicases: caretakers of the genome. Nat. Rev. Cancer. 2003;3:169–178. doi: 10.1038/nrc1012. [DOI] [PubMed] [Google Scholar]
- 123.Wu L. Role of the BLM helicase in replication fork management. DNA Repair (Amst) 2007;6:936–944. doi: 10.1016/j.dnarep.2007.02.007. [DOI] [PubMed] [Google Scholar]
- 124.Machwe A, Xiao L, Lloyd RG, Bolt E, Orren DK. Replication fork regression in vitro by the Werner syndrome protein (WRN): holliday junction formation, the effect of leading arm structure and a potential role for WRN exonuclease activity. Nucleic Acids Res. 2007;35:5729–5747. doi: 10.1093/nar/gkm561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Harrigan JA, Bohr VA. Human diseases deficient in RecQ helicases. Biochimie. 2003;85:1185–1193. doi: 10.1016/j.biochi.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 126.Opresko PL, Cheng WH, Bohr VA. Junction of RecQ helicase biochemistry and human disease. J. Biol. Chem. 2004;279:18099–18102. doi: 10.1074/jbc.R300034200. [DOI] [PubMed] [Google Scholar]
- 127.Wu L, Hickson ID. The Bloom's syndrome helicase suppresses crossing over during homologous recombination. Nature. 2003;426:870–874. doi: 10.1038/nature02253. [DOI] [PubMed] [Google Scholar]
- 128.Bugreev DV, Yu X, Egelman EH, Mazin AV. Novel pro- and anti-recombination activities of the Bloom's syndrome helicase. Genes Dev. 2007;21:3085–3094. doi: 10.1101/gad.1609007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kanagaraj R, Saydam N, Garcia PL, Zheng L, Janscak P. Human RECQ5beta helicase promotes strand exchange on synthetic DNA structures resembling a stalled replication fork. Nucleic Acids Res. 2006;34:5217–5231. doi: 10.1093/nar/gkl677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Wang W, Seki M, Narita Y, Nakagawa T, Yoshimura A, Otsuki M, Kawabe Y, Tada S, Yagi H, Ishii Y, et al. Functional relation among RecQ family helicases RecQL1, RecQL5, and BLM in cell growth and sister chromatid exchange formation. Mol. Cell Biol. 2003;23:3527–3535. doi: 10.1128/MCB.23.10.3527-3535.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hu Y, Lu X, Barnes E, Yan M, Lou H, Luo G. Recql5 and Blm RecQ DNA helicases have nonredundant roles in suppressing crossovers. Mol. Cell Biol. 2005;25:3431–3442. doi: 10.1128/MCB.25.9.3431-3442.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Carles-Kinch K, George JW, Kreuzer KN. Bacteriophage T4 UvsW protein is a helicase involved in recombination, repair and the regulation of DNA replication origins. EMBO J. 1997;16:4142–4151. doi: 10.1093/emboj/16.13.4142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Nelson SW, Benkovic SJ. The T4 phage UvsW protein contains both DNA unwinding and strand annealing activities. J. Biol. Chem. 2007;282:407–416. doi: 10.1074/jbc.M608153200. [DOI] [PubMed] [Google Scholar]
- 134.Webb MR, Plank JL, Long DT, Hsieh TS, Kreuzer KN. The phage T4 protein UvsW drives Holliday junction branch migration. J. Biol. Chem. 2007;282:34401–34411. doi: 10.1074/jbc.M705913200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Long DT, Kreuzer KN. Fork regression is an active helicase-driven pathway in bacteriophage T4. EMBO Rep. 2009;10:394–399. doi: 10.1038/embor.2009.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kerr ID, Sivakolundu S, Li Z, Buchsbaum JC, Knox LA, Kriwacki R, White SW. Crystallographic and NMR analyses of UvsW and UvsW.1 from bacteriophage T4. J. Biol. Chem. 2007;282:34392–34400. doi: 10.1074/jbc.M705900200. [DOI] [PubMed] [Google Scholar]
- 137.Heyer WD, Li X, Rolfsmeier M, Zhang XP. Rad54: the Swiss Army knife of homologous recombination? Nucleic Acids Res. 2006;34:4115–4125. doi: 10.1093/nar/gkl481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Torres-Ramos CA, Prakash S, Prakash L. Requirement of RAD5 and MMS2 for postreplication repair of UV-damaged DNA in Saccharomyces cerevisiae. Mol. Cell Biol. 2002;22:2419–2426. doi: 10.1128/MCB.22.7.2419-2426.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Blastyak A, Pinter L, Unk I, Prakash L, Prakash S, Haracska L. Yeast Rad5 protein required for postreplication repair has a DNA helicase activity specific for replication fork regression. Mol. Cell. 2007;28:167–175. doi: 10.1016/j.molcel.2007.07.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Johnson RE, Prakash S, Prakash L. Yeast DNA repair protein RAD5 that promotes instability of simple repetitive sequences is a DNA-dependent ATPase. J. Biol. Chem. 1994;269:28259–28262. [PubMed] [Google Scholar]
- 141.Flaus A, Martin DM, Barton GJ, Owen-Hughes T. Identification of multiple distinct Snf2 subfamilies with conserved structural motifs. Nucleic Acids Res. 2006;34:2887–2905. doi: 10.1093/nar/gkl295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Singleton MR, Dillingham MS, Wigley DB. Structure and mechanism of helicases and nucleic acid translocases. Annu. Rev. Biochem. 2007;76:23–50. doi: 10.1146/annurev.biochem.76.052305.115300. [DOI] [PubMed] [Google Scholar]
- 143.Yusufzai T, Kadonaga JT. HARP is an ATP-driven annealing helicase. Science. 2008;322:748–750. doi: 10.1126/science.1161233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Meetei AR, Medhurst AL, Ling C, Xue Y, Singh TR, Bier P, Steltenpool J, Stone S, Dokal I, Mathew CG, et al. A human ortholog of archaeal DNA repair protein Hef is defective in Fanconi anemia complementation group M. Nat. Genet. 2005;37:958–963. doi: 10.1038/ng1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Mathew CG. Fanconi anaemia genes and susceptibility to cancer. Oncogene. 2006;25:5875–5884. doi: 10.1038/sj.onc.1209878. [DOI] [PubMed] [Google Scholar]
- 146.Gari K, Decaillet C, Stasiak AZ, Stasiak A, Constantinou A. The Fanconi anemia protein FANCM can promote branch migration of Holliday junctions and replication forks. Mol. Cell. 2008;29:141–148. doi: 10.1016/j.molcel.2007.11.032. [DOI] [PubMed] [Google Scholar]
- 147.Gari K, Decaillet C, Delannoy M, Wu L, Constantinou A. Remodeling of DNA replication structures by the branch point translocase FANCM. Proc. Natl Acad. Sci. USA. 2008;105:16107–16112. doi: 10.1073/pnas.0804777105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Sun W, Nandi S, Osman F, Ahn JS, Jakovleska J, Lorenz A, Whitby MC. The FANCM ortholog Fml1 promotes recombination at stalled replication forks and limits crossing over during DNA double-strand break repair. Mol. Cell. 2008;32:118–128. doi: 10.1016/j.molcel.2008.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Li Z, Lu S, Hou G, Ma X, Sheng D, Ni J, Shen Y. Hjm/Hel308A DNA helicase from Sulfolobus tokodaii promotes replication fork regression and interacts with Hjc endonuclease in vitro. J. Bacteriol. 2008;190:3006–3017. doi: 10.1128/JB.01662-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lusetti SL, Cox MM. The bacterial RecA protein and the recombinational DNA repair of stalled replication forks. Annu. Rev. Biochem. 2002;71:71–100. doi: 10.1146/annurev.biochem.71.083101.133940. [DOI] [PubMed] [Google Scholar]
- 151.Cassuto E, West SC, Mursalim J, Conlon S, Howard-Flanders P. Initiation of genetic recombination: homologous pairing between duplex DNA molecules promoted by recA protein. Proc. Natl Acad. Sci. USA. 1980;77:3962–3966. doi: 10.1073/pnas.77.7.3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Cunningham RP, DasGupta C, Shibata T, Radding CM. Homologous pairing in genetic recombination: recA protein makes joint molecules of gapped circular DNA and closed circular DNA. Cell. 1980;20:223–235. doi: 10.1016/0092-8674(80)90250-0. [DOI] [PubMed] [Google Scholar]
- 153.Chow SA, Chiu SK, Wong BC. RecA protein-promoted homologous pairing and strand exchange between intact and partially single-stranded duplex DNA. J. Mol. Biol. 1992;223:79–93. doi: 10.1016/0022-2836(92)90717-x. [DOI] [PubMed] [Google Scholar]
- 154.Seigneur M, Ehrlich SD, Michel B. RuvABC-dependent double-strand breaks in dnaBts mutants require recA. Mol. Microbiol. 2000;38:565–574. doi: 10.1046/j.1365-2958.2000.02152.x. [DOI] [PubMed] [Google Scholar]
- 155.Courcelle J, Hanawalt PC. RecQ and RecJ process blocked replication forks prior to the resumption of replication in UV-irradiated Escherichia coli. Mol. Gen. Genet. 1999;262:543–551. doi: 10.1007/s004380051116. [DOI] [PubMed] [Google Scholar]
- 156.Robu ME, Inman RB, Cox MM. RecA protein promotes the regression of stalled replication forks in vitro. Proc. Natl Acad. Sci. USA. 2001;98:8211–8218. doi: 10.1073/pnas.131022698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Kadyrov FA, Drake JW. UvsX recombinase and Dda helicase rescue stalled bacteriophage T4 DNA replication forks in vitro. J. Biol. Chem. 2004;279:35735–35740. doi: 10.1074/jbc.M403942200. [DOI] [PubMed] [Google Scholar]
- 158.Yoon D, Wang Y, Stapleford K, Wiesmuller L, Chen J. p53 inhibits strand exchange and replication fork regression promoted by human Rad51. J. Mol. Biol. 2004;336:639–654. doi: 10.1016/j.jmb.2003.12.050. [DOI] [PubMed] [Google Scholar]
- 159.Sung P, Krejci L, Van Komen S, Sehorn MG. Rad51 recombinase and recombination mediators. J. Biol. Chem. 2003;278:42729–42732. doi: 10.1074/jbc.R300027200. [DOI] [PubMed] [Google Scholar]
- 160.Grompone G, Ehrlich D, Michel B. Cells defective for replication restart undergo replication fork reversal. EMBO Rep. 2004;5:607–612. doi: 10.1038/sj.embor.7400167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Friedberg EC, Wagner R, Radman M. Specialized DNA polymerases, cellular survival, and the genesis of mutations. Science. 2002;296:1627–1630. doi: 10.1126/science.1070236. [DOI] [PubMed] [Google Scholar]
- 162.Hishida T, Kubota Y, Carr AM, Iwasaki H. RAD6-RAD18-RAD5-pathway-dependent tolerance to chronic low-dose ultraviolet light. Nature. 2009;457:612–615. doi: 10.1038/nature07580. [DOI] [PubMed] [Google Scholar]
- 163.Dillingham MS, Kowalczykowski SC. RecBCD enzyme and the repair of double-stranded DNA breaks. Microbiol. Mol. Biol. Rev. 2008;72:642–671. doi: 10.1128/MMBR.00020-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Flores MJ, Bierne H, Ehrlich SD, Michel B. Impairment of lagging strand synthesis triggers the formation of a RuvABC substrate at replication forks. EMBO J. 2001;20:619–629. doi: 10.1093/emboj/20.3.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Michel B, Grompone G, Flores MJ, Bidnenko V. Multiple pathways process stalled replication forks. Proc. Natl Acad. Sci. USA. 2004;101:12783–12788. doi: 10.1073/pnas.0401586101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Mimitou EP, Symington LS. Sae2, Exo1 and Sgs1 collaborate in DNA double-strand break processing. Nature. 2008;455:770–774. doi: 10.1038/nature07312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Zhu Z, Chung WH, Shim EY, Lee SE, Ira G. Sgs1 helicase and two nucleases Dna2 and Exo1 resect DNA double-strand break ends. Cell. 2008;134:981–994. doi: 10.1016/j.cell.2008.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Gravel S, Chapman JR, Magill C, Jackson SP. DNA helicases Sgs1 and BLM promote DNA double-strand break resection. Genes Dev. 2008;22:2767–2772. doi: 10.1101/gad.503108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Cotta-Ramusino C, Fachinetti D, Lucca C, Doksani Y, Lopes M, Sogo J, Foiani M. Exo1 processes stalled replication forks and counteracts fork reversal in checkpoint-defective cells. Mol. Cell. 2005;17:153–159. doi: 10.1016/j.molcel.2004.11.032. [DOI] [PubMed] [Google Scholar]
- 170.Lilley DM, White MF. The junction-resolving enzymes. Nat. Rev. Mol. Cell Biol. 2001;2:433–443. doi: 10.1038/35073057. [DOI] [PubMed] [Google Scholar]
- 171.Ip SC, Rass U, Blanco MG, Flynn HR, Skehel JM, West SC. Identification of Holliday junction resolvases from humans and yeast. Nature. 2008;456:357–361. doi: 10.1038/nature07470. [DOI] [PubMed] [Google Scholar]
- 172.Bianco PR, Brewer LR, Corzett M, Balhorn R, Yeh Y, Kowalczykowski SC, Baskin RJ. Processive translocation and DNA unwinding by individual RecBCD enzyme molecules. Nature. 2001;409:374–378. doi: 10.1038/35053131. [DOI] [PubMed] [Google Scholar]
- 173.Anderson DG, Kowalczykowski SC. The translocating RecBCD enzyme stimulates recombination by directing RecA protein onto ssDNA in a chi-regulated manner. Cell. 1997;90:77–86. doi: 10.1016/s0092-8674(00)80315-3. [DOI] [PubMed] [Google Scholar]
- 174.Carles-Kinch K, Kreuzer KN. RNA-DNA hybrid formation at a bacteriophage T4 replication origin. J. Mol. Biol. 1997;266:915–926. doi: 10.1006/jmbi.1996.0844. [DOI] [PubMed] [Google Scholar]
- 175.Doan PL, Belanger KG, Kreuzer KN. Two types of recombination hotspots in bacteriophage T4: one requires DNA damage and a replication origin and the other does not. Genetics. 2001;157:1077–1087. doi: 10.1093/genetics/157.3.1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Rafferty JB, Sedelnikova SE, Hargreaves D, Artymiuk PJ, Baker PJ, Sharples GJ, Mahdi AA, Lloyd RG, Rice DW. Crystal structure of DNA recombination protein RuvA and a model for its binding to the Holliday junction. Science. 1996;274:415–421. doi: 10.1126/science.274.5286.415. [DOI] [PubMed] [Google Scholar]