


Abstract
The prediction of transcription factor binding sites in genomic sequences is in principle very useful to identify upstream regulatory factors. However, when applying this concept to genomes of multicellular organisms such as mammals, one has to deal with a large number of false positive predictions since many transcription factor genes are only expressed in specific tissues or cell types. We developed TS-REX, a database/software system that supports the analysis of tissue and cell type-specific transcription factor-gene networks based on expressed sequence tag abundance of transcription factor-encoding genes in UniGene EST libraries. The use of expression levels of transcription factor-encoding genes according to hierarchical anatomical classifications covering different tissues and cell types makes it possible to filter out irrelevant binding site predictions and to identify candidates of potential functional importance for further experimental testing. TS-REX covers ESTs from H. sapiens and M. musculus, and allows the characterization of both presence and specificity of transcription factors in user-specified tissues or cell types. The software allows users to interactively visualize transcription factor-gene networks, as well as to export data for further processing. TS-REX was applied to predict regulators of Polycomb group genes in six human tumor tissues and in human embryonic stem cells.
INTRODUCTION
The state of a normal or diseased cell is determined by external signals and by its intrinsic gene expression pattern. Transcription factors (TFs) are major regulators of gene expression, typically controlling more than one gene and acting in concert. Such TF-gene interactions can be described as networks, which are crucial to understand hierarchies of gene expression regulation. The identification of TF binding sites (TFBSs) in genomic DNA sequences has played an important role in predicting transcriptional networks. A large number of TFs binds to specific DNA sequence stretches with a length of 5–25 bp (1), which have been experimentally determined and have served to define motifs that are often used for in silico binding site prediction. This is typically done by searching gene sequences for DNA stretches appearing more often than expected based on a background DNA model (2).
A fundamental problem in building TF-gene networks based on binding motifs in DNA sequences of putative target genes is the rate of false positive predictions of TFBSs (3). Different errors account for such false positive predictions. One error is based on the probability of occurrence of short sequence motifs in large stretches of genomic DNA. To reduce this error, the motif search can be restricted to genomic DNA sequences which are conserved among species. This strategy is based on the assumption that DNA stretches playing a crucial biological role may be evolutionarily conserved (4). Therefore, several methods for the prediction of TFBSs only consider conserved DNA sequence blocks (2,5–7). A second error is based on the fact that although a TFBS might be correctly predicted, binding of the respective factor might only occur in certain cells or tissues. This is either because the factor itself is not expressed or because additionally required co-factors are missing. The error that is related to the absence of a TF in a given tissue or cell type can be reduced by filtering predicted TFs based on their expression in the tissue or cell type of interest.
Both tissue specificity of gene expression profiles and the combination of TFBS information with gene expression data are the focus of a number of databases and software tools. The closest to TS-REX in terms of objectives and scope are MatInspector (8,9), the Promoter Analysis Pipeline (10,11) and the method described by Jeffery et al. (12). MatInspector allows TFBS prediction based on position weight matrix families, and includes tissue associations of the respective TFs. The focus of the other two tools is to identify TFBSs that are associated with co-regulated genes. Jeffery and colleagues particularly aim to find differences in TFBS occurrence between sets of differentially expressed genes.
However, none of the above-mentioned tools combines knowledge of TF expression levels with binding site prediction tools in order to allow extraction of tissue-specific portions of predicted TF-gene networks. The value of our approach stems both from its potential to address the problem of reducing false positive rates in in silico binding site prediction as previously mentioned, and from its prospective contribution to efforts aiming at a systems-level characterization of transcriptional regulation via a combination of network-based analytical techniques with anatomical annotations (13,14).
We have therefore developed a new resource, called Tissue-Specific Regulatory Network Extractor (TS-REX), consisting of a TF tissue database and a client–server software tool for the visualization of tissue and cell type-specific TF-gene networks based on TF expression levels. TS-REX extracts and displays tissue and cell type specificity of TF-gene networks. In addition to the availability of a direct interface to TFBS prediction tools such as TOUCAN (2,7), one distinctive feature of TS-REX is the richness of its anatomical classification, namely a newly established order of UniGene expressed sequence tag (EST) libraries, which allows a fine-grained dissection of TF-gene networks based on quantitative information about TF expression on an anatomical basis.
The TS-REX database comprises both quantitative estimates of tissue specificity obtained from UniGene EST library data and a comprehensive manually refined hierarchical anatomical classification. ESTs from both H. sapiens and M. musculus are covered.
The TS-REX software visualizes TF-gene networks and allows users to select anatomical structures such as tissues and cell types from the TS-REX hierarchical classification or from a user-provided input file containing tissue annotations, in order to highlight those TFs that are present in or specific to tissues or cell types of interest. To facilitate the assessment of different degrees of tissue or cell type specificity of TFs, the software also provides the user with a significant amount of flexibility in terms of parameter choice, as well as with the possibility to export data for further analysis.
TS-REX is a web-based tool with the database maintained at Lund University. Users can upload their input files to the TS-REX server via a graphical user interface, and queries to the database are transparently performed by the software. An overview is provided in Figure 1, together with an indication of the way the system is supposed to be used.
Figure 1.

Overview of TS-REX. Users have access to a graphical user interface that communicates with the server-side component of the TS-REX software, which in turn performs queries to the database at Lund University. The user-provided primary input file, which can be either an output file from MotifScanner or a tab delimited text file containing a correspondence between target genes and TRANSFAC® identifiers of predicted TF proteins, is processed by the network viewer module. Two alternatives are then available: the software uses either the TS-REX database to identify TFs present in or specific to tissues or cell types of interest, or an auxiliary user-provided file reporting genes that are known to be expressed in different tissues. The latter option allows the extraction of expressed TF genes from the user's own data on rare tissues or cells that are not contained in the TS-REX database. The tissue browser makes it possible to select tissues and cell types of interest, and to export files for subsequent analysis.
The TS-REX client can be launched from http://kundera.thep.lu.se:8080/TSREX/TSREX.jnlp (Java 1.6 is required, which currently makes TS-REX available under the Windows and Linux platforms).
MATERIALS AND METHODS
Database
The following sections describe the individual steps that led to the generation of the TS-REX database.
Establishment of a hierarchical classification based on UniGene EST libraries
The TS-REX hierarchical classification associates each EST library identifier with a library group based on which tissue or cell type that library corresponds to.
A two-step procedure was followed to build the classification. Perl scripts were used to parse the online tissue-related information that UniGene provides for a number of EST libraries, in order to extract available anatomical annotations. The data generated by this automated procedure subsequently underwent an extensive manual revision and editing process, in order to have each library associated with refined annotations. This second step included organizing tissue-related information into a hierarchical, comprehensive, fine-grained classification, comprising multiple categories corresponding to different aggregation levels, namely pathologies, systems, organs, tissues and cell types.
All those EST libraries that (i) were available via the UniGene Library Browser, (ii) contained at least one EST sequence and (iii) were considered as biologically relevant were included in the TS-REX classification, excluding normalized and subtracted libraries. This corresponds to 604 murine and 5708 human libraries.
Assignment of EST counts to library identifiers
UniGene data (Hs.data and Mm.data files from the NCBI FTP website corresponding to UniGene builds #201 and #162, respectively) were downloaded and processed in order to extract lists of EST sequence identifiers associated with the various genes in the different EST libraries. The murine database comprises 82 105 UniGene clusters and 4 029 157 EST sequence identifiers, while the human one contains 123 993 UniGene clusters and 6 509 336 EST sequence identifiers. For each library identifier, an EST count was obtained for every gene as the number of EST sequences corresponding to that gene in that library.
Calculation of p-values and transcripts-per-million of UniGene clusters for different library groups
P-values were determined using a binomial model to calculate the probability for a given gene to be associated with the observed number of ESTs or more (15), corresponding to a given entry in the anatomical hierarchy (e.g. a given organ, tissue or cell type) under the hypothesis that the gene considered is unspecific. Namely, the p-value corresponding to gene g and entry i in the hierarchy was calculated according to:
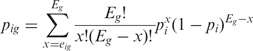 |
1 |
where eig is the sum of the EST counts for gene g in all libraries associated with anatomical structure i, Eg is the total number of EST sequences in the database for gene g, and pi is the fraction of EST sequences corresponding to i. If si represents the total number of EST sequences corresponding to i, i.e. if 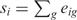 , then
, then
| 2 |
For instance, if the items considered in the hierarchical classification are cell types, each p-value in Equation (1) represents the probability for gene g to correspond to a number of EST sequences in cell type i at least equal to the observed one under the assumption that all cell types are equivalent, i.e. that g does not exhibit any cell type specificity. If the p-value for the encoding gene of a TF in a given cell type according to Equation (1) is lower than a given threshold, the corresponding TF protein is considered to be specific to that cell type.
While the binomial approach presented above can be used to determine tissue specificity, it is not suited to estimate whether the number of EST sequences of a TF-encoding gene is high enough for the corresponding TF to be considered as present in the selected anatomical structures. Instead EST counts were used to calculate the number of transcripts-per-million (TPM) for gene g corresponding to anatomical structure i, according to:
| 3 |
For instance, a TF is classified by TS-REX as present in a given cell type if the TPM score of its TF-encoding gene corresponding to that cell type exceeds a user-specified threshold.
Generation of the TS-REX encoding datasets
The correspondence between TFs and their encoding genes is a crucial component of TS-REX. The TS-REX encoding datasets incorporate this information for all TFs reported in TRANSFAC® (1,16) for both H. sapiens and M. musculus. TS-REX allows users to authenticate both to the publicly available version of TRANSFAC®, for which access is provided via free registration at www.gene-regulation.com, and to the licensed version of TRANSFAC®. Users who authenticate to the latter can make use of the complete TS-REX encoding datasets; otherwise, TS-REX uses a subset of the encoding datasets corresponding to those TFs that are reported in the public version of TRANSFAC®. The publicly available and the licensed versions of TRANSFAC® will be referred to as TRANSFAC® Public and TRANSFAC® Professional in the following.
The TS-REX encoding datasets were obtained using information from TRANSFAC® Professional 11.1, and from the Hs.data and Mm.data files provided by UniGene at www.ncbi.nlm.nih.gov. Namely, the encoding datasets were obtained according to the procedure detailed as follows:
TRANSFAC® data were used to extract an association between TFs reported in TRANSFAC® (for H. sapiens and M. musculus) and the RefSeq identifiers of the corresponding encoding genes. Subsequently, Hs.data and Mm.data from UniGene were parsed, and the previously found RefSeq identifiers were used to extract the UniGene identifier(s) of the encoding gene. This matching procedure was necessary due to UniGene identifiers being periodically revised and changing over time: the UniGene identifiers that are internally used by TS-REX correspond to UniGene builds #201 and #162 for H. sapiens and M. musculus, respectively (‘TS-REX UniGene IDs’ in the following).
For those TRANSFAC® TFs for which no RefSeq-based match to UniGene could be found, the TS-REX UniGene ID(s) of the corresponding encoding gene was (were) retrieved by searching the TS-REX naming dataset (see description below) for the gene symbol provided by TRANSFAC®. Only UniGene clusters containing at least one RefSeq identifier were reported for the TF-encoding genes, unless this resulted in no TF-encoding genes being provided.
The TS-REX UniGene IDs of the encoding genes of those TFs that did not have any RefSeq identifiers in TRANSFAC® were also retrieved based on gene symbol, as described above. As in step (ii), only UniGene clusters associated with at least one RefSeq identifier were included for the TF-encoding genes, unless this resulted in no TF-encoding genes being provided.
Generation of the TS-REX naming dataset
TS-REX uses an internal representation for genes and TFs that is based on TS-REX UniGene IDs and TRANSFAC® TF identifiers. However, the software also displays gene and factor names, as previously described. In order for this to be possible, TS-REX includes a mapping between TS-REX UniGene IDs or TRANSFAC® TF identifiers and the corresponding gene symbols or TF names from UniGene and TRANSFAC®, respectively, for both H. sapiens and M. musculus.
Software
A combination of technologies including Java™, Perl and SQL was used for the development of the TS-REX software. Additional notes on the implementation of the TS-REX software are provided at TS-REX startup.
Use of TS-REX is not recommended on computers with less than 1GB RAM. Performance, in particular with reference to interactive network visualization, is also affected by network size, depending on computer specifications.
Polycomb group gene application
Human and murine DNA sequences for Enhancer of zeste homolog 2 (EZH2), Suppressor of zeste 12 homolog (SUZ12), and Polycomb complex protein BMI-1 (BMI1) corresponding to conserved regions between the two species of ∼2000 bp around the transcriptional start site were retrieved using the VISTA Browser [http://pipeline.lbl.gov, (17)] and the UCSC Genome Browser [http://genome.ucsc.edu, (18)]: Human Mar. 2006 chr7:148211087-148212876 and Mouse Feb. 2006 chr6:47523460-47525135 for EZH2; Human Mar. 2006 chr10: 22649733-22651591 and Mouse Feb. 2006 chr2: 18594320-18596071 for BMI1; Human Mar. 2006 chr17:27287892-27289964 and Mouse Feb. 2006 chr11: 79809025-79811009 for SUZ12. We applied MotifScanner 3.1.1 (http://homes.esat.kuleuven.be/~thijs/Work/MotifScanner.html) for binding site prediction using matrix files from TRANSFAC® Professional 11.1 (for H. sapiens and M. musculus separately), as well as murine and human conserved non-coding sequences as a background model (namely, the file hsmmCNS_ens36_3.bg was used). The MotifScanner parameter choice corresponded to a double-stranded search with p set to 0.5 (p is a parameter between 0 and 1, higher values allowing higher motif degeneracy). A detailed documentation of MotifScanner is available at http://homes.esat.kuleuven.be/~thijs/help/help_motifscanner.html.
Cells and culture conditions
The human cervix carcinoma cell line Hela was grown in Dulbecco's Modified Eagle's Medium and the human colon adenocarcinoma cell line SW480 in RPMI-1640 medium (Invitrogen) at 37°C in 5% CO2 in air. Media were supplemented with 10% heat-inactivated FBS and 1% penicillin/streptomycin. To activate expression of early growth response 1 (EGR1), cells were treated with 10 ng/ml phorbol 12-myristate-13-acetate (PMA; Sigma) for 2 h.
Quantitative PCR
RNA was isolated using an RNA preparation kit (QIAGEN) according to the manufacturer's instructions. For cDNA synthesis, 2 µg of total RNA were treated with 2 U RNase free DNase (Promega) to prevent DNA contamination followed by reverse-transcription at 37°C for 90 min with 2 µl M-MLV Reverse Transcriptase (Promega, 200 U/µl) and 2 µl oligo-dT-primer (TIB Molbiol, 20 pmol/µl) in a volume of 40 µl. After cDNA synthesis, samples were diluted to 100 µl. Quantitative PCR (qPCR) was performed using the iQ5 Real-Time PCR system (Bio-Rad). A 20 μl reaction contained 1 μl cDNA, 0.1 μM of the forward and reverse primers and 10 μl of the Power SYBR Green PCR Master Mix (Applied Biosystems). Reactions were done in triplicates.
The following primers were used for target gene expression analysis:
EZH2 forward (5′-AGGACGGCTCCTCTAACCAT-3′),
EZH2 reverse (5′-CTTGGTGTTGCACTGTGCTT-3′),
SUZ12 forward (5′-CAGCTCATTTGCAGCTTACG-3′), SUZ12 reverse (5′-CGGGTTTTGTTTGATTGAGG-3′), BMI1 forward (5′-ATGCCCAGCAGCAATGAC-3′), and BMI1 reverse (5′-CTCCAGCATTTGTCAGTCCA-3′).
Primers for EGR1 transcript level measurements were:
EGR1 forward (5′-AGCCCTACGAGCACCTGAC-3′), and EGR1 reverse (5′-AGCGGCCAGTATAGGTGATG-3′).
For normalization, expression analyses of different housekeeping genes, Beta-2-microglobulin (B2M) and TATA box-binding protein (TBP) were performed using the primers B2M forward (5′-CGAGACATGTAAGCAGCATCA-3′), B2M reverse (5′-CAAACATGGAGACAGCACTCA-3′), TBP forward (5′-ACAACAGCCTGCCACCTTAC-3′), and TBP reverse (5′-GCCTTTGTTGCTCTTCCAAA-3′).
The reaction profile was as follows: 10 min at 95°C, 40 cycles of 15 s at 95°C and 60 s at 60°C. CT values were determined by the Bio-Rad iQ5 software (version 2.0). Relative expression levels were evaluated using the ΔΔCT method comparing CT values of the target gene with CT values of the housekeeping genes.
Transfection
For over-expression studies, 2 × 105 cells were plated onto six-well tissue culture plates and transiently transfected with 2 µg EGR1 expression vectors (pCMV-EGR1) (19) or with empty vectors (pCMV5) as negative control using Lipofectin (Invitrogen). After 48-h cultivation, cells were harvested.
Western blot analysis
Fifty micrograms of whole cell lysates were separated by SDS–PAGE and transferred to a nitrocellulose membrane (Macherey-Nagel, Düren, Germany). Membranes were blocked with 5% milk powder (Roth, Karlsruhe, Germany) and probed with rabbit polyclonal antibodies against EGR1 (Santa Cruz) or β-Actin (Sigma). Goat peroxidase-coupled anti-rabbit IgG (Promega) was used as secondary antibody.
Chromatin immunoprecipitation (ChIP)
Cells were treated with 1% formaldehyde for 10 min at room temperature to cross-link DNA to chromatin-associated proteins. Reactions were terminated by adding glycine (final concentration 0.125 M). Cells were harvested and resuspended in SDS lysis buffer (1% SDS, 10 mM EDTA, 50 mM Tris–HCl pH 8.1) and protease inhibitors (1 mM PMSF, 1 µg/ml aprotinin and 1 µg/ml leupeptin). To shear DNA, cells were sonicated three times for 30 s at 20% amplitude (Branson Sonifier W-250D) and centrifugated at 12 000 × g for 10 min at 4°C. The supernatants were diluted in 10× ChIP dilution buffer (0.01% SDS, 1% Triton X-100, 2 mM EDTA, 16.7 mM Tris–HCl pH 8.1 and 167 mM NaCl) and protease inhibitors as above. Samples were incubated with 2 µg anti-EGR1, IgG or no antibody at 4°C overnight with rotation. Thirty microliters of salmon sperm DNA/protein A-agarose slurry (Sigma) was added and incubated at 4°C for 1 h. The immune complex-binding agarose beads were collected by centrifugation and washed sequentially. DNA was eluted with 1% SDS, 0.1 M NaHCO3 and reverse cross-linked by heating at 65°C for 4 h after the addition of NaCl (final concentration 0.2 M). Before immunoprecipitation, a small chromatin-protein sample was excluded and used as input sample for a positive PCR control. DNA was purified using QIAquick purification kit (Qiagen) according to the manufacturer's instructions. The immunoprecipitated DNA was analyzed by semiquantitative PCR with an annealing temperature of 58°C for 36 cycles using primers spanning EGR1-binding motifs:
EZH2 AE forward (5′-CTCCACTGCCTTCTGAGTCC-3′), EZH2 AE reverse (5′-GGGCCAAATAAAAGCGATG-3′), SUZ12 AF forward (5′-GTGACTGACGGGGGAATC-3′), SUZ12 AF reverse (5′-AGGGAAGGAGGGAGGAAAA-3′), SUZ12 Q forward (5′-CGAGCGGTTGGTATTGCAG-3′), SUZ12 Q reverse (5′-AGGAGGAGGCCGAGTAACTG-3′), BMI1 AB forward (5′-CCCCCACAGCAACTATGAAA-3′), BMI1 AB reverse (5′-GCGCGATCGGTTTTATTCT-3′), BMI1 AC forward (5′-CTTGGCTCGCATTCATTTTC-3′), and BMI1 AC reverse (5′-CTACGTACCCGGAAAGAGCA-3′).
All PCR reactions were performed in triplicates and quantified by densitometric evaluation of signal intensity using the NIH image software.
RESULTS
Establishment of a database containing tissue and cell type-specific gene expression information based on mouse and human UniGene EST libraries
We generated a database containing quantitative estimates of gene expression information at different anatomical levels, starting from murine and human EST library data provided by UniGene. The processing of these primary data, together with the development of an extensive anatomical classification and with the corresponding annotation of all relevant UniGene EST libraries, led to the establishment of a database quantifying gene expression on a fine-grained hierarchical anatomical basis. Expression level information is stored in the TS-REX database for each UniGene cluster that is associated with at least one EST sequence in an EST library included in the hierarchical classification.
This classification is the result of a manual editing process aimed at (i) grouping anatomical structures into a fine-grained ontology in terms of pathologies, systems, organs, non-tumor tissues or tumor types (‘tissues’ in the following) and cell types, and (ii) mapping relevant UniGene EST libraries to the corresponding entry in the classification. Normalized and subtracted EST libraries were excluded. Each of the two pathology-related categories that are represented in the classification, namely ‘non-tumor’ and ‘tumor’, is the root of a hierarchy that spans four different aggregation levels, the most fine-grained structures corresponding to cell types.
An extract from the human hierarchical classification is displayed in Figure 2.
Figure 2.

Extract from the TS-REX human hierarchical classification. Individual UniGene EST library identifiers (column 1) are associated with anatomical categories specified in terms of pathologies, systems, organs, tissues and cell types whenever possible (columns 2–6).
Since the focus of TS-REX is tissue or cell-based dissection of TF-gene networks, the TS-REX software only makes use of the portion of the TS-REX database that corresponds to those genes that are reported to encode at least one TF in TRANSFAC®.
Most TRANSFAC® TF identifiers are covered by the TS-REX database (Supplementary Figure 1A and B), namely 1443 out of 1539 for H. sapiens, and 1080 out of 1128 for M. musculus.
Table 1 summarizes the total numbers of EST sequences and UniGene clusters included in the TS-REX database for H. sapiens and M. musculus, as well as the numbers of available pathologies, systems, organs, tissues and cell types in the hierarchical classification.
Table 1.
Summary of the total numbers of EST sequences and UniGene clusters included in the TS-REX database, and of the numbers of pathologies, systems, organs, tissues and cell types included in the TS-REX hierarchical classification
| H. sapiens | M. musculus | |
|---|---|---|
| Total number of ESTs | 6 509 336 | 4 029 157 |
| Total number of UniGene clusters | 123 993 | 82 105 |
| Number of pathologies (non-tumor, tumor) | 2 | 2 |
| Number of systems | 16 | 16 |
| Number of organs | 62 | 48 |
| Number of tissues | 155 | 75 |
| Number of cell types | 44 | 64 |
We designed TS-REX to allow a quantitative characterization of both presence and specificity of TFs relative to a given anatomical selection. For this reason, we stored different estimates of gene expression in the TS-REX database to be used with regards to either presence or specificity according to the user's parameter choice. More precisely, TS-REX quantifies presence of TFs in a set of anatomical structures in terms of transcripts per million (20), while specificity is characterized based on p-values calculated from the EST counts of the corresponding encoding genes, as detailed in ‘Materials and Methods’.
Extraction of tissue and cell type-specific TF networks through a web-based software
The TS-REX software is meant to be used in conjunction with existing TFBS prediction tools, for example TOUCAN, in order to highlight those TFs that are present in or specific to anatomical structures of interest, such as tissues or cell types, in the context of predicted TF-gene networks. Alternatively, other datasets of TF-target gene interactions can be used (see ‘Data input formats’ below).
A combination of technologies was used to develop the TS-REX software, both in order to meet specific requirements (e.g. for development of graphical user interfaces, for network visualization, and for string processing) and in order to base the system on cross-platform technologies.
The workflow is divided into the following steps:
Choose whether to use the public or the licensed version of the TRANSFAC® database, and log in from TS-REX.
Provide an input file containing TFBS prediction results, which subsequently gets uploaded to the TS-REX server for further processing.
Visualize the corresponding TF-gene network.
Use the TS-REX tissue browser to select anatomical structures of interest.
Visually present those predicted TFs that satisfy the selection criteria specified at step 4.
Export data for further analysis.
TS-REX also includes a demonstration feature that allows users to visualize a network from the Polycomb group gene (PcG) analysis without authentication to TRANSFAC®. In this case, a TF-gene network corresponding to TF proteins present in glioma and/or in neuroblastoma is used, and only TFs reported in TRANSFAC® Public can be highlighted.
Authentication to TRANSFAC®
This section describes the authentication procedure to TRANSFAC® from the TS-REX software. At application startup, the user is required to choose between the public and the licensed version of the TRANSFAC® database. TRANSFAC® contains information about TFs, including the corresponding encoding genes and statistical descriptions of the nucleotide composition of their binding sites. It is often used in conjunction with TFBS prediction tools. TS-REX incorporates information about TFs reported in TRANSFAC® Professional 11.1 for H. sapiens and M. musculus. In order for the system to make use of these data without any restrictions, users must log into the online version of TRANSFAC® Professional via the TS-REX authentication module. Authentication to TRANSFAC® Public is also possible, and results in the TS-REX software retrieving information from the TS-REX database for the encoding genes of those TFs that correspond to publicly available TRANSFAC® records only. Authentication to either TRANSFAC® Public or TRANSFAC® Professional is required in order to use TS-REX, with the exception of the demonstration feature. The development of the authentication module of TS-REX was inspired by a similar functionality available in the PAINT software (21). The file formats that TS-REX accepts as input are described below.
Data input formats
TS-REX requires the user to provide an input file in one of two possible forms:
A file exported from TOUCAN, containing the results of a TFBS prediction using MotifScanner with TRANSFAC® as motif database (2,7,22,23), or
A tab delimited text file containing two columns, namely (a) a target gene list, and (b) a list of the TRANSFAC® identifiers of the corresponding TFs that were predicted to regulate those genes using other binding site prediction tools.
When an input file of the first type is provided, it is automatically processed by the TS-REX software and converted into an internal data representation equivalent to an input file of the second type. This process is completely transparent to the user. An input file of either type will be referred to as ‘primary input file’ in the following.
As an alternative to using the TF gene expression information that is stored in the TS-REX database according to a hierarchical classification of anatomical structures, TS-REX also allows the user to provide an additional tab delimited text file (‘auxiliary file’ in the following). This option can be used in case users are interested in TF-gene networks of rare tissues or cell types that are not represented in the TS-REX database but for which gene expression information can be provided by the users themselves. TS-REX identifies TF-encoding genes in a list of genes (auxiliary file) that are expressed in such tissues or cell types. The auxiliary file can contain up to five gene lists, each of them reporting the UniGene gene symbols of genes that are known to be expressed in the corresponding tissue or cell type. Each of these lists can include up to 20 000 gene symbols. When the system is operated in this mode, the TS-REX database is bypassed, and TS-REX identifies TF-encoding genes among all genes present in the auxiliary file, and those are then highlighted in the TF-gene network. An example of a TS-REX auxiliary file is provided in Supplementary Figure 2.
TS-REX input files that correspond to the Polycomb group gene analysis (see ‘Discussion’) are provided in the Supplementary Data:
Supplementary File 1 contains data obtained using MotifScanner. This information corresponds to TFBS prediction results for human EZH2, SUZ12 and BMI1 as described in ‘Materials and Methods’.
Supplementary File 2 is an alternative input file that reports target genes and TRANSFAC® identifiers of predicted TFs as a two-column tab delimited text file. This makes it possible to provide TFBS prediction results obtained using another method than MotifScanner as input to TS-REX.
Network viewer
The network viewer is the TS-REX module that parses the user-provided primary input file and visualizes the corresponding TF-gene network before and after the selection of tissues or cell types. Target genes, TF genes and TF proteins are represented as nodes with different shapes and colors (Supplementary Figure 3). Network links correspond to relations between TF-encoding genes and encoded TF proteins, and to relations between predicted TF proteins and target genes. The concepts corresponding to a TF and to a network node representing a TF will be used interchangeably in the following. The network viewer is automatically started once the input file has been uploaded to the TS-REX server.
Node labels are obtained by merging the UniGene gene symbol (for either TF-encoding or target genes) or the TF name from TRANSFAC® with the corresponding UniGene or TRANSFAC® TF identifier. This choice was driven by the need to use distinct labels for different nodes in the network, since multiple UniGene identifiers may be associated with the same gene symbol, and multiple TRANSFAC® TF identifiers may correspond to the same TF protein. Moreover, displaying both factor names and TRANSFAC® TF identifiers makes it possible for the user to retrieve information for factors of interest from TRANSFAC® while preserving the readability of the TF-gene network.
Zooming and panning are possible, as is the search for nodes whose label contains a user-specified string, in order to allow users to quickly spot the position of genes or TFs of interest. Factors satisfying user-specified selection criteria, e.g. those that are present in a given tissue or cell type, are highlighted by coloring them red (see section below on ‘Tissue browser’).
An overview of the functionalities that TS-REX provides in terms of network visualization is given in Supplementary Figure 3, which displays the TS-REX network viewer: node shapes and colors are different for target genes (pink squares), predicted TF proteins (light-blue circles), and TF-encoding genes (green squares). The text area allowing the user to search for genes or factors whose name contains a given string is also visible, as well as buttons for zoom and pane operations. The figure demonstrates the use of the gene/factor search functionality after zooming in and translating the main network view to a region on the right-hand side of the overall network. Nodes corresponding to search results are colored yellow.
The CPU performance of the algorithm depends on the size of the dataset. When the number of binding site predictions in the primary input file exceeds a given threshold (which was set to 10 000 as a result of tests under different conditions), TS-REX does not allow visualization of the corresponding TF-gene network, which would be too large to be interactively rendered without significantly degrading software performance. However, the communication between software and database, the identification of TFs present in or specific to given anatomical structures and the possibility to export results to files are still functional. Data exported in tabular format can then be used as a refined input file (thus replacing the original input file by one containing only predicted TFs that satisfy a given anatomical selection), whose size would be suitable for interactive visualization.
Tissue browser: selection of tissue and cell type groups
The tissue browser is the TS-REX module that allows the user to explore the TS-REX hierarchical classification and to choose anatomical structures to be used to identify tissue-specific TFs in the TF-gene network that corresponds to the user-provided primary input file. It is started after the network viewer by using the ‘Tissue filter’ button. TS-REX subsequently pinpoints which of the predicted TFs in the analyzed dataset meet the specified selection criteria. Users can choose whether they want TS-REX to identify predicted TFs present in or specific to the selected anatomical categories; transcripts-per-million (TPM) or p-values from the TS-REX database are used by the system according to this choice, as described in ‘Materials and Methods’. For the sake of completeness, TS-REX also allows identification of those TFs that are not present in the selected anatomical structures.
In general, both options (presence and specificity) are available. However, when an auxiliary file is provided, the tissue browser does not allow identification of tissue-specific TFs, but only considers the presence of TF-encoding genes in this file. Each column in the auxiliary file must contain a header reporting the name of the corresponding tissue or cell type; this name is then displayed in the tissue browser in order to allow the user to select tissues of interest (Supplementary Figure 2).
TS-REX allows selection of pathologies, systems, organs, tissues or cell types, or combinations of those. When more than one item in the hierarchy is selected, the search in the TS-REX database is carried out corresponding to a logical OR of those categories, e.g. those TFs that are present in or specific to at least one of the selected tissues or cell types are identified and are highlighted in the TF-gene network.
The tissue browser includes a set of buttons that allow the user (i) to highlight the relevant subnetwork, i.e. change the graphical appearance of those nodes that correspond to TFs satisfying the selection criteria, (ii) to restore the default state of the graphical representation of the network or of the tree corresponding to the hierarchical classification or (iii) to export data to different files.
Figure 3 demonstrates the use of the tissue browser to identify TF proteins present in given anatomical structures with reference to the human classification. In this example, glioma, glioblastoma and oligodendroglioma are selected as tissues of interest. The number of EST libraries corresponding to this selection is displayed in red, in order to allow the user to critically evaluate the reliability of the results.
Figure 3.

Use of the tissue browser window to select anatomical structures, in this case human glioma, glioblastoma and oligodendroglioma tissues. More generally, users can select anatomical structures belonging to different levels in the hierarchy (1). Whenever more than one item is selected in the tissue browser, those TFs that meet the selection criteria for either of them are identified by the software and the corresponding nodes are highlighted in the network. In general, it is possible to identify TFs that are either present in or specific to given tissues or cell types (2); in both cases, a threshold level (TPM in case of presence, p-value in case of specificity) has to be specified by the user (3). The number of EST libraries in the TS-REX database that correspond to the anatomical selection performed is also indicated (4). A set of buttons allows the user to highlight network nodes that correspond to those TFs that meet the selection criteria, or to export data to different files (5).
TS-REX makes it possible to fine-tune the TPM or p-value threshold used for the identification of which TFs meet the selection criteria (see ‘Materials and Methods’), in order to allow users to change the definition of presence or specificity according to particular requirements. For instance, this additional layer of control could be used to investigate varying degrees of tissue specificity of different TFs in a TF-gene network. In general, the identification of TFs specific to a given cell type may lead to a set of factors still exhibiting different degrees of specificity, a piece of information that would be lost if only one fixed p-value threshold could be used. On the other hand, selecting lower p-value thresholds makes it possible to pinpoint which factors are more specific among those that were initially identified. The results provided by TS-REX in terms of which TFs are present in or specific to a given tissue or cell type always depend on a user-specified threshold, and choices of transcript-per-million and p-value thresholds are independent from each other.
Selection of TFs based on their enrichment in specific tissues or cell types
The quantitative estimates stored in the TS-REX database (TPMs or p-values) or, alternatively, tissue-related information contained in the user-provided auxiliary file are used to identify which TFs in the TF-gene network under investigation meet the selection criteria specified by the user. The corresponding network nodes are colored red in order to facilitate visual inspection, as exemplified in Figure 4A, where TFs present in human glioma, glioblastoma or oligodendroglioma in the Polycomb group TF-gene network are identified. Figure 4B displays the network containing only the highlighted TF nodes from Figure 4A, and was obtained using one of the files exported from TS-REX using the ‘Export selected’ functionality (see ‘Data export’), similar to the one that is exemplified in Supplementary File 5 (see ‘Data export’). This file contains a correspondence between target genes and TRANSFAC® identifiers of predicted TFs, restricted to highlighted TFs.
Figure 4.

View of TF-gene networks from the Polycomb group gene analysis. (A) Portion of the Polycomb group gene network after the identification of those TFs that are present in human glioma, glioblastoma or oligodendroglioma (TPM threshold = 100). Nodes corresponding to those TFs that meet the selection criteria are colored red. Squares and circles correspond to genes and TFs, respectively. Target and TF-encoding genes can be distinguished based on their color: the former are pink, the latter are green. (B) Portion of the corresponding subnetwork including only TF proteins present in glioma, glioblastoma or oligodendroglioma. This sub-network was obtained using one of the TS-REX exported files corresponding to the network in Figure 4A (see Results), namely a file that is similar to Supplementary File 5.
Data export
Data export is a crucial component of TS-REX that allows users to save data to different files in order to (i) inspect those TPMs or p-values that underlie the identification of relevant TFs as graphically displayed by the network viewer and to (ii) use data for further analysis. This allows TS-REX to be used in the context of user-defined analytical pipelines, downstream from existing binding site prediction tools, and upstream of subsequent processing. An example could be the generation of summarizing statistics on those TFs that are present in or specific to a given cell type in the context of a predicted TF-gene network for target genes of interest.
TS-REX includes two distinct export procedures, both related to those TFs that were highlighted as relevant to the selection performed, e.g. present in or specific to a given tissue or cell type: (i) ‘Export selected’ and (ii) ‘Export p-values/TPMs’ (Figure 3). Examples of files exported from TS-REX are provided as supplementary data.
Option (i) allows users to save the following three files: (a) an extract from the primary input file restricted to highlighted TFs, including lists of TRANSFAC® matrix identifiers corresponding to predicted binding sites (Supplementary File 3; this information is only available when the primary input file is an output file from MotifScanner), (b) a list of the UniGene EST library identifiers underlying the current selection, including an indication of which TF-encoding genes are represented in which libraries (Supplementary File 4) and (c) a file containing the correspondence between target genes and TRANSFAC® identifiers of predicted TFs, restricted to highlighted TFs (Supplementary File 5). The latter file can be used as input to TS-REX in order to visualize that portion of the original TF-gene network that includes highlighted TFs only.
Option (ii) allows users to save those TPMs or p-values that underlie the identification of which TFs meet the selection criteria specified, i.e. those numerical values that were internally used by the system in order to highlight TFs in the network (Supplementary File 6).
Prediction of TFs regulating genes, which encode Polycomb group proteins in different tumor types and embryonic stem cells
We used TS-REX to support the identification of so far unknown TFs controlling the expression of the Polycomb group protein encoding genes EZH2, SUZ12 and BMI1 in seven different human tissues, for which an over-expression and/or functional importance of these genes has previously been shown: breast cancer, prostate cancer, colon cancer, glioma, neuroblastoma, lymphoma and human embryonic stem cells. The use of TS-REX allowed rejection of up to 92% of the TF candidates originally predicted using TOUCAN, as detailed in Table 2, where the numbers of selected EST libraries for the different tissues are also indicated.
Table 2.
Summary of false positive rejection rates achieved with TS-REX in the PcG analysis
| Tissue | Number of selected EST librariesa | Target gene | Number of TFs predicted with TOUCANb | Number of false positives excluded by TS-REXc | Rejection rate (%)d | Average rejection rate (%)e |
|---|---|---|---|---|---|---|
| BC | 594 | BMI1 | 246 | 212 | 86 | 85 |
| EZH2 | 300 | 253 | 84 | |||
| SUZ12 | 346 | 298 | 86 | |||
| CC | 680 | BMI1 | 246 | 190 | 77 | 80 |
| EZH2 | 300 | 248 | 83 | |||
| SUZ12 | 346 | 281 | 81 | |||
| ES | 7 | BMI1 | 246 | 220 | 89 | 90 |
| EZH2 | 300 | 273 | 91 | |||
| SUZ12 | 346 | 315 | 91 | |||
| GL | 26 | BMI1 | 246 | 170 | 69 | 73 |
| EZH2 | 300 | 220 | 73 | |||
| SUZ12 | 346 | 266 | 77 | |||
| LY | 12 | BMI1 | 246 | 167 | 68 | 68 |
| EZH2 | 300 | 199 | 66 | |||
| SUZ12 | 346 | 243 | 70 | |||
| NB | 13 | BMI1 | 246 | 220 | 89 | 90 |
| EZH2 | 300 | 268 | 89 | |||
| SUZ12 | 346 | 320 | 92 | |||
| PC | 139 | BMI1 | 246 | 175 | 71 | 72 |
| EZH2 | 300 | 217 | 72 | |||
| SUZ12 | 346 | 248 | 72 |
BC: breast cancer; CC: colon cancer; ES: embryonic stem cell; GL: glioma; LY: lymphoma; NB: neuroblastoma; PC: prostate cancer.
aThe numbers of EST libraries corresponding to each tissue selection.
bFor each tissue and for each target gene, the initial numbers of TFs predicted using MotifScanner.
cThe numbers of false positives excluded by TS-REX based on a TPM threshold of 100.
dPercentage of rejected TFs with TPM counts lower than 100.
eAverage rejection rates of predicted TFs upstream of BMI1, EZH2, and SUZ12 for individual tissues.
In a first step, we identified conserved genomic DNA sequences of approximately 2000 bp around the transcriptional start site of the three genes in human and mouse using VISTA (17) and then determined TF-binding sites within these sequences using MotifScanner version 3.1.1 (2,7,22,23).
Binding sites present both in the murine and in the human sequence of each gene were identified, the resulting data were fed into TS-REX, and predicted TFs present in the six human tumor tissues and in human ES cells were identified using a TPM threshold of 100. The complete results, including the number of binding sites for all TFs predicted to regulate EZH2, SUZ12 and BMI1 in the different tissues, are provided as individual tab delimited text files for each tissue (breast cancer, Supplementary File 7; colorectal cancer, Supplementary File 8; ES cells, Supplementary File 9; glioma, Supplementary File 10; lymphoma, Supplementary File 11; neuroblastoma, Supplementary File 12; prostate cancer, Supplementary File 13).
Summarizing statistics on predicted TFs upstream of EZH2, SUZ12 and BMI1 in the different tissues are reported in Table 3 and in Supplementary Table 1. For each target gene, Supplementary Table 1 lists all predicted TFs, together with the corresponding numbers of TFBSs; occurrences of individual TRANSFAC® matrices are separately counted and reported as comma-separated sequences. The tissues in which each TF is present are also included. In order to present the most frequent TFs, Table 3 shows those TF proteins that are present in at least three out of the seven tissues of interest in this analysis, and for which at least three TRANSFAC® matrix identifiers are predicted in promoters of at least two target genes.
Table 3.
Summarizing statistics on predicted TFs upstream of EZH2, SUZ12 and BMI1 in the different tissues of interest in this analysis
| Transcription factor | BMI1 | EZH2 | SUZ12 | Present in |
|---|---|---|---|---|
| AhR | 2,2 | 2,2 | 1 | GL,LY,PC |
| DEC1 | 2 | 1 | BC,CC,GL,LY,PC | |
| DP-1 | 2,2,2 | 2,2,2,2,2 | 2,2,2,2,2 | BC,CC,GL,LY,PC |
| E12 | 2 | 2,2,2,1 | CC,GL,LY,PC | |
| E47 | 2 | 2,2,1 | CC,GL,LY,PC | |
| Egr-1 | 1,2,2 | 2,2 | 2,1 | CC,GL,PC |
| Fra-2 | 1 | 1,2,2 | CC,GL,LY | |
| HIF-1alpha | 2,1,2 | 1,2,2 | 1,1,1 | BC,CC,ES,GL,LY,NB,PC |
| HIF-1alpha-isoform1 | 2,1,2 | 1,2,2 | 1,1,1 | BC,CC,ES,GL,LY,NB,PC |
| HMG I | 1 | 1 | 2,2 | BC,CC,ES,GL,LY,NB,PC |
| HMG-Y | 1 | 1 | 2,2 | BC,CC,ES,GL,LY,NB,PC |
| HTF4 | 2 | 2,1 | BC,CC,GL,NB,PC | |
| HTF4gamma | 2 | 2,1 | BC,CC,GL,NB,PC | |
| IRF-1 | 1,1,1 | 2,2 | CC,LY,PC | |
| ITF-1 | 2 | 2,1 | CC,GL,LY,PC | |
| ITF-2 | 2 | 2,1 | GL,LY,NB,PC | |
| MAZ | 2 | 2 | 1 | BC,CC,ES,GL,LY,NB,PC |
| MRF-2-isoform1 | 2 | 2,1 | CC,GL,LY,PC | |
| NF-YC-3 | 1 | 2 | 2,2 | BC,ES,GL,LY |
| PEA3 | 2 | 1,2 | CC,ES,GL | |
| POU2F1 | 2 | 2,1,2,1,1 | GL,LY,NB | |
| RAR-alpha | 2 | 2,2 | 2 | BC,GL,NB,PC |
| RAR-alpha1 | 2 | 2,2 | 2 | BC,GL,NB,PC |
| RXR-beta | 2 | 2,2 | 2 | GL,LY,PC |
| SEF2-1B | 2 | 2,1 | GL,LY,NB,PC | |
| STAT1 | 2,1 | 2,2 | 2,2,2 | BC,CC,GL,LY,PC |
| STAT3 | 2 | 2 | 2,2 | BC,CC,GL,PC |
| STAT6 | 1,1 | 2,2 | 2,2,2 | BC,CC,LY,PC |
| Smad2 (437 AA) | 2 | 1 | GL,LY,PC | |
| Smad2-L | 2 | 2,2 | 2,1 | GL,LY,PC |
| Smad3 | 2 | 2,2 | 2,1 | CC,ES,GL |
| Smad4 | 2 | 2,2 | 2,1 | BC,ES,LY |
| Smad4delta3 | 2 | 1 | BC,ES,LY | |
| Smad4delta4-6 | 2 | 1 | BC,ES,LY | |
| Smad4delta4-7 | 2 | 1 | BC,ES,LY | |
| Smad4delta5-6 | 2 | 1 | BC,ES,LY | |
| Smad4delta6 | 2 | 1 | BC,ES,LY | |
| Sp1 | 2,2,2 | 2,2,2 | 1,2,2 | CC,ES,LY |
| T3R-alpha | 2 | 2 | 2 | CC,GL,NB,PC |
| T3R-alpha1 | 2 | 2 | 2 | CC,GL,NB,PC |
| T3R-alpha2 | 2 | 2 | 2 | CC,GL,NB,PC |
| USF2 | 2,2,2 | 2,2,1 | 1 | GL,LY,PC |
| USF2a | 2,2,2 | 2,2,1 | 1 | GL,LY,PC |
| USF2b | 2,2 | 2,1 | GL,LY,PC | |
| YY1 | 1,1,2,2 | 1,1,1 | 1 | CC,LY,PC |
| c-Ets-2 | 1,2 | 2,2 | CC,GL,LY | |
| c-Fos | 1,1 | 1,1,1,2,2 | BC,CC,GL,LY,NB,PC | |
| c-Jun | 1,1 | 1,1,1,2,2 | CC,GL,PC | |
| c-Myb | 1,1,1 | 2,2,2 | 2,2,2 | BC,LY,PC |
| c-Myb-isoform1 | 1,1,1 | 2,2,2 | 2,2,2 | BC,LY,PC |
| c-Myc | 1,1,2,2,1,2 | 2,1 | 1,1 | CC,GL,LY,PC |
| p53 | 2 | 2 | BC,CC,GL,LY,NB,PC | |
| p53-isoform-1 | 2 | 2 | BC,CC,GL,LY,NB,PC |
This list is restricted to those TFs that are present in at least three out of the seven tissues analyzed, and for which at least three TRANSFAC® matrix identifiers are predicted in promoters of at least two target genes. For each TF, numbers of predicted TFBSs are indicated, separating different TRANSFAC® matrices and reporting the corresponding numbers of occurrences as comma-separated sequence, together with the list of tissues in which that TF is present. Complete statistics are provided in Supplementary Table 1. BC: breast cancer; CC: colon cancer; ES: embryonic stem cell; GL: glioma; LY: lymphoma; NB: neuroblastoma; PC: prostate cancer. TF names are derived from TRANSFAC®.
We tested one of these novel predictions, namely the binding of EGR1 to EZH2, SUZ12 and BMI1 promoter sequences. In addition, we investigated whether over-expressing EGR1 affects the expression of the three target genes in a colorectal cancer cell line (SW480) and in a cervical cancer cell line (Hela). EGR1 EST counts of human colorectal cancer tissues were much higher (TPM = 222.33) than those of human cervical carcinoma tissues (TPM = 25.01). SW480 and Hela cells are not represented in the TS-REX database for reasons explained in ‘Materials and Methods’ with reference to the database generation process, but qPCR experiments showed a comparable difference in EGR1 transcript levels (Figure 5A). This expression difference was less considerable at the protein level (Figure 5B). Chromatin immunoprecipitation experiments revealed that EGR1 binds to genomic regions of all three target genes in which EGR1-binding motifs are present (Figure 5C and D). EGR1 binding to the three target genes is enhanced by phorbol 12-myristate-13-acetate (PMA) stimulation and by EGR1 over-expression. However, transient over-expression of this single TF did not lead to a significant change of expression of the three genes in SW480 and Hela cells as judged by qPCR measurements (data not shown), suggesting that additional factors act in combination with EGR1, with several candidate TFs being presented in this study.
Figure 5.

(A) EGR1 transcript levels in SW480 and Hela cells determined by qPCR. The vertical axis shows EGR1 expression differences relative to the reference gene B2M. Mean values and the standard deviation of three measurements are shown. (B) Detection of human EGR1 protein (82 kDa) in Hela and SW480 cells transfected with empty vector (CMV) or with EGR1 expression vector (EGR1) using western blot analysis. β-Actin levels served as loading control (bottom). (C, D) EGR1 binding to EZH2, SUZ12 and BMI1 promoter sequences. SW480 and Hela cells were (C) transiently transfected with empty vector (CMV) or with EGR1 expression vector (EGR1), or alternatively (D) treated with PMA or left untreated. Chromatin was precipitated using antibodies against EGR1, IgG or no antibody (no Ab), and analyzed by PCR. Different primer pairs for EZH2 (AE), SUZ12 (AF, Q) and BMI1 (AB, AC) are indicated. Results are presented as the ratio of immunoprecipitate (IP) over total input DNA utilized. Error bars show the standard deviations of three different PCR reactions.
DISCUSSION
We have developed a novel database/software resource for the tissue and cell type-specific dissection of TF-gene networks based on a newly established hierarchical anatomical classification of human and mouse UniGene ESTs and on quantitative estimates of gene expression levels.
Our approach offers distinct advantages in comparison to similar tools, which combine TFBS information with gene expression data. Even though MatInspector (8,9) integrates TFBS prediction functionalities with tissue associations of TFs, these associations do not represent quantitative expression levels, as they are based on information from published article abstracts, and they are not as fine grained and structured as the TF gene expression information provided by TS-REX. The Promoter Analysis Pipeline (10,11) and the method by Jeffery et al. (12) predict TFBSs in genomic regions of target genes, which share similar expression patterns. In contrast, TS-REX focuses on the tissue and cell type-specific expression of predicted TF genes to extract those TFs, which match the expression of their target genes. Although Jeffery and colleagues (12) demonstrate the integration of TF gene expression data, such data are not accessible to the user and are not structured as they are in TS-REX. Cytoscape (24) has a broader and different scope than TS-REX, which makes a direct comparison difficult. However, even though the modular architecture of Cytoscape allows it to make use of different external data sources and annotation datasets, one characteristic feature of TS-REX is the fact that its novel anatomical classification, its EST abundance database, and its interface with TF-binding site prediction functionalities provided by TOUCAN are all part of a unified design.
The TS-REX database and software resource was used to predict regulators of Polycomb group genes in six human tumor tissues and in human embryonic stem cells. Since TS-REX enables a tissue and cell type-specific screen based on TF gene expression levels, it was possible to filter out a large number of irrelevant predictions (up to 92%), which allowed us to focus on those that are of potential functional importance and represent interesting candidates for further experimental testing. Polycomb group proteins form complexes of different composition, which primarily act as negative regulators of gene expression by means of chromatin alterations (25–29). Besides the important role of PcG proteins in maintaining spatial patterns of gene silencing during development, they are critically involved in regulating mammalian stem and cancer cells (30,31). Within the cancer research field, an increasing number of reports supports a role of PcG proteins, in particular EZH2, SUZ12 and BMI1, in tumor development and in the maintenance of tumor-initiating cells. These genes are frequently over-expressed in cancer as compared to normal tissue, high expression levels correlate with poor patient prognosis in certain cancer types, and gene perturbation experiments have revealed their involvement in the generation and maintenance of tumorigenic cells (27,31,32).
Furthermore, recent findings led investigators to suggest that PcG target genes, including several known tumor suppressor genes, might be particularly prone to stable silencing in stem cells as a critical step during cancer development (33–35). These results were also interpreted as support for a stem cell origin of cancer, for which experimental evidence has so far been provided in certain cancer types (36,37).
The molecular mechanisms of PcG gene over-expression in different tumor types remain largely unresolved except for some cases of gene amplification and few upstream acting factors identified in certain cell types. Although several published and ongoing studies in the field of stem cell and cancer research focus on identifying PcG target genes, comprehensive knowledge about upstream TFs acting in different tissues and cell types is missing.
Our study revealed interesting novel predictions besides already known upstream acting factors, thus validating the applied approach (Table 3 and Supplementary Table 1). Factors that are known to regulate one or more of the three target genes are E2F TFs (38–43), p53 (44), TFs of the GLI-Krüppel family to which YY1 belongs (45,46), MYC family TFs (47–49) and TCF4 (50). Several newly predicted conserved binding sites in the studied genomic sequences of EZH2, SUZ12 and BMI1, which are bound by TFs present in many of the six tumor tissues and embryonic stem cells, are particularly interesting (see Table 3 and Supplementary Table 1). EGR1, HIF1A, DEC1 and ATF4 are hypoxia-inducible TFs, and accumulating experimental evidence supports a role for hypoxia in regulating normal stem cell and cancer cell functions (for review see 51). In this study, we demonstrate the binding of EGR1 to all three tested Polycomb group gene promoters, which is enhanced by PMA stimulation and EGR1 over-expression. Our results suggest that Polycomb group protein genes could be involved in hypoxia-mediated regulation of stem cell and cancer cell functions, which could be investigated more thoroughly in future studies.
In summary, we have presented a tool that allows extraction and visualization of TF-gene interactions, with a focus on identifying those TFs that are present in tissues or cell types of interest, in order to reduce false positive rates in TFBS prediction.
Its flexibility (i) in identifying TFs either present in or specific to tissues or cell types of interest using a fine-grained hierarchical anatomical classification, (ii) in fine-tuning tissue selection parameters and (iii) in exporting TPMs/p-values to text files for further analysis, makes TS-REX a valuable novel resource for analysis and visualization of tissue and cell type-specific TF-gene interactions. TF gene expression levels are currently only based on EST counts, and individual cell types or cell lines might not be sufficiently covered by EST libraries in UniGene or might not be included in the TS-REX database for reasons mentioned in ‘Materials and Methods’. However, the modular architecture of TS-REX enables a future integration of additional datasets, e.g. microarray and chromatin immunoprecipitation data.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Swedish Foundation for Strategic Research; the Swedish Cancer Foundation; the Swedish Pediatric Cancer Foundation; the Swedish Research Council; the Governmental Funding of Clinical Research within the National Health Services; the Mary Béves Foundation; the Berta Kamprad foundation, the Gunnar Nilsson cancer foundation, the Thorsten and Elsa Segerfalk foundation and the EU FP6 project Genostem. Funding for open access charge: Funds from Lund University Faculty of Medicine.
Conflict of interest statement. None declared.
Supplementary Material
ACKNOWLEDGEMENTS
We thank Jari Häkkinen for setting up the Java Web Start interface of TS-REX, Morten Krogh and Yingchun Liu for useful discussions and valuable comments, and Markus Ringnér for pointing us to the PAINT software. We are grateful to Stein Aerts, Gert Thijs and Peter Van Loo for their support with TOUCAN, and we thank Raj Vadigepalli for kindly providing the PAINT login scripts to TRANSFAC®.
REFERENCES
- 1.Wingender E, Dietze P, Karas H, Knuppel R. TRANSFAC: a database on transcription factors and their DNA binding sites. Nucleic Acids Res. 1996;24:238–241. doi: 10.1093/nar/24.1.238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Aerts S, Thijs G, Coessens B, Staes M, Moreau Y, DeMoor B. Toucan: deciphering the cis-regulatory logic of coregulated genes. Nucleic Acids Res. 2003;31:1753–1764. doi: 10.1093/nar/gkg268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kel AE, Gossling E, Reuter I, Cheremushkin E, Kel-Margoulis OV, Wingender E. MATCH: a tool for searching transcription factor binding sites in DNA sequences. Nucleic Acids Res. 2003;31:3576–3579. doi: 10.1093/nar/gkg585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sauer T, Shelest E, Wingender E. Evaluating phylogenetic footprinting for human-rodent comparisons. Bioinformatics. 2006;22:430–437. doi: 10.1093/bioinformatics/bti819. [DOI] [PubMed] [Google Scholar]
- 5.Lenhard B, Wasserman WW. TFBS: computational framework for transcription factor binding site analysis. Bioinformatics. 2002;18:1135–1136. doi: 10.1093/bioinformatics/18.8.1135. [DOI] [PubMed] [Google Scholar]
- 6.Sandelin A, Wasserman WW, Lenhard B. ConSite: web-based prediction of regulatory elements using cross-species comparison. Nucleic Acids Res. 2004;32:W249–W252. doi: 10.1093/nar/gkh372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aerts S, Van Loo P, Thijs G, Mayer H, de Martin R, Moreau Y, De Moor B. TOUCAN 2: the all-inclusive open source workbench for regulatory sequence analysis. Nucleic Acids Res. 2005;33:W393–W396. doi: 10.1093/nar/gki354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Quandt K, Frech K, Karas H, Wingender E, Werner T. MatInd and MatInspector: new fast and versatile tools for detection of consensus matches in nucleotide sequence data. Nucleic Acids Res. 1995;23:4878–4884. doi: 10.1093/nar/23.23.4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cartharius K, Frech K, Grote K, Klocke B, Haltmeier M, Klingenhoff A, Frisch M, Bayerlein M, Werner T. MatInspector and beyond: promoter analysis based on transcription factor binding sites. Bioinformatics. 2005;21:2933–2942. doi: 10.1093/bioinformatics/bti473. [DOI] [PubMed] [Google Scholar]
- 10.Chang LW, Fontaine BR, Stormo GD, Nagarajan R. PAP: a comprehensive workbench for mammalian transcriptional regulatory sequence analysis. Nucleic Acids Res. 2007;35:W238–W244. doi: 10.1093/nar/gkm308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chang LW, Nagarajan R, Magee JA, Milbrandt J, Stormo GD. A systematic model to predict transcriptional regulatory mechanisms based on overrepresentation of transcription factor binding profiles. Genome Res. 2006;16:405–413. doi: 10.1101/gr.4303406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jeffery IB, Madden SF, McGettigan PA, Perriere G, Culhane AC, Higgins DG. Integrating transcription factor binding site information with gene expression datasets. Bioinformatics. 2007;23:298–305. doi: 10.1093/bioinformatics/btl597. [DOI] [PubMed] [Google Scholar]
- 13.Deplancke B, Mukhopadhyay A, Ao W, Elewa AM, Grove CA, Martinez NJ, Sequerra R, Doucette-Stamm L, Reece-Hoyes JS, Hope IA, et al. A gene-centered C. elegans protein-DNA interaction network. Cell. 2006;125:1193–1205. doi: 10.1016/j.cell.2006.04.038. [DOI] [PubMed] [Google Scholar]
- 14.Vermeirssen V, Barrasa MI, Hidalgo CA, Babon JA, Sequerra R, Doucette-Stamm L, Barabasi AL, Walhout AJ. Transcription factor modularity in a gene-centered C. elegans core neuronal protein-DNA interaction network. Genome Res. 2007;17:1061–1071. doi: 10.1101/gr.6148107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yu X, Lin J, Zack DJ, Qian J. Computational analysis of tissue-specific combinatorial gene regulation: predicting interaction between transcription factors in human tissues. Nucleic Acids Res. 2006;34:4925–4936. doi: 10.1093/nar/gkl595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wingender E, Chen X, Fricke E, Geffers R, Hehl R, Liebich I, Krull M, Matys V, Michael H, Ohnhauser R, et al. The TRANSFAC system on gene expression regulation. Nucleic Acids Res. 2001;29:281–283. doi: 10.1093/nar/29.1.281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mayor C, Brudno M, Schwartz JR, Poliakov A, Rubin EM, Frazer KA, Pachter LS, Dubchak I. VISTA: visualizing global DNA sequence alignments of arbitrary length. Bioinformatics. 2000;16:1046–1047. doi: 10.1093/bioinformatics/16.11.1046. [DOI] [PubMed] [Google Scholar]
- 18.Kent WJ, Sugnet CW, Furey TS, Roskin KM, Pringle TH, Zahler AM, Haussler D. The human genome browser at UCSC. Genome Res. 2002;12:996–1006. doi: 10.1101/gr.229102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Thiel G, Schoch S, Petersohn D. Regulation of synapsin I gene expression by the zinc finger transcription factor zif268/egr-1. J. Biol. Chem. 1994;269:15294–15301. [PubMed] [Google Scholar]
- 20.Kodzius R, Matsumura Y, Kasukawa T, Shimokawa K, Fukuda S, Shiraki T, Nakamura M, Arakawa T, Sasaki D, Kawai J, et al. Absolute expression values for mouse transcripts: re-annotation of the READ expression database by the use of CAGE and EST sequence tags. FEBS Lett. 2004;559:22–26. doi: 10.1016/S0014-5793(04)00018-3. [DOI] [PubMed] [Google Scholar]
- 21.Vadigepalli R, Chakravarthula P, Zak DE, Schwaber JS, Gonye GE. PAINT: a promoter analysis and interaction network generation tool for gene regulatory network identification. Omics. 2003;7:235–252. doi: 10.1089/153623103322452378. [DOI] [PubMed] [Google Scholar]
- 22.Thijs G, Moreau Y, De Smet F, Mathys J, Lescot M, Rombauts S, Rouze P, De Moor B, Marchal K. INCLUSive: integrated clustering, upstream sequence retrieval and motif sampling. Bioinformatics. 2002;18:331–332. doi: 10.1093/bioinformatics/18.2.331. [DOI] [PubMed] [Google Scholar]
- 23.Coessens B, Thijs G, Aerts S, Marchal K, De Smet F, Engelen K, Glenisson P, Moreau Y, Mathys J, De Moor B. INCLUSive: a web portal and service registry for microarray and regulatory sequence analysis. Nucleic Acids Res. 2003;31:3468–3470. doi: 10.1093/nar/gkg615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cline MS, Smoot M, Cerami E, Kuchinsky A, Landys N, Workman C, Christmas R, Avila-Campilo I, Creech M, Gross B, et al. Integration of biological networks and gene expression data using Cytoscape. Nat. Protoc. 2007;2:2366–2382. doi: 10.1038/nprot.2007.324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gil J, Bernard D, Peters G. Role of polycomb group proteins in stem cell self-renewal and cancer. DNA Cell Biol. 2005;24:117–125. doi: 10.1089/dna.2005.24.117. [DOI] [PubMed] [Google Scholar]
- 26.Hormaeche I, Licht JD. Chromatin modulation by oncogenic transcription factors: new complexity, new therapeutic targets. Cancer Cell. 2007;11:475–478. doi: 10.1016/j.ccr.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 27.Rajasekhar VK, Begemann M. Concise review: roles of polycomb group proteins in development and disease: a stem cell perspective. Stem Cells. 2007;25:2498–2510. doi: 10.1634/stemcells.2006-0608. [DOI] [PubMed] [Google Scholar]
- 28.Kohler C, Villar CB. Programming of gene expression by Polycomb group proteins. Trends Cell Biol. 2008;18:236–243. doi: 10.1016/j.tcb.2008.02.005. [DOI] [PubMed] [Google Scholar]
- 29.Schwartz YB, Pirrotta V. Polycomb complexes and epigenetic states. Curr. Opin. Cell Biol. 2008;20:266–273. doi: 10.1016/j.ceb.2008.03.002. [DOI] [PubMed] [Google Scholar]
- 30.Pietersen AM, van Lohuizen M. Stem cell regulation by polycomb repressors: postponing commitment. Curr. Opin. Cell Biol. 2008;20:201–207. doi: 10.1016/j.ceb.2008.01.004. [DOI] [PubMed] [Google Scholar]
- 31.Sauvageau M, Sauvageau G. Polycomb group genes: keeping stem cell activity in balance. PLoS Biol. 2008;6:e113. doi: 10.1371/journal.pbio.0060113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sparmann A, van Lohuizen M. Polycomb silencers control cell fate, development and cancer. Nat. Rev. Cancer. 2006;6:846–856. doi: 10.1038/nrc1991. [DOI] [PubMed] [Google Scholar]
- 33.Balch C, Nephew KP, Huang TH, Bapat SA. Epigenetic “bivalently marked” process of cancer stem cell-driven tumorigenesis. Bioessays. 2007;29:842–845. doi: 10.1002/bies.20619. [DOI] [PubMed] [Google Scholar]
- 34.Ohm JE, McGarvey KM, Yu X, Cheng L, Schuebel KE, Cope L, Mohammad HP, Chen W, Daniel VC, Yu W, et al. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat. Genet. 2007;39:237–242. doi: 10.1038/ng1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Widschwendter M, Fiegl H, Egle D, Mueller-Holzner E, Spizzo G, Marth C, Weisenberger DJ, Campan M, Young J, Jacobs I, et al. Epigenetic stem cell signature in cancer. Nat. Genet. 2007;39:157–158. doi: 10.1038/ng1941. [DOI] [PubMed] [Google Scholar]
- 36.Wang JC, Dick JE. Cancer stem cells: lessons from leukemia. Trends Cell Biol. 2005;15:494–501. doi: 10.1016/j.tcb.2005.07.004. [DOI] [PubMed] [Google Scholar]
- 37.Ailles LE, Weissman IL. Cancer stem cells in solid tumors. Curr. Opin. Biotechnol. 2007;18:460–466. doi: 10.1016/j.copbio.2007.10.007. [DOI] [PubMed] [Google Scholar]
- 38.Muller H, Bracken AP, Vernell R, Moroni MC, Christians F, Grassilli E, Prosperini E, Vigo E, Oliner JD, Helin K. E2Fs regulate the expression of genes involved in differentiation, development, proliferation, and apoptosis. Genes Dev. 2001;15:267–285. doi: 10.1101/gad.864201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weinmann AS, Bartley SM, Zhang T, Zhang MQ, Farnham PJ. Use of chromatin immunoprecipitation to clone novel E2F target promoters. Mol. Cell Biol. 2001;21:6820–6832. doi: 10.1128/MCB.21.20.6820-6832.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bracken AP, Pasini D, Capra M, Prosperini E, Colli E, Helin K. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. Embo. J. 2003;22:5323–5335. doi: 10.1093/emboj/cdg542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Oberley MJ, Farnham PJ. Probing chromatin immunoprecipitates with CpG-island microarrays to identify genomic sites occupied by DNA-binding proteins. Methods Enzymol. 2003;371:577–596. doi: 10.1016/S0076-6879(03)71043-X. [DOI] [PubMed] [Google Scholar]
- 42.Bieda M, Xu X, Singer MA, Green R, Farnham PJ. Unbiased location analysis of E2F1-binding sites suggests a widespread role for E2F1 in the human genome. Genome Res. 2006;16:595–605. doi: 10.1101/gr.4887606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nowak K, Kerl K, Fehr D, Kramps C, Gessner C, Killmer K, Samans B, Berwanger B, Christiansen H, Lutz W. BMI1 is a target gene of E2F-1 and is strongly expressed in primary neuroblastomas. Nucleic Acids Res. 2006;34:1745–1754. doi: 10.1093/nar/gkl119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tang X, Milyavsky M, Shats I, Erez N, Goldfinger N, Rotter V. Activated p53 suppresses the histone methyltransferase EZH2 gene. Oncogene. 2004;23:5759–5769. doi: 10.1038/sj.onc.1207706. [DOI] [PubMed] [Google Scholar]
- 45.Leung C, Lingbeek M, Shakhova O, Liu J, Tanger E, Saremaslani P, Van Lohuizen M, Marino S. Bmi1 is essential for cerebellar development and is overexpressed in human medulloblastomas. Nature. 2004;428:337–341. doi: 10.1038/nature02385. [DOI] [PubMed] [Google Scholar]
- 46.Liu S, Dontu G, Mantle ID, Patel S, Ahn NS, Jackson KW, Suri P, Wicha MS. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006;66:6063–6071. doi: 10.1158/0008-5472.CAN-06-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Guney I, Wu S, Sedivy JM. Reduced c-Myc signaling triggers telomere-independent senescence by regulating Bmi-1 and p16(INK4a) Proc. Natl Acad. Sci. USA. 2006;103:3645–3650. doi: 10.1073/pnas.0600069103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Guo WJ, Datta S, Band V, Dimri GP. Mel-18, a polycomb group protein, regulates cell proliferation and senescence via transcriptional repression of Bmi-1 and c-Myc oncoproteins. Mol. Biol. Cell. 2007;18:536–546. doi: 10.1091/mbc.E06-05-0447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen X, Xu H, Yuan P, Fang F, Huss M, Vega VB, Wong E, Orlov YL, Zhang W, Jiang J, et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells. Cell. 2008;133:1106–1117. doi: 10.1016/j.cell.2008.04.043. [DOI] [PubMed] [Google Scholar]
- 50.Kirmizis A, Bartley SM, Farnham PJ. Identification of the polycomb group protein SU(Z)12 as a potential molecular target for human cancer therapy. Mol. Cancer Ther. 2003;2:113–121. [PubMed] [Google Scholar]
- 51.Keith B, Simon MC. Hypoxia-inducible factors, stem cells, and cancer. Cell. 2007;129:465–472. doi: 10.1016/j.cell.2007.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.