


Abstract
Advances in sequencing technologies have accelerated the sequencing of new genomes, far outpacing the generation of gene and protein resources needed to annotate them. Direct comparison and alignment of existing cDNA sequences from a related species is an effective and readily available means to determine genes in the new genomes. Current spliced alignment programs are inadequate for comparing sequences between different species, owing to their low sensitivity and splice junction accuracy. A new spliced alignment tool, sim4cc, overcomes problems in the earlier tools by incorporating three new features: universal spaced seeds, to increase sensitivity and allow comparisons between species at various evolutionary distances, and powerful splice signal models and evolutionarily-aware alignment techniques, to improve the accuracy of gene models. When tested on vertebrate comparisons at diverse evolutionary distances, sim4cc had significantly higher sensitivity compared to existing alignment programs, more than 10% higher than the closest competitor for some comparisons, while being comparable in speed to its predecessor, sim4. Sim4cc can be used in one-to-one or one-to-many comparisons of genomic and cDNA sequences, and can also be effectively incorporated into a high-throughput annotation engine, as demonstrated by the mapping of 64 000 Fagus grandifolia 454 ESTs and unigenes to the poplar genome.
INTRODUCTION
Advances in DNA sequencing will bring a significant growth in the number and diversity of available genomes over the next few years. More than 100 animals and 50 plants have been sequenced to various degrees of completion and more are slated to be sequenced (http://www.ncbi.nlm.nih.gov/genomes/leuks.cgi; http://www.genome.gov/10002154). With costs falling sharply and increased accessibility to sequencing technologies, it will soon become feasible for individual investigators to sequence their species of interest. To be useful to researchers, these genomes will need to be analyzed to determine genes and other functional elements. While new genome sequencing projects are progressing at a fast pace, however, the generation of expressed DNA (cDNA, EST, mRNA) and protein sequences needed to annotate them has been slow (1). Moreover, sequencing of full-length mRNA sequences critical for annotation quality has focused on a handful of high-priority species (2–5). An economical and increasingly popular approach is to generate mixed collections of resources from multiple closely related organisms and share them across several projects (http://www.fagaceae.org). Mapping gene information already available in databases provides an efficient means to annotate the new genomes, one which requires fast and accurate alignment tools that can be readily used, with little or no human intervention, for a variety of comparisons.
Tools for aligning cDNA and genomic sequences typically have been designed for high sequence similarity and lose power in comparisons across species, or are too slow to handle large annotation tasks. Indeed, programs such as sim4 (6), Spidey (7), BLAT (8), MgAlignIt (9), ESTmapper (10) and GMAP (11) use heuristic alignment methods to align sequences of the same species efficiently and with high accuracy, but their performance drops significantly as the sequence similarity decreases. Only a few of these programs have been adapted to aligning sequences cross-species. For instance, BLAT translates both the query and the database into protein sequences before matching and GMAP uses an adjusted parameter set, but the quality of output is below what is required for automated annotation. Other tools, such as GeneSeqer (12), EST_GENOME (13) and EXALIN (14), employ probabilistic or exact dynamic programming methods and are capable of aligning sequences cross-species, but lack the speed required for whole-genome annotation and are still limited in the range of evolutionary distances they address.
Computationally, aligning a cDNA with a genomic sequence containing that gene entails partitioning the cDNA into exons and the genomic sequence into exons and introns, such that exons are similar between the two sequences except for a few differences caused by sequencing errors and polymorphisms. Additionally, introns must start and end with specific splice signals (GT–AG is the most common). In comparisons between species, evolutionary mutation and gap patterns compound the differences, increasing the difficulty of alignment. Thus, a cross-species spliced alignment tool must be able to handle sequence differences arising from a variety of sources and to correctly identify the splice junctions, and it must do so efficiently and without user intervention to allow application to large automated genome annotation projects. By far the main challenge that confronts existing cross-species alignment tools is their low sensitivity, leading in turn to incomplete gene models and poor splice junction accuracy. Further challenges arise from differences in gene models caused by evolutionary block insertion and deletion events.
Among the most important factors for program sensitivity is the match pattern used to identify exact or near-exact word matches between the sequences, called the seed. The traditional blast (15) seed required an exact match of 11 contiguous positions (11111111111), and is called continuous. This seed has been adopted by most alignment algorithms until its limitations have recently been revealed (16). To improve sensitivity, spaced seeds allow mismatches at specified positions in the seed pattern. Judiciously chosen spaced seeds that take into account the characteristics of the alignment achieve significantly higher sensitivity than continuous seeds (17,18), and some have already been successfully implemented into whole-genome alignment programs such as PatternHunter (16) and blastz (19). Alignments of gene sequences have characteristics that differentiate them from genomic alignments, including higher order dependencies between positions (20), transition-transversion biases (21) and 3-periodicity due to their codon structure. We recently incorporated these features into new mathematical models and were able to design improved seeds for cross-species cDNA-to-genome alignment (22). An additional practical consideration for developing alignment tools, especially as the number of species-to-species comparisons increases, is their applicability range. Designing seeds for even one comparison is computationally expensive. An economical alternative is to identify a small number of program parameters that perform well on a large number of comparisons and thus can be seamlessly used without regard to the species compared. We recently characterized and identified such seeds, which we termed universal, for a large number of vertebrate comparisons (23,24), and incorporate them into our program sim4cc.
Starting from the design principles above, we developed an algorithm and software tool, called sim4cc (sim4 for cross-species comparisons), for aligning cDNA and genomic sequences between species at various evolutionary distances. Sim4cc is built on the foundation of our earlier program sim4 (6), one of the earliest spliced-alignment tools, but has incorporated significant changes to adapt it to cross-species comparisons, including universal spaced seeds designed for a wide range of species comparisons, more sophisticated splice site models and evolutionarily-aware alignment algorithms. Like its predecessor, sim4cc is designed to align a cDNA with a genomic region containing a homolog of that gene, but it can be incorporated easily into a high-throughput genome annotation engine. Moreover, with its small memory footprint and user-friendly interface, it is well suited for use by individual researchers who wish to analyze their genomic sequence of interest on their local computer. Source code for the program is available free of charge from our web site http://www.cbcb.umd.edu/software/sim4cc.
MATERIALS AND METHODS
We first describe the foundation of sim4cc, followed by improvements.
Basic Sim4cc algorithm
Like sim4 (6), sim4cc has the following stages.
Determining ‘exon cores’: A blast-like search detects seed matches between the sequences and extends them to gap-free local alignments (MSPs), scoring 1 for a nucleotide match, −1 for a transition and −3 for a transversion. A maximally scoring subset of the MSPs is selected using sparse dynamic programming, and neighboring MSPs are merged to form ‘exon cores’. This initial coarse gene structure is then gradually refined in the following stages.
Exon refinement: Gaps between consecutive ‘exon cores’ are filled with two alignment procedures: the first, a fast greedy sequence-alignment algorithm that extends the exons toward each other; and the second, a new blast-like search invoked with more permissive parameters to find new, previously missed exons.
Intron refinement: Intron boundaries are repositioned, if necessary, to better conform to the consensus splice signals (GT–AG). The best intron position is chosen based on a score that combines splice signal strength with the quality of alignment in the surrounding exons. The procedure allows for non-canonical splice signals.
Nucleotide-level alignment: Homologous exons between the two sequences are aligned with a greedy algorithm, and exon alignments are then concatenated via long intron gaps to produce the final spliced alignment of the two sequences.
Several limitations become apparent when comparing sequences cross-species, including loss of sensitivity, a decrease in splice junction accuracy, and local inaccuracies in the nucleotide-level alignment that arise from evolutionary mutations. The following sections describe algorithmic improvements to address these problems.
Universal spaced seeds
Spaced seeds have recently emerged as a mechanism to increase alignment sensitivity. Unlike continuous seeds, which require an exact match of k contiguous bases and are represented as vectors of 1s, spaced seeds allow for some positions in the seed pattern to vary, for instance the seed 101100001011 has the wildcard positions 2, 5, 6, 7, 8 and 10. The number of 1s in the seed is called the seed weight and controls the specificity (22). The length of the seed is called span. Alignments are also represented as (0, 1) strings, where 1s represent matches and 0s mismatches, drawn from a probabilistic model, for instance a Bernoulli or Markov chain. Given a seed and an alignment model, the seed sensitivity can be estimated as the probability that it will detect a random alignment string of length L = 64 (16) generated from the alignment model. Optimal seeds can then be determined mathematically using dynamic programming (17,18,22).
To better suit the characteristics of coding sequences, including but not limited to higher-order dependencies between positions (20), specific transition-transversion biases and codon-based periodicity, we recently extended the models to incorporate these features, most notably by introducing a new wildcard symbol (x) that allows transitions but not transversions, and by using a higher order 3-periodic Markov model of alignment. With these extensions we were able to derive more sensitive seeds than previously reported that also had better sensitivity-specificity tradeoffs in practice (22). These considerations are important for fine tuning sensitivity and specificity when developing alignment tools.
Furthermore, to accommodate the multitude and variety of new genomes, we characterized and designed seeds that work well for a wide range of comparisons, so called universal seeds. Our approach was founded on two principles: first, that similar alignment models induce similar behavior of seeds (23), and second, that with high probability seeds optimized for more distant comparisons perform well on the closer ones (24). We recently designed and validated such seeds for a comprehensive set of 66 pairwise comparisons of twelve vertebrate species (human, chimp, macaque, mouse, rat, cow, dog, opossum, chicken, frog, fugu and zebrafish). Using the Kullback–Leibler Divergence (25) as a measure of comparison closeness, the comparisons were clustered into four groups with remarkable evolutionary significance, largely corresponding to different depths in the phylogenetic tree of the species, and seeds were optimized for each group. In the end, seeds optimized for the most distant set of comparisons, between frog or fishes and the rest of the sampled species, were selected to be implemented in sim4cc (23). The default seed in stage i. is 1xx1011011011xx11, and alternative seeds, such as those reported in (23,24), can be specified by the user via the command line.
Splice junction models
When comparing sequences within the same species, relatively simple strategies are sufficient to refine the exon edges in stage iii. In cross-species comparisons, as the distance between the predicted and true exon ends increases, more sophisticated splice junction strategies are needed. We tested and incorporated two probabilistic splice models to augment the simple 4 × 4 GT–AG position weight matrix (PWM) implemented in sim4: the Glimmer model (26) and the GeneSplicer model (27).
The Glimmer fixed Interpolated Context (IC) splice-site model
The Glimmer method (26) was used to produce a probabilistic model of splice signals from a training set of fixed-length strings by creating a separate, non-periodic ICM model for each position i in the string. The probability of the entire length-n string is then the product of the probabilities of each character i, 1 ≤ i ≤ n, with the appropriate ICM model for each position. The position-i model represents the probability of the character at position i conditioned by the Markov context window consisting of characters 1 … (i – 1) that precede it in the string. Because there will practically never be enough training data to learn a model that uses all the characters in this context window, a subset of them is selected by computing the pairwise mutual information between each position k (from 1 to i –1) and the predicted position i. The position k with the maximum mutual information is chosen and the training data set is partitioned based on the character occurring at position k. Within each partition subset the process is repeated until a maximum number of positions are reached, or the size of the training set drops below a minimum value. To score splice sites, a positive model is learned from a training set of true splice sites, and a negative model is learned from a training set of non-splice sites. The score of a given test string is the log-odds ratio of these two models for that string.
The GeneSplicer Interpolated Markov (IM) splice-site model
The splice site scores produced by GeneSplicer (27) reflect the strength of the splice site signal in a fixed window around the splice site consensus (GT, AG), weighted by the probability that there is a switch between the coding and non-coding sequences at that position. Thus, the score of a fixed-length sequence f predicted to contain a splice site is given by:
where Ps denote probabilities, S5′ (f) and S3′ (f) represent the upstream and downstream regions of the splice site consensus contained in f, respectively, and 𝒞 and 𝒞 represent the coding and non-coding complementary events that characterize a region according to the type of splice site (e.g. 𝒞 is ‘coding’ for donor sites and ‘non-coding’ for acceptor sites). The probability P(f |splicesite) is estimated with the Maximum Dependence Decomposition method (20), while all the other probabilities are estimated using coding and non-coding content sensors represented by two interpolated Markov models built on all coding and non-coding regions in the training data, respectively.
The models were trained on an aggregate data set consisting of 10 000 positive and 50 000 negative examples from twelve vertebrate genomes (human, chimp, macaque, mouse, rat, cow, dog, opossum, chicken, frog, fugu and zebrafish) (Supplementary Data S3). The final splice site scores in stage (iii) of the sim4cc algorithm are then determined by a linear combination of the quality of alignment in the two 30-bp windows surrounding the start and the end of the intron, and the splice signal strength determined by the consensus PWM and by either of the GeneSplicer or Glimmer models, as specified by the user. Specifically, if i, i +1, j1, j2 (between 1 and 30) represent the current endpoints of the two adjacent exons in the cDNA and genomic windows, respectively, then the splice configuration score is [modified from (28)]:
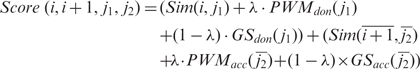 |
where Sim(i, j) (Sim(i, j)) is the percent sequence identity of the best sequence alignment ending at positions(i, j) in the two windows, respectively, PWM(·) is the original dinucleotide splice site score implemented in sim4, GS(·) is either the GeneSplicer or Glimmer splice signal score, and λ is a constant between 0 and 1, calibrated by iteratively testing values in 0.01 increments in sim4cc. The configuration with the maximum score is chosen as the splice junction.
Nucleotide-level and exon-level alignment
Evolutionary changes, both point mutations and insertions or deletions of exonic blocks, can produce significant differences between sequences that will confound alignment programs. To account for local sequence variations, we implemented affine-gap penalty greedy alignment algorithms (29). Apart from substitutions and small indels, large deletions or insertions of exonic material cause differences in gene structure between the species that are more challenging to detect and interpret. Based on empirical analyses, we set a minimum intron size of 50 bp. Lastly, some spurious exons are likely to occur at alignment ends. Most of these are eliminated without significant loss in sensitivity by removing short terminal exons that are farther than 20 kb from the alignment core.
Construction of vertebrate reference data sets
To evaluate the performance of sim4cc and several other spliced alignment programs, reference annotation sets were constructed for four types of comparisons: between human and mouse, dog, chicken and zebrafish, respectively. To create an initial annotation, in a first stage human RefSeq (3) mRNA sequences were aligned to the human genome version hg18 (http://www.genome.ucsc.edu) using the high-throughput software ESTmapper (10). Alignments were then refined by comparison with the expert curated VEGA annotation (30). On human chromosome 1, of the 2554 RefSeq ESTmapper alignments 1808 had exon-intron structure identical to the VEGA annotation and did not require adjustment. Another 114 differed by less than 10 bp at one or several exon boundaries. Manual inspection of this set resolved 24 of these in favor of the VEGA annotation and 87 in favor of ESTmapper, while the remaining three genes were ambiguous. Furthermore, 287 alignments showed significant differences in gene structure between the two annotations, possibly due to splice variations, and the remaining 325 did not have a VEGA match. These two latter categories were therefore excluded from our selection. In the end, the procedure produced 1922 human gene annotations on chromosome 1.
In stage two, these initial annotations were trimmed to contain only regions in common to the two species. For this purpose, orthologs of human mRNAs in three of the four species (mouse, dog and chicken) were determined from homoloGene (build 54, April 2007) (1), and by blast reciprocal matches (coverage ≥0.6 and E < 10−6) for the zebrafish species not yet represented in homoloGene. Sequences in each pair were then aligned with the program blastz (19) to find gene regions in common to the two species. Lastly, the common regions were projected onto the human annotation via the ESTmapper mRNA-genomic alignments to determine a final set of gene annotations on human chromosome 1 for evaluating the tools.
Data sets for the annotation of plant genomes
To test whether sim4cc is suitable for the annotation of plant genomes, 64 237 American beech ESTs produced by 454 pyrosequencing and 8163 unigenes of these sequences generated with the software SeqManPro (Lasergene) were downloaded from the web site of The Fagaceae Project (http://www.fagaceae.org). The poplar genome (version 1.1) and annotation were obtained from the web site of the Joint Genome Institute (http://genome.jgi-psf.org/Poptr1_1/Poptr1_1.download.html). All cDNA sequences were mapped to the Populus trichocarpa genome using the high-throughput tools GMAP, with the cross-species option ‘–X’, and ESTmapper, considering only the best alignment per query. The filter parameters for ESTmapper were modified as follows: for ESTs, greater than 50% coverage, 70% or higher sequence identity and at least 100 bp of aligned sequence were required, and for unigenes, more than 70% sequence identity and at least 100 bp of aligned sequence. In stage two, sim4cc was applied to the genomic regions identified earlier, extended by 50 kb in each direction. For each query sequence, a ‘genomic coverage’ value was computed as the fraction of the gene's bases contained in alignments. Similarly, the ‘gene coverage’ was determined as the fraction of bases overlapping the reference annotation. The numbers of sequences with coverage x or higher, before and after the application of sim4cc, when varying x, were plotted and used for evaluation.
To determine novel ‘genes’ not present in the published poplar annotation, unigene spliced alignments with more than 80% genomic coverage and less than 20% gene coverage were identified for each method. For each query sequence (‘gene’) in the set, potential paralogous matches elsewhere on the genome were detected based on two criteria: at least 50% genomic coverage and higher or similar sequence identity, or at least 50% gene coverage. The remaining sequences after eliminating these potential paralogs were deemed new ‘genes’ (Supplementary Table S5).
RESULTS
We start by evaluating the performance of sim4cc and several spliced alignment programs on four types of comparisons between species at diverse evolutionary distances: human–dog, human–mouse, human–chicken and human–zebrafish. For simplicity, we will refer to these by the second species. For each comparison, a gold reference was generated consisting of annotations of genes in the human genome based on sequence homology at gene (mRNA) level between the species. During evaluation, the output of each tool is directly compared with the reference annotation to determine its sensitivity and specificity at exon, nucleotide and splice junction (intron) level.
Construction of vertebrate reference data sets
An accurate and comprehensive ‘gold reference’ is essential to evaluate the programs' performance. The four vertebrate comparisons above were selected because the species are richly represented in the GenBank curated repositories, and because they sample a sufficiently diverse range of evolutionary distances to allow observations on the possible uses and limitations of the tools. An ideal reference data set should capture all and only orthologous gene regions between the species, to evaluate the completeness of alignments produced by the programs, and should contain accurate representations of the gene structures on the reference genome, to evaluate the correctness of the gene models produced.
To generate an accurate representation of the genes along the reference genome, we start from an initial set of spliced alignments of human mRNAs on the human genome and compare them with the curated set of annotations in the Vertebrate Gene Annotation (VEGA) database, using manual analysis to reconcile any discrepancies (see Methods section). The procedure generates 1922 gene annotations on human chromosome 1. Furthermore, to produce a complete reference set for each comparison, homologous regions between mRNA orthologs in the two species are projected onto the curated human gene annotations produced earlier to determine the final ‘gold reference’. In most cases, alignments of mRNA orthologs were contiguous, evidence that the core gene structure has been preserved between the species. A small number of alignments showed gaps greater than 50 bp (three in dog, 47 in mouse, six in chicken and three in zebrafish) in either sequence, likely caused by evolutionary insertions or deletions. Annotations of these genes were modified to remove the species-specific regions. This procedure produced 46 (dog), 818 (mouse), 158 (chicken) and 232 (zebrafish) gene pairs. The average sequence identities between orthologs for the four comparisons were 84.1%, 79.8%, 74.1% and 68.2%, respectively.
Since only the human gene annotation is used during the evaluation process, other evolutionary events such as merging or splitting of exons in either species do not affect the evaluation outcome. The evaluation scheme only determines whether the conserved regions are contained in the alignment and whether the gene structure produced on the human genome agrees with the reference annotation.
Comparative evaluation
We evaluated the performance of sim4cc and seven other programs on the four reference sets. The seven programs include the same-species tools BLAT (8), sim4 (6) and GMAP (11), and the cross-species programs EXALIN (14), GeneSeqer (12), Exonerate (31), GMAP with the cross-species option ‘-X’ (GMAPX) and translated BLAT (tBLAT). BLAT and GMAP are designed for high-throughput genome-wide operation, whereas the remaining programs are intended to align cDNAs to genomic regions. Except for the cross-species options, the default parameters were used. We use each program to map the 818 mouse, 46 dog, 158 chicken and 232 zebrafish RefSeq mRNA sequences against their homologous genomic regions on human chromosome 1, then compare the outcome against the reference annotation. Thus, genes that do not have an a priori determined homolog on human chromosome 1 are not mapped. GMAP and GMAPX produced the same output in our settings, and therefore only one is reported. We apply standard sensitivity and specificity measures [Sn = TP/(TP + FN), Sp = TP/(TP + FP)] (32) to measure the accuracy at the nucleotide, exon and splice junction levels. The values for all programs are listed in Table 1. When only the protein coding regions are considered, while the performance of all programs improves due to the higher sequence similarity, the trends are similar (Supplementary Table S1).
Table 1.
Performance of spliced alignment programs on the four vertebrate reference data sets
| Method | Nucleotide |
Exon |
Splice junction |
Time | |||
|---|---|---|---|---|---|---|---|
| Sn | Sp | Sn | Sp | Sn V = 0(=10) | Sp V = 0(=10) | ||
| Mouse: 818 genes, 8264 exons, 7408 introns | |||||||
| sim4 | 0.690 | 0.996 | 0.899 | 0.993 | 0.710 (0.741) | 0.660 (0.689) | 28.6 s |
| BLAT | 0.656 | 0.987 | 0.831 | 0.945 | 0.070 (0.517) | 0.047 (0.352) | 2 min 33.3 s |
| tBLAT | 0.774 | 0.985 | 0.942 | 0.985 | 0.183 (0.814) | 0.142 (0.634) | 8 min 55.7 s |
| GMAP | 0.719 | 0.996 | 0.785 | 0.995 | 0.758 (0.763) | 0.952 (0.958) | 1 min 22.3s |
| Exonerate | 0.849 | 0.984 | 0.870 | 0.996 | 0.811 (0.828) | 0.920 (0.939) | 20 min 19.4 s |
| GeneSeqer | 0.603 | 0.988 | 0.647 | 0.921 | 0.574 (0.582) | 0.829 (0.840) | 4 h 24 min 54.8 s |
| EXALIN | 0.846 | 0.997 | 0.948 | 0.996 | 0.926 (0.941) | 0.942 (0.957) | 6 h 33 min 29.3 s |
| sim4cc | 0.934 | 0.995 | 0.973 | 0.997 | 0.932 (0.944) | 0.939 (0.951) | 57.3 s |
| Dog: 46 genes, 419 exons, 370 introns | |||||||
| sim4 | 0.818 | 0.995 | 0.936 | 0.980 | 0.795 (0.816) | 0.770 (0.791) | 1.5 s |
| BLAT | 0.778 | 0.988 | 0.866 | 0.939 | 0.059 (0.608) | 0.048 (0.488) | 7.6 s |
| tBLAT | 0.869 | 0.981 | 0.932 | 0.950 | 0.162 (0.824) | 0.141 (0.716) | 30.4 s |
| GMAP | 0.875 | 0.996 | 0.861 | 0.989 | 0.849 (0.854) | 0.978 (0.984) | 4.0 s |
| Exonerate | 0.959 | 0.983 | 0.943 | 0.997 | 0.878 (0.900) | 0.931 (0.954) | 38.3 s |
| GeneSeqer | 0.677 | 0.995 | 0.671 | 0.941 | 0.600 (0.603) | 0.914 (0.918) | 11 min 55.0 s |
| EXALIN | 0.940 | 0.996 | 0.972 | 0.984 | 0.965 (0.973) | 0.960 (0.968) | 14 min 7.4 s |
| sim4cc | 0.972 | 0.988 | 0.965 | 0.976 | 0.941 (0.951) | 0.961 (0.972) | 2.1s |
| Chicken: 156 genes, 1624 exons, 1462 introns | |||||||
| sim4 | 0.414 | 0.992 | 0.589 | 0.987 | 0.287 (0.304) | 0.428 (0.452) | 6.3 s |
| BLAT | 0.347 | 0.978 | 0.433 | 0.881 | 0.017 (0.132) | 0.023 (0.178) | 29.1s |
| tBLAT | 0.739 | 0.986 | 0.834 | 0.975 | 0.142 (0.653) | 0.143 (0.658) | 1 min 50.7s |
| GMAP | 0.315 | 0.989 | 0.257 | 0.991 | 0.214 (0.216) | 0.932 (0.940) | 18.4 s |
| Exonerate | 0.424 | 0.945 | 0.530 | 0.988 | 0.425 (0.438) | 0.851 (0.873) | 2 min 50.2 s |
| GeneSeqer | 0.451 | 0.987 | 0.431 | 0.915 | 0.372 (0.384) | 0.810 (0.835) | 30 min 15.6 s |
| EXALIN | 0.762 | 0.998 | 0.825 | 0.996 | 0.788 (0.806) | 0.954 (0.975) | 1 h 14 min 23.5 s |
| sim4cc | 0.872 | 0.982 | 0.879 | 0.993 | 0.799 (0.816) | 0.872 (0.890) | 9.5 s |
| Zebrafish: 232 genes, 2549 exons, 2315 introns | |||||||
| sim4 | 0.101 | 0.984 | 0.196 | 0.991 | 0.029 (0.031) | 0.161 (0.171) | 7.6 s |
| BLAT | 0.064 | 0.966 | 0.083 | 0.798 | 0.001 (0.008) | 0.007 (0.067) | 39.4 s |
| tBLAT | 0.573 | 0.984 | 0.628 | 0.960 | 0.086 (0.376) | 0.129 (0.568) | 2 min 31.6 s |
| GMAP | 0.057 | 0.993 | 0.023 | 1.000 | 0.010 (0.010) | 0.958 (0.958) | 14.8 s |
| Exonerate | 0.298 | 0.890 | 0.244 | 0.990 | 0.145 (0.148) | 0.812 (0.829) | 3 min 34.5s |
| GeneSeqer | 0.143 | 0.989 | 0.128 | 0.940 | 0.116 (0.117) | 0.871 (0.877) | 9 min 19.4 s |
| EXALIN | 0.509 | 0.997 | 0.539 | 0.994 | 0.480 (0.495) | 0.954 (0.984) | 1 h 20 min 31.5 s |
| sim4cc | 0.701 | 0.970 | 0.732 | 0.985 | 0.546 (0.567) | 0.757 (0.785) | 18.8 s |
All programs were run with their default parameters. Columns represent sensitivity and specificity values at nucleotide, exon and splice junction (intron) level, the latter when allowing for a margin V (0 or 10) of error around the splice site. Sensitivity was calculated as Sn = TP/(TP + FN) and specificity as Sp = TP/(TP + FP). Run times were averaged over 10 executions of the program on a Dell workstation with 3.2 GHz Intel CPUs and 2 GB RAM.
The eight programs have very similar specificity, but vary significantly in their sensitivity.
As expected, programs for same-species comparisons (sim4, BLAT) perform the worst in all cases, by all criteria. Cross-species programs reach 72–97% sensitivity at the nucleotide level depending on the comparison and excluding the outliers, namely GeneSeqer for all comparisons, and GMAP and Exonerate for chicken and zebrafish. It should be noted that among all programs tested, GeneSeqer has been designed for the analysis of plant genomes (Supplementary Table S2) and therefore its performance on vertebrate comparisons may not be representative. Sim4cc has the highest nucleotide sensitivity for all four comparisons, achieving between 2% (dog) and 19% (zebrafish) higher sensitivity compared to the closest competitor, either Exonerate (dog, mouse) or EXALIN (chicken, zebrafish), and between 10% (dog) and 16% (zebrafish) more than the next closest program, tBLAT. The gains are more pronounced as the evolutionary distance increases. When the gene model is considered, sim4cc has better or comparable accuracy with EXALIN, and both programs are significantly better than the rest of the methods. In a comparison between the two, sim4cc's intron specificity is somewhat lower for distant comparisons, due partly to the larger number of internal unmatched cDNA regions caused by dissimilar sequences, but its run time is 200–400 times faster for all comparisons. Indeed, the advantages of sim4cc are even better showcased when the tradeoff between speed and accuracy is considered. Sim4cc has the highest accuracy while being comparable in speed with its predecessor sim4 and also, for this specific task, with the high-throughput programs GMAP and BLAT. Thus we believe sim4cc is the program best suited for comparative gene annotation.
Performance with sequence similarity level
We further sought to assess the programs’ performance based on species-independent factors, by grouping alignments based on their percent sequence identity. The 1254 alignments were divided into five-percentage point intervals and performance statistics were determined for each group. Nucleotide sensitivity values of different programs for different sequence similarity levels are plotted in Figure 1, and other statistics are shown in Supplementary Figure S4. The lowest sequence similarity levels, between 50–65%, had only a handful of alignments and thus were not representative, and were omitted. The results confirm our previous observations. As the plots indicate, all programs perform well for highly similar sequences, but cross-species programs gain increasingly over their same-species competitors as sequence similarity decreases. Sim4cc, GMAP and EXALIN performed the best at 85–90% sequence identity, followed relatively closely by Exonerate, tBLAT and, surprisingly, sim4. As the sequence identity falls below 85%, significant differences in performance emerge between the programs, with GMAP and sim4 losing power sharply, and more gradual decreases for the other tools. Sim4cc consistently shows the best performance, for all sequence identity levels, with significant performance gains (e.g. a 7% gain for the 75–80% interval and 11% gain for the 65–70% interval) over its closest competitor, EXALIN. The contrast between the top performers, sim4cc and EXALIN, and the rest of the programs is even starker when splice junctions are considered (Supplementary Figure S4). While this experiment can serve as a general guide in selecting a program to use depending on the expected sequence similarity, it should be used with caution, since significant differences in mutation patterns between species, such as proportion of gaps versus point mutations, may alter the performance of the tools.
Figure 1.

Performance of spliced alignment programs (nucleotide sensitivity, vertical axis) with varying sequence identity levels (horizontal axis). The numbers of gene pairs for each sequence identity level in decreasing order from 90–95% to 65–70% are: 40, 135, 266, 281, 211 and 156.
Detailed analyses of Sim4cc-spliced alignments
To observe sim4cc's advantages and limitations more closely, we examined the 46 dog–human alignments and the first 100 alignments from each of the mouse-human and chicken-human alignment sets produced with sim4cc.
Of the 46 dog–human alignments, 29 were in full concordance with the reference annotation, an additional three missed the last or first exons and two more had internal cDNA gaps but without contradicting the gene structure. The remaining 17 alignments collectively harbored seven incorrect splice junctions and six cases of alternative placements of exons, of which three involved relatively short internal exons (21 bp, 21 bp and 35 bp respectively) and the other three were short spurious marginal exons.
Similarly, the 100 mouse-human alignments contained 77 that were in agreement with the reference annotation: 64 fully agreed with the annotation, five were incomplete at either end, and eight differed from the annotation only by cDNA gaps. Two of the cDNA gaps were caused by missed exons contained in very long introns (80 kb and 110 kb), which are very infrequent and are a well-known challenge to spliced alignment programs. The remaining 23 alignments collectively harbored two cDNA gaps, 19 inaccurate splice junctions, and seven cases of alternative placement of exons, of which four involve short internal exons of lengths 5–30 bp. Such short exons have long confounded alignment programs (33) and appear over-represented in our data set.
As expected, the accuracy of gene models is lower for human–chicken alignments. Although, again, 60 alignments do not contradict the gene structure in the reference annotation, only 38 of these fully agreed with the annotation, five missed only terminal exons, and 17 also contained cDNA gaps. Of the remaining 40 alignments, 25 had relatively small differences in their gene structure due to cDNA gaps and/or inaccurate splice junctions (≤30 bp off), while 15 had more significant differences, including misplaced exons or portions of exons. The extent to which these inconsistencies affected the gene content is reflected in the evaluation statistics: the sensitivity and specificity at the splice junction level are smaller than for the closer comparisons, however, only a small fraction of bases are missed or aligned incorrectly. It should be noted, also, that many of the observed discrepancies appear to be genuine evolutionary differences that had not been identified by our stringent criteria (>50 bp).
Application to the large-scale annotation of a plant genome
Recent years have seen an unprecedented growth in the resources generated for plant genomics, including but not limited to genomic sequences, cDNAs and expression profiling resources, which are expected to continue to develop over the coming years. There are currently 13 plant genomes completed or close to completion (http://www.ncbi.nlm.nih.gov/genomes/leuks.cgi?p3=11:Plants&taxgroup=11:Plants|12%3A), several others in progress, and many more expected to be sequenced. The plant cDNA resources already available in databases or being generated by the application of new sequencing technologies represent a tremendous resource for annotating these genomes.
To test the applicability of sim4cc to a whole-genome annotation project for the growing repertoire of plant species, we applied it to the poplar genome. The genome of Populus trichocarpa has recently been sequenced and annotated (34), and is the first forest tree to be sequenced. With its 485.5 million bases of genomic sequence split into 22 010 scaffolds varying in length between 35.6 million bp and 1001 bp, with an average of 22 056 bp and a median of 1534 bp, the poplar genome is a good model for future tree sequencing efforts. The gene annotation project produced 45 555 gene variants.
Several cDNA repositories for plant species exist and more are being created. The Fagaceae Project (http://www.fagaceae.org/) is a collaborative effort to develop genomic resources and tools for the Fagaceae family of trees, including chestnuts, oaks and beeches, with the goal to build genetic and physical maps of these species. Complementary DNA libraries were built and used to produce ESTs for five of these species, using a combination of conventional and 454 sequencing, which will be used in the comparative mapping of the species. This collection of sequences thus represents an excellent resource for testing our program.
We mapped 64 237 454 ESTs and 8163 unigenes of these sequences generated with the software SeqManPro (Lasergene) to the poplar genome, in two stages. First, a high-throughput program, either GMAP (11) or ESTmapper (10), was run to locate matching regions. The parameters of the programs, number of reported regions, and statistics of the mapped sequences are listed in Table 2. There is little bias in the length of cDNAs between the mapped and unmapped sets, barring the 100 bp threshold, indicating that it is not the length but rather the lack of sequence similarity that is the primary reason why roughly 60% of the sequences failed to map with any of the methods. GMAP appears to be less sensitive than ESTmapper in detecting matching regions, but it is likely that both programs suffer from their choice of seed and stringent matching criteria. To take full advantage of the repertoire of cross-species resources, more sensitive tools are needed. In stage two, sim4cc was used to generate a more complete alignment of the cDNA against the genomic regions extended in both directions by 50 kb.
Table 2.
Characteristics and mapping statistics of Fagus grandifolia cDNA sequences on the poplar genome
| Data | All | Mapped |
|
|---|---|---|---|
| EM | GMAPX | ||
| 454 ESTs | |||
| Sequences | 64 237 | 24 810 | 19 034 |
| Length (avg) | 229 bp | 242 bp | 225 bp |
| Regions | n/a | 85 806 | 64 742 |
| sim4cc-alignment statistics | |||
| Sequence id. (avg) | n/a | 94.19 | 90.81 |
| Coverage (avg) | n/a | 97.26 | 93.00 |
| 454 unigenes | |||
| Sequences | 8163 | 2887 | 1625 |
| Avg length | 359 bp | 397 bp | 449 bp |
| Regions | n/a | 3243 | 2643 |
| sim4cc-alignment statistics | |||
| Sequence id. (avg) | n/a | 83.29 | 83.10 |
| Coverage (avg) | n/a | 89.31 | 87.11 |
454 ESTs were mapped with the tool ESTmapper (EM) at ≥50% coverage and ≥70% sequence identity and unigenes at ≥70% sequence identity, retaining only alignments longer than 100 bases. Only the ‘best’ alignment for each query was selected to determine matching regions (note: if indistinguishable from each other, several best alignments may be retained). GMAP was used in cross-species mode ‘−X’, and all other parameters as set by default. Alignment statistics were averaged over all regions. n/a = Not applicable.
We sought to quantify the improvement in cDNA alignments when sim4cc is used compared to the initial high-throughput generated alignments. Figure 2 shows the numbers of sequences, either 454 ESTs (A) or unigenes (B), with more than a fraction x of their bases aligned to the poplar genome before and after the sim4cc application, when varying the threshold x. The advantage of using sim4cc is clear, with unigenes gaining on average roughly 13% and 454 ESTs adding 6% of aligned sequence.
Figure 2.

Number of Fagus grandifolia (A) 454 EST sequences (out of 64 237) and (B) unigenes of these sequences (out of 8163) that can be aligned to the poplar genome at varying coverage cutoffs (horizontal axis), both before and after the application of sim4cc. Only those ESTmapper (EM) alignments covering more than 50% of the input sequence were retained.
Similarly, we also wanted to analyze the enrichment in aligned sequence against a known reference annotation. The 45 555 annotated transcripts were generated by an independent effort and represent a suitable data set for our analysis, albeit not yet curated and likely incomplete. Figure 3 shows the numbers of sequences with more than a fraction x of their bases overlapping the existing annotation, both before and after the application of sim4cc. Again, the increase in sensitivity is clear, with genes gaining 12% (unigenes) and 9% (ESTs), respectively, of aligned sequence on average. When comparing the outcome of the two evaluation methods, against the genome versus against the reference annotation, there is good concordance between the mapping curves, however the absolute mapping rates against the annotation are smaller. Such differences can be explained by new genes or portions of genes that are sampled by the ESTs, or may be false positives. The phenomenon is more pronounced for ESTs, which are a more abundant source of gene variation, including alternative splicing events.
Figure 3.

Number of Fagus grandifolia (A) 454 EST sequences (out of 64 237) and (B) unigenes of these sequences (out of 8163) sequences that overlap the gene annotation of the poplar genome at varying coverage cutoffs, both before and after the application of sim4cc. Only those ESTmapper (EM) alignments covering more than 50% of the input sequence were retained.
Comparative mapping of other species’ cDNAs to a target genome can be used to enrich the existing annotation. We sought unigene sequences that are not yet represented in the poplar annotation and thus are potential new genes. Using a conservative rule, we looked for sequences with high (≥80%) coverage on the genome, but with little (≤ 20%) or no overlap with the existing annotation. Depending on the high-throughput method used, this procedure identified 58 (GMAPX) and 97 (ESTmapper) new ‘genes’, respectively, for a combined 109 new ‘genes’ for both methods (Supplementary Table S5). Blast searches against NCBI's non-redundant databases of proteins and nucleotide sequences (1) showed several matches to plant ribosomal RNAs or other repeats, but also matches to genes such as putative receptor protein kinase, transcription regulator and numerous hypothetical or unnamed proteins.
DISCUSSION
Comparative gene annotation by mapping foreign cDNAs to a target genome has been instrumental in finding genes in many eukaryotic genomes (34–39). Its importance for gene annotation only grows as new species are being sequenced, which may not have the necessary native cDNA and protein resources to annotate them. A comparative strategy will particularly benefit the large number of draft genomes, many of them fragmented, and niche genomes that are of interest to small research communities. A broad potential application area is plant genomics, where concerted efforts are made to leverage genomic information across species (40,41).
Most spliced alignments programs were designed to align cDNAs against the genome of the same or a very closely related species and are inadequate for cross-species comparisons. The critical factor is poor alignment sensitivity, which in turn drives lower splice junction accuracy. Additional design considerations include speed and robustness with different types of species-to-species comparisons or sequence data. We developed a new program, sim4cc, which overcomes difficulties in existing spliced alignment tools. To increase alignment sensitivity, it uses spaced seeds that allow the detection of approximate matches according to mathematically optimized match-mismatch patterns (22). Additionally, our seeds were specifically designed to work well for a wide range of sequence comparisons, at various evolutionary distances, and therefore can be seamlessly used regardless of the species compared (23,24). Critical to designing our ‘universal’ seeds has been sampling from a variety of species-to-species comparisons, especially from the more distant ones. Optimizing seeds requires specific technical expertise to train the alignment models, compare and cluster comparisons into groups and mathematically derive good seeds for each group, and is computationally very expensive (e.g. designing seeds for a single species-to-species comparison took two months on a 10 CPU computer cluster). In our earlier publications (23,24) we determined and reported seeds for cDNA-to-genome comparisons among vertebrates for various degrees of sequence divergence and for a variety of seed weights or, equivalently, specificity levels (22), which can be readily used by both program users and program developers. While we do plan to develop similar models and seeds for plant comparisons, and will release them as soon as they become available, due to the inherent robustness of spaced patterns we expect that the seeds trained on the vertebrate set will work well on those applications as well. Lastly, to increase the accuracy of sim4cc we further improved gene models by introducing complex splice site models (26,27) and by post-processing alignments to take into account likely evolutionary events. As a result, sim4cc outperforms existing methods when accuracy and speed are considered. In particular, it has >10% higher sensitivity than the closest competitor for some comparisons, while being two orders of magnitude faster. Importantly, while it performs well on relatively close comparisons, it also works reasonably well for distant species as much as >250 Myrs apart, such as human and chicken, thus extending the evolutionary range typically targeted by cDNA-to-genome alignment programs. Similarity-based programs are expected to perform better on coding regions compared to UTRs, due to the higher sequence similarity in those regions. Delimiting UTRs is an important step in annotating genes and a precursor to answering many downstream biological questions, from identifying alternative and dual promoters, to finding alternative polyadenylation sites and binding sites of microRNAs to their mRNA targets (42,43). Most importantly, cDNA-to-genome alignment programs are the only source of UTR information for gene annotation, which cannot be provided by either de novo gene finders or protein alignments (44). Notably, sim4cc seems to better capture alignments in UTR regions compared to other heuristic programs and even some exact dynamic programming-based tools (Table 1 and Supplementary Table S1), likely owing to the robustness of the spaced seed patterns and the alignment algorithms used to extend and bridge seed matches. Lastly, we tested sim4cc on EST reads obtained with the new 454 sequencing technology, which are likely to be generated in the future. Of course, sim4cc is not a silver bullet and will not be applicable to all types of comparisons, for instance when sequencing errors and high mutation rates compound. In these cases, protein alignments may be a better alternative.
Sim4cc has a small memory footprint and can be compiled and run on most platforms. Given its accuracy, parameter independence and speed, it can be effectively integrated into a whole-genome annotation pipeline. It is also user friendly, with a small parameter list and the same input and output format as its predecessor, sim4, and therefore well suited for use by individual researchers.
Although the improvement over existing tools is clear, further refinements are needed to improve the performance and to test the limits of nucleotide based alignments. For instance, short exons are hard to detect and are often split between the adjacent exons owing to the longer seed span and low sequence identity (Supplementary Table S6). Richer splice context models and an additional exon refinement stage could be later implemented to detect short exons (33,45). Moreover, evolutionary insertions and deletions of genetic material can alter the gene structure. Although most of these are located in the UTRs and can be identified from the output when using the open reading frame program option, some occur in the protein domain. These regions will appear as matching, albeit weakly, and will be erroneously reported as part of the gene, and thus will remain a weakness of comparative methods based purely on sequence alignments. Lastly, sim4cc is not a tool for whole-genome comparison, but can easily become one by combination with a search engine that pre-identifies likely matching regions. We have tested two such high-throughput programs, ESTmapper and GMAP, but it is apparent that more sensitive approaches, which incorporate spaced seeds and better match filters, are needed. We will test such extensions for future distributions of the program.
Sim4cc is distributed free of charge from http://www.cbcb.umd.edu/software/sim4cc/.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Seed optimizations were performed on the ‘Herd’ Scientific Computing Cluster at the George Washington University (NSF grant CLS20163A). Sloan Research Fellowship (to L.F.); National Institutes of Health grant R01-LM006845 (to Steven L. Salzberg). Funding for open access charge: R01-LM006845 (to Steven L. Salzberg).
Conflict of interest statement. None declared.
Supplementary Material
REFERENCES
- 1.Wheeler DL, Barrett T, Benson DA, Bryant SH, Canese K, Chetvernin V, Church DM, Dicuccio M, Edgar R, Federhen S, et al. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2008;36:D13–D21. doi: 10.1093/nar/gkm1000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Okazaki Y, Furuno M, Kasukawa T, Adachi J, Bono H, Kondo S, Nikaido I, Osato N, Saito R, Suzuki H, et al. Analysis of the mouse transcriptome based on functional annotation of 60,770 full-length cDNAs. Nature. 2002;420:563–573. doi: 10.1038/nature01266. [DOI] [PubMed] [Google Scholar]
- 3.Pruitt KD, Tatusova T, Maglott DR. NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2007;35:D61–D65. doi: 10.1093/nar/gkl842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Strausberg RL, Feingold EA, Grouse LH, Derge JG, Klausner RD, Collins FS, Wagner L, Shenmen CM, Schuler GD, Altschul SF, et al. Generation and initial analysis of more than 15,000 full-length human and mouse cDNA sequences. Proc. Natl Acad. Sci. USA. 2002;99:16899–16903. doi: 10.1073/pnas.242603899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gerhard DS, Wagner L, Feingold EA, Shenmen CM, Grouse LH, Schuler G, Klein SL, Old S, Rasooly R, Good P, et al. The status, quality, and expansion of the NIH full-length cDNA project: the Mammalian Gene Collection (MGC) Genome Res. 2004;14:2121–2127. doi: 10.1101/gr.2596504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Florea L, Hartzell G, Zhang Z, Rubin GM, Miller W. A computer program for aligning a cDNA sequence with a genomic DNA sequence. Genome Res. 1998;8:967–974. doi: 10.1101/gr.8.9.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wheelan SJ, Church DM, Ostell JM. Spidey: a tool for mRNA-to-genomic alignments. Genome Res. 2001;11:1952–1957. doi: 10.1101/gr.195301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kent WJ. BLAT—the BLAST-like alignment tool. Genome Res. 2002;12:656–664. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee BT, Tan TW, Ranganathan S. MGAlignIt: a web service for the alignment of mRNA/EST and genomic sequences. Nucleic Acids Res. 2003;31:3533–3536. doi: 10.1093/nar/gkg561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Florea L, Di Francesco V, Miller J, Turner R, Yao A, Harris M, Walenz B, Mobarry C, Merkulov GV, Charlab R, et al. Gene and alternative splicing annotation with AIR. Genome Res. 2005;15:54–66. doi: 10.1101/gr.2889405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu TD, Watanabe CK. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics. 2005;21:1859–1875. doi: 10.1093/bioinformatics/bti310. [DOI] [PubMed] [Google Scholar]
- 12.Usuka J, Zhu W, Brendel V. Optimal spliced alignment of homologous cDNA to a genomic DNA template. Bioinformatics. 2000;16:203–211. doi: 10.1093/bioinformatics/16.3.203. [DOI] [PubMed] [Google Scholar]
- 13.Mott R. EST_GENOME: a program to align spliced DNA sequences to unspliced genomic DNA. Comput. Appl. Biosci. 1997;13:477–478. doi: 10.1093/bioinformatics/13.4.477. [DOI] [PubMed] [Google Scholar]
- 14.Zhang M, Gish W. Improved spliced alignment from an information theoretic approach. Bioinformatics. 2006;22:13–20. doi: 10.1093/bioinformatics/bti748. [DOI] [PubMed] [Google Scholar]
- 15.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J. Mol. Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 16.Ma B, Tromp J, Li M. PatternHunter: faster and more sensitive homology search. Bioinformatics. 2002;18:440–445. doi: 10.1093/bioinformatics/18.3.440. [DOI] [PubMed] [Google Scholar]
- 17.Buhler J, Keich U, Sun YN. Designing seeds for similarity search in genomic DNA. J. Comput. Sys. Sci. 2005;70:342–363. [Google Scholar]
- 18.Keich U, Li M, Ma B, Tromp J. On spaced seeds for similarity search. Discrete Appl. Mathematics. 2004;138:253–263. [Google Scholar]
- 19.Schwartz S, Kent WJ, Smit A, Zhang Z, Baertsch R, Hardison RC, Haussler D, Miller W. Human-mouse alignments with BLASTZ. Genome Res. 2003;13:103–107. doi: 10.1101/gr.809403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burge C, Karlin S. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 1997;268:78–94. doi: 10.1006/jmbi.1997.0951. [DOI] [PubMed] [Google Scholar]
- 21.Nei M, Kumar S. Molecular evolution and phylogenetics. Oxford, NY: Oxford University Press; 2000. [Google Scholar]
- 22.Zhou L, Florea L. Designing sensitive and specific spaced seeds for cross-species mRNA-to-genome alignment. J. Comput. Biol. 2007;14:113–130. doi: 10.1089/cmb.2006.0130. [DOI] [PubMed] [Google Scholar]
- 23.Zhou L, Mihai I, Florea L. Effective cluster-based seed design for cross-species sequence comparisons. Bioinformatics. 2008;24:2926–2927. doi: 10.1093/bioinformatics/btn547. [DOI] [PubMed] [Google Scholar]
- 24.Zhou L, Stanton J, Florea L. Universal seeds for cDNA-to-genome comparison. BMC Bioinformatics. 2008;9:36. doi: 10.1186/1471-2105-9-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cover TM, Thomas JA. Elements of information theory. NY: Wiley; 1991. [Google Scholar]
- 26.Delcher AL, Bratke KA, Powers EC, Salzberg SL. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics. 2007;23:673–679. doi: 10.1093/bioinformatics/btm009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pertea M, Lin X, Salzberg SL. GeneSplicer: a new computational method for splice site prediction. Nucleic Acids Res. 2001;29:1185–1190. doi: 10.1093/nar/29.5.1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Florea L. Detecting functional regions in DNA with sequence comparison methods: program development and evaluation. University Park: Penn State University; 2000. Ph.D. Thesis. [Google Scholar]
- 29.Chao KM, Zhang J, Ostell J, Miller W. A tool for aligning very similar DNA sequences. Comput. Appl. Biosci. 1997;13:75–80. doi: 10.1093/bioinformatics/13.1.75. [DOI] [PubMed] [Google Scholar]
- 30.Wilming LG, Gilbert JG, Howe K, Trevanion S, Hubbard T, Harrow JL. The vertebrate genome annotation (Vega) database. Nucleic Acids Res. 2008;36:D753–D760. doi: 10.1093/nar/gkm987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Slater GS, Birney E. Automated generation of heuristics for biological sequence comparison. BMC Bioinformatics. 2005;6:31. doi: 10.1186/1471-2105-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Burset M, Guigo R. Evaluation of gene structure prediction programs. Genomics. 1996;34:353–367. doi: 10.1006/geno.1996.0298. [DOI] [PubMed] [Google Scholar]
- 33.Volfovsky N, Haas BJ, Salzberg SL. Computational discovery of internal micro-exons. Genome Res. 2003;13:1216–1221. doi: 10.1101/gr.677503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tuskan GA, Difazio S, Jansson S, Bohlmann J, Grigoriev I, Hellsten U, Putnam N, Ralph S, Rombauts S, Salamov A, et al. The genome of black cottonwood, Populus trichocarpa (Torr. & Gray) Science. 2006;313:1596–1604. doi: 10.1126/science.1128691. [DOI] [PubMed] [Google Scholar]
- 35.Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- 36.Gibbs RA, Weinstock GM, Metzker ML, Muzny DM, Sodergren EJ, Scherer S, Scott G, Steffen D, Worley KC, Burch PE, et al. Genome sequence of the Brown Norway rat yields insights into mammalian evolution. Nature. 2004;428:493–521. doi: 10.1038/nature02426. [DOI] [PubMed] [Google Scholar]
- 37.Holt RA, Subramanian GM, Halpern A, Sutton GG, Charlab R, Nusskern DR, Wincker P, Clark AG, Ribeiro JM, Wides R, et al. The genome sequence of the malaria mosquito Anopheles gambiae. Science. 2002;298:129–149. doi: 10.1126/science.1076181. [DOI] [PubMed] [Google Scholar]
- 38.Consortium THGS. Insights into social insects from the genome of the honeybee Apis mellifera. Nature. 2006;443:931–949. doi: 10.1038/nature05260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Warren WC, Hillier LW, Marshall Graves JA, Birney E, Ponting CP, Grutzner F, Belov K, Miller W, Clarke L, Chinwalla AT, et al. Genome analysis of the platypus reveals unique signatures of evolution. Nature. 2008;453:175–183. doi: 10.1038/nature06936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Jackson S, Rounsley S, Purugganan M. Comparative sequencing of plant genomes: choices to make. Plant Cell. 2006;18:1100–1104. doi: 10.1105/tpc.106.042192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhu W, Buell CR. Improvement of whole-genome annotation of cereals through comparative analyses. Genome Res. 2007;17:299–310. doi: 10.1101/gr.5881807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kan Z, Rouchka EC, Gish WR, States DJ. Gene structure prediction and alternative splicing analysis using genomically aligned ESTs. Genome Res. 2001;11:889–900. doi: 10.1101/gr.155001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115:787–798. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- 44.Zhang MQ. Computational prediction of eukaryotic protein-coding genes. Nat. Rev. Genet. 2002;3:698–709. doi: 10.1038/nrg890. [DOI] [PubMed] [Google Scholar]
- 45.Dogan RI, Getoor L, Wilbur WJ, Mount SM. Features generated for computational splice-site prediction correspond to functional elements. BMC Bioinformatics. 2007;8:410. doi: 10.1186/1471-2105-8-410. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.