

Abstract
The pathogenesis of Alzheimer's disease (AD) is thought to be related to the accumulation of amyloid β (Aβ) in amyloid deposits and toxic oligomeric species. Immunomodulation is emerging as an effective means of shifting the equilibrium from Aβ accumulation to clearance; however, excessive cell mediated inflammation and cerebral microhemorrhages are two forms of toxicity which can occur with this approach. Vaccination studies have so far mainly targeted the adaptive immune system. In the present study, we have stimulated the innate immune system via the Toll-like receptor 9 (TLR9) with cytosine-guanosine-containing DNA oligodeoxynucleotides in Tg2576 AD model transgenic mice. This treatment produced a 66% and 80% reduction in the cortical (p = 0.0001) and vascular (p = 0.0039) amyloid burden, respectively, compared with nontreated AD mice. This was in association with significant reductions in Aβ42, Aβ40, and Aβ oligomer levels. We also show that treated Tg mice performed similarly to wild-type mice on a radial arm maze. Our data suggest that stimulation of innate immunity via TLR9 is highly effective at reducing the parenchymal and vascular amyloid burden, along with Aβ oligomers, without apparent toxicity.
Keywords: Alzheimer's disease, amyloid β, transgenic, immunity, behavior, antibody, histochemistry
Introduction
The deposition of amyloid β (Aβ) peptides in the CNS in the form of amyloid plaques is one of the hallmarks of Alzheimer's disease (AD) (Jellinger, 2008b). Several lines of evidence favor the conclusion that Aβ accumulation destroys neurons in the brain, leading to deficits in cognitive abilities (Hardy, 2006; Wisniewski and Konietzko, 2008).
One intervention which has a significant impact on Aβ-related pathology and associated behavioral deficits is vaccination. However when this approach was tried in humans, in contrast to results in AD animals models, encephalitis emerged as a significant form of toxicity in some patients (Morgan, 2006; Brody and Holtzman, 2008; Wisniewski and Konietzko, 2008). The immunotherapeutic approaches attempted so far have targeted stimulation of the adaptive immune system. All vertebrates have two complementary immune responses: innate and adaptive. The adaptive immune response, which takes time to specifically target a foreign protein and provides a memory response upon subsequent re-exposure, is mediated by the more recently evolved T and B cells. Over the past several years, stimulation of innate immune responses has emerged as an effective method both for activating phagocytes, including macrophages/microglia and dendritic cells (DCs), and for inducing specific humoral immune responses (Krieg, 2006; Crack and Bray, 2007). Thus, our research group predicted that innate immune stimulation would be useful for AD prevention and therapy by inducing prevention or removal of brain amyloid deposits, with no associated toxicity.
One of the most potent methods to stimulate the innate immune system is via the Toll-like receptors (TLRs). TLRs are a family of innate immune mediators that are expressed by a variety of immune and nonimmune cells (Crack and Bray, 2007). In vertebrates, the TLRs evolved to function primarily for recognizing invading microbial pathogens, including bacteria, viruses, fungi and protozoans, and activating appropriate signaling pathways to effectively clear the threat. There are 13 distinct TLR family members currently known in mammals, of which the pathogen specificities of 10 (TLR1–9 and 11) have been identified (Crack and Bray, 2007).
Our recent results in another group of conformational neurodegenerative disorders, the prion diseases, suggested Toll-like receptor 9 (TLR9) to be an attractive candidate to exploit for AD prevention and therapy (Spinner et al., 2007). The function of TLR9 is to specifically bind DNA that contain unmethylated cytosine-guanosine (CpG) sequences, which are commonly found in the genomes of prokaryotes (bacteria) and viruses and underrepresented in those of eukaryotes. Various CpG DNA drugs that are TLR9 agonists are safe in humans and rodents (Krieg, 2006; Crack and Bray, 2007). Clinical trials are currently in progress for their utilization for diseases such as cancer, viral infection and asthma/allergy (Krieg, 2006).
In our proof of principle studies we used type B CpG oligodeoxynucleotides (ODNs) to stimulate the innate immune system in Tg2576 AD model mice to evaluate whether this approach could reduce amyloid deposition and lead to behavioral improvements, with no induced toxicity.
Materials and Methods
Animals and treatment
The studies were performed in the heterozygous Tg2576 mouse model (Hsiao et al., 1996). These mice over-express a 695 amino acid splice form (Swedish mutation K670N M671I) of the human amyloid β precursor protein (APP) and show a rapid increase in Aβ levels at ∼6 months of age with Aβ deposition developing in the subsequent months although extensive amyloid burden is usually not observed before two years of age (Hsiao et al., 1996). The Tg2576 mice used were bred internally on a C57B6 × SJL F1 background. These mice carry the recessive retinal degeneration (rd) mutation due to the SJL strain. Mice homozygous for the mutation have impaired vision and were excluded from this study. Also to reduce any confounds in the behavioral testing due to impaired vision, albino mice were excluded from this study. The animals used in this study were maintained on a 12 h light/dark cycle. All mouse care and experimental procedures were approved by the Institutional Animal Care and Use Committee at the New York University School of Medicine. Female Tg2576 mice were injected intraperitoneally with either the TLR9 agonist CpG ODN 1826 (2.5 mg/kg body weight, ∼63 μg) or vehicle (HBSS) beginning at the age of 6 weeks, and once a month thereafter for a total of 14 injections. Unless specifically designed to be methylated, CpG-containing ODNs synthesized in the laboratory or purchased from suppliers are unmethylated, and therefore can be used to activate TLR9. CpG ODN 1826 [5′-TCC ATG ACG TTC CTG ACG TT-3′ (CpG motifs underlined)], with a complete phosphorothioate backbone, was purchased from Integrated DNA Technologies. The dose of CpG ODN 1826 used was the same as in our prior study, in which we stimulated the innate immune system in mice to enhance a response to 139A scrapie associated fibrils (Spinner et al., 2007). Controls were non-Tg C57BL/6 × SJL mice injected with HBSS on the same schedule. During the treatment animals were closely monitored for signs of toxicity, such as differences in total body weight, and after death their organs were examined for any signs of pathology. No toxicity was evident in the CpG ODN-treated group.
Behavioral testing
Before cognitive testing, the mice were subjected to locomotor activity tests. This measurement of locomotor behavioral was performed to verify that any CpG ODN treatment-related effects observed in the cognitive tasks could not be explained by differences in locomotor activity. The behavioral study was performed in 24 CpG ODN-treated Tg animals. Twenty age-matched vehicle-treated Tg mice and 25 non-Tg age-matched littermates were used as controls.
Locomotor activity.
A Hamilton-Kinder Smart-Frame Photobeam System was used to make a computerized recording of animal activity over a designated period of time, as we have previously described (Sigurdsson et al., 2004; Sadowski et al., 2006; Scholtzova et al., 2008). Exploratory locomotor activity was recorded in a circular open field activity chamber measuring (70 × 70 cm). A video camera mounted above the chamber automatically recorded horizontal movements in the open field in each dimension (i.e., x, y, and two z planes). Total distance was measured in centimeters (cm) traveled and is defined as sequential movement interruptions of the animal measured relative to the background. The duration of the behavior was timed for 15 min. Results are reported based on distance traveled (cm), mean resting time, and velocity (average and maximum) of the animal.
Radial arm maze.
Spatial learning (working memory) was evaluated using an eight-arm radial maze with a water well at the end of each arm, as we have previously reported (Sigurdsson et al., 2004; Asuni et al., 2006; Sadowski et al., 2006;). Clear Plexiglas guillotine doors, operated by a remote pulley system, controlled access to the arms from a central area from which the animals entered and exited the apparatus. After 3–4 d of adaptation, water-restricted mice (2 h daily access to water) were given one training session per day for 12 consecutive days. For each session, all arms were baited with 0.1% saccharine solution, and animals were permitted to enter all arms until the eight rewards had been consumed. The number of errors (entries to previously visited arms) and time to complete each session were recorded. The behavioral testing was performed by an individual blinded to the animal's treatment status.
Aβ autoantibody response
The autoantibody levels were determined at 1:200 dilutions of plasma using ELISA as described previously (Sigurdsson et al., 2004; Asuni et al., 2006; Sadowski et al., 2006) in which 0.5 μg per well of the Aβ40 or Aβ42 peptide was coated onto microtiter wells (Immulon 2HB; Thermo Electron Corporation). The antibodies in plasma were detected by a goat anti-mouse IgG linked to a horseradish peroxidase conjugate (Sigma; A8786) at 1:3000 dilution. Tetramethyl benzidine (TMB; Pierce) was the development substrate.
Histological studies
Following completion of behavioral testing at 17 months of age, the mice were anesthetized with sodium pentobarbital (150 mg/kg, i.p.) and perfused transaortically with 0.1 m PBS, pH 7.4. The brains were removed and the right hemisphere was immersion-fixed in periodate-lysine-paraformaldehyde (PLP), whereas the left hemisphere was snap-frozen for measurement of Aβ oligomers and Aβ levels. After fixation, brains were placed in 2%DMSO/20%glycerol in PBS and stored until sectioned. Serial coronal brain sections (40 μm) were cut and eight series of sections at 0.32 mm intervals saved for histological and immunohistochemical analysis of staining with: (1) antibodies 6E10/4G8, (2) Thioflavin-S, (3) anti-GFAP antibody, (4) anti-CD11b antibody, and (5) anti-CD45 antibody stained sections. Aβ deposits were stained either with a mixture of monoclonal antibodies 6E10/4G8 or Thioflavin-S for fibrillar amyloid. GFAP is a component of the glial intermediate filaments that forms part of the cytoskeleton and is found predominantly in astrocytes. We used two different markers to identify microglia: CD11b [member of β-integrin family of adhesion molecules; also known as MAC-1 or complement receptor 3 (CR3)] and CD45 (protein-tyrosine phosphatase) which are commonly used markers for the microglial activation at the earliest and later stages of plaque development, respectively (Morgan et al., 2005). The remaining series were placed in ethylene glycol cryoprotectant (30% sucrose/30% ethylene glycol in 0.1 mol/L phosphate buffer) and stored at −20°C until used. Immunostaining with 6E10/4G8, or antibodies to GFAP, CD45 and CD11b was performed as previously described (Sadowski et al., 2006). Briefly, free-floating sections were incubated in solutions containing both anti-Aβ antibodies 6E10 and 4G8 (Signet), at a 1:1000 dilution for 3 h. A mouse on mouse immunodetection kit (Vector Laboratories) was used with the biotinylated anti-mouse IgG secondary antibody reacted for 1 h at a 1:1000 dilution. Antibody staining was revealed with 3,3′-diaminobenzidine (DAB; Sigma-Aldrich) and nickel ammonium sulfate intensification. GFAP staining (polyclonal, 1:1000; 3 h, Dako) was performed with a primary antibody diluent composed of 0.3% Triton X-100, 0.1% sodium azide, 0.01% bacitracin, 1% bovine serum albumin (BSA), and 10% normal goat serum in PBS, and secondary biotinylated goat anti-rabbit antibody (Vector) reacted for 1 h at 1:1000 dilution. CD45 (rat antimouse, 1:1000; 3 h, AbD Serotec) and CD11b immunohistochemistry (rat antimouse, 1:500; 3 h, Serotec) were performed similarly to that for GFAP staining except that the secondary antibody was goat anti-rat (Vector) diluted 1:1000. Selected series were double-stained for Thioflavin-S and CD45. Thioflavin-S staining was performed on mounted sections, as published previously (Sadowski et al., 2006). Perl's Prussian blue staining for ferric iron in hemosiderin (degradation product of hemoglobin) was performed on another set of sections to detect cerebral bleeding. Equally spaced sections were mounted and stained in a solution containing 10% potassium ferrocyanide and 20% hydrochloric acid for 45 min. For the hemosiderin stain, 10–15 sections were examined and the average number of iron positive profiles per section was calculated.
Image analysis
Immunostained tissue sections were quantified with a Bioquant stereology semiautomated image analysis system (R&M Biometrics) using a random unbiased hierarchical sampling scheme, as published previously (Sadowski et al., 2006; Scholtzova et al., 2008). Seven sections were analyzed per animal. All procedures were performed by an individual blinded to the experimental condition of the study. Total Aβ burden (defined as the percentage of test area occupied by Aβ) was quantified for the cortex and for the hippocampus on coronal plane sections stained with the monoclonal antibodies 6E10 and 4G8. Intensification with nickel ammonium sulfate resulted in black Aβ with minimal background staining that facilitated threshold detection. The cortical area was dorsomedial from the cingulate cortex and extended ventrolaterally to the rhinal fissure within the right hemisphere. Test areas (640 μm × 480 μm) were randomly selected by applying a grid (800 μm × 800 μm) over the traced contour. Hippocampal measurements (600 μm × 600 μm) were performed similarly to the cortical analysis (Sadowski et al., 2006; Scholtzova et al., 2008). Total fibrillar Aβ burden (parenchymal and vascular) and cerebral amyloid angiopathy (CAA) burden (Aβ burden in the vasculature) were evaluated separately in sections stained with Thioflavin-S, using methods described previously (Asuni et al., 2006; Sadowski et al., 2006). The CD45 microglia burden (the percentage of area in the measurement field occupied by CD45 immunoreactive microglia) was quantified in a manner analogous to that used to measure the Aβ burden.
Rating of microgliosis
The assessment of the CD11b immunostained sections was based on a semiquantitative analysis of the extent of microgliosis (0, a few resting microglia; 1, a few ramified and/or phagocytic microglia; 2, moderate number of ramified/phagocytic microglia; 3, numerous ramified/phagocytic microglia), as we have previously reported (Asuni et al., 2006).
Rating of astrocytosis
Reactive astrocytosis was rated on a scale of 0.5–3. The rating was based on a semiquantitative analysis of the extent of GFAP immunoreactivity (number of GFAP immunoreactive cells and complexity of astrocytic branching), as we have previously published (Asuni et al., 2006).
Tissue homogenization and sandwich ELISA for Aβ levels
Before extraction of Aβ from brain tissue, 10% (w/v) homogenates were prepared in tissue homogenization buffer (20 mm Tris base, pH 7.4, 250 mm sucrose, 1 mm EDTA, 1 mm EGTA) with 100 mm phenylmethylsulphonyl fluoride and protease inhibitors [protease inhibitors cocktail (Complete, Roche Diagnostic) plus pepstatin A] added immediately before homogenization, as we have previously published (Asuni et al., 2006; Sadowski et al., 2006; Scholtzova et al., 2008). For extraction of soluble Aβ, brain homogenates were thoroughly mixed with an equal volume of 0.4% diethylamine (DEA)/100 mm NaCl, then spun at 135,000 × g for 1 h at 4°C, and subsequently neutralized with 1/10 volume of 0.5 m Tris, pH 6.8. The samples were then aliquoted, flash-frozen on dry ice, and stored at −80°C until loaded onto ELISA plates. Similarly for extraction of the total Aβ, homogenates (200 μl) were added to 440 μl of cold formic acid (FA) and sonicated for 1 min on ice. Subsequently, 400 μl of this solution was spun at 100,000 × g for 1 h at 4°C. Then, 210 μl of the resulting supernatant was diluted into 4 ml of FA neutralization solution (1 m Tris base, 0.5 M Na2HPO4, 0.05% NaN3), aliquoted, flash-frozen on dry ice, and stored at −80°C until used for Aβ measurements.
The total and soluble Aβ levels were measured using a combination of mouse monoclonal antibody 6E10 (specific to an epitope present on amino acid residues 1–16 of Aβ) and two different rabbit polyclonal antibodies specific for Aβ40 (R162) and Aβ42 (R165), in a double-antibody sandwich ELISA as described previously (Sadowski et al., 2006). The optical density (OD) was measured at 450 nm. The relationship between OD and Aβ peptide concentration was determined by a four-parameter logistic log function. Nonlinear curve fitting was performed with the KinetiCalc program (Biotek Instruments) to convert OD of plasma to estimated concentrations. The assay was performed by an investigator blinded to group assignment. The levels of Aβ species are presented as μg of Aβ per g of wet brain, taking into account dilution factors introduced by multiple steps throughout the assay (brain homogenization and extraction procedures).
Western blot analysis of Aβ oligomers
For Western immunoblot analysis, 10% (w/v) brain homogenates were centrifuged at 25,000 × g for 10 min at 4°C, and the supernatants were transferred to clean tubes and stored as previously described (Sadowski et al., 2006). The total protein concentration in the supernatant was determined using the Bicinchoninic acid assay (BCA; Pierce). Samples (40 μg of total protein), mixed with an equal volume of Tricine sample buffer, were electrophoresed on 12.5% Tris-tricine polyacrylamide gels (under nonreducing conditions) and transferred to nitrocellulose membranes. The blots were blocked with 5% nonfat dry milk in Tris-buffered saline Tween 20 (TBS-T) for 2 h at room temperature. Oligomer-specific A11 polyclonal antibody (Biosource) was diluted (1:1000) in 0.1%BSA/TBS-T and incubated with the blots for 2 h at room temperature. Bound antibody was visualized with horseradish peroxidase-conjugated goat anti-rabbit IgG (1:8000; 1 h, Pierce) and the ECL detection system (Pierce). The specificity of A11 staining was confirmed by probing the membrane with anti-Aβ monoclonal antibodies 6E10 or 4G8 (Sadowski et al., 2006). Densitometric analysis of A11 immunoreactive oligomer specific bands was performed with NIH ImageJ version 1.34 software.
Statistical analysis
Data from the radial arm maze were analyzed by two-way repeated-measures ANOVA followed by a Neuman–Keuls post hoc test. Differences between groups in object recognition test, amyloid burden, Aβ levels within the brain, levels of oligomers, CD45, CD11 activated microglia, and GFAP astrogliosis were analyzed using a Student's unpaired two-tailed t test. Assessment of brain microhemorrhages was analyzed using a one-tailed t test.
Correlation was determined by calculating the Pearson r correlation coefficient. All data were analyzed with Statistica, version 6.1 (StatSoft).
Results
Behavioral studies
After the treatment, at the age of 16 months, the mice were subjected to behavioral testing. The behavioral analysis consisted of both a cognitive assessment and measurement of exploratory locomotor activity (Fig. 1). The latter test was included to verify that performance on cognitive tests was not influenced by locomotor abnormalities. No statistical differences between groups were discerned in any of the locomotor parameters measured (Fig. 1). In addition to locomotor evaluation the mice underwent two cognitive tests. Working memory was evaluated using the radial arm maze (Fig. 2). The overall performance (number of errors) of the mice differed significantly between Tg groups (two-way repeated-measures ANOVA, group (treatment) effect, p = 0.019; days effect, p < 0.0001; interaction (group vs days), p = 0.144). The CpG ODN-treated group was better at navigating the maze than the vehicle-treated Tg group. A significant difference was observed, with CpG ODN-treated Tg mice performing comparably to Wt littermates (Newman–Keuls post hoc test, Tg-CpG vs Tg-vehicle, p = 0.026; Tg-CpG vs Wt, p = 0.814.). Vehicle-treated Tg mice made significantly more working memory errors than Wt animals (Newman–Keuls post hoc test, p = 0.039).
Figure 1.

Locomotor activity. A–D, At 16 months of age (posttreatment), both Tg groups and their wild-type (Wt) littermates did not differ in any of the locomotor parameters measured: maximum speed (A), distance traveled (B) average speed (C) and resting time (D). The error bars show the SEM. This applies also to all subsequent figures.
Figure 2.

Working memory improvement in CpG ODN-treated Tg2576 mice. Tg2576 mice treated with CpG ODN navigated the radial arm maze with significantly fewer errors than control Tg mice, and their performance was similar to that of their Wt age-matched littermates [two-way repeated-measures ANOVA, group (treatment) effect, p = 0.019; days effect, p < 0.0001; interaction (group vs days), p = 0.144, Newman–Keuls multiple comparison post hoc testing showed Tg-CpG vs Tg-vehicle, p = 0.026; Tg- vehicle vs Wt, p = 0.039; Tg-CpG vs Wt, p = 0.814].
Amyloid burden
The mice were killed at 17 months of age after behavioral testing and the brains were processed for histology and subsequent stereological analysis, as described previously (Asuni et al., 2006; Sadowski et al., 2006). Histological observation in APP Tg2576 mice indicated that CpG ODN-treated mice had fewer plaques compared with vehicle-Tg mice as visualized by Thioflavin-S staining (fibrillar Aβ burden), and immunostaining (6E10/4G8) (Fig. 3A–I).
Figure 3.

Treatment with CpG ODN decreased cortical and hippocampal amyloid plaque burden in APP Tg2576 mice. A, B, F, G, Histological analysis of APP Tg mice depict the difference in Aβ burden. Aβ immunostaining (6E10/4G8) showed greater Aβ accumulation in cortical (A) and hippocampal (F) sections of vehicle-treated APP mice, compared with sections from CpG ODN- treated APP mice (B, G). C, D, H, I, Similarly, Thioflavin-S cortical and hippocampal staining also revealed differences between vehicle-treated (C, H) and CpG ODN-treated (D, I) APP mice. Stereological analysis of total amyloid burden (Aβ load) showed a significant reduction in APP-Tg mice treated with CpG ODN compared with age-matched Tg control mice treated with vehicle. E, J, There was a 66% reduction in cortical (E) amyloid burden (***p = 0.0001) and a 59% reduction in hippocampal (J) amyloid burden (**p = 0.002) as quantified using unbiased random sampling scheme and semiautomated image analysis system. The scale bar in B corresponds to cortical images A-D. The scale bar in G corresponds to hippocampal images F-I.
Quantitative analysis of total amyloid burden was determined by stereological techniques, using random unbiased sampling on the immunostained serial sections evenly spaced along the entire-rostrocaudal axis of the brain (Fig. 3). Peripheral administration of TLR9 agonist CpG ODN led to 66% (two-tailed t test, p = 0.0001) reduction in total cortical amyloid burden (Fig. 3E) and 59% (p = 0.002) reduction in hippocampal amyloid burden (Fig. 3J) compared with age-matched control Tg animals that received vehicle only. Quantitative assessment of total cortical fibrillar amyloid burden also revealed a significant 74% (two-tailed t test, p = 0.0001) reduction. A 78% reduction of the total fibrillar amyloid burden was observed in the hippocampus (two-tailed t test, p < 0.0001). When analyzed separately, an 80% (p = 0.0039) reduction in the CAA burden of cortical vessels was noted in the CpG ODN-treated animals (Fig. 4A–C). Brain microhemorrhages were detected in low numbers in Tg2576 mouse brain sections stained with Perl's stain. However, following treatment with CpG ODN we observed a significant decrease in the extent of cerebral microhemmorhages (Fig. 4D) (one-tailed t test, p = 0.029).
Figure 4.

Aβ burden in the vasculature (CAA burden) and brain microhemorrhages. A–C, Thioflavin-S staining (A, B) revealed a visible reduction in the CAA burden of the penetrating cortical vessels (white arrowhead). There was an 80% decrease (C) in the burden of CAA in CpG ODN-treated Tg2576 mice (Tg-CpG vs Tg-vehicle, **p = 0.0039). D, Quantification of CAA-associated microhemorrhages (Perl's stain) also revealed a significant reduction of iron positive profiles per brain section in CpG-treated group (Tg-CpG vs Tg-vehicle, *p = 0.029).
Assessment of Aβ levels and Aβ oligomers in the brain
ELISA measurements revealed a statistically significant decrease in the levels of total (FA extracted) Aβ40 and Aβ42 by 59% (two-tailed t test, p = 0.019) and 56% (p = 0.026), respectively, after the CpG ODN treatment (Fig. 5A). The levels of soluble (DEA extracted) Aβ40 and Aβ42 fractions were significantly reduced by 75% (two-tailed t test, p = 0.003) and 74% (p = 0.0019), respectively, in CpG-treated mice (Fig. 5B). In addition, the measurements of total Aβ levels and total Aβ burden in the cortex (Aβ40, p < 0.0001, r2 = 0.75; Aβ42, p < 0.0001, r2 = 0.83) and hippocampus (Aβ40, p = 0.0025, r2 = 0.39; Aβ42, p = 0.0014, r2 = 0.43) correlated well and indicated a similar percentage reduction in the treated mice. No differences in the level of expression of human APP were found between CpG ODN-treated and vehicle-treated Tg mice (data not shown). CpG ODN treatment is know to affect gene expression of numerous proteins, APP is not among these (Gao et al., 2002; Klaschik et al., 2007; Nagarajan et al., 2007).
Figure 5.

Aβ levels in brain. A, B, Treatment with CpG ODN significantly decreased total (A) and soluble (B) brain Aβ levels in Tg2576 mice. A, Aβ40, 59% reduction, *p = 0.019; Aβ42, 56% reduction,*p = 0.026. B, Aβ40, 75% reduction, **p = 0.003; Aβ42, 74% reduction, **p = 0.0019.
Soluble oligomeric Aβ ligands (also known as ADDLs) may account for memory loss and AD neuropathology, thus presenting a significant therapeutic target. The levels of pathogenic Aβ oligomers in the brain homogenates were assessed by Western-blot using the A11 oligomer-specific antibody (Fig. 6). CpG ODN treatment led to a significant decrease in the levels of A11 immuno reactive (56 kDa) oligomers (two-tailed t test, p = 0.033).
Figure 6.

Western blot detection and densitometric analysis of A11 immunoreactive oligomer-specific bands. A, B, Western blot (A) of brain homogenates stained with A11 oligomer-specific polyclonal antibody and densitometric analysis (B) of oligomer-specific (56 kDa) band showed significant difference between CpG ODN-treated and vehicle-treated Tg animals (*p = 0.033).
Furthermore, there was a correlation between the levels of 56 kDa Aβ assemblies and Aβ levels, with total Aβ levels correlating better than soluble Aβ levels (total Aβ40, p = 0.0507, r2 = 0.186; total Aβ42, p = 0.047, r2 = 0.192; data not shown).
Associated histopathology
In addition to the analysis of Aβ burden, we evaluated the treatment effect of CpG ODNs on microglial activation in APP Tg2576 mice. Subsequent immunohistochemical staining for the adhesion receptor CD11b, a well established microglial and mononuclear phagocyte marker was performed. The assessment of microglial marker CD11b was based on semiquantitative analysis of the extent of microgliosis. CpG ODN treatment resulted in overall cortical (two-tailed t test, p = 0.0001) and hippocampal (data not shown) reduction in CD11b immunoreactivity (Fig. 7). Although CD11b microglial expression was also found in our non-Tg animals, the staining intensity of CD11b marker was very low (data not shown). In addition, we confirmed the CD11b immunohistochemistry results by staining the brains with another commonly used microglial and macrophage marker CD45, which is typically expressed in association with more mature plaques (Morgan et al., 2005). At 17 months of age, quantitative stereological analysis revealed an overall reduction in CD45 immunoreactivity. The CpG ODN-treated mice demonstrated a 71% reduction in cortical (Fig. 8A–C) and a 73% reduction in hippocampal CD45 reactive microglia burden (Fig. 8D–F). Despite a reduction in CD11b immunoreactivity and overall numbers of activated microglia labeled with CD45 antibody, there was a significant increase in activated microglia around the few remaining plaques in the CpG-treated group. Semiquantitation of CD45 immunoreactivity surrounding the plaques, measuring between 5 and 50 μm in diameter, was evaluated on a separate set of sections which were immunolabeled with CD45 antibody and Thioflavin S stained to visualize amyloid plaques (two-tailed t test, p = 0.047) (Fig. 9). No difference in CD11b immunoreactivity around the remaining plaques was found in sections double stained with CD11b antibody and Thioflavin S comparing the CpG-treated and vehicle Tg groups (data not shown). Astrocytes were detected using the astrocyte-specific marker GFAP. Semiquantitative rating of astroglial staining in cortex indicated fewer astrocytes in the CpG ODN-treated group (two-tailed t test, Tg-CpG vs Tg-vehicle, p = 0.006) (Fig. 10).
Figure 7.

CpG ODNs reduced overall cortical and hippocampal CD11b immunoreactivity in APP Tg2576 mice. A–C, Immunostaining (A, B) with CD11b microglial marker followed by semiquantitative analysis (C) revealed significant reduction in cortical and hippocampal (data not shown) microgliosis in CpG ODN-treated (A) compared with vehicle-treated (B) Tg animals (Tg-CpG vs Tg-vehicle, ***p = 0.0001). The degree of microgliosis was graded on a scale from 0 to 3.
Figure 8.

Reduction in cortical and hippocampal CD45 immunoreactivity (CD45 load) in CpG ODN-treated Tg2576 mice. Cortical (A, B) and hippocampal (D, E) CD45 immunohistochemistry also indicated an overall reduction in microglial activity in CpG ODN- treated mice. Quantitative stereological analysis within the cortex revealed a 71% reduction (***p < 0.001) in CD45 immunoreactivity in CpG ODN-treated Tg mice compared with control Tg mice (C). Likewise, CD45 immunoreactivity within the hippocampus was reduced by 73% (***p < 0.001) in Tg-CpG group compared with Tg-vehicle group (F). The scale bars in B and E correspond to cortical and hippocampal images, respectively.
Figure 9.
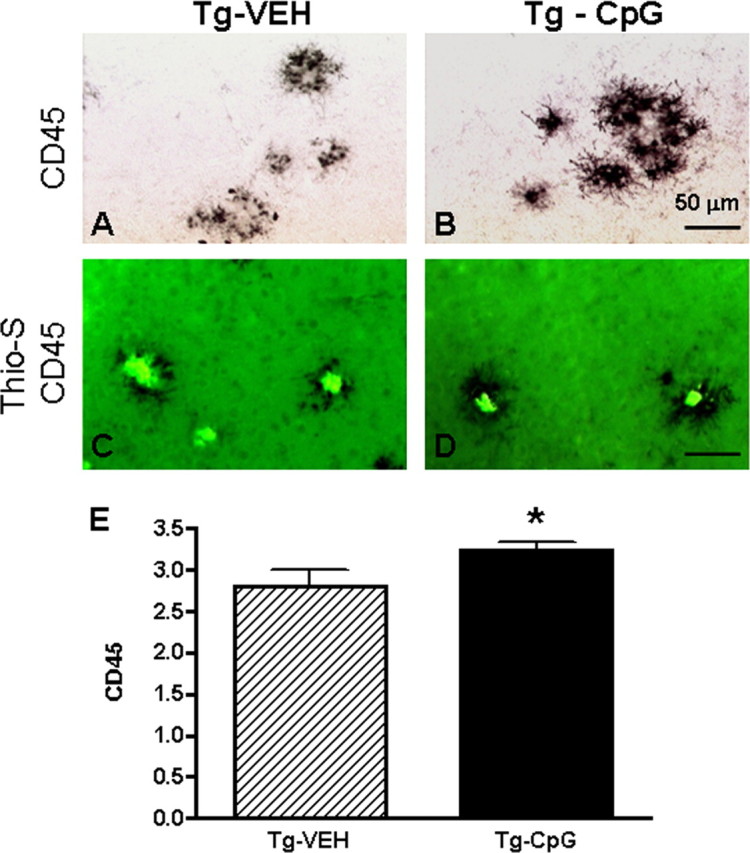
Microglial reactivity around the plaques. A–E, Immunostaining of CD45 (A, B) double-stained with Thioflavin-S (C, D) followed by semiquantitative analysis (E) demonstrated an increase in CD45 immunoreactivity around remaining plaques in the CpG-treated group (Tg-CpG vs Tg-vehicle, *p = 0.047). Representative sections are shown in A–D. Scale bar, 50 μm.
Figure 10.

Treatment with CpG ODNs reduced cortical GFAP reactive astrocytosis in APP Tg2576 mice. A–D, GFAP immunostaining (A–C) followed by semiquantitative analysis (D) revealed fewer activated astrocytes in CpG ODN-treated Tg animals compared with vehicle-treated animals (Tg-CpG vs Tg-vehicle, **p = 0.006; Tg-vehicle vs Wt, ***p = 0.0005; Tg-CpG vs Wt, p < 0.054). Reactive astrocytosis was rated on a scale of 0.5–3.
In evaluating the efficacy of CpG ODN administration in AD mice model, we found that stimulation of TLR9 signaling led to a remarkable reduction of amyloid burden which was paralleled by a reduction in the numbers of activated microglia and astrocytes.
Furthermore, since antigen presenting cells including microglia are activated by TLR ligands, humoral immunity to Aβ may be induced. To determine whether the CpG ODN amyloid removal effect correlated with the production of antibody, the autoantibody response toward Aβ40 and Aβ42 was assessed periodically. No group differences were observed in the levels of autoantibodies in animals at 12 months of age. However, plasma obtained at the end of the study (at 17 months) contained significantly higher antibody levels against Aβ40 (p = 0.017) in the CpG ODN-treated group when compared with vehicle-treated Tg controls while no significant difference was noted in CpG ODN-treated mice against Aβ42 (p = 0.09) (Fig. 11).
Figure 11.

Levels of autoantibodies. A, B, At 17 months of age there was a significantly higher autoantibody response toward Aβ40 (A) and a trend for a higher response to Aβ42 (B) in CpG ODN-treated Tg mice when compared with vehicle-treated Tg mice. The Tg-vehicle mice did not differ from the wild-type controls. A, Tg-CpG versus Tg-vehicle, *p = 0.017; Tg- vehicle versus Wt, p = 0.24; Tg-CpG versus Wt, *p = 0.042. B, Tg-CpG versus Tg-vehicle, p = 0.09; Tg- vehicle versus Wt, p = 0.44; Tg-CpG versus Wt, p = 0.15. No apparent differences were observed between the groups in 12-month-old animals (data not shown).
Discussion
Vaccination was the first treatment approach shown to have genuine impact on AD-related pathology, at least in animal models of AD (Brody and Holtzman, 2008). The striking biological effect of vaccination in preclinical testing and the apparent lack of side effects in AD Tg mice prompted Elan/Wyeth to launch clinical trials with a vaccine containing preaggregated Aβ1–42 and QS21 as an adjuvant. The phase II of trial was prematurely terminated when 6% of vaccinated patients manifested symptoms of acute meningoencephalitis (Wisniewski and Konietzko, 2008). Autopsies performed on a limited number of trial patients suggested that significant Aβ clearance of parenchymal plaques had occurred, similar to what had been reported in the animal studies, confirming the validity of this approach for amyloid clearance in humans (Ferrer et al., 2004; Masliah et al., 2005; Bombois et al., 2007; Boche and Nicoll, 2008). Some of these patients also had T-cell infiltration around cerebral blood vessels suggesting a deleterious excessive Th1 immune response. Neuropsychiatric testing of vaccinated patients, who mounted an antibody response, showed a modest but statistically significant cognitive benefit (Hock et al., 2003; Gilman et al., 2005). These data indicated that vaccination may be beneficial for human AD patients, but the design of the vaccine must be modified.
We postulated stimulation of the innate immune system as a possible alternative method for modulating amyloid-related pathology, without associated toxicity. Our prior work in prion disease, suggested this could be accomplished through TLR9 activation (Spinner et al., 2007), which induces both innate and adaptive immunity. In rodents, TLR9 is expressed in all myeloid cells (including macrophages/microglia, and DCs), plasmacytoid DCs (pDCs), and B cells (among others), which are activated upon its ligation (Krieg, 2006). TLR9 is localized to the endosomal compartment where it can recognize unmethylated CpG DNA from internalized bacteria and viruses. There also exist different classes of TLR9 stimulating CpG DNAs (classes A, B, and C), each of which has a slightly different profile of cells activated and signaling pathways stimulated (Krieg, 2006). The type B CpG ODN that we used potently stimulates phagocyte activation, DC maturation, and B cell proliferation, enhancing microbial/antigen clearance, antigen presentation and cross-presentation, and specific humoral immune responses (Crack and Bray, 2007; Spinner et al., 2007).
Our results indicate that stimulation of TLR9 by CpG ODNs dramatically reduces amyloid burden in a mouse model of AD. There was a 66% reduction in cortical amyloid burden and a 59% reduction in hippocampal amyloid burden. This reduction in amyloid was associated with cognitive improvement as determined by radial arm maze testing. In the behavioral studies, we verified that any differences between the groups are not attributable to differences in locomotor activity. Behavioral improvements are likely related to reduction in 56 kDa Aβ oligomers, which are linked more closely to functional deficits in AD model mice than fibrillar Aβ deposits (Cleary et al., 2005). Our studies document that immune stimulation targeting TLR9, while highly effective at reducing Aβ deposition, is not associated with any apparent CNS toxicity. Our analysis of brain microglia reactivity shows a marked reduction in both the cortex and hippocampus, as assessed by CD11b and CD45 immunoreactivity, reflecting the overall reduction of the amyloid burden in the CpG ODN-treated mice. In addition, as assessed by GFAP immunoreactivity, CNS astrocytosis was also markedly reduced. Hence there was no evidence of encephalitis in the brains of treated mice.
An additional potential complication of immunomodulation in the clearance of amyloid deposits is the occurrence of cerebral microhemorrhages. Several reports have shown an increase in microhemorrhages in different AD mouse models following passive intraperitoneal immunization with various monoclonal antibodies having high affinities for Aβ plaques and CAA (Pfeifer et al., 2002; Wilcock et al., 2004; Racke et al., 2005). Microhemorrhages following active immunization in animal models have been reported in at least one study (Wilcock et al., 2007). Early autopsies from the AN1792 trial indicated no clearance of vascular amyloid. In one of these cases numerous cortical bleeds, which are typically rare in AD patients, were evident suggesting that these may have been related to the immunization (Ferrer et al., 2004). This is an important issue since CAA is present in virtually all AD cases, with ∼20% of AD patients having “severe” CAA (Jellinger, 2008a). Furthermore, CAA is present in ∼33% of cognitively normal elderly populations (Zhang-Nunes et al., 2006). Hence it is important that in the present study, we show that stimulation of the innate immune system with CpG ODNs reduces the CAA burden by 80%; while not producing any evidence of increased cerebral microhemorrhages.
To speculate on the possible mechanisms of action of intraperitoneally administered CpG ODNs one must consider the known pharmacodynamics. In humans, CpG ODNs administered peripherally, but not intravenously, are known to distribute throughout tissues that include mainly liver, kidneys and spleen (Krieg, 2006). In addition, CpG ODNs do not pass through the intact blood–brain-barrier (BBB) (Krieg, 2006; Crack and Bray, 2007). Direct TLR ligation in microglia is known to enhance their ability to degrade Aβ (Iribarren et al., 2005; Majumdar et al., 2007). However since the BBB is likely intact at early stages of AD-like pathology, direct penetration of CpG ODNs into the brain, and therefore direct stimulation of microglia, is unlikely during the prophylactic treatment we performed. Direct action of CpG ODNs on cells in the brain is not the likely mechanism by which this TLR9 agonist reduces Aβ plaque at early stages of disease.
Early in the disease process, ameliorative mechanisms of TLR9 stimulation likely involve direct targets in the periphery. A probable candidate in rodents is peripheral macrophages. Bone marrow-derived macrophages, which are known to express TLR9, have been found to enter the brain in mouse AD models and limit the accumulation of Aβ in plaques (Stalder et al., 2005; Simard et al., 2006; Soulet and Rivest, 2008). Thus, the effect of CpG ODNs on peripheral macrophages and myeloid DCs may be to induce heightened levels of surveillance and activity by these cells, and thus increased influx into the brain and clearance of Aβ. Alternatively, activation of immune cells in the periphery may elicit the secretion of cytokines and chemokines that enter the CNS and induce either direct Aβ clearance by resident microglia, or induce CNS cell signaling leading to recruitment of peripheral macrophages capable of clearing Aβ. That CpG ODN treatment enhances clearance of deposited Aβ through recruitment of peripheral macrophages to the CNS, is supported by increased CD45 immunoreactive microglia around the few remaining plaques in the CpG ODN-treated group (Fig. 9), without associated increases in CD11b immunoreactivity. CD45 labeled microglia have been suggested to have a more likely peripheral origin (Guillemin and Brew, 2004).
At later stages of AD, when penetration of CpG ODNs into the CNS may be possible, microglia may be a direct target of the treatment. Mechanisms by which direct TLR ligation on microglia, macrophages and other APCs enhance antigen presentation, and subsequent adaptive immune responses have been described in detail (Blander and Medzhitov, 2006). Such mechanisms appear to involve induction of both phagocytic activation and enhanced antigen presentation (Majumdar et al., 2007). If direct activation of microglia does occur in the current model, the clearance of Aβ by microglia is likely very rapid, since at the 17 month time-point at which we evaluated pathology, both microgliosis and Aβ deposition in the CNS is low. This would suggest that after the Aβ is mostly cleared, microgliosis largely subsides.
An alternative possibility, which is not mutually exclusive, is that stimulation of TLR9 by CpG ODNs leads to secondary activation of adaptive immunity with the production of autoantibodies against Aβ. Support for this hypothesis is that there were higher levels of antibodies against Aβ40 in CpG ODN-treated mice at 17 months of age. However, stimulation of autoantibodies is not likely to have played an important role in our model system since the elevation of anti-Aβ40 antibodies in the 17-month-old CpG ODN-treated group, although statistically significant, was very modest and at no time were levels of anti-Aβ42 autoantibodies significantly elevated. It is Aβ42 that is thought to be the most pathogenic of the Aβ peptides (Walsh and Selkoe, 2004).
The effect of induction of specific TLR signaling has been examined previously in mouse AD models. Knock-out of TLR2 or TLR4 in AD model mice was shown to accelerate Aβ deposition (Tahara et al., 2006; Richard et al., 2008). Accordingly, a single intracranial administration of the TLR4 ligand lipopolysaccharide (LPS) in AD model Tg2576 mice significantly reduces Aβ deposition within 7 d, an effect requiring microglial activation (Herber et al., 2007). Studies in which large doses of the LPS were administer to mice intraperitoneally, however reported deleterious effects, including the exacerbation of amyloid deposition and cognitive declines and/or increased neuroinflammation and neuronal death (Qiao et al., 2001; Cunningham et al., 2005; Lee et al., 2008). These findings contrast to ours in which peripheral administration of CpG ODNs is clearly beneficial leading to reductions in both amyloid deposition and cognitive decline. Differences between the effects of peripheral LPS and CpG ODNs may be reconciled by the fact that these two TLR agonists trigger different signaling pathways, leading to different cytokine and gene activation profiles (Gao et al., 2002). In our own in vitro studies, we have observed that even low doses of LPS lead to cytokine responses in macrophages much greater than those observed at high doses of CpG ODNs (unpublished data).
Our results indicate that stimulation of the innate immune system through TLR9 with CpG ODNs is an effective and apparently nontoxic method to reduce the amyloid burden in AD model mice. Significantly, use of CpG ODNs has been shown to be safe in normal human volunteers (Vicari et al., 2007). Furthermore, we found that amyloid reduction was associated with significant cognitive benefits in an AD mouse model. This approach has significant implications for future human immunomodulatory approaches to prevent AD in humans.
Footnotes
This work was supported by National Institutes of Health Grants AG20245 and AG15408, as well as by Alzheimer's Association Grant IIRG-06-26434. Support was also provided by the Alzheimer's Association/Stranahan Foundation, NIRG-04-1162. We also thank Emily Petrus for technical assistance and Ausma Rabe for helpful discussions.
References
- Asuni AA, Boutajangout A, Scholtzova H, Knudsen E, Li YS, Quartermain D, Frangione B, Wisniewski T, Sigurdsson EM. Aβ derivative vaccination in alum adjuvant prevents amyloid deposition and does not cause brain microhemorrhages in Alzheimer's model mice. Eur J Neurosci. 2006;24:2530–2542. doi: 10.1111/j.1460-9568.2006.05149.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blander JM, Medzhitov R. On regulation of phagosome maturation and antigen presentation. Nat Immunol. 2006;7:1029–1035. doi: 10.1038/ni1006-1029. [DOI] [PubMed] [Google Scholar]
- Boche D, Nicoll JA. The role of the immune system in clearance of Abeta from the brain. Brain Pathol. 2008;18:267–278. doi: 10.1111/j.1750-3639.2008.00134.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bombois S, Maurage CA, Gompel M, Deramecourt V, Mackowiak-Cordoliani MA, Black RS, Lavielle R, Delacourte A, Pasquier F. Absence of beta-amyloid deposits after immunization in Alzheimer disease with Lewy body dementia. Arch Neurol. 2007;64:583–587. doi: 10.1001/archneur.64.4.583. [DOI] [PubMed] [Google Scholar]
- Brody DL, Holtzman DM. Active and passive immunotherapy for neurodegenerative diseass. Annu Rev Neurosci. 2008;31:175–193. doi: 10.1146/annurev.neuro.31.060407.125529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cleary JP, Walsh DM, Hofmeister JJ, Shankar GM, Kuskowski MA, Selkoe DJ, Ashe KH. Natural oligomers of the amyloid-beta protein specifically disrupt cognitive function. Nat Neurosci. 2005;8:79–84. doi: 10.1038/nn1372. [DOI] [PubMed] [Google Scholar]
- Crack PJ, Bray PJ. Toll-like receptors in the brain and their potential roles in neuropathology. Immunol Cell Biol. 2007;85:476–480. doi: 10.1038/sj.icb.7100103. [DOI] [PubMed] [Google Scholar]
- Cunningham C, Wilcockson DC, Campion S, Lunnon K, Perry VH. Central and systemic endotoxin challenges exacerbate the local inflammatory response and increase neuronal death during chronic neurodegeneration. J Neurosci. 2005;25:9275–9284. doi: 10.1523/JNEUROSCI.2614-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I, Boada RM, Sánchez Guerra ML, Rey MJ, Costa-Jussá F. Neuropathology and pathogenesis of encephalitis following amyloid-beta immunization in Alzheimer's disease. Brain Pathol. 2004;14:11–20. doi: 10.1111/j.1750-3639.2004.tb00493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao JJ, Diesl V, Wittmann T, Morrison DC, Ryan JL, Vogel SN, Follettie MT. Regulation of gene expression in mouse macrophages stimulated with bacterial CpG-DNA and lipopolysaccharide. J Leukoc Biol. 2002;72:1234–1245. [PubMed] [Google Scholar]
- Gilman S, Koller M, Black RS, Jenkins L, Griffith SG, Fox NC, Eisner L, Kirby L, Rovira MB, Forette F, Orgogozo JM. Clinical effects of Aβ immunization (AN1792) in patients with AD in an interupted trial. Neurology. 2005;64:1553–1562. doi: 10.1212/01.WNL.0000159740.16984.3C. [DOI] [PubMed] [Google Scholar]
- Guillemin GJ, Brew BJ. Microglia, macrophages, perivascular macrophages, and pericytes: a review of function and identification. J Leukoc Biol. 2004;75:388–397. doi: 10.1189/jlb.0303114. [DOI] [PubMed] [Google Scholar]
- Hardy J. A hundred years of Alzheimer's disease research. Neuron. 2006;52:3–13. doi: 10.1016/j.neuron.2006.09.016. [DOI] [PubMed] [Google Scholar]
- Herber DL, Mercer M, Roth LM, Symmonds K, Maloney J, Wilson N, Freeman MJ, Morgan D, Gordon MN. Microglial activation is required for Abeta clearance after intracranial injection of lipopolysaccharide in APP transgenic mice. J Neuroimmune Pharmacol. 2007;2:222–231. doi: 10.1007/s11481-007-9069-z. [DOI] [PubMed] [Google Scholar]
- Hock C, Konietzko U, Streffer JR, Tracy J, Signorell A, Müller-Tillmanns B, Lemke U, Henke K, Moritz E, Garcia E, Wollmer MA, Umbricht D, de Quervain DJ, Hofmann M, Maddalena A, Papassotiropoulos A, Nitsch RM. Antibodies against β-amyloid slow cognitive decline in Alzheimer's disease. Neuron. 2003;38:547–554. doi: 10.1016/s0896-6273(03)00294-0. [DOI] [PubMed] [Google Scholar]
- Hsiao K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Aβ elevation and amyloid plaques in transgenic mice. Science. 1996;274:99–102. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- Iribarren P, Chen K, Hu J, Gong W, Cho EH, Lockett S, Uranchimeg B, Wang JM. CpG-containing oligodeoxynucleotide promotes microglial cell uptake of amyloid beta 1–42 peptide by up-regulating the expression of the G-protein-coupled receptor mFPR2. FASEB J. 2005;19:2032–2034. doi: 10.1096/fj.05-4578fje. [DOI] [PubMed] [Google Scholar]
- Jellinger KA. Morphologic diagnosis of “vascular dementia” - a critical update. J Neurol Sci. 2008a;270:1–12. doi: 10.1016/j.jns.2008.03.006. [DOI] [PubMed] [Google Scholar]
- Jellinger KA. Neuropathological aspects of Alzheimer disease, Parkinson disease and frontotemporal dementia. Neurodegener Dis. 2008b;5:118–121. doi: 10.1159/000113679. [DOI] [PubMed] [Google Scholar]
- Klaschik S, Gursel I, Klinman DM. CpG-mediated changes in gene expression in murine spleen cells identified by microarray analysis. Mol Immunol. 2007;44:1095–1104. doi: 10.1016/j.molimm.2006.07.283. [DOI] [PubMed] [Google Scholar]
- Krieg AM. Therapeutic potential of Toll-like receptor 9 activation. Nat Rev Drug Discov. 2006;5:471–484. doi: 10.1038/nrd2059. [DOI] [PubMed] [Google Scholar]
- Lee JW, Lee YK, Yuk DY, Choi DY, Ban SB, Oh KW, Hong JT. Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J Neuroinflammation. 2008;5:37. doi: 10.1186/1742-2094-5-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar A, Cruz D, Asamoah N, Buxbaum A, Sohar I, Lobel P, Maxfield FR. Activation of microglia acidifies lysosomes and leads to degradation of Alzheimer amyloid fibrils. Mol Biol Cell. 2007;18:1490–1496. doi: 10.1091/mbc.E06-10-0975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masliah E, Hansen L, Adame A, Crews L, Bard F, Lee C, Seubert P, Games D, Kirby L, Schenk D. Aβ vaccination effects on plaque pathology in the absence of encephalitis in Alzheimer disease. Neurology. 2005;64:129–131. doi: 10.1212/01.WNL.0000148590.39911.DF. [DOI] [PubMed] [Google Scholar]
- Morgan D. Immunotherapy for Alzheimer's disease. J Alzheimers Dis. 2006;9:425–432. doi: 10.3233/jad-2006-9s348. [DOI] [PubMed] [Google Scholar]
- Morgan D, Gordon MN, Tan J, Wilcock D, Rojiani AM. Dynamic complexity of the microglial activation response in transgenic models of amyloid deposition: implications for Alzheimer therapeutics. J Neuropathol Exp Neurol. 2005;64:743–753. doi: 10.1097/01.jnen.0000178444.33972.e0. [DOI] [PubMed] [Google Scholar]
- Nagarajan G, Kuo CC, Liang CM, Chen CM, Liang SM. Effects of CpG-B ODN on the protein expression profile of swine PBMC. Vet Res. 2007;38:795–808. doi: 10.1051/vetres:2007032. [DOI] [PubMed] [Google Scholar]
- Pfeifer M, Boncristiano S, Bondolfi L, Stalder A, Deller T, Staufenbiel M, Mathews PM, Jucker M. Cerebral hemorrhage after passive anti-Aβ immunotherapy. Science. 2002;298:1379. doi: 10.1126/science.1078259. [DOI] [PubMed] [Google Scholar]
- Qiao X, Cummins DJ, Paul SM. Neuroinflammation-induced acceleration of amyloid deposition in the APPV717F transgenic mouse. Eur J Neurosci. 2001;14:474–482. doi: 10.1046/j.0953-816x.2001.01666.x. [DOI] [PubMed] [Google Scholar]
- Racke MM, Boone LI, Hepburn DL, Parsadainian M, Bryan MT, Ness DK, Piroozi KS, Jordan WH, Brown DD, Hoffman WP, Holtzman DM, Bales KR, Gitter BD, May PC, Paul SM, DeMattos RB. Exacerbation of cerebral amyloid angiopathy-associated microhemorrhages in amyloid precursor protein transgenic mice by immunotherapy is dependent on antibody recognition of deposited forms of amyoid β. J Neurosci. 2005;25:629–636. doi: 10.1523/JNEUROSCI.4337-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richard KL, Filali M, Préfontaine P, Rivest S. Toll-like receptor 2 acts as a natural innate immune receptor to clear amyloid β1–42 and delay the cognitive decline in a mouse model of Alzheimer's disease. J Neurosci. 2008;28:5784–5793. doi: 10.1523/JNEUROSCI.1146-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sadowski M, Pankiewicz J, Scholtzova H, Mehta PD, Prelli F, Quartermain D, Wisniewski T. Blocking the apolipoproteinE/Amyloid β interaction reduces the parenchymal and vascular amyloid-β deposition and prevents memory deficit in AD transgenic mice. Proc Natl Acad Sci U S A. 2006;103:18787–18792. doi: 10.1073/pnas.0604011103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scholtzova H, Wadghiri YZ, Douadi M, Sigurdsson EM, Li YS, Quartermain D, Banerjee P, Wisniewski T. A NMDA receptor antagonist leads to behavioral improvement and amyloid reduction in Alzheimer's disease model transgenic mice shown by micro-magnetic resonance imaging. J Neurosci Res. 2008;86:2784–2791. doi: 10.1002/jnr.21713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdsson EM, Knudsen E, Asuni A, Fitzer-Attas C, Sage D, Quartermain D, Goni F, Frangione B, Wisniewski T. An attenuated immune response is sufficient to enhance cognition in an Alzheimer's disease mouse model immunized with amyloid-β derivatives. J Neurosci. 2004;24:6277–6282. doi: 10.1523/JNEUROSCI.1344-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard AR, Soulet D, Gowing G, Julien JP, Rivest S. Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron. 2006;49:489–502. doi: 10.1016/j.neuron.2006.01.022. [DOI] [PubMed] [Google Scholar]
- Soulet D, Rivest S. Bone-marrow-derived microglia: myth or reality? Curr Opin Pharmacol. 2008;8:508–518. doi: 10.1016/j.coph.2008.04.002. [DOI] [PubMed] [Google Scholar]
- Spinner DS, Kascsak RB, Lafauci G, Meeker HC, Ye X, Flory MJ, Kim JI, Schuller-Levis GB, Levis WR, Wisniewski T, Carp RI, Kascsak RJ. CpG oligodeoxynucleotide-enhanced humoral immune response and production of antibodies to prion protein PrPSc in mice immunized with 139A scrapie-associated fibrils. J Leukoc Biol. 2007;81:1374–1385. doi: 10.1189/jlb.1106665. [DOI] [PubMed] [Google Scholar]
- Stalder AK, Ermini F, Bondolfi L, Krenger W, Burbach GJ, Deller T, Coomaraswamy J, Staufenbiel M, Landmann R, Jucker M. Invasion of hematopoietic cells into the brain of amyloid precursor protein transgenic mice. J Neurosci. 2005;25:11125–11132. doi: 10.1523/JNEUROSCI.2545-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahara K, Kim HD, Jin JJ, Maxwell JA, Li L, Fukuchi K. Role of toll-like receptor signalling in Abeta uptake and clearance. Brain. 2006;129:3006–3019. doi: 10.1093/brain/awl249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicari AP, Schmalbach T, Lekstrom-Himes J, Morris ML, Al-Adhami MJ, Laframboise C, Leese P, Krieg AM, Efler SM, Davis HL. Safety, pharmacokinetics and immune effects in normal volunteers of CPG 10101 (ACTILON), an investigational synthetic toll-like receptor 9 agonist. Antivir Ther. 2007;12:741–751. [PubMed] [Google Scholar]
- Walsh DM, Selkoe DJ. Deciphering the molecular basis of memory failure in Alzheimer's disease. Neuron. 2004;44:181–193. doi: 10.1016/j.neuron.2004.09.010. [DOI] [PubMed] [Google Scholar]
- Wilcock DM, Rojiani A, Rosenthal A, Subbarao S, Freeman MJ, Gordon MN, Morgan D. Passive immunization against Abeta in aged APP-transgenic mice reverses cognitive deficits and depletes parenchymal amyloid deposits in spite of increased vascular amyloid and microhemorrhage. J Neuroinflammation. 2004;1:24. doi: 10.1186/1742-2094-1-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcock DM, Jantzen PT, Li Q, Morgan D, Gordon MN. Amyloid-beta vaccination, but not nitro-nonsteroidal anti-inflammatory drug treatment, increases vascular amyloid and microhemorrhage while both reduce parenchymal amyloid. Neuroscience. 2007;144:950–960. doi: 10.1016/j.neuroscience.2006.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski T, Konietzko U. Amyloid-β immunization for Alzheimer's disease. Lancet Neurol. 2008;7:805–811. doi: 10.1016/S1474-4422(08)70170-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang-Nunes SX, Maat-Schieman ML, van Duinen SG, Roos RA, Frosch MP, Greenberg SM. The cerebral beta-amyloid angiopathies: hereditary and sporadic. Brain Pathol. 2006;16:30–39. doi: 10.1111/j.1750-3639.2006.tb00559.x. [DOI] [PMC free article] [PubMed] [Google Scholar]