

Abstract
Herein we report the synthesis of symmetrical C-linked and pseudo-symmetrical O-linked disaccharides structurally related to Araf motifs present in the cell wall of MTB. Their activity in a competition-based arabinosyltransferase assay using [14C]-DPA as the glycosyl donor is also presented. In addition, in vitro inhibitory activity for the disaccharides was determined in a colorimetric broth microdilution assay system against MTB H37Ra and Mycobacterium avium.
The development of multi-drug resistant strains of Mycobacterium tuberculosis (MTB)1,2 and co-infection with HIV3 has limited TB treatment options, initiating a worldwide effort to discover new biochemical targets and selective inhibitors. The mycobacterial cell wall remains an excellent target, focusing discovery efforts on new proteins involved in the biogenesis of this critical bacterial barrier.4 The cell wall of MTB allows the bacterium to elude cellular defenses and thrive within macrophages of the host. Particular components of that barrier, arabinofuranose (Araf), galactofuranose (galf) and rhamnopyranose (rhaf), and several of the attendant synthetic enzymes are not found in humans, and they offer the potential for development of highly selective and potent new drugs.
The major components of the mycobacterial integument are the mycolyl arabinogalactan – peptidoglycan complexes (mAGPs) and lipoarabinomannan (LAM) associated lipoglycans.5 The arabinan portion of the cell wall is composed of Araf homopolymers with different linkages viz. α(1→5), α(1→2) and α(1→3), and requires several different sugar processing enzymes, or arabinosyltransferases (AraT’s), for its complete genesis.5,6 Several synthetic O- and S-alkyl arabinofuranoside acceptors have been prepared for the development of arabinosyl-transferase assays7 and other biological and structural studies based on the unbranched and highly branched polysaccharides of AG and LAM.8–10
As part of ongoing programs to find carbohydrate-based antimycobacterial agents targeting biogenesis of the mycobacterial cell wall polysaccharides, we and others have synthesized several analogs of α(1→5) Araf disaccharides.9b,11,12 We have reported the synthesis and antimycobacterial activity of analogs (Figure 1) with substitution at the non-reducing terminus (ring B).
Figure 1.

5′-substituted Araf α(1→5) Araf disaccharides
We designed O-linked and C-linked disaccharide analogs (3 and 4) that also possess C-substitution at the anomeric center of the reducing terminus. This substitution is based on the N,N-dicyclohexyl-methylamino substitution of 2 at the 5′-position that showed the best activity among reported derivatives.11 C-sugars and C-linked disaccharides offer advantages as enzyme probes and inhibitors, and as drugs. Most importantly, this linkage prevents glycosidase-mediated cleavage. We report the synthesis and inhibitory data of pseudo-symmetrical and symmetrical disaccharides 3 and 4 respectively.
In our study of the Araf 1-5 linkages, it was noted that the simple O-linked disaccharide core is pseudo-symmetrical around the central -O-C- bond. Hence, we targeted 3 and 4 to incorporate the N,N-dicyclohexyl group of the active compound 2 at both ends of the targets to take advantage of this symmetry to potentially increase binding efficiency. Compound 4 contains a true C2-symmetry axis as does the intermediate 17 that may also offer the advantage of reduced degrees of freedom around the disaccharide linkage that could further improve binding efficiency.
Several groups have reported C-linked glycoside analogs of the α-D-Araf-(1→5)-α-D-Araf motif found in the cell wall of mycobacteria using various approaches.13–16 We describe the synthesis of targets 3 and 4 through coupling of a 5-azidoarabinosyl donor with a 1-azido D-mannitol derivative and C–C bond formation by Wittig olefination14 respectively. These syntheses began with 2,5-anhydro-1-azido-1-deoxy-D-mannitol (5) prepared from D-glucosamine hydrochloride by diazotization-mediated ring contraction and selective monotosylation followed by introduction of the azido group using NaN3.17 Compound 5 was persilylated to produce 6 which on selective desilylation at the 6-position using a trifluoroacetic acid-water mixture (1:1) in dry THF at −4°C produced 7 as shown in Scheme 1.
Scheme 1.

(a) TBDMSCl, imidazole, DMF, 50 °C, 18 h; (b) TFA-Water (1:1), THF, −4 °C, 4 h, 85% (in 2 steps).
The synthesis of disaccharide 3 was achieved by coupling of p-thiotolyl 1,5-dideoxy-5-azido-2,3-di-O-acetyl-α-D-arabinofuranoside (8),11 with 2,5-Anhydro-1-azido-3,4-di-O-tert-butyldimethylsilyl-1-deoxy-D-mannitol (7) in the presence of NIS and the Lewis acid promoter triflic acid to produce 9 (Scheme 2). After several column purifications, a slight trace of unreacted 8 was present necessitating deacetylation to yield the diazido disaccharide 10 as a pure, colorless oil. N,N-dialkylation of 10 via reductive alkylation with cyclohexane carboxyaldehyde in MeOH over 10% Pd/C at room temperature under H2 atmosphere gave 11 in 74% yield as a colorless oil after purification. Desilylation of 11 in a trial reaction with Bu4N+F− produced only a poor yield (48%) of 3. The synthesis of 3 was achieved by desilylation of 11 using Et4N+F− in THF in 88% yield after purification.
Scheme 2.

Reagents and conditions. (a) NIS, TfOH, CH2Cl2, −20 °C, 15 min; (b) 7N NH3/MeOH, MeOH, rt, overnight, 85% (in 2 steps); (c) C6H11CHO, 10% Pd/C, MeOH, rt, 4h, 74%; (d) Et4N+F−, THF, rt, overnight, 88%.
The attempted synthesis of C-linked disaccharide 4 via Wittig olefination18 to give a C-linked diazido disaccharide similar to 9 was unsuccessful. Disaccharide 4 was produced using Wittig olefination as shown in Scheme 3. Azido saccharide 7 was first converted to the N,N-dicyclohexylmethylamino derivative 12, ultimately yielding 14 and 15 for coupling. Compound 12 was converted to 6-iodomannitol derivative 13 by heating with iodine, PhP3, and imidazole. After purification, 13 was heated with neat Ph3P to produce the triphenylphosphonium iodide 14. Compound 12 was alternatively oxidized with PCC in CH2Cl2 to aldehyde 15 that was used directly in the coupling without purification.19 The two saccharides 14 and 15 were coupled in the presence of BuLi at −30°C to give a mixture of olefin 16 (E/Z ratio 96:4 by NMR) after purification. Deblocking of 16 with Et4N+F− in THF and purification produced 17 in 86% yield as an E/Z mixture. Reduction of 17 produced the symmetrical C-linked disaccharide 4 in 62% yield.
Scheme 3.

Reagents & Conditions. (a) C6H11CHO, Pd/C, MeOH, rt, 4h, 95%; (b) I2, imidazole, Ph3P, toluene, 80 °C, 1 h, 56%; (c) PCC, CH2Cl2, rt, 4h, 85%; (d) PPh3, 120 °C, 4 h, 67%; (e) THF-HMPA, BuLi, −30 °C, 2 h, 55%; (f) Et4N+F−, THF, rt, overnight, 86%; (g) Pd(OH)2, H2, EtOAc-MeOH (1:1), rt, 4h, 62%.
All compounds were characterized by ESIMS analysis and 1H NMR spectroscopy.20 nOe, and D2O exchange experiments were performed as needed to confirm NMR assignments.
Activity was determined in the cell-free enzymatic arabinosyltransferase acceptor assay7 in the presence of membranes and is based on inhibition of [14C]Araf incorporation from [14C]DPA by the control α(1→5)-linked 1-O-octyl arabinofuranosyl disaccharide.11 Disaccharide analogs 3, 4, and 17 showed inhibitory activity at a concentration of 3.6 mM (with control acceptor at 0.4 mM) and specific IC50 values were 2.80, 3.44 and 4.15 mM respectively. Bacterial growth inhibition was determined versus MTB H37Ra (ATCC 25177) and Mycobacterium avium (NJ 211) at the initial concentrations of 1.28 and 12.8 μg/mL.21 Initial activity was confirmed using half-log dilutions at 16, 8, and 4 ug/mL to determine an MIC as reported.21 Ethambutol showed a MIC in the range 2 – 4 μg/mL. Compounds 3 and 17 showed a modest MIC of 8 μg/mL, 4 and 10 gave an MIC of 16 μg/mL and 11 a MIC of >12.8 μg/mL against MTB. Against M. avium, however, compound 17 showed a MIC of 8 μg/mL, and 3 and 4 a MIC of 16 μg/mL. The blocked analogs 10 and 11 were inactive at 12.8 μg/mL. In conclusion, we report efficient syntheses of O- and Clinked disaccharides 3, 4 and 17 and their inhibitory activity against MTB.
Figure 2.
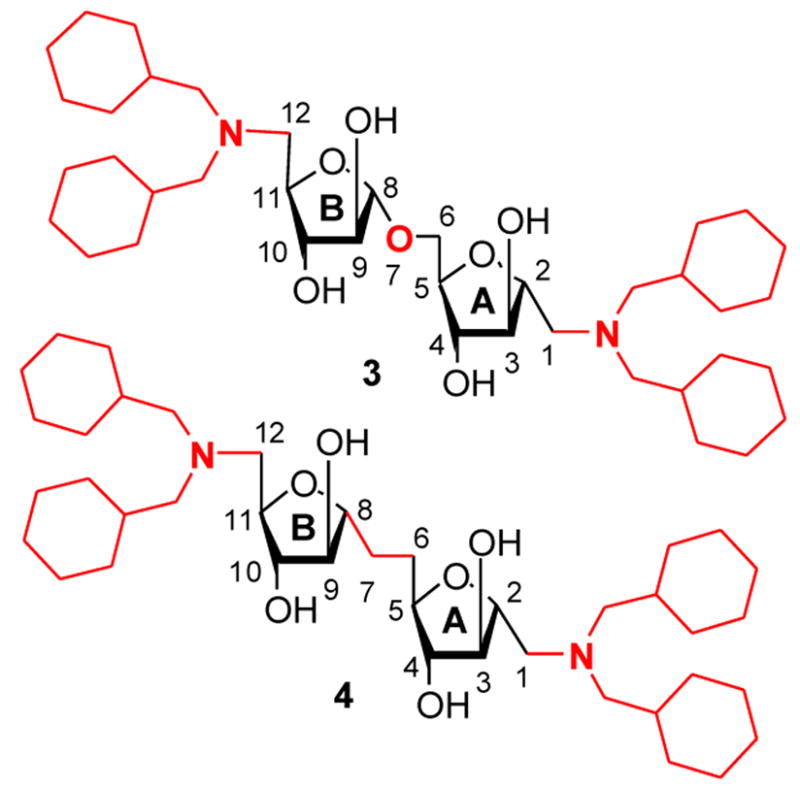
Target O- and C-linked Disaccharides
Acknowledgments
RCR acknowledges NIH/NIAID grant R01AI45317. GSB acknowledges support from Mr. James Bardrick, The Wellcome Trust and MRC.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.WHO. http://www.who.int/tb/en/
- 2.Collins FM. Crit Rev Microbiol. 1993;19:1. doi: 10.3109/10408419309113520. [DOI] [PubMed] [Google Scholar]
- 3.Dolin PJ, Raviglione MC, Kochi A. Bull World Health Org. 1994;72:213. [PMC free article] [PubMed] [Google Scholar]
- 4.Basso LA, Blanchard JS. Resolving the Antibiotic Paradox. In: Rosen, Mobashery, editors. Resistance to Antitubercular Drugs. Plenum Publishers; New York: 1998. p. 115. [Google Scholar]
- 5.a) Brennan PJ. Annu Rev Biochem. 1995;64:29. doi: 10.1146/annurev.bi.64.070195.000333. [DOI] [PubMed] [Google Scholar]; b) Chatterjee D. Curr Opin Chem Biol. 1997;1:579. doi: 10.1016/s1367-5931(97)80055-5. [DOI] [PubMed] [Google Scholar]; c) Daffe M, Draper P. Adv Microb Physiol. 1998;39:131. doi: 10.1016/s0065-2911(08)60016-8. [DOI] [PubMed] [Google Scholar]
- 6.Lee RE, Mikusova K, Brennan PJ, Besra GS. J Am Chem Soc. 1995;117:11829. [Google Scholar]
- 7.Lee RE, Brennan PJ, Besra GS. Glycobiology. 1997;7:1121. doi: 10.1093/glycob/7.8.1121. [DOI] [PubMed] [Google Scholar]
- 8.a) Lowary TL. J Carbohydr Res. 2000;21:691. doi: 10.1016/s0008-6215(99)00296-7. and references therein. [DOI] [PubMed] [Google Scholar]; b) Sanchez S, Bamhaoud T, Prandi J. Tetrahedron Lett. 2000;41:7447. [Google Scholar]
- 9.a) Pathak AK, Pathak V, Khare NK, Maddry JA, Reynolds RC. Carbohydr Lett. 2001;4:117. [PubMed] [Google Scholar]; b) Pathak AK, Pathak V, Maddry JA, Suling WJ, Gurcha SS, Besra GS, Reynolds RC. Bioorg Med Chem. 2001;9:3145. doi: 10.1016/s0968-0896(01)00180-8. [DOI] [PubMed] [Google Scholar]
- 10.a) Pathak AK, Pathak V, Bansal N, Maddry JA, Reynolds RC. Tetrahedron Lett. 2001;42:979. [Google Scholar]; b) Pathak AK, Pathak V, Gurcha SS, Besra GS, Reynolds RC. Bioorg Med Chem Lett. 2002 ;12:2749. doi: 10.1016/s0960-894x(02)00536-x. [DOI] [PubMed] [Google Scholar]
- 11.Pathak AK, Pathak V, Kulshrestha M, Kinnaird D, Suling WJ, Gurcha SS, Besra GS, Reynolds RC. Tetrahedron. 2003;59:10239. [Google Scholar]
- 12.Cociorva OM, Lowary TL. Carbohydr Res. 2004;339:853 – 865. doi: 10.1016/j.carres.2003.12.015. and reference therein. [DOI] [PubMed] [Google Scholar]
- 13.Gurjar MK, Nagaprasad R, Ramana CV. Tetrahedron Lett. 2002;43:7577. [Google Scholar]
- 14.Dondoni A, Marra A. Tetrahedron Lett. 2003;44:4067. [Google Scholar]
- 15.Aslam T, Fuchs MGG, Le Formal A, Wightman RH. Tetrahedron Lett. 2005;46:3249. [Google Scholar]
- 16.Chang GX, Lowary TL. Tetrahedron Lett. 2006;47:4561. [Google Scholar]
- 17.Reynolds RC, Bansal N, Rose J, Friedrich J, Suiling WJ, Maddry JA. Carbohydr Res. 1999;317:164. doi: 10.1016/s0008-6215(99)00069-5. [DOI] [PubMed] [Google Scholar]
- 18.Dondoni A, Zuurmond HM, Boscarato A. J Org Chem. 1997;62:8114. doi: 10.1021/jo971177n. [DOI] [PubMed] [Google Scholar]
- 19.Compound formation was monitored by ESI-MS that showed m/z at 583 [M+H]+.
- 20.Selected data: Compound 6. 1H NMR (CDCl3): δ 4.13 (s, 1H, H-3), 3.97 – 4.04 (m, 3H, H-2, H-4, H-5), 3.66 (d, 2H, J = 7.4 Hz H2-6), 3.46 (dd, 1H, J = 7.0, 12.3 Hz, H-1a), 3.32 (dd, 1H, J = 6.7, 12.3 Hz, H-1b), 0.893 (s, 9H, 3xCH3), 0.890 (s, 9H, 3xCH3), 0.89 (s, 9H, 3xCH3), 0.10 (12H, s, 4xCH3), 0.05 (6H, s, 2xCH3). ESIMS: m/z calcd for C24H53N3O4Si3 532.3422, found 532.3418 [M+H]+. Compound 7. 1H NMR (CDCl3): δ 4.10 – 4.06 (m, 2H, H-2, H-5), 4.05 – 4.00 (m, 2H, H-3, H-4), 3.71 (dd, 2H, J = 4.5, 5.7 Hz, H2-6), 3.50 (dd, 1H, J = 7.0, 12.4 Hz, H-1a), 3.36 (dd, 1H, J = 6.8, 12.4 Hz, H-1b), 2.38 (t, 1H, J = 5.7 Hz, 6-OH), 0.91, 0.90 (s, 9H, 3xCH3), 0.90 (s, 9H, 3xCH3), 0.12 (3H, s, CH3), 0.11 (3H, s, CH3), 0.10 (3H, s, CH3), 0.09 (3H, s, CH3). ESI-MS: m/z calcd for C18H39N3O4Si2 418.2557, found 418.2551 [M+H]+. Compound 11. 1H NMR (CDCl3): δ 5.01 (1H, s, H-1′), 4.76 (1H, d, J = 1.4 Hz, H-1), 4.17 (1H, br s, H-4′), 4.03 – 3.97 (3H, m, H-2, H-4, H-2′), 3.83 – 3.76 (3H, m, H-3, H-5a, H-3′), 3.68 – 3.58 (1H, m, OCH2), 3.59 (1H, dd, J = 3.2 Hz, J = 10.4 Hz, H-5b), 3.35 – 3.28 (1H, m, OCH2), 2.71 – 2.60 (2H, m, H2-5′), 2.44 (2H, dd, J = 7.7, 12.6 Hz, NCH2), 2.16 (2H, dd, J = 5.2, 12.6 Hz, NCH2), 1.87 – 1.60 (4H, m, cyclohexyl CH2′s), 1.57 –1.50 (10H, m, cyclohexyl), 1.46 – 1.41 (2H, m, CH2), 1.37 (10H, br s, 5xCH2), 1.20 – 1.08 (8H, m, cyclohexyl), 0.90 – 0.86 (21H, m, 7xCH3), 0.09, 0.07, 0.06 (each s, 4xCH3). ESIMS: m/z calcd. for C51H98N2O7Si2 907.6990, found 907.6998 [M+H]+. Compound 3. 1H NMR (CD3OD): δ 4.95 (1H, dd, J = 1.6 Hz, H-1′), 4.83 (1H, d, J = 1.7 Hz, H-1), 4.04 (1H, ddd, J = 3.5, 6.2, 6.6 Hz, H-4′), 4.00 (1H, dd, J = 1.6 Hz, J = 3.8 Hz, H-2′), 4.03 – 3.99 (1H, m, H-4), 3.94 (1H, dd, J = 1.7, 3.9 Hz, H-2), 3.87 (1H, dd, J = 3.8, 6.6 Hz, H-3′), 3.86 – 3.79 (2H, m, H-3, H-5a), 3.73 – 3.65 (1H, m, OCH2), 3.65 (1H, dd, J = 3.6, 11.1 Hz, H-5b), 3.50 (1H, dd, J = 3.3, 13.3 Hz, H-5′a), 3.44 – 3.36 (2H, m, OCH2), 3.37 (1H, dd, J = 6.2, 13.3 Hz, H-5′b), 1.62 – 1.53 (2H, m, CH2), 1.30 (10H, br s, 5xCH2), 0.92 – 0.87 (3H, m, CH3). ESIMS: m/z calcd. for C18H33N3O8Na 442.2165, found 442.2176 [M+Na]+. Compound 12. 1H NMR (CDCl3): δ 4.00 (3H, m, H-3, H-4, H-5), 3.96 (1H, dd, J = 5.5, 8.1 Hz, H-2), 3.68 (1H, d, J = 4.1 Hz, H-6a), 2.72 (1H, dd, J = 8.1, 13.6 Hz, H-1a), 2.43 (1H, dd, J = 5.5, 13.6 Hz, H-1b), 2.18 (2H, m, NCH2), 1.93 – 1.10 (22H, m, Cyclohexyl), 0.90, 0.89 (each 9H, s, 6xCH3), 0.12 (3H, s, CH3), 0.11 (6H, s, 2xCH3), 0.80 (3H, s, CH3). ESIMS: m/z calcd. for C32H65NO4Si2 584.4530, found 584.4534 [M+H]+. Compound 13. 1H NMR (CDCl3): δ 4.24 (1H, s, H-4), 4.14 (1H, dd, J = 5.3, 10.4 Hz, H-5), 4.04 (1H, dd, J = 5.1, 8.5 Hz, H-2), 3.96 (1H, s, H-3), 3.44 (1H, dd, J = 9.4, 10.4 Hz, H-6a), 3.22 (1H, dd, J = 5.3, 9.4 Hz, H-6b), 2.67 (1H, dd, J = 8.5, 13.4 Hz, H-1a), 2.39 (1H, dd, J = 5.1, 13.4 Hz, H-1b), 2.19 (1H, dd, J = 8.1, 12.6 Hz, NCH2), 2.12 (1H, dd, J = 5.7, 12.8 Hz, NCH2), 1.85 – 1.10 (20H, m, cyclohexyl) 0.92 – 0.76 (4H, m, cyclohexyl), 0.91 (9H, s, 3xCH3), 0.89 (9H, s, 3xCH3), ESIMS: m/z calcd. for C32H64INO3Si2 694.3549, found 694.3539 [M+H]+. Compound 14. 1H NMR (CDCl3): δ 7.89 – 7.82 (4H, m, Ar), 7.76 – 7.74 (2H, m, Ar), 7.71 – 7.62 (6H, m, Ar), 7.55 – 7.52 (1H, m, Ar), 7.49 – 7.43 (2H, m, Ar), 4.68 (1H, m, H-6a), 4.42 (1H, s, H-4), 4.29 (1H, m, H-5), 3.39 (1H, s, H-3), 3.85 (1H, m, H-6b), 3.71 (1H, dd, J = 5.6, 7.1 Hz, H-2), 2.46 (1H, dd, J = 7.1, 13.8 Hz, H-1a), 2.39 (1H, dd, J = 5.6, 13.8 Hz, H-1b), 1.95 (1H, dd, J = 6.3, 12.0 Hz, NCH2), 2.46 (1H, dd, J = 7.4, 12.0 Hz, NCH2), 1.71 – 1.56 (10H, m, cyclohexyl), 1.31 – 1.03 (10H, m, cyclohexyl), 0.93 (9H, s, 3xCH3), 0.82 (9H, s, 3xCH3), 0.71 – 0.61 (2H, m, cyclohexyl), 0.23 (3H, s, CH3), 0.21 (3H, s, CH3), 0.18 (6H, s, 2xCH3). ESIMS: m/z calcd. for C50H79NO3IPSi2 825.5335, found 825.5330 [M−I]+. Compound 16. 1H NMR (CDCl3): δ 5.66 (2H, dd, J = 2.0, 6.6 Hz, H-6, H-7), 4.54 (2H, d, J = 6.6, 13.2 Hz, H-5, H-8), 3.99 (2H, s, H-3, H-10), 3.97 (2H, dd, J = 4.5, 8.7 Hz, H-2, H-11), 3.91 (2H, s, H-4, H-9), 2.76 (2H, dd, J = 8.7, 13.4 Hz, H-1a, H-12a), 2.34 (2H, dd, J = 4.6, 13.4 Hz, H-1b, H-12b), 2.20 (2H, dd J = 8.6, 12.6 Hz, NCH2), 2.12 (2H, dd J = 5.7, 12.6 Hz, NCH2), 1.91 – 1.11 (40H, m, cyclohexyl), 0.93 (18H, s, 6xCH3), 0.87 (18H, s, 6xCH3), 0.92 – 0.75 (4H, m, cyclohexyl), 0.11 (6H, s, 2xCH3), 0.10 (6H, s, 2xCH3), 0.073 (6H, s, 2xCH3), 0.070 (6H, s, 2xCH3). ESIMS: m/z calcd. for C64H126N2O6Si4 1131.8765, found 1131.8756 [M+H]+. Compound 17. 1H NMR (CDCl3): δ 5.84 (2H, dd, J = 1.1, 4.7 Hz, H-6, H-7), 4.59 (2H, dd, J = 4.7, 13.4 Hz, H-5, H-8), 3.99 (2H, dd, J = 3.9, 6.7 Hz, H-2, H-11), 3.98 (4H, s, H-3, H-4, H-9, H-10), 2.67 (2H, dd, J = 6.7, 13.2 Hz, H-1a, H-12a), 2.56 (2H, dd, J = 2.9, 13.2 Hz, H-1b, H-12b), 2.28 (2H, dd, J = 8.3, 12.8 Hz, NCH2), 2.14 (2H, dd, J = 5.4, 12.8 Hz, NCH2), 1.82 – 1.09 (44H, m, cyclohexyl, 4xOH), 0.91 – 0.78 (4H, m, cyclohexyl). ESIMS: m/z calcd. for C40H70N2O6 675.5306, found 675.5317 [M+H]+. Compound 4. 1H NMR (DMSO-d6, D2O exchanged): δ 3.75 (8H, m, H-2, H-3, H-4, H-5, H-8, H-9, H-10, H-11), 2.53 (2H, m, H-1a, H-12a), 2.35 (2H, dd, J = 7.6, 13.6 Hz, H-1b, H-12b), 2.20 (2H, dd, J = 8.2, 12.5 Hz, NCH2), 2.12 (2H, dd, J = 6.6, 12.5 Hz, NCH2), 1.85 – 1.08 (40H, m, cyclohexyl), 0.88 – 0.71 (4H, m, cyclohexyl). ESIMS: m/z calcd. for C40H72N2O6 677.5463, found 677.5466 [M+H]+.
- 21.Suling WJ, Reynolds RC, Barrow EW, Wilson LN, Piper JR, Barrow WW. J Antimicrob Chemother. 1998;42:811. doi: 10.1093/jac/42.6.811. [DOI] [PubMed] [Google Scholar]