

Abstract
We sequenced all mitochondrial tRNA genes in a 61-year-old man with chronic progressive external ophthalmoplegia and mitochondrial myopathy but without mtDNA rearrangements, and identified a heteroplasmic m.3244G>A mutation in the tRNALeu (UUR) gene. This mutation had been previously associated with the MELAS phenotype, but not described in any detail.
The mutation load in muscle was 84% and COX-negative fibers harbored greater levels of mutant genomes than COX-positive fibers. The m.G3244G>A mutation affects a highly conserved nucleotide in the dihydrouridine loop and has been associated with a wobble modification deficiency of the mutant tRNA.
Keywords: mtDNA, CPEO, tRNALeu(UUR), mutation
1. Introduction
Point mutations in mitochondrial DNA (mtDNA) cause a wide spectrum of disorders and frequently affect mitochondrial transfer RNA (tRNA) genes. Distinguishing pathogenic tRNA point mutations from neutral polymorphisms is often difficult, although pathogenic mutations are usually heteroplasmic and affect highly conserved DNA sites. Currently, more than 90 different tRNA mutations are listed as pathogenic (MITOMAP: A human Mitochondrial genome Database, www.mitomap.org), but there is no strict correlation between mutations and clinical phenotypes [1].
Chronic progressive external ophthalmoplegia (CPEO), one of the most frequent manifestations of mitochondrial disease, is often caused by single or multiple large-scale rearrangements of mtDNA. However, mtDNA point mutations have also been associated with CPEO, most notably the m.3243A>G MELAS mutation [2]. Here, we report a case of CPEO without large-scale mtDNA rearrangements but harboring instead a heteroplasmic m.3244G>A mutation in the tRNALeu(UUR) gene. This mutation was mentioned as part of “unpublished work” in a paper comparing the taurine-containing modification (5-taurinomethyluridine) at the anticodon wobble position in two sets of different tRNALeu(UUR) mutations [3]. The authors stated that the m.3244G>A mutation was associated with MELAS, but gave no clinical or pathological details on the patient (or patients) harboring this mutation. As it is generally accepted that the pathogenicity of a given nucleotide change is not conclusively established until it is reported in at least two different families, we report this patient to (i) confirm the pathogenicity of the m.3244G>A mutation (together with single fiber PCR); and (ii) to illustrate its clinical heterogeneity.
2. Case Report
This 61-year-old man had droopy eyelids since adolescence, initially without visual difficulties. In school, he participated in athletic activities and had no cognitive difficulties. At the age of 53, after undergoing blepharoplasty, he still had both ptosis and ophthalmoparesis and was referred to our neuromuscular center for further evaluation. The patient reported occasional cramping of the calves and three episodes of unexplained syncope in the past few years. There was no history of neuromuscular disease in the family.
He also carried the diagnoses of chronic lymphatic leukemia (CLL), for which he never had any therapy, hypercholesterolemia, and gastroesophageal reflux. The patient’s mother and two siblings were also diagnosed with CLL but had no treatment. His 63-year-old brother was examined and showed 4/5 weakness of iliopsoas muscles and had difficulty getting up from a squatting position.
The patient’s neurological examination showed bilateral exotropia and inability to adduct or abduct either eye past the midline. His upper gaze was also limited, more severely in the right eye. There was moderate ptosis bilaterally, with the eyelids covering half of the irises. Saccadic movements were slow. Motor examination showed 4/5 weakness of neck flexors and both iliopsoas muscles, but all other muscle groups had normal strength. Mental status, sensory examination, coordination, and tendon reflexes were normal. Gait was normal but the patient had slight difficulty getting up from a squatting position with his arms folded. Tendon reflexes were absent in the arms and diminished in the legs.
Laboratory studies showed elevated arterial lactate (2.5 mM/L, normal 0.5–1.60) and mildly elevated serum CK (368 U/l, normal <294) but normal pyruvate. The rest of the routine blood chemistry was normal.
Histochemical analysis of a left quadriceps muscle biopsy showed COX-negative ragged red fibers (RRF) as well as non-ragged red COX-negative fibres. In cross sections, roughly 5% of all fibers were COX-negative, more than expected in a 61-year-old person. These findings were consistent with a primary mitochondrial disease, although biochemical analysis showed normal activities of respiratory chain complexes (data not shown).
Southern blot analysis of the patient’s skeletal muscle mtDNA excluded the presence of large-scale rearrangements. However, direct sequencing of all 22 tRNAs revealed a novel m.3244G>A transition in the tRNALeu (UUR) gene (Figure 1A), which disrupts a highly conserved G base in the dihydrouridine loop (DHU loop; Figure 1B and C). PCR/RFLP analysis (using two different restriction analysis strategies) showed that the mutation was heteroplasmic in the patient’s muscle (84%, Figure 2) and urinary sediment (31%, not shown), but it was undetectable in blood. The patient’s brother also carried the mutation in blood (12%) but not in the urinary sediment. The mutation was undetectable both in blood and in urinary sediment from the patient’s sister (data not shown).
Figure 1.
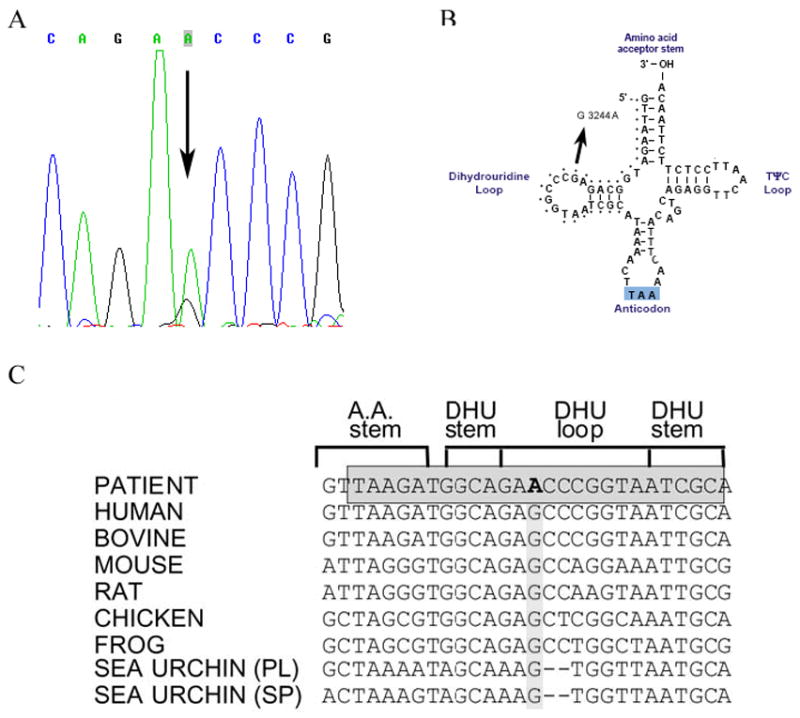
A) Sequence of the mitochondrial tRNALeu (UUR) gene in DNA isolated from the patient’s muscle. The arrow indicates the m.3244G>A transition. B) Schematic representation of the tRNALeu (UUR) cloverleaf structure showing the mutation reported here. C) Evolutionary conservation of the tRNALeu (UUR) DHU loop. The grey box shows the strict degree of conservancy of the sequences in this position in different eukaryotic species. The m.3244G>A transition is shown in bold.
Figure 2.
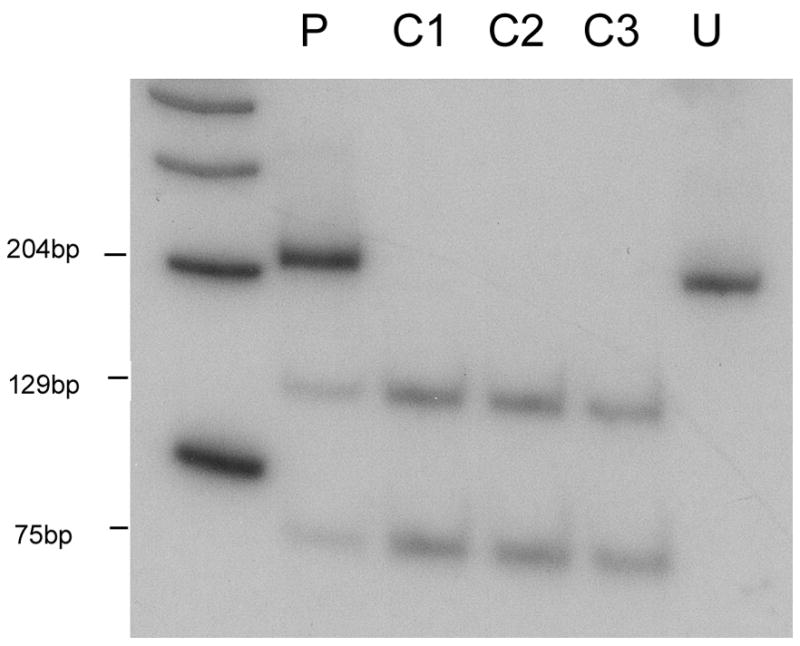
PCR-RFLP analysis. Radiolabeled fragments, after digestion with BanII, were separated on a gel, visualized and quantitated using a BioRad phosphorimager. BanII cuts the wild-type DNA into a 75 bp and a 129bp fragment, whereas mutated DNA is uncut (204 bp). P, patient; C1-C3, controls; U, uncut PCR product.
Single-fibre PCR analysis showed that COX-negative fibres had higher levels of mutant mtDNA (85.4%, SD ± 9%; n=9) than COX-positive fibres (64.09%, SD ± 14.6%; n=9). This difference was statistically significant (p<0.01) (Figure 3).
Figure 3.
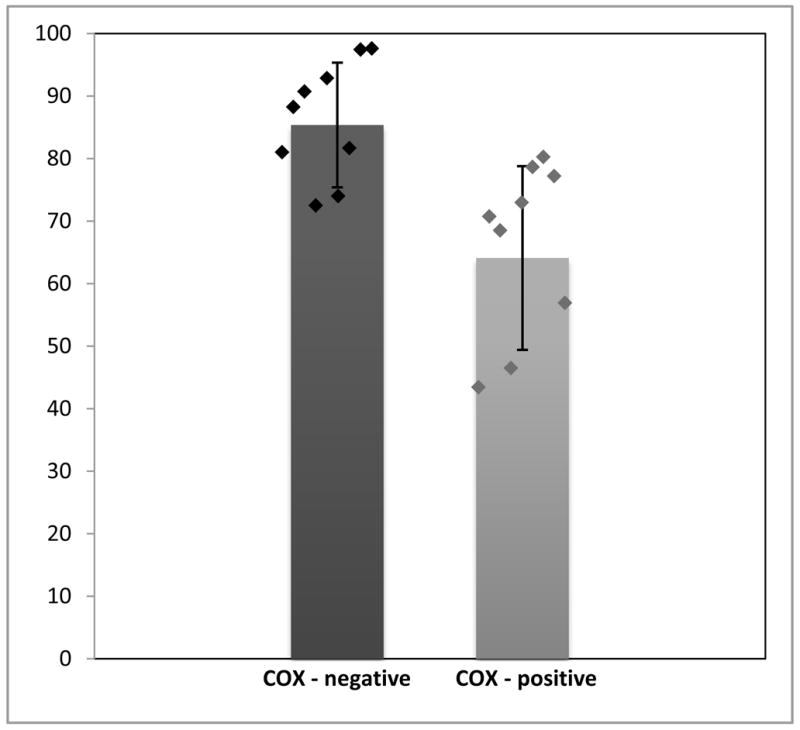
Percentage of mutant mtDNA in COX-positive and COX-negative muscle fibers demonstrated by single-fiber PCR analysis (Means ± SD are shown by bars; individual values are shown by diamonds).
5. Discussion
The mitochondrial tRNALeu (UUR) gene is a recognized hot spot: to date, 20 pathogenic point mutations have been documented, associated with clinical phenotypes as diverse as mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS), cardiomyopathy, CPEO, diabetes mellitus, deafness, and Leber hereditary optic neuropathy (LHON) [4].
Our patient had juvenile onset of ptosis and CPEO, which, together with the histochemical findings in the muscle biopsy, suggested a mtDNA mutation. After excluding large-scale rearrangements by Southern blot analysis, we identified a novel m.3244G>A transition in the DHU loop of the tRNA Leu (UUR) gene. This transition has been attributed only once before to at least one patient with presumed typical MELAS [3].
A number of features suggests that the m.3244G>A mutation is pathogenic and not a neutral polymorphism. Firstly, the mutation was associated with typical histochemical muscle changes. Second, the mutation has not been reported as a neutral polymorphism in the mitochondrial genome databases (http://www.mitomap.org, http://www.genpat.uu.se/mtDB) and we did not encounter it in 100 muscle biopsies from normal subjects or mitochondrial disease controls. Third, in the affected tissue, skeletal muscle, the mutation was heteroplasmic, which is a common feature of deleterious mutations, and its load was high enough to explain the symptoms. Fourth, single-fiber PCR analysis showed a statistically significant correlation between histochemically COX-negative fibers and mutation load. Fifth, this base change disrupts an evolutionarily highly conserved region of the gene. Sixth, the novel m.3244G>A mutation, which is adjacent to the most common MELAS mutation in the DHU loop, cause hypomodification of the tRNA, which presumably results in aminoacylation deficiency, and either mistranslation of UUR codons or translational frame shift [3]. Five mutations in the tRNALeu(UUR) gene, including m.3244G>A, disrupt the normal addition of 5-taurinomethyluridine to the anticodon wobble position, albeit perhaps to different degrees [3, 5]. The degree of translational defect imposed by each mutation may be a key factor that determines the severity of the clinical phenotype. Although the m.3244G>A mutation was associated with the MELAS phenotype [3], the milder phenotype in our patient is not contradictory because the more common m.3243A>G mutation, which disrupts the 5-taurinomethyluridine modification, can also cause MELAS or CPEO.
Mitochondrial CPEO is most commonly associated with large-scale single or multiple deletions of mtDNA, but several tRNA point mutations, both de novo and maternally inherited, have also been described. The apparently sporadic occurrence of PEO in our patient and the lack of symptoms in his mother suggested at first that this was a de novo mutation. However, the extremely mild weakness in his brother, who had detectable levels of the mutation in blood, suggests that the mutation was present in the mother (and the sister) at subthreshold levels. Alternatively, the mutation arose de novo in the mother’s oocytes and did not reach detectable levels in the sister’s accessible tissues.
Mutations in the tRNALeu (UUR) gene display remarkable clinical variability, ranging from pure myopathy with or without PEO to multisystem disorders such as MELAS [6]. Six of them lie in the DHU loop of the gene and have been associated with MELAS, mitochondrial myopathy, or myoclonic epilepsy and ragged red fibers (MERRF) [7, 8], but not with CPEO.
Considering the extensive role of mitochondria in ATP production, free radical generation, and regulation of apoptosis, mutations in mtDNA are likely to affect cellular energy, increase oxidative stress, and affect the apoptotic pathway. A plethora of mtDNA somatic mutations have been identified in cancer cells, but it is not clear how mutations arise and whether the mutation rate is accelerated or slowed at different stages of malignant progression [9, 10]. The relationship, if there is any, between the m.3244A>G mutation and the CLL in this family remains unclear.
Our study confirms the pathogenicity of the m.3244G>A mutation, provides the first clinical description of a patient, and suggests that not all mutations lacking the normal taurine-containing modification at the anticodon wobble position express the MELAS phenotype.
We also underscore the importance of sequencing all 22 mtDNA tRNA genes in sporadic patients with CPEO and RRF but without large-scale mtDNA rearrangements.
Acknowledgments
This work has been supported by NIH Grant HD32062 and the Marriott Mitochondrial Disorders Clinical Research Fund (MMDCRF).
Footnotes
Conflict of interest: None of the authors has any conflict of interest or financial disclosure to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.DiMauro S, Davidzon G. Mitochondrial DNA and disease. Ann Med. 2005;37:222–32. doi: 10.1080/07853890510007368. [DOI] [PubMed] [Google Scholar]
- 2.Moraes CT, Ciacci F, Silvestri G, et al. Atypical clinical presentations associated with the MELAS mutation at position 3243 of human mitochondrial DNA. Neuromusc Disord. 1993;3:43–50. doi: 10.1016/0960-8966(93)90040-q. [DOI] [PubMed] [Google Scholar]
- 3.Kirino Y, Goto YI, Campos Y, Arenas J, Suzuki T. Specific correlation between the wobble modification deficiency in mutant tRNAs and the clinical features of a human mitochondrial disease. Proc Nat, Acad Sci USA. 2005;102:7127–32. doi: 10.1073/pnas.0500563102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hutchinson WM, Thyagarajan D, Poulton J, et al. Clinical and molecular features of encephalomyopathy due to the A3302G mutation in the mitochondrial tRNALeu(UUR) gene. Arch Neurol. 2005;62:1920–3. doi: 10.1001/archneur.62.12.1920. [DOI] [PubMed] [Google Scholar]
- 5.Pulkes T, Hanna MG. Human mitochondrial DNA disease. Adv Drug Deliv Rev. 2001;49:27–43. doi: 10.1016/s0169-409x(01)00124-7. [DOI] [PubMed] [Google Scholar]
- 6.Schon EA, Bonilla E, DiMauro S. Mitochondrial DNA mutations and pathogenesis. J Bioenerg Biomembr. 1997;29:131–49. doi: 10.1023/a:1022685929755. [DOI] [PubMed] [Google Scholar]
- 7.Nishigaki Y, Tadesse S, Bonilla E, et al. A novel mitochondrial tRNALeu(UUR) mutation in a patient with features of MERRF and Kearns-Sayre syndrome. Neuromusc. Disord. 2003;13:334–40. doi: 10.1016/s0960-8966(02)00283-3. [DOI] [PubMed] [Google Scholar]
- 8.Yasukawa T, Suzuki T, Ishii N, Ueda T, Ohta S, Watanabe K. Defect in modification at the anticodon wobble nucleotide of mitochondrial tRNALys with the MERRF encephalomyopathy pathogenic mutation. FEBS Lett. 2000;467:175–8. doi: 10.1016/s0014-5793(00)01145-5. [DOI] [PubMed] [Google Scholar]
- 9.Yao YG, Ogasawara Y, Kajigaya S, et al. Mitochondrial DNA sequence variation in single cells from leukemia patients. Blood. 2007;109:756–62. doi: 10.1182/blood-2006-01-011007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carew JS, Zhou Y, Albitar M, Carew JD, Keating MJ, Huang P. Mitochondrial DNA mutations in primary leukemia cells after chemotherapy: clinical significance and therapeutic implications. Leukemia. 2003;17:1437–47. doi: 10.1038/sj.leu.2403043. [DOI] [PubMed] [Google Scholar]