

Abstract
Intracellular polyamine synthesis is regulated by the enzyme ornithine decarboxylase (ODC), and its inhibition by α-difluromethylornithine (DFMO), confers resistance to apoptosis. We have previously shown that DFMO leads to the inhibition of de novo polyamine synthesis, which in turn rapidly activates Src, STAT3 and NF-κB via integrin-β3 in intestinal epithelial cells. One mechanism to explain these effects involves the activation of upstream growth factor receptors, such as the epidermal growth factor receptor (EGFR). We therefore hypothesized that EGFR phosphorylation regulates the early response to polyamine-depletion. DFMO increased EGFR phosphorylation on tyrosine residues 1173 (pY1173) and 845 (pY845) within 5min. Phosphorylation declined after 10min and was prevented by the addition of exogenous putrescine to DFMO-containing medium. Phosphorylation of EGFR was concomitant with the activation of ERK1/2. Pretreatment with either DFMO or EGF for 1h protected cells from TNF-α/CHX-induced apoptosis. Exogenous addition of polyamines prevented the protective effect of DFMO. In addition, inhibition of integrin β3 activity (with RGDS), Src activity (with PP2), or EGFR kinase activity (with AG1478), increased basal apoptosis and prevented protection conferred by either DFMO or EGF. Polyamine-depletion failed to protect B82L fibroblasts lacking the EGFR (PRN) and PRN cells expressing either a kinase dead EGFR (K721A) or an EGFR (Y845F) mutant lacking the Src phosphorylation site. Conversely, expression of WT-EGFR (WT) restored the protective effect of polyamine depletion. Fibronectin activated the EGFR, Src, ERKs and protected cells from apoptosis. Taken together, our data indicate an essential role of EGFR kinase activity in MEK/ERK-mediated protection, which synergizes with integrin beta-3 leading to Src-mediated protective responses in polyamine-depleted cells.
Keywords: Integrin, Src, putrescine, EGF, DFMO, ERK, RGDS, IEC-6
1. Introduction
The mucosa of the intestinal tract is one of the fastest growing and rapidly turning over tissues in the body [1, 2]. Proliferation occurs in undifferentiated stem cells located in the crypts of the small intestine. Proliferation is balanced by cell loss through exfoliation at the surface leading to a steady state cell population. The cells move from the crypt to the apex of the villus where they exfoliate within 2-3 days [3, 4]. Exfoliation of cells involves apoptosis. Apoptosis may also be responsible for the elimination of extra stem cells and excess cells from the villus tip. Thus, spontaneous apoptosis plays an important role in regulating the number of stem cells in the epithelium of the small intestine and the number of cells exiting the crypt and migrating onto villi [5-7]. Many damaging agents including ionizing radiation, chemicals, chemotherapeutic agents, and food products induce apoptosis of intestinal epithelia [8, 9]. Furthermore, activation of death receptor-mediated pathways also results in a physiologic apoptotic response. Radiation and chemotherapy target cancer cells as well as normal proliferating cells. Bone marrow and intestinal epithelia are the foremost targets of these therapies. The damage to mucosal cells results in diarrhea, dehydration, and secondary infections. These side effects often impose limits to the effective therapy and compromise the quality of life for the patient. Therefore, efforts to decrease the severity of side effects on the mucosa of the intestinal tract may provide promising and effective therapeutic strategies.
The polyamines putrescine, spermidine, and spermine are abundant in eukaryotic cells [10, 11]. They are largely bound to negatively charged molecules such as DNA, RNA, and proteins [12]. Polyamines play crucial roles in cell proliferation [11,13], migration [14,15], transformation [16], and apoptosis [11,17]. ODC (ornithine decarboxylase) is a key regulatory enzyme of polyamine biogenesis. Augmentation of ODC activity is associated with oncogenic Ras-mediated neoplastic transformation [18], while v-Src- [19], activated RhoA- [20], overexpression of eukaryotic initiation factor 4E-mediated transformation [21] and the inhibition of ODC activity reverse the transformed phenotype. Overexpression of ODC-antizyme induced the degradation of ODC and prevented apoptosis in fibroblasts [22]. Thus, ODC activity as well as polyamine levels are tightly regulated. Studies in various cell systems have shown a rapid and significant elevation of ODC activity during apoptosis. And we have shown that inhibition of polyamine synthesis prevents apoptosis [23].
The current concept of polyamine depletion involves long-term exposure to α-difluromethylornithine (DFMO). Cells are grown in the presence of DFMO for 4 days during which intracellular putrescine disappears within 24 h, and spermidine within 48h, and the spermine content decreases to 40% by 96 h [23]. In different cell systems, duration of treatment may vary but the levels of intracellular polyamines are depleted to a similar extent. In almost all cell systems studied, inhibition of ODC using the highly specific inhibitor DFMO and the subsequent depletion of polyamines inhibits apoptosis. Although, polyamine depletion has been shown to activate antiapoptotic pathways, the molecular switch regulated by polyamines is yet to be identified. We have shown that polyamines modulate src-mediated survival signaling via integrin β3 (24). Interestingly, Src and ERK1/2 were activated independently of each other within 30 min of DFMO treatment, and addition of putrescine along with DFMO prevented Src and ERK1/2 activation [24].
In the present study, we show that inhibition of polyamine synthesis modulates the membrane proximal epidermal growth factor receptor (EGFR) and integrin signaling leading to the activation of the antiapoptotic signaling cascade. Furthermore, our results showing rapid effects of DFMO on EGFR and integrin β3 signaling prompted us to revisit the concept of polyamine depletion.
2. Materials and Methods
2.1. Reagents
Disposable cell culture ware was purchased from Corning Glass works (Corning, NY). Media and other cell culture reagents were obtained from Invitrogen. Dialyzed fetal bovine serum (dFBS) was purchased from Sigma (St Louis, MO). Recombinant rat TNF-α and EGF was obtained from BD PharMingen International (San Diego, CA). The Enhanced Chemiluminescence (ECL) Western Blot detection system was purchased from Perkin Elmer (Boston, MA). DFMO was a gift from ILEX Oncology™ Inc, (San Antonio, TX). ERK1/2 and Akt (Phospho and total), and cleaved (active) caspase-3 (Asp175) antibodies were from Cell Signaling (Beverly, MA). Total and p-Tyr 418 Src, p-Tyr 785 β3 integrin antibodies were obtained from Biosource (Camarillo, CA). Total-EGFR and phospho-EGFR antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA) and BD Biosciences (San Diego, CA) respectively. PP2 (Src family tyrosine kinase inhibitor dissolved in DMSO) and AG1478 (EGFR kinase inhibitor) were purchased from Calbiochem, EMD Biosciences (La Jolla, CA) and Biomol International (Plymouth Meeting, PA) respectively. RGDS (Arg-Gly-Asp-Ser) peptide fragment was from the American Peptide Company (Sunnyvale, CA, U.S.A.). The Cell Death Detection ELISA Plus kit was purchased from Roche Diagnostics Corp. (Indianapolis, IN). Fibronectin-coated cell culture plates were purchased from BD Biosciences (Bedford, MA). B82L, EGFR knockout (PRN), B82L expressing wild-type-EGFR (WT), B82L expressing EGFRK721A (K721A), and B82L expressing EGFRY845F (Y845F) cells were provided by Dr. Paul Bertics (University of Wisconsin, MADISON, WI) and Dr. Sally Parson (University of Virginia, Charlottesville, VA) respectively. The IEC-6 cell line (ATCC CRL 1592) was obtained from American Type Culture Collection (Rockville, MD) at passage 13. The cell line was derived from normal rat intestine and was developed and characterized by Quaroni et al [25]. IEC-6 cells originate from intestinal crypt cells as judged by morphological and immunologic criteria. They are nontumorigenic and retain the undifferentiated character of epithelial stem cells. Tests for mycoplasma were always negative. All chemicals were of the highest purity commercially available.
2.2. Cell culture
Cell stocks were maintained in T-150 flasks in a humidified, 37 °C incubator in an atmosphere of 10% CO2. The medium consisted of Dulbecco's Modified Eagle Medium (DMEM) with 5% heat inactivated FBS and 10μg insulin and 50μg gentamicin sulfate per ml. The stock flask was passaged weekly, fed 3 times per week, and passages 15-22 were used. For experiments, the stock cells were harvested with 0.05% trypsin and 0.53 mM EDTA and counted using a Beckman Coulter Counter (Model Z1). For all experiments, cells were grown for 4 days in control, 5mM DFMO or DFMO plus 10μM putrescine (PUT)-containing DMEM with 5% dFBS. Cells were fed on day 2 and serum-starved with control, DFMO, or DFMO/PUT containing medium for 24 h (on day 3). On day 4, experimental treatments were carried out in the respective serum-free medium followed by harvesting. This 4-day scheme was used based on our previous findings that maximal polyamine depletion occurs after 4 days of treatment with 5 mM DFMO [14]. Exogenous PUT added along with DFMO served as a control to indicate that all results were due to the depletion of polyamines and not to DFMO itself.
2.3. Apoptosis studies
The DNA fragmentation assay was carried out using a cell death detection ELISA kit as described earlier [26-28].
2.4. Western blot analysis
The protocol for western blots has been described earlier (26-28). The cell lysates were centrifuged at 14,000 × g for 10 min at 4° C followed by SDS-PAGE. Proteins were transferred overnight to Immobilon-P membranes (Millipore Bedford, MA, USA) and probed with the indicated antibodies at 1:1000 dilution overnight at 4° C in TBS buffer containing 0.1% Tween-20 and 5% non-fat dry milk (blotting grade, Biorad). Membranes were subsequently incubated with horseradish peroxidase-conjugated secondary antibodies at room temperature for 1 h and the immunocomplexes were visualized by an ECL detection system (Perkin Elmer). The membranes were stripped and probed for the actin to confirm equal loading and normalization.
2.5. Statistical analysis
All data are expressed as means ± SE. Experiments were repeated three times, with triplicate samples for each. Analysis of variance and appropriate post-hoc testing determined the significance of the differences between means. Values of p<0.05 were regarded as significant.
3. Results
3.1. Effect of short-term DFMO treatment on apoptosis
We have shown that polyamine depletion protects cells against apoptosis and that DFMO increases ERK and integrin β3-mediated Src activation within 30 min [24]. In order to determine whether rapid activation of these signaling pathways in response to DFMO protects cells against apoptosis confluent IEC-6 cells were pretreated with DFMO (5 mM) or DFMO (5mM) + putrescine (10μM) for 1h in serum free medium and exposed to TNF-α/CHX for 3h and DNA fragmentation was measured. DNA fragmentation was significantly decreased in cells pretreated with DFMO compared to untreated cells (Fig.1). The protective effect of DFMO was evident as early as 1h. However, addition of putrescine along with DFMO prevented the protection conferred by DFMO. These results imply that decreased levels of putrescine are responsible for the protective effects of DFMO and suggest that putrescine may regulate initial signaling events during apoptosis. Since, DFMO increased ERK1/2 and Src activity within 30 min [24], we predicted that the decreased putrescine might influence EGFR activation, an early membrane proximal event. EGFR immunoprecipitated from cells treated with DFMO (5 mM) or DFMO (5mM) + putrescine (10μM) or DFMO + spermine (5μM) were probed with phospho-specific EGFR antibodies. DFMO treatment increased overall tyrosine phosphorylation (data not shown) and site-specific phosphorylation of Tyr-1173 and Tyr-845 within 2 min and which remained elevated for 10 min. Exogenous addition of either putrescine or spermine along with DFMO prevented the phosphorylation of EGFR (Fig. 1B). Since p-Tyr1173 and p-Tyr845 represent EGFR kinase and Src-mediated phosphorylation domains respectively, we determined whether inhibition of EGFR kinase activity by AG1478 prevented EGFR activation induced by the inhibition of polyamine synthesis. DFMO-induced phosphorylation of EGFR (pY1173)) was completely inhibited by pretreatment of cells with AG1478 (30 min), while pY845 phosphorylation was inhibited to a lesser extent (Fig. 1B).
Fig.1. Effect of DFMO on apoptosis and EGFR activation.
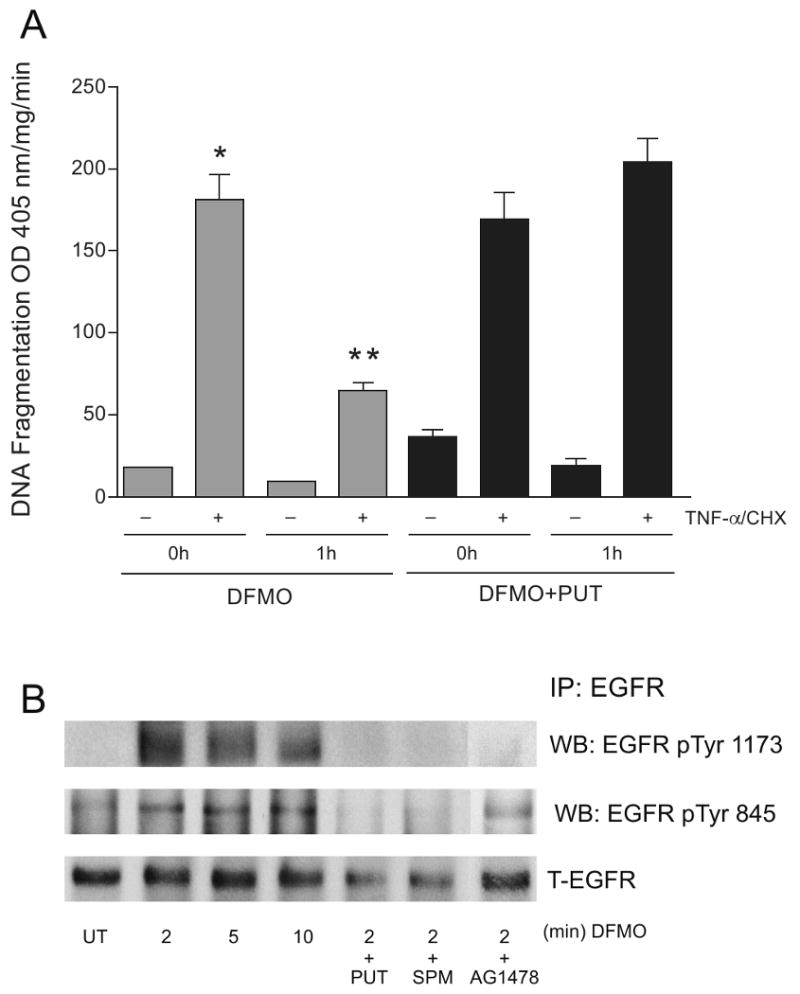
(A) Confluent serum starved cells were treated with 5 mM DFMO or DFMO (5mM) +putrescine (10 μM) for 1h followed by TNF-α/CHX for 3h. Cells were washed and DNA fragmentation was measured as described in the methods section. (mean ± SE, n=3, p<0.05considered significant). *, significantly different from TNF-α/CHX untreated, **, significantly different from TNF-α/CHX treated 0h. (B) Confluent serum starved cells were treated with 5 mM DFMO or DFMO (5mM) +putrescine (10μM), DFMO (5mM) + spermine (10μM), or DFMO+AG1478 for indicated time period and were washed and lysed using lysis buffer containing protease and phosphatase inhibitors. Whole cell lysates were subjected to immunoprecipitation using an EGFR specific antibody. The immunoprecipitates were washed 3 times with lysis buffer, subjected to SDS-PAGE and the membranes were probed with EGFR pY1173 and pY845 specific antibodies. The membranes were stripped and probed with EGFR specific antibody. Representative blots from 3 observations are shown.
3.2. Role of EGFR and Src kinases in apoptotic signaling
TNF-α/CHX-induced apoptosis as judged by DNA fragmentation was significantly decreased in cells treated with DFMO (D+TNF-α/CHX) compared to untreated cells (UT+TNF-α/CHX). Inhibition of EGFR kinase activity completely eliminated the protective effects of DFMO and further augmented both basal and TNF-α/CHX-induced apoptosis (Figure 2A). Inhibition of Src kinase (D+PP2+TNF-α/CHX) also eliminated the protective effects of DFMO (D+TNF-α/CHX) and restored DNA fragmentation to control levels (UT+TNF-α/CHX). Inhibition of EGFR and Src kinase by AG1478 and PP2 respectively increased basal apoptosis (minus TNF-α/CHX) to levels similar to those of control cells (UT+TNF-α/CHX). Putrescine prevented the effects of DFMO and restored TNF-α/CHX-induced apoptosis to control levels (UT+ TNF-α/CHX). Furthermore, modulation of polyamine levels also influenced the downstream signaling pathways (Figure 2B). DFMO increased ERK1/2 and AKT activation within 2 min, and activities remained elevated for 10 min. Addition of putrescine or spermine along with DFMO prevented maximal ERK1/2 and AKT activation observed at 5 min. Preincubation of cells with AG1478 for 30 min (D+AG) completely inhibited ERK1/2 and AKT activation induced in response to DFMO. Pretreatment of cells with PP2, an inhibitor of Src kinase or RGDS, an antagonist integrin β3 peptide, followed by DFMO for 5 min (DFMO+PP2 and DFMO +RGDS) had no effect on ERK1/2 activation stimulated by DFMO. However, PP2 completely abolished the AKT activation observed in the DFMO group. RGDS decreased AKT activation to a lesser extent compared to AG1478 and PP2. Putrescine and spermine decreased AKT activation induced in response to DFMO to an extent similar to that observed with RGDS. Inhibition of EGFR and Src kinases decreased ERK2 protein to a greater extent than ERK1. Total AKT protein levels were relatively unaltered. Furthermore, Src activation remained higher compared to untreated cells throughout the time of exposure to DFMO. AG1478, PP2, putrescine, and spermine prevented the increases in Src activation observed in response to DFMO. DFMO increased phosphorylation of integrin β3 (pY785) which began within 5 min and remained elevated thereafter. Both AG1478 and RGDS blocked the phosphorylation of integrin β3 (pY785), however, the effect of AG1478 was more prominent. These results indicate that polyamines regulate integrin β3-mediated Src activation via the EGFR.
Fig.2. DFMO inhibits apoptosis by activating ERK1/2, integrin β3, Src and AKT.
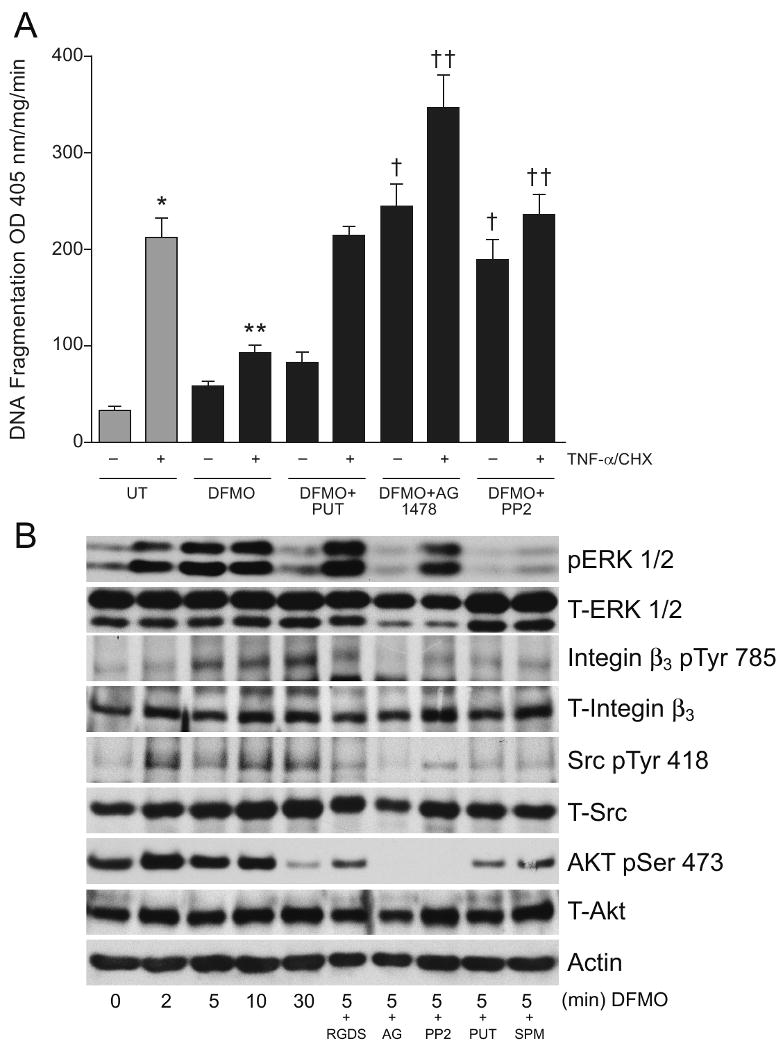
(A) Confluent serum starved cells were left untreated or pretreated with AG1478 (10 μM) or PP2 (10 μM) for 30 mins followed by DFMO (5 mM) or DFMO +putrescine (10μM) for 1h. These groups were then exposed to TNF-α/CHX for 3h. Cells were washed, and DNA fragmentation was measured as described in the methods section. (mean ± SE, n=3, p<0.05considered significant). *, significantly different from minus TNF-α/CHX UT, **, significantly different from TNF-α/CHX treated UT, †, significantly different from TNF-α/CHX treated DFMO group, ††, significantly different from TNF-α/CHX treated DFMO group or TNF-α/CHX treated UT group.
(B) Confluent serum starved cells were treated with 5 mM DFMO for the indicated time period. A second group of cells pretreated with RGDS or AG1478 or PP2 were treated with DFMO for 5 min. A third group of cells were treated with DFMO (5mM) +putrescine (10μM) or DFMO (5mM) + spermine (10μM) for 5 min. Cells were washed and lysed using lysis buffer containing protease and phosphatase inhibitors. Whole cell lysates were subjected to SDS-PAGE and western blot analysis using phospho-specific ERK1/2, Src, AKT, and integrin β3 antibodies. The membranes were stripped and probed with respective antibodies recognizing total protein. The membranes were also stripped and probed with β-actin antibody. Representative blots from 3 observations are shown.
3.3. Role of EGFR and Src kinases in EGF-mediated protection against apoptosis
ERK1/2 and Src activation play an important role in antiapoptotic signaling in polyamine depleted cells, and these pathways are activated by both short-term-DFMO and EGF [24, 28]. Therefore, we determined whether EGF protects cells against apoptosis and studied the downstream signaling pathways. Results depicted in figure 3A show that EGF significantly decreased TNF-α/CHX-induced DNA fragmentation compared to untreated cells. Cells pretreated for 30 min with AG1478 or PP2 and exposed to EGF for 30 min followed by the addition of TNF-α/CHX (EGF+AG or EGF+PP2) had significantly higher levels of DNA fragmentation compared to cells exposed to EGF (EGF + TNF-α/CHX). Pretreatment of cells with AG1478 or PP2 significantly increased DNA fragmentation in the absence of the apoptotic stimulus (EGF+PP2 or EGF+AG minus TNF-α/CHX). Since EGF protected cells against TNF-α/CHX-induced apoptosis, we investigated the signaling cascade initiated by EGF (Fig. 3B). EGF treatment resulted in extensive and time dependent phosphorylation of EGFR in its autophosphorylation (pY1173) and Src phosphorylation (pY845) domains with a concomitant decrease in the level of the receptor. EGFR activation increased ERK, Src, and AKT activities. Pretreatment of cells with AG1478 completely prevented EGFR activation as judged by the absence of pY1173- and pY845-EGFR phosphorylation. Inhibition of EGFR kinase by AG1478 prevented EGFR internalization as evidenced by the higher levels of EGFR compared to untreated cells. Furthermore, AG1478 completely inhibited ERK1/2 and AKT activation and decreased the activation of Src. Inhibition of Src by PP2 decreased EGF-induced phosphorylation of EGFR at pY1173 and pY845. However, the levels of EGFR protein increased with EGF exposure in the presence of PP2. Unlike AG1478, PP2 had no effect on EGF-induced ERK1/2 activation. Interestingly, in PP2 pretreated cells, EGF-mediated AKT activation (AKT-pSer473) increased at 5 min followed by nearly complete loss of AKT-pSer473. These results suggest that EGFR activation is essential for the initiation of antiapoptotic signaling in IEC-6 cells.
Fig.3. EGF protects cells against apoptosis via EGFR-mediated signaling.
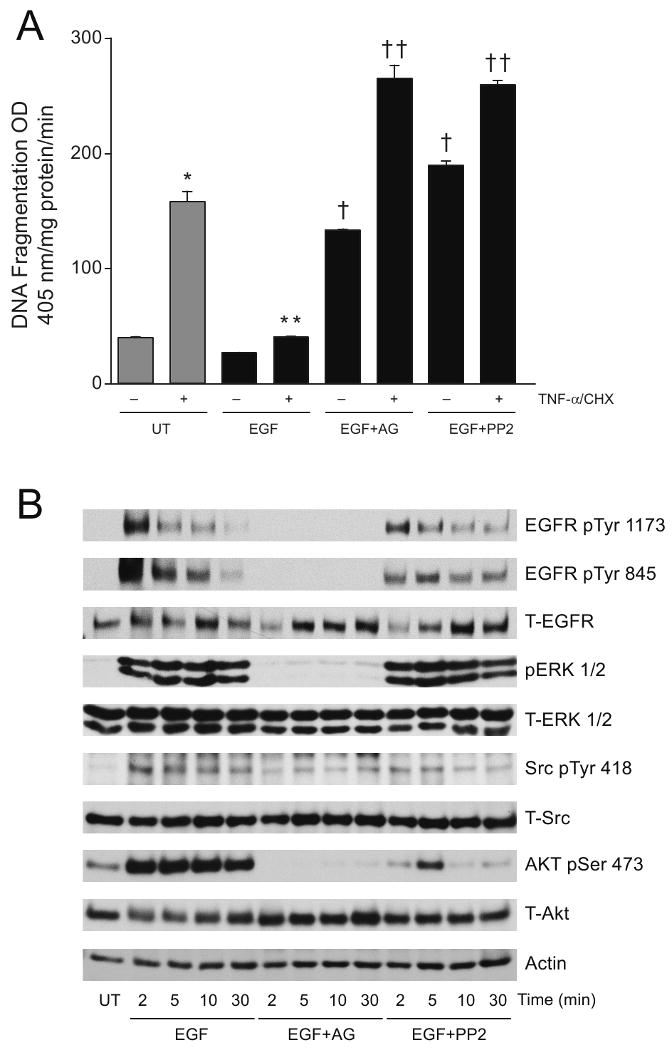
(A) Confluent serum starved cells left untreated (UT) or pretreated with AG1478 (10 μM) or PP2 (10 μM) for 30 mins were exposed to EGF (10 ng/ml) for additional 30 mins and incubated with TNF-α/CHX for 3h. Cells were washed and DNA fragmentation was measured as described in the methods section. (mean ± SE, n=3, p<0.05considered significant). *, significantly different from minus TNF-α/CHX UT, **, significantly different from TNF-α/CHX treated UT, †, significantly different from TNF-α/CHX treated EGF group, ††, significantly different from TNF-α/CHX treated EGF group and TNF-α/CHX treated UT group.
(B) Confluent serum starved cells left untreated (UT) or pretreated with AG1478 (10 μM) or PP2 (10 μM) were exposed to EGF (10 ng/ml) for the indicated time period. Cells were washed and lysed using lysis buffer containing protease and phosphatase inhibitors. Whole cell lysates were subjected to SDS-PAGE and western blot analysis using phospho-specific ERK1/2, Src, and AKT antibodies. The membranes were stripped and probed with respective antibodies recognizing total protein. The membranes were also stripped and probed with an actin antibody. Representative blots from 3 observations are shown.
3.4. EGFR-mediated signaling during apoptosis
From the effects of DFMO and EGF on apoptotic signaling, it is evident that the signaling is mediated through EGFR. Therefore, we used EGFR knockout (PRN), PRN transfected with wild-type EGFR (WT), PRN transfected with a kinase dead mutant EGFR (K721A), and PRN transfected with mutant EGFR deficient in Src mediated phosphorylation (Y845F) cell lines to establish the role of EGFR in apoptotic signaling. Although TNF-α/CHX-induced DNA fragmentation in PRN-cells (183.7±24.9) appeared higher than in wild-type-EGFR cells (130.5±13.5), the difference was not significant. EGF significantly decreased TNF-α/CHX-induced DNA fragmentation in WT cells (72.7±4.6) compared to untreated cells (130.5±13.5). Unlike WT-cells, EGF had no effect on TNF-α/CHX-induced apoptosis in PRN cells suggesting the suitability of these cells for further study. Results in figure 4A (inset panel) show that PRN cells did not express EGFR, and that WT and K721A expressed robust amounts of EGFR. EGFR expression was relatively low in Y845F cells compared to WT and K712A cells. These results confirm the status of EGFR in these cell systems. Results in figure 4A show that DNA fragmentation was slightly higher in Y845F cells compared to WT and it increased significantly in K721A cells. DFMO treatment completely prevented TNF-α/CHX-induced apoptosis in WT cells. Y845F and K721A cells grown in the presence of DFMO showed significantly higher TNF-α/CHX-induced DNA fragmentation compared to their respective control groups. We also analyzed cell extracts from this experiment for active caspase 3 (Fig. 4B). TNF-α/CHX induced caspase 3 activation in all cell types compared to undetectable levels in untreated cells. Active caspase 3 was undetectable in WT cells grown in the presence of DFMO. However, DFMO had no effect on the levels of caspase 3 induced by TNF-α/CHX in the other three cell types.
Fig.4. EGFR expression and apoptosis.
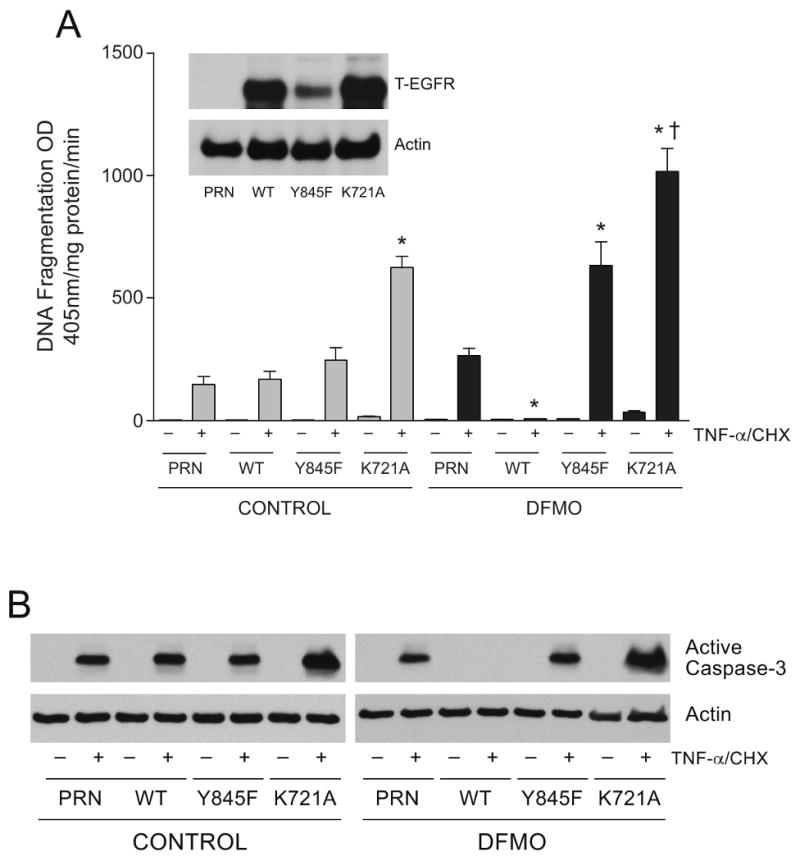
(A) Cell lysates from NIH 3T3 EGFR knockout cells (PRN), PRN-cells expressing wild-type EGFR (WT), PRN-cells expressing K721A mutant EGFR (K721A), PRN-cells expressing Y845F mutant EGFR (Y845F) were subjected to SDS-PAGE and western blot analysis. The membrane was probed with EGFR antibody. The membrane was stripped and probed with β actin antibody. Representative blots from 3 observations are shown (Inset panel). NIH 3T3 EGFR knockout cells (PRN), PRN-cells expressing wild-type EGFR (WT), PRN-cells expressing K721A mutant EGFR (K721A), PRN-cells expressing Y845F mutant EGFR (Y845F) were grown in control and DFMO containing medium for 3 days and incubated for 24 h in the respective serum free medium. Cells were then exposed to TNF-α/CHX for 3h, washed, and DNA fragmentation was measured as described in the methods section. (mean ± SE, n=3, p<0.05considered significant). *, significantly different from TNF-α/CHX treated WT and PRN, †, significantly different from TNF-α/CHX treated control K721A.
(B) The cell extracts from the above experiment were subjected to SDS-PAGE and western blot analysis. The membranes were probed with an antibody recognizing active caspase 3. The membranes were stripped and probed with β actin antibody. Representative blots from 3 observations are shown.
WT and PRN cells were treated with EGF or DFMO for 3 min, and cell extracts were subjected to western blot analysis to determine the phosphorylation of signaling proteins that were activated in response to EGF and DFMO in IEC-6 cells. WT cells treated with either DFMO or EGF had high levels of activated EGFR, ERK1/2, Src, and AKT as judged by the levels of their phosphorylated forms (Fig. 5). These data indicate that DFMO and EGF elicit almost identical signaling pathways originating at the EGFR and suggest an important role for EGFR kinase activity and Src-mediated EGFR phosphorylation in the survival of these cells.
Fig.5. EGFR-mediated signaling in response to EGF and DFMO.
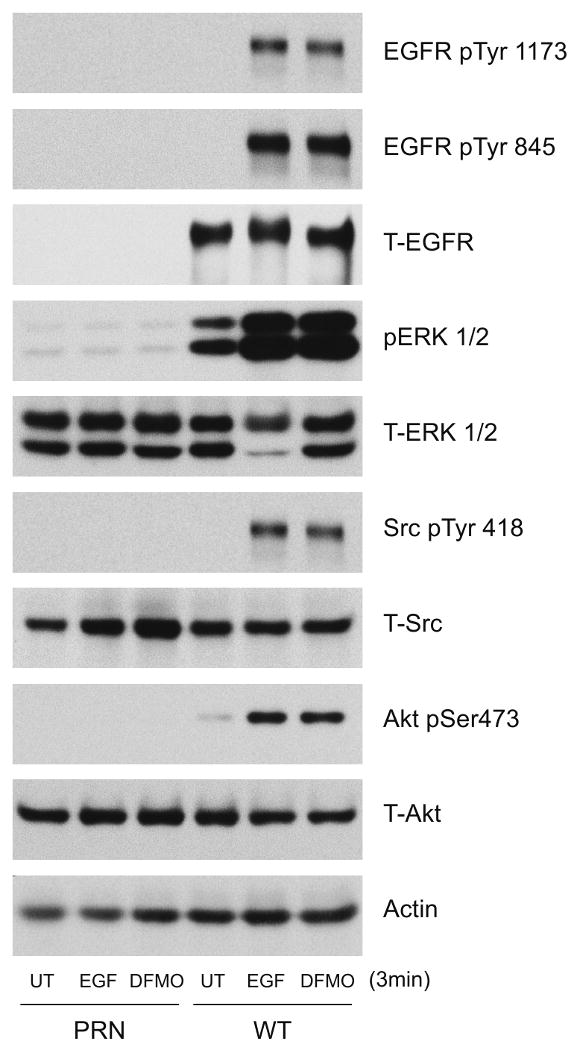
Extracts prepared from confluent serum starved NIH 3T3 EGFR knockout cells (PRN) and PRN-cells expressing the wild-type EGFR (WT) cells left untreated or treated with EGF (10 ng/ml) or DFMO (5mM) for 3 mins were subjected to SDS-PAGE and western blot analysis. The membranes were probed with phospho specific EGFR, ERK1/2, Src, and AKT antibodies. The membranes were stripped and probed with antibodies recognizing respective total proteins and β actin. Representative blots from 3 observations are shown.
3.5. Role of the EGFR in integrin β3-mediated survival signaling
Since both EGF and integrin β3 activate Src, we determined whether the EGFR was involved in integrin-mediated survival signaling. EGFR phosphorylation (pY1173 and pY845) increased in a time dependent manner during attachment of cells on fibronectin (FN)-coated plates (Fig. 6A). Total EGFR decreased during the first two hours and was restored thereafter. Activation of ERK1/2, Src, and integrin β3 increased in a time dependent manner during attachment on FN-coated plates compared to plastic (PL) as judged by the phosphorylation of the respective proteins (Fig. 6A). Addition of putrescine or spermine during attachment prevented all of the responses observed with FN-coated plates. Additionally, IEC-6 cells plated on plastic (PL)- and FN-coated plates in the presence and absence of the EGFR kinase inhibitor AG1478 were allowed to attach and cell extracts were analyzed by western blot. AG1478 completely prevented attachment of cells on PL (data not shown). FN induced EGFR phosphorylation at pY1173 with a concomitant increase in ERK1/2 activation, and these effects were prevented by AG1478 (Fig. 6B). In addition, FN failed to induce ERK1/2 and Src activation in cells lacking the EGFR (PRN). However, restoration of EGFR expression in WT cells allowed integrin β3-mediated EGFR, ERK1/2, and Src activation (Fig. 6C). Finally, we compared the effect of FN and DFMO on TNF-α-induced apoptosis in IEC-6 cells (Fig. 7). As expected DFMO significantly decreased DNA fragmentation in cells grown on PL compared to untreated cells. However, cells grown on FN-coated plates had significantly less DNA fragmentation compared to the untreated group and the DFMO group of cells grown on PL. These results indicate that the functional EGFR plays an important role in integrin β 3-mediated survival signaling.
Fig.6. Fibronectin-induced Integrin β3 activation mediates signaling via the EGFR.
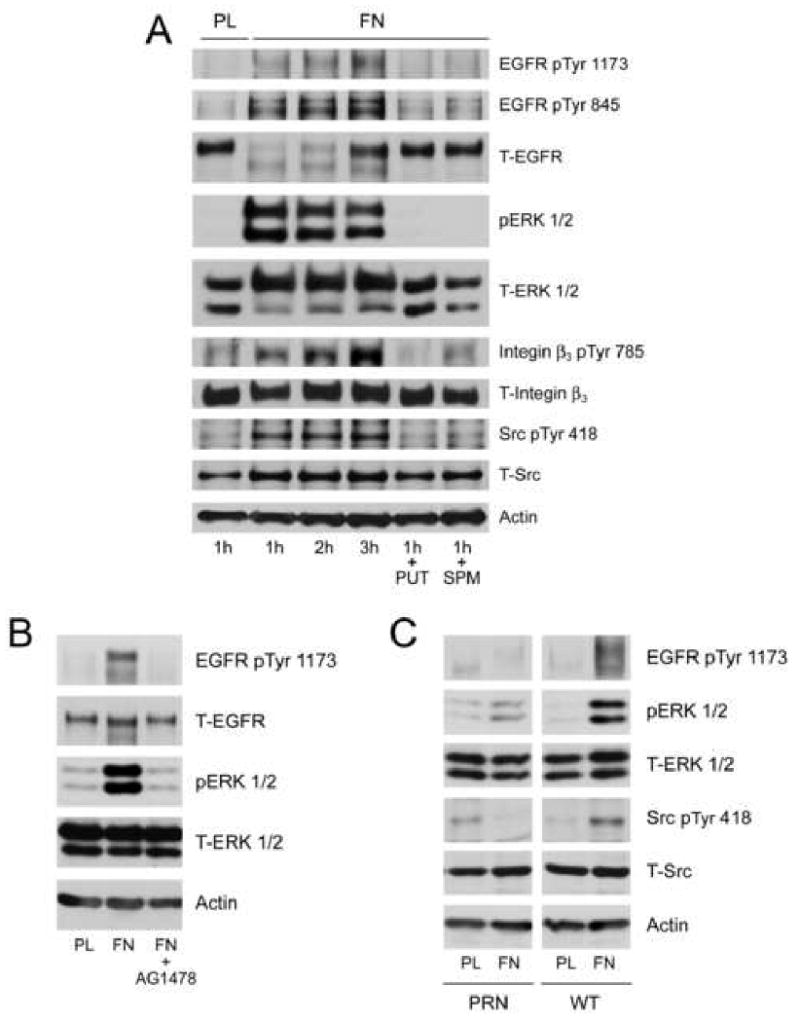
IEC-6 cells seeded on plastic (PL) or fibronectin (FN)-coated plates were allowed to attach for the indicated time periods. Additional groups of cells plated on FN were exposed to putrescine (10μM) or spermine (5 μM) for 1h. Plates were washed with DPBS, and cells were lysed using lysis buffer containing protease and phosphatase inhibitors. The cell extracts were subjected to SDS-PAGE and western blot analysis. The membranes were probed with phospho specific EGFR, ERK1/2, integrin β3, Src, and AKT specific antibodies. The membranes were stripped and probed with antibodies recognizing respective total proteins and β actin (A). Representative blots from 3 observations are shown (A). IEC-6 (B), PRN, and WT (C) cells were allowed to attach on plastic or fibronectin-coated plates for 1h. One group of IEC-6 cells on FN plates was exposed to AG1478 (10μM) during attachment. Plates were washed with DPBS and cells were lysed using lysis buffer containing protease and phosphatase inhibitors. The cell extracts were subjected to SDS-PAGE and western blot analysis. The membranes were probed with phospho specific EGFR, ERK1/2, and Src, antibodies. The membranes were stripped and probed with antibodies recognizing respective total proteins and β actin. Representative blots from 3 observations are shown.
Fig.7. FN-mediated signaling protects cells against apoptosis.

Confluent IEC-6 cells grown on plastic or FN-coated plates were left untreated or were treated with DFMO and exposed to TNF-α/CHX for 3h. Cells were washed, and DNA fragmentation was measured as described in the methods section (A). (mean ± SE, n=3, p<0.05considered significant). **, significantly different from TNF-α/CHX treated plastic group, †, significantly different from TNF-α/CHX untreated plastic.
4. Discussion
Increased EGFR kinase, integrin β3, and Src kinase activities after 5 min and decreased apoptosis after 1h of DFMO exposure (Figs. 1 and 2) allowed us to predict that changes in the levels of polyamines modulated the upstream signaling events during apoptosis. Because DFMO inhibits ODC activity, which catalyzes the conversion of ornithine to putrescine, the earliest effect of DFMO might be a decrease in putrescine levels in close proximity to the plasma membrane. Increased phosphorylation of EGFR at tyrosine 1173, which is localized in the intracellular domain (Fig. 1B), in response to DFMO suggests that under basal conditions putrescine binds to the EGFR and modulates its autophosphorylation in response to signaling cues. DFMO failed to protect cells lacking EGFR, while expression of wild-type EGFR restored protective responses suggesting an essential role of the EGFR in preventing apoptosis (Fig. 4). Furthermore, AG1478 prevented EGFR activation in response to DFMO and EGF and increased apoptosis in IEC-6 cells (Figs. 1B and 2A) suggesting that EGFR kinase activity is crucial for downstream signaling. Increased apoptosis in kinase defective EGFR cells (K721A) and loss of protection by DFMO in these cells further confirm the essential role of EGFR during apoptotic signaling (Fig.4). The activation of EGFR is manifested by autophosphorylation, which creates a docking site for downstream signal transducers [29]. These interactions are often mediated by specific sequences at the Src homology 2 (SH2) domains on target proteins [30]. Biscardi et al. [31] and Tice et al., [32] found that c-Src-dependent EGFR phosphorylation leads to hyper-activation of receptor kinase activity and identified Tyr845 and Tyr 1101 as c-Src-dependent sites of phosphorylation. Our data also show that both EGF and DFMO increase Src-pY418 and EGFR-pY845 phosphorylation indicative of the activation of EGFR by Src (Figs. 1B and 3B). The cells expressing mutant EGFR lacking the Src phosphorylation site showed increased apoptosis (Fig. 4). We have previously shown that DFMO activates Src within 30 min via integrin β3 [24]. Results in figure 2B show that DFMO increased integrin β3 and Src phosphorylation and that RGDS prevented it. The inhibition of EGFR and Src kinases prevented integrin β3 and Src activation in response to DFMO, effects similar to those observed with putrescine or spermine (Fig. 2B). Additionally, FN increased integrin β3 phosphorylation accompanied by pY1173 and pY845 phosphorylation of EGFR and all of these effects were prevented by putrescine and spermine (Fig. 6). Together, these results indicate that EGFR activates Src and integrin β3, and Src also regulates activation of integrin β3 in a polyamine dependent manner. Although, from these data it is not clear whether EGFR activates Src directly or through integrin β3, it is evident that putrescine plays an important role in modulating the upstream signaling cascade. Since putrescine prevented integrin β3 and EGFR kinase activation in response to DFMO and FN, we propose that putrescine may bind the EGFR and prevent its interaction with integrin β3 and Src or bind integrin β3 and Src to prevent their interaction with the EGFR (Fig. 8).
Fig.8. Schematic representation of polyamine-mediated signaling.

Under basal conditions putrescine binds to the EGFR, integrin β3 and Src. Decreased putrescine during inhibition of ODC increases EGFR kinase and EGFR-mediated integrin β3 activities leading to activation of two major antiapoptotic signaling pathways MEK/ERK and Src. The stimulation of Src activates PI3K/AKT and JAK/STAT3 pathways. Together these pathways prevent TNF-α/CHX-induced apoptosis in IEC-6 cells.
Many intracellular signaling molecules are activated by integrin engagement, including the Ras/Raf/MEK/ERK pathway, the phosphatidylinositol 3′-kinse (PI3K/AKT) pathway, Src and Abl tyrosine kinases, focal adhesion kinase (FAK), and myosin light chain kinase (MLCK) [33]. The possibility that integrins can coordinate their activities with other receptors is supported by several recent findings showing interdependence and cross talk between various classes of cellular receptors [34, 35]. Recently, Bill et al., 2004 [36] showed that integrin-mediated adhesion of epithelial cells to extracellular matrix (ECM) induces prolonged tyrosine phosphorylation and partial activation of the EGFR in an integrin-dependent and EGFR ligand-independent manner. Our data demonstrate that integrin engagement by FN increases integrin β3 and Src phosphorylation along with EGFR phosphorylation at Tyr 1173 and Tyr 845 leading to downstream activation of ERK1/2 and AKT pathways (Fig. 6A).
We have previously shown that polyamine depletion by DFMO prevents apoptosis by activating MEK1/ERK1/2-dependent Bad phosphorylation and integrin β3-mediated Src/PI3K/AKT and Src/JAK activation [26-28, 37]. These Src-mediated pathways activate transcription factors NF-κB and STAT3 respectively, leading to increased synthesis of antiapoptotic proteins Bcl-2, cIAP2, and Mcl1. Together, these proteins prevent JNK activation, cytochrome C release from mitochondria, inhibit caspase 9 and caspase 3 activation and prevent apoptosis [26-28]. Thus, the absence of polyamines in these cells prevents apoptosis by either inactivating pro-apoptotic proteins and/or activating anti-apoptotic proteins. Since the inhibition of MEK/ERK and Src increases apoptosis irrespective of the intracellular polyamine levels, we predicted that polyamines influence the activities of the upstream regulators of MEK and Src. It is important to note that the addition of putrescine to polyamine depleted cells during TNFα/CHX treatment does not restore apoptosis (data not shown). However, addition of putrescine or spermine along with DFMO during the growth period (4 days) maintains apoptosis at control levels [23, 24, 26-28]. These results imply that polyamines per se do not directly influence the activities of downstream target proteins involved in apoptosis. Furthermore, activation of signaling pathways leading to protection from apoptosis was similar whether cells were treated with DFMO for 4 days or for 2 min. This affirms the importance of initial signaling events in the protection of cells rather than the levels of polyamines. Moreover, the failure of DFMO to activate ERK1/2 and Src/AKT signaling in cells lacking EGFRs also indicates that the modulation of EGFR-mediated signaling by polyamines is crucial in apoptosis. Although, these observations indicate that polyamines play an active role in the regulation of signaling pathways, they raise important questions concerning the nature and significance of polyamine depletion.
Several studies including our own using IEC-6 and other cell types have employed incubation of cells with DFMO for 4-16 days and have shown that polyamine depletion decreases the expression or activities of various proteins and that these effects are prevented by exogenous polyamines (putrescine or spermidine) added along with DFMO [38-40], indicating that the effects are due to polyamine depletion and not to DFMO. Since cells treated with DFMO + putrescine have never undergone polyamine depletion, it is not clear whether exogenous polyamines prevent the effects observed due to polyamine depletion. Together our previous studies, carried out using 4 day and short-term DFMO treatment models strongly suggest that rather than depletion of polyamines, a change in the level of the localized, compartmentalized pool of free polyamines initiates a signaling cascade which is probably sustained due to continued decrease in polyamine pools with increased length of DFMO exposure.
Several growth factors have been shown to protect enterocytes from apoptosis. EGFR expression begins during embryonic development and is retained throughout adulthood in the normal gastrointestinal tract. The EGFR is present in parietal cells, in the proliferative zone of the crypt/villus axis, and along the horizontal axis of the epithelium of small intestine and colon [41, 42]. In polarized epithelial cells including gastrointestinal epithelial cells, EGFR are mainly localized on the basolateral membrane [43-46]. For example, in polarized Caco2 cells, the EGFR concentration is about 15-fold higher in basolateral versus apical membranes [47]. A single amino acid change in a naturally occurring mutation in the EGFR (waved-2 mutation) increased enterocyte apoptosis during partial small bowel resection and decreased proliferation. Furthermore, EGFR null pups demonstrated several epithelial tissue defects such as attenuated DNA synthesis and short intestinal villi [48, 49]. Thus, EGFR signaling plays a critical role in the homeostasis of intestinal epithelium. Since polyamines have been shown to play an important role in the regulation of proliferation, migration and apoptosis in rat models and in cultured intestinal epithelial cells, we propose that polyamines modulate the assembly of the signaling scaffold at the level of the EGFR, integrin β3, and Src leading to the activation of downstream antiapoptotic signaling cascades.
Acknowledgments
This work was supported by National Institute of Diabetes and Digestive and Kidney Disease (NIDDK) grants DK-16505 and DK-52784 and by the Thomas A. Gerwin Endowment. We gratefully acknowledge Mary Jane Viar and Rebecca West for technical assistance. We sincerely acknowledge Gregg Short and Danny Morse for help in preparing the figures.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cheng H, Leblond CP. Am J Anat. 1974;141:537. doi: 10.1002/aja.1001410407. [DOI] [PubMed] [Google Scholar]
- 2.Schmidt GH, Winton DJ, Ponder BAJ. Development. 1988;103:785. doi: 10.1242/dev.103.4.785. [DOI] [PubMed] [Google Scholar]
- 3.Potten CS, Loeffler MA. J Theor Biol. 1987;127:381. doi: 10.1016/s0022-5193(87)80136-4. [DOI] [PubMed] [Google Scholar]
- 4.Wright NA, Irwin M. Cell Tissue Kinet. 1982;15:595. doi: 10.1111/j.1365-2184.1982.tb01066.x. [DOI] [PubMed] [Google Scholar]
- 5.Grossman J, Walther K, Artinger M, Kiessling S, Schlomerich J. Cell Growth Differ. 2001;12:147. [PubMed] [Google Scholar]
- 6.Hall PA, Coates PJ, Ansari B, Hopwood D. j Cell Sci. 1994;107:3569. doi: 10.1242/jcs.107.12.3569. [DOI] [PubMed] [Google Scholar]
- 7.Mayhew TM, Myklebust R, Whybrow A, Jenkin R. Histol Histopathol. 1999;14:257. doi: 10.14670/HH-14.257. [DOI] [PubMed] [Google Scholar]
- 8.Ijiri K, Potten CS. Br J Cancer. 1983;47:175. doi: 10.1038/bjc.1983.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Potten CS. Int J radiat Biol. 1990;58:925. doi: 10.1080/09553009014552281. [DOI] [PubMed] [Google Scholar]
- 10.Pegg AE, McCann PP. Am J Physiol. 1982;243:C212. doi: 10.1152/ajpcell.1982.243.5.C212. [DOI] [PubMed] [Google Scholar]
- 11.Pegg AE. Biochem J. 1986;234:249. doi: 10.1042/bj2340249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Seiler N, Raul F. J Cell Mol Med. 2005;9:623. doi: 10.1111/j.1582-4934.2005.tb00493.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tabor CW, Tabor H. Annu Rev Biochem. 1984;53:749. doi: 10.1146/annurev.bi.53.070184.003533. [DOI] [PubMed] [Google Scholar]
- 14.Mccormack SA, Viar MJ, Johnson LR. Am J Physiol Gastrointest Liver Physiol. 1993;264:G367. doi: 10.1152/ajpgi.1993.264.2.G367. [DOI] [PubMed] [Google Scholar]
- 15.Wang JY, Johnson LR. Gastroenterology. 1991;100:333. doi: 10.1016/0016-5085(91)90200-5. [DOI] [PubMed] [Google Scholar]
- 16.Auvinen M, Paasinen A, Andersson LC, Holtta E. Nature. 1992;360:355. doi: 10.1038/360355a0. [DOI] [PubMed] [Google Scholar]
- 17.Schipper RG, Penning LS, Verhofstad AJ. Semin Cancer Biol. 2000;10:55. doi: 10.1006/scbi.2000.0308. [DOI] [PubMed] [Google Scholar]
- 18.Holtta E, Sistonen L, Alitalo K. J Biol Chem. 1988;263:4500. [PubMed] [Google Scholar]
- 19.Holtta E, Auvinen M, Andersson LC. J Cell Biol. 1993;122:903. doi: 10.1083/jcb.122.4.903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shantz LM, Pegg AE. Cancer Res. 1998;58:2748. [PubMed] [Google Scholar]
- 21.Shantz LM, Coleman CS, Pegg AE. Cancer Res. 1996;56:5136. [PubMed] [Google Scholar]
- 22.Tewari M, Hamid QA, Tuncay OC, Tewari DS. Oral Oncol. 1998;34:538. doi: 10.1016/s1368-8375(98)00044-x. [DOI] [PubMed] [Google Scholar]
- 23.Ray RM, Viar MJ, Yuan Q, Johnson LR. Am J Physiol Cell Physiol. 2000;278:C480. doi: 10.1152/ajpcell.2000.278.3.C480. [DOI] [PubMed] [Google Scholar]
- 24.Bhattacharya S, Ray RM, Johnson LR. Biochem J. 2006;397:437. doi: 10.1042/BJ20060256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Quaroni A, Wands J, Trelstad RL, Isselbacher KJ. J Cell Biol. 1979;80:248. doi: 10.1083/jcb.80.2.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bhattacharya S, Ray RM, Johnson LR. Apoptosis. 2005;10:759. doi: 10.1007/s10495-005-2943-3. [DOI] [PubMed] [Google Scholar]
- 27.Bhattacharya S, Ray RM, Johnson LR. Biochem J. 2005;392:335. doi: 10.1042/BJ20050465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bhattacharya S, Ray RM, Johnson LR. Am J Physiol Gastrointest Liver Physiol. 2004;286:G479. doi: 10.1152/ajpgi.00342.2003. [DOI] [PubMed] [Google Scholar]
- 29.Ullrich A, Schlessinger J. Cell. 1990;61:203. doi: 10.1016/0092-8674(90)90801-k. [DOI] [PubMed] [Google Scholar]
- 30.Pawson T, Schlessinger J. Curr Biol. 1993;3:434. doi: 10.1016/0960-9822(93)90350-w. [DOI] [PubMed] [Google Scholar]
- 31.Biscardi JS, Maa MC, Tice DA, Cox E, Leu TH, Parsons SJ. J Biol Chem. 1999;274:8335. doi: 10.1074/jbc.274.12.8335. [DOI] [PubMed] [Google Scholar]
- 32.Tice DA, Biscardi JS, Nickels AL, Parson SJ. Proc Natl Acad Sci. 1999;96:1415. doi: 10.1073/pnas.96.4.1415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schoenwaelder SM, Burridge K. Curr Opin Cell Biol. 1999;11:274. doi: 10.1016/s0955-0674(99)80037-4. [DOI] [PubMed] [Google Scholar]
- 34.Carpenter G. J Cell Biol. 1999;146:697. doi: 10.1083/jcb.146.4.697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schwartz MA, Baron V. Curr Opin Cell Biol. 1999;11:197. doi: 10.1016/s0955-0674(99)80026-x. [DOI] [PubMed] [Google Scholar]
- 36.Bill HM, Knudsen B, Moores SL, Muthuswamy SK, Rao VR, Brugg JS, Miranti CK. Mol Cell Biol. 2004;24:8586. doi: 10.1128/MCB.24.19.8586-8599.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ray RM, Bhattacharya S, Johnson LR. J Biol Chem. 2005;280:31091. doi: 10.1074/jbc.M503041200. [DOI] [PubMed] [Google Scholar]
- 38.Wang JY, McCormack SA, Viar MJ, Wang H, Tzen CY, Scott RE, Johnson LR. Am J Physiol. 1993;265:G331. doi: 10.1152/ajpgi.1993.265.2.G331. [DOI] [PubMed] [Google Scholar]
- 39.Patel AR, Wang JY. Am J Physiol. 1997;273:C1020. doi: 10.1152/ajpcell.1997.273.3.C1020. [DOI] [PubMed] [Google Scholar]
- 40.Celano P, Baylin SB, Giardiello FM, Nelkin BD, Casero RA. J Biol Chem. 1988;263:5491. [PubMed] [Google Scholar]
- 41.Playford RJ, Hanby AM, Gschmeissner S, Peiffer LP, Wright NA, McGarrity T. Gut. 1996;39:262. doi: 10.1136/gut.39.2.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Visco V, Belleudi F, Marchese C, Leone L, Aimati L, Cardinali G, Kovacs D, Frati L, Torris MR. J Cell Physiol. 2004;200:31. doi: 10.1002/jcp.10385. [DOI] [PubMed] [Google Scholar]
- 43.Mori S, Morishita Y, Sakai K, Kurimoto S, Okamoto M, Kawamoto T, Kuroki T. Acta Pathol Jpn. 1987;37:1909. doi: 10.1111/j.1440-1827.1987.tb03305.x. [DOI] [PubMed] [Google Scholar]
- 44.Thompson JF, Van den Berg M, Stokkers PC. Gastroenterology. 1994;107:1278. doi: 10.1016/0016-5085(94)90528-2. [DOI] [PubMed] [Google Scholar]
- 45.Scheving LA, Shiurba RA, Nguyen TD, Gray GM. J Biol Chem. 1989;264:1735. [PubMed] [Google Scholar]
- 46.He C, Hobert M, Friend L, Carlin C. J Biol Chem. 2002;277:38284. doi: 10.1074/jbc.M104646200. [DOI] [PubMed] [Google Scholar]
- 47.Bishop WP, Wen JT. Am J Physiol. 1994;267:G892. doi: 10.1152/ajpgi.1994.267.5.G892. [DOI] [PubMed] [Google Scholar]
- 48.Luetteke NC, Phillips HK, Qiu TH, Copeland NG, Earp HS, Jenkin NA, Lee DC. Genes Dev. 1994;8:399. doi: 10.1101/gad.8.4.399. [DOI] [PubMed] [Google Scholar]
- 49.Miettinen PJ, Berger JE, Meneses J, Phung Y, Pedersen RA, Web Z, Derynck R. Nature. 1995;376:337. doi: 10.1038/376337a0. [DOI] [PubMed] [Google Scholar]