

Abstract
Background and Purpose
To evaluate the effect of APOE genotype and the feasibility of administering an apoE-mimetic therapeutic to modify outcomes in a murine model of intracerebral hemorrhage (ICH).
Methods
ICH was induced via stereotactic injection of 0.1 U Clostridial collagenase into the left basal ganglia of wild-type (WT) and apolipoprotein-E targeted-replacement (APOETR) mice, consisting of either homozygous 3/3 (APOE3TR) or 4/4 (APOE4TR) genotypes. Animals were randomized to receive either vehicle or apoE-mimetic peptide. Outcomes included functional neurological tests (21-point neuroseverity score and rotorod latency) over the initial 7 d after injury, radiographic and histological hemorrhage size at 3 and 7 d, brain water content for cerebral edema at 24 h, and q-PCR for inflammatory markers at 6, 24, and 48 h.
Results
APOE3TR animals demonstrated superior neuroseverity scores and rotorod latencies over the first 3 d after ICH, decreased cerebral edema at 24 h, and reduced up-regulation of IL-6 and eNOS at 6 h when compared to their APOE4TR counterparts. Following intravenous administration of 1 mg/kg apoE-mimetic peptide, both WT and APOE4TR animals exhibited improved functional outcomes over 7 d after ICH, less edema at 24 h and reduced up-regulation of IL-6 and eNOS when compared to mice that did not receive the peptide.
Conclusions
Our data indicate that APOE genotype influences neurological outcome after ICH in a murine model. In particular APOE4 is associated with poor functional outcome and increased cerebral edema. Additionally, this outcome can be modified by the addition of an apoE mimetic-peptide, COG1410.
Keywords: Intracerebral hemorrhage, Apolipoprotein E, Mouse, Inflammation, Gene therapy
INTRODUCTION
Apolipoprotein E (apoE) is a 34 kD protein known for its role in cholesterol metabolism; however, there is emerging evidence suggesting a singularly important role for apoE in the injured central nervous system (CNS). 1 There are three common human isoforms of apoE, designated apoE2, apoE3, and apoE4 (APOE = gene; apoE = protein) that differ by single amino acid substitutions at residues 112 and 158. Presence of the APOE4 allele is associated with poor prognosis in a variety of acute and chronic neurological diseases, including intracerebral hemorrhage (ICH). 2 Although a full understanding of the mechanism(s) by which apoE affects the CNS response to injury remains elusive, it is clear that presence of the APOE4 allele is an independent risk factor for ICH, 3, 4 and there is increasing evidence supporting its role in down-regulating endogenous inflammatory responses in an isoform-specific fashion, which is consistent with its known immunomodulatory properties. 5 Recent observations demonstrating the effects of apoE in modifying secretion of inflammatory mediators and generation of cerebral edema have been shown following closed head injury 6 and systemic inflammation responses. 7 Furthermore, ICH is associated with glial activation and release of inflammatory mediators, which contribute to breakdown of the blood brain barrier, enhancement of secondary neuronal injury, and development of cerebral edema. 8 Therefore, the isoform-specific effect of apoE on neuroinflammation and acute injury responses may be particularly relevant in modifying outcomes after ICH, and clinical studies have implicated the presence of APOE4 with poor outcome in this setting. 2, 9, 10
Given the importance of apoE in modifying acute brain injury responses after ICH, one possible therapeutic strategy might be to administer an exogenous lipoprotein in an effort to improve neurological outcomes. Unfortunately, the native protein does not cross the blood brain barrier, effectively precluding its use as an intervention. 11 However, recent observations suggest apoE effects inflammation via specific receptor interactions, and that small peptides derived from the receptor-binding region can maintain the bioactivity of the holoprotein. 12 In particular, peptides comprising apoE residues 133–149 compete with the holoprotein for receptor-binding, 13, 14 microglial suppression, 12 and neuroprotective properties of the native 299-amino acid holoprotein. 15 Thus the administration of apoE-mimetic peptides may represent a novel pharmacogenomic therapeutic strategy in ICH.
In the current study, we extend clinical observations implicating an isoform-specific role for apoE in modifying outcome after ICH to a murine model of collagenase-induced basal ganglia hemorrhage in targeted replacement (TR) “knock-in” APOE mice (APOETR) created by replacing only the coding regions of mouse APOE with human APOE allele-specific coding sequences, without disturbing any known regulatory regions. These animals express human apoE protein isoforms at physiological levels in a temporal and spatial pattern similar to that observed in humans, allowing us to then test whether administration of an apoE-mimetic peptide can improve functional and histological outcomes in this clinically relevant, experimental ICH paradigm. Further, to test for potential pharmacogenomic interactions between endogenous APOE background and the apoE-based therapeutic, we assess differential therapeutic responses in WT, APOE3TR, and APOE4TR animals after administration of the mimetic peptide.
MATERIALS AND METHODS
Transgenic animals
All animal procedures were designed to minimize animal discomfort and numbers, conformed to international guidelines on the use of animals, and were approved by the Duke University Institutional Animal Care and Use Committee. APOETR mice were created by gene targeting of a human APOE3 or APOE4 genomic construct into E14TG2a embryonic stem cells derived from 129P2/OlaHsd mice. 16 The targeted embryonic stem cells were injected into C57BL6/J blastocytes, and the resulting chimeras were bred to wild-type (WT) C57BL6/J mice and backcrossed to C57BL6/J mice for eight generations. This resulted in TR mice created by replacing only the coding regions of mouse APOE with human APOE allele-specific coding sequences, without disturbing any known regulatory regions, thus resulting in animals that express human apoE protein at physiological levels in both a temporal and spatial pattern similar to primates and humans. These targeted replacement mice have no trace of the native murine apoE, as demonstrated by Western blotting. Northern blot analysis has demonstrated apoE levels in the skin, spleen, kidney, small intestine, heart, testes, muscle, lung, liver, and brain, which are expressed at normal physiological levels when compared to native apoE in WT animals. Moreover, the levels of human apoE protein in hippocampus and frontal cortex were similar between targeted replacement mice and non-demented human tissue. 17 The colony was maintained by homozygous matings, and genotypes were confirmed prior to each experiment.
Intracerebral hemorrhage model
Our murine injury model was adapted from a previously described model of ICH in rats. 18 Sixteen to 20 week-old male WT, APOE3TR, and APOE4TR mice were used in these experiments to avoid potential interaction of vascular deterioration due to advanced age. Prior to injury, mice were prerandomized to treatment or vehicle groups. The trachea was intubated after anesthesia induction with 4.6% isoflurane and the lungs were mechanically ventilated with 1.6% isoflurane in 30% O2/70% N2. Rectal temperature was maintained at 37 ± 0.5 °C by underbody warming system. The animal’s head was secured in a stereotactic frame, local anesthetic injected, and the scalp incised. After exposure of the skull, a burr hole was created 2 mm left lateral to bregma, and a 0.5 μl syringe needle (Hamilton, Reno, NV, USA) was advanced to a depth of 3 mm from cortex. Type IV-S Clostridial collagenase (Sigma, St. Louis, MO, USA) was injected over 5 min (0.1 U in 0.4 μl NS). The incision was then closed, and animals were allowed to recover spontaneous ventilation with subsequent extubation.
Synthesis and administration of peptide
Peptides were synthesized by NeoMPS (San Diego, CA) to a purity of 95%. COG1410 is acetyl-AS-Aib-LRKLAib-KRLL-amide, which is derived from apoE residues 138–149 with Aib (amino isobutyric acid) substitutions at positions 140 and 145. For all experiments, peptides were dissolved in sterile saline immediately prior to use and injected via the tail vein with 100 μl of 250 μg/ml COG1410 peptides or 100 μl of sterile saline vehicle as control.
Testing of neurological deficits
An automated rotorod (Ugo Basile, Comerio, Italy) was used to assess vestibulomotor function. 19 On the day prior to hemorrhage induction, mice underwent two consecutive conditioning trials at a set rotational speed (16 revolutions/min) for 60 s followed by three additional trials with an accelerating rotational speed. The average time to fall from the rotating cylinder in the latter three trials was recorded as baseline latency. After injury, mice underwent consecutive daily testing with three trials of accelerating rotational speed (inter-trial interval of 15 min). Average latency to fall from the rod was recorded. Mice unable to grasp the rotating rod were given a latency of 0 s.
Neuroseverity scoring was assessed after injury by using a neurobehavioral examination (scoring scale 7 – 21) as previously described. (Table 1) 20 Motor score (4 – 12) was derived from spontaneous activity, symmetry of limb movements, climbing, balance and coordination. Sensory score (3 – 9) was derived from body proprioception, vibrissae, visual, and tactile responses. Sensory tests examined function from both cerebral hemispheres.
Table 1.
Neuroseverity Scoring.
| Motor | Score | Scoring | Protocol | Sensory | Score | Scoring | Protocol |
|---|---|---|---|---|---|---|---|
| Spontaneous Activity | 1 | Does not rise up | Place mouse in empty cage and observe | Body Proprioception | 1 | No reaction | Push mouse from side of neck with blunt cotton swab |
| 2 | Does not approach all cage sides | 2 | Slowed head turning | ||||
| 3 | Normal exploration | 3 | Normal | ||||
| Motor | 1 | Minimal movement of one side | Hold mouse by tail and observe limb extension | Vibrissae | 1 | No reaction | From behind touch whiskers with elongated cotton swab |
| 2 | Abnormal walking. Mild weakness | 2 | Slowed reaction | ||||
| 3 | Normal symmetry | 3 | Normal | ||||
| Climbing | 1 | Hang for seconds & falls | Place mouse on mesh and flip upside down | Tactile Response | 1 | No reaction | Place mouse on mesh & touch palmar area of forepaw with sharp needle |
| 2 | Hangs, but cannot displace mesh | 2 | Slowed reaction | ||||
| 3 | Hangs & displaces mesh | 3 | Normal | ||||
| Balance & Coordination | 1 | Holds but falls | Place mouse sideways on 2 cm diameter pole | ||||
| 2 | Holds, but unable to walk | ||||||
| 3 | Holds & walk | ||||||
Histology
Mice were anesthetized and euthanized on Day 3 or 7 following injury. Brains were removed and frozen at −20 °C. Coronal sections of 20 μm thickness were taken at 320 μm intervals over the rostral-caudal extent of the lesion. Sections were stained with hematoxylin and eosin, and lesion volume measured by digitally sampling stained sections with an image analyzer. Lesion volumes (mm3) were computed as running sums of lesion area multiplied by the known interval (e.g., 320 μm) between sections over the extent of the lesion expressed as an orthogonal projection.
Magnetic Resonance Imaging
Magnetic resonance (MR) imaging was performed using a 7T Bruker MRI (Bruker Biospin). Animals were anesthetized via inhalation of 1.5% isofluorane in room air and cardiopulmonary parameters were monitored continuously. Core temperature was maintained at 37 ± 0.5 °C via a circulating water bath. Each MR imaging session lasted 20 minutes, which was equivalent across animal groups. Images were acquired at a 256 ×256 matrix and a 4-cm2 field of view. A RARE sequence was performed for both T1 (TE/TR = 7.5/1,300) and T2 (TE/TR 12/4200) weighted imaging. Eighteen slices at 1 mm thickness were performed for entire brain coverage.
Assessment of cerebral edema
To coincide with the beginning of the period of maximal inflammatory effect, 21 mice were anesthetized, euthanized at 24 h after injury, and perfused with 30 ml of phosphate-buffered saline (PBS) via transcardiac puncture. Brains were sectioned mid-sagittally with removal of cerebellum and brainstem, and each hemisphere weighed immediately (‘wet’ weight). Hemispheres were allowed to dehydrate over 24 h at 100 °C and then re-weighed (‘dry’ weight). To compare across genotypes, cerebral edema was expressed as water content calculated as a percentage of wet weight (wet weight − dry weight)/(wet weight) × 100. 22
Quantification of mRNA for TNF-α, IL-6, and eNOS
Mice were anesthetized, euthanized, and perfused with 30 ml PBS via transcardiac puncture at baseline, 6, 24, or 48 h after injury. Brains were sectioned mid-sagittally, flash frozen at −20 °C, and stored at −80 °C. RNA was extracted from pulverized frozen hemispheres using the PerfectPure RNA Tissue Kit (5 PRIME, Gaithersburg, MD) according to manufacturer’s instructions. RNA was quantified using a Bio-Rad SmartSpec 3000 Spectrophotometer (Bio-Rad, Hercules, CA) and was reverse transcribed to cDNA using the High Capacity cDNA Archive Kit(Applied Biosystems, Foster City, CA) with MultiScribe reverse transcriptase and random primers. The levels of mRNA expression weredetermined by quantitative real-time PCR (q-PCR) performedon a ABI 7300 Sequence Detection Software system (Applied Biosystems), using 100 ng cDNA per reaction, adding Taqman Universal PCRmaster mix and TaqMan Assays-on-DemandGene Expression primer/probe sets (all from Applied Biosystems) for the murine cytokines. Relative mRNA quantification was calculated using the 2−ΔΔCT method 23 The threshold cycle number (Ct) of the target. gene for each sample is normalized using a housekeeping gene (endogenous 18S), and calibrated to the control samples (uninjured animals). Final results were expressed as fold changes over uninjured animals.
Statistical analysis
Rotorod performance and neuroseverity scoring were compared with repeated measures analysis of variance (ANOVA) with time as the repeated variable. Hemorrhage size, interhemispheric water weight difference, and q-PCR fold changes were compared using student’s t-test. Statistical significance was assumed with p < 0.05, and values expressed as mean ± S.E.M.
RESULTS
Effects of Endogenous APOE polymorphism
To evaluate the endogenous effects of APOE polymorphism after ICH, collagenase-induced hemorrhage was performed in the left basal ganglia of APOETR mice. Hematoxylin/eosin staining revealed the characteristic development of a well-circumscribed hematoma and the development of cerebral edema. (Figure 1) Seventy-two hours after injury, hematoma volumes were comparable when measured in APOE4TR and APOE3TR animals (n=8/group, 23.6 ± 1.41 in APOE4TR v. 24.4 ± 2.03 mm3 in APOE3TR). Additionally, MRI was performed on a separate cohort of animals (n=5/group) given 0.075 U of collagenase to assess correlation with volumetric measurement by H&E staining. Hemorrhage volume was assessed by MRI at 2, 24 and 72 h and by H&E histology at 72 h in the same set of animals. MR imaging revealed a stable hematoma volume in all animals (10.2 ± 2.65 at 2 hrs v. 12.8 ± 4.01 at 24 hrs v. 11.3 ± 3.16 mm3 at 72 h) without evidence of expansion but with transition from homogenous signal present with blood to a more heterogeneous lesion with the formation of iron breakdown products. (Figure 2) Furthermore, assessment of volume by MRI at each of the time-points did not demonstrate any differences between the APOETR groups (2 h: 10.8 ± 5.24 in APOE3TR v. 9.4 ± 2.68 in APOE4TR; 24 h: 12.7 ± 7.11 in APOE3TR v. 12.9 ± 5.46 in APOE4TR; 72 h: 11.0 ± 5.39 in APOE3TR v. 11.5 ± 4.55 mm3 in APOE4TR). Finally, hemorrhage volumes measured by MR at 72 h correlated well with histology at 72 h after injury in the same set of animals (72 h: 11.3 ± 3.16 in MRI v. 13.4 ± 3.19 mm3 in H&E, r2 = 0.98). Although there was no difference in hemorrhage volume as a function of apoE-isoform, APOE4TR animals did have a significant increase in brain water content in the injured hemisphere at 24 h as compared to their APOE3TR counterparts (80.0 ± 0.4 in APOE4TR v. 75.1 ± 1.0 % in APOE3TR, p < 0.01).
Figure 1.
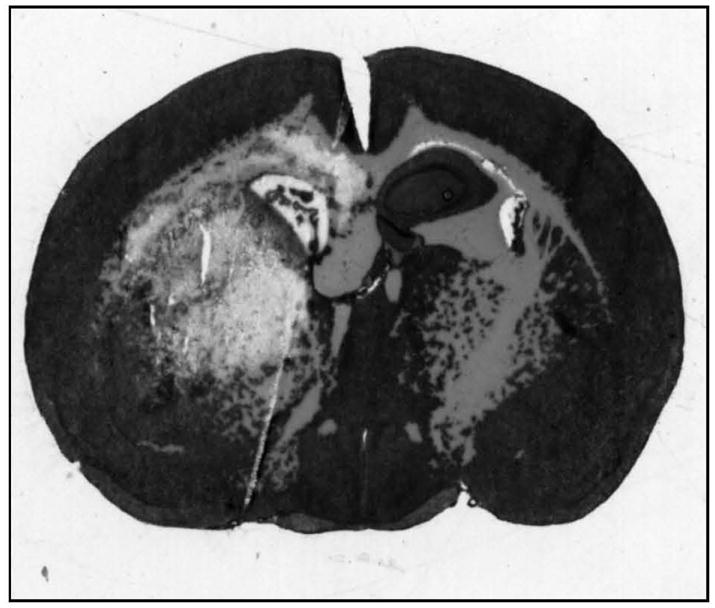
H&E stain of ICH in APOETR3 mice at 72 h after injury. Note pericollosal edema, significant midline shift, and expansion of the ipsilateral lateral ventricle presumably due to ‘trapping’ secondary to inflammatory brain swelling.
Figure 2.

A–C. MRI of APOE4TR mouse after collagenase-induced ICH at 2 h (A), 24 h (B), and 72 h (C) after injury. T2-weighted RARE spin echo images at 2 h (A) show predominantly low signal hematoma within the right basal ganglia, consistent with deoxyhemoglobin and intracellular methemoglobin. At 72 h (C), there is conversion to predominantly high signal, consistent with extracellular methemoglobin.
To assess whether apoE might directly influence early inflammatory events in the CNS in an isoform-specific fashion, quantitative q-PCR was performed for the inflammatory cytokines IL-6, eNOS, and TNF-α at 6, 24, and 48 h (n = 5/group/timepoint) after ICH induction. RNA levels for all proteins were increased at 6 h in the injured hemisphere compared to the uninjured hemisphere (TNF-α: 15.05 ± 2.64 in injured, 0.86 ± 0.26 in uninjured; Figure 3A; eNOS: 1.11 ± 0.2 in injured, 0.64 ± 0.22 in uninjured; IL-6: 22.88 ± 0.87 in injured, 1.22 ± 0.45 in uninjured; p < 0.01) with return to baseline within 48 h after injury. Although IL-6 and eNOS were up-regulated in all animals, these cytokines were significantly elevated in APOE4TR animals as compared to APOE3TR at 6 h (IL-6: 22.81 ± 0.87 in APOE4TR v. 3.93 ± 1.13 in APOE3TR; eNOS: 1.11 ± 0.19 in APOE4TR v. 0.57 ± 0.06 in APOE3TR; p < 0.01; Figure 3B & D); additionally TNF-α RNA up-regulation was not significant (p < 0.09; Figure 3C). Collectively, these results suggest that, although APOE genotype does not appear to have a primary effect on hematoma formation, presence of the APOE4 allele is associated with a greater degree of inflammation, leading to increased cerebral edema.
Figure 3.

A–CA–D. Quantitative q-PCR of inflammatory markers after ICH in APOETR mice. At 6, 24, and 48 h, IL-6 peaks and returns to normal levels in the injured hemisphere on APOE4TR mice when compared to the uninjured hemisphere (A). IL-6 (B) and eNOS (D) are significantly reduced in the injured hemispheres of APOE3TR mice compared to their APOE4TR counterparts. TNF-α was not significant(C).
Finally, to determine whether increases in inflammation and cerebral edema in the APOE4TR animals were associated with worsened functional deficit, behavioral testing with serial rotorod and neuroseverity assessment was performed daily for 3 d following injury on a separate cohort of APOE3TR (n=9) and APOE4TR (n=11) animals. APOE4TR animals had a greater degree of functional deficit as compared to the APOE3TR mice, as quantified by shorter rotorod latencies (p < 0.01; Figure 4A) and poorer neuroseverity scores (p < 0.01; Figure 4B) over the three days tested; furthermore, these functional outcome measures appeared to mirror each other in terms of recovery. It should be noted that sham operated animals of both genotypes (APOE3TR and APOE4TR) did not exhibit significant functional neurological deficit by either rotorod latency or neuroseverity scoring over any of the days tested. Together these data suggest the clinical relevance of this model of ICH given that endogenous APOE genotype influences functional neurological outcomes and that the presence of an APOE4 allele results in increased cerebral edema with poorer recovery.
Figure 4.

A &B. Rotorod latencies (A) and neuroseverity scoring (B) over 3 consecutive days following ICH induction in APOETR mice.
Effects of Exogenous ApoE-mimetic peptide
To demonstrate whether the beneficial effects of endogenous apoE3 could be harnessed as a therapeutic strategy, vehicle or COG1410 (1 mg/kg) was injected intravenously in C57-BL/6 mice at 30 min and 4 h after ICH induction (n = 8/group). Although there was no difference in hemorrhage volume assessed at 7 d (24.3 ± 1.17 in vehicle treated vs. 23.5 ± 3.02 mm3 in COG1410 treated), animals treated with doses of apoE-mimetic peptide demonstrated a durable improvement in functional outcomes, extended to 7 d to assess the possibility of a ‘ceiling effect’ for COG1410 by rotorod latencies (Figure 5A, p < 0.01) and neuroseverity scores (p < 0.01) that persisted over the course of testing. Additionally, sham-operated animals did not demonstrate any functional deficits after injury. This was associated with a reduction in the brain water content in the injured hemisphere due to cerebral edema at 24 h after injury (n = 5/group; 78.7 ± 0.9 in vehicle treated v. 75.5 ± 0.5 % in COG1410 treated; p < 0.05). Finally, to evaluate the role of inflammation in the development of cerebral edema and its influence on functional outcomes, we performed quantitative q-PCR in a separate cohort of animals (n = 5/group) at 6 h after ICH induction for the inflammatory cytokines eNOS, IL-6, and TNF-α. Hemorrhage was again associated with an up-regulation of all cytokines at 6 h in the injured hemisphere of WT mice (TNF-α: 13.86 ± 1.86, injured v. 4.03 ± 0.81, uninjured; IL-6: 22.77 ± 3.24, injured v. 3.87 ± 1.02, uninjured; eNOS: 1.21 ± 0.08, injured v. 0.99 ± 0.07, uninjured; p < 0.01). However, when given the apoE-mimetic peptide both IL-6 and eNOS were significantly reduced (IL-6: 22.77 ± 3.24 in vehicle treated v. 9.52 ± 2.95 in COG1410 treated; eNOS: 1.52 ± 0.05 in vehicle treated v. 1.21 ± 0.08 in COG1410 treated; p <0.01). These data suggest that the administration of an apoE-mimetic peptide influences neurological behavioral outcomes by down-regulating inflammation resulting in less post-hemorrhagic cerebral edema.
Figure 5.
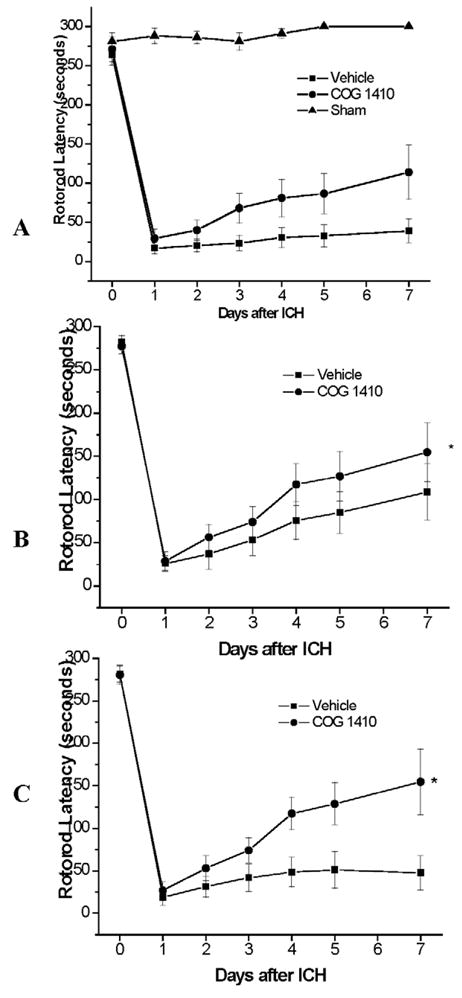
A–C. Rotorod latencies after ICH in different APOE genotypes (WT: A, APOE3TR: B, APOE4TR: C) after administration of mimetic peptide or saline via tail vein injection 30 min and 4 h after injury.
To further assess the degree of pharmacogenomic interaction between the apoE peptide and endogenous APOE genotype, the experiment was repeated in APOE3TR and APOE4TR animals. In both of these lines, although apoE-mimetic peptide administration did not affect hemorrhage size (n = 8/group; APOE4TR: 23.4 ± 2.58 in vehicle treated v. 24.2 ± 1.46 mm3 in COG1410 treated; APOE3TR: 23.3 ± 2.06 in vehicle treated v. 24.5 ± 1.56 mm3 in COG1410 treated), administration of COG1410 improved functional outcomes over the 7 d tested without demonstrable ‘ceiling effect’ (p < 0.05; Figure 5B–C), with the most robust therapeutic effect in the APOE4TR animals. Additionally brain water content in the injured hemisphere was also significantly reduced in the APOE4TR animals treated with apoE-mimetic peptide at 24 hrs after injury (n = 6/group, 79.7 ± 0.6 in vehicle treated v. 75.0 ± 0.5 % in COG1410 treated, p<0.01). In aggregate, these data demonstrate that administration of an apoE-mimetic peptide represents a potentially viable therapeutic strategy that may improve outcome independent of the endogenous apoE isoform.
DISCUSSION
In the current study, we extend clinical observations implicating a role for APOE polymorphism in modifying functional outcomes after supratentorial ICH using a transgenic murine model of collagenase-induced basal ganglia hemorrhage. Consistent with the clinical literature, we found that presence of the human APOE4 gene and its apoE4 products was associated with poor functional outcome, independent of any direct effect on hematoma volume. Moreover, we demonstrate that treatment with an apoE-mimetic therapeutic peptide, whose sequence does not encompass any of holoprotein’s polymorphic residues, improved durable functional outcome in WT, APOE3TR and APOE4TR animals.
Although originally described in the context of cholesterol metabolism, clinical observations suggest that APOE polymorphism plays a uniquely important role in modifying the CNS response to acute and chronic injury. 1, 24 In particular, presence of the APOE4 allele has been associated with poor neurological outcome after ICH 2 and traumatic brain injury. 25 Although the mechanism(s) by which apoE affects the CNS response to injury remains poorly defined, one unifying hypothesis is that apoE modifies neuroinflammatory responses in an isoform-specific fashion by modulating microglial activation. This function of apoE in the generation of inflammation in the brain has been well described in a number of in vitro and preclinical models, 26 and has recently been translated into the clinical setting, where APOE4 is associated with more robust inflammatory responses following ex vivo stimulation of human monocyte derived macrophages, 27 cardiopulmonary bypass, 28 and in the critically ill population. 29
In the current study, we demonstrate that the expression of APOE4 is associated with enhanced neuroinflammatory responses and impaired functional outcomes independent of hematoma volume following ICH. CNS inflammation, characterized by glial activation, release of inflammatory mediators, and breakdown of the blood brain barrier may be particularly relevant in the development of cerebral edema and secondary neuronal injury following ICH. One possible mechanism by which apoE3 may confer a neuroprotective effect is via down-regulation of microglial activation, an effect which has been demonstrated in vitro 30 and in vivo. 5 This effect is evidenced by a decrease in inflammatory mediators such as IL-6 and eNOS following ICH in our model. Therefore, it appears that our murine model of ICH is clinically relevant as the isoform-specific effects of apoE that we observed in the model are mirrored in the human condition. Our results are consistent with the hypothesis that apoE exerts an isoform-specific effect on neuroinflammatory responses independent of any direct effect on hematoma volume, as presence of an APOE4 allele was associated with increased cerebral edema in the company of up-regulated markers of inflammation, with similar findings being published after ICH in humans. It is, therefore, logical to assume that our clinically relevant murine model of ICH should be utilized to evaluate potential, targeted pharmacogenomic interactions.
In addition to providing mechanistic insight into the role of APOE polymorphism in acute brain injury, a more complete understanding of the function of apoE in the injured CNS may also raise the possibility of developing novel therapeutic strategies. Although apoE does not cross the blood brain barrier, many of its adaptive properties appear to be mediated by signaling cascades initiated by specific interaction with cell surface receptors. 31 Based on these receptor interactions, a series of peptides derived from the receptor-binding domain of apoE were created and demonstrated to exert many of the same adaptive functional effects as the intact holoprotein. These studies have been extended to preclinical models of traumatic brain injury, where a peptide derived from the receptor-binding region, apoE(133–149), was well tolerated, crossed the BBB, and was associated with improved functional and histological outcomes. 6 The peptide used in the current study, COG1410, represents a second-generation apoE-based therapeutic in which the helicity and anti-inflammatory potency of the peptide was enhanced by the introduction of two non-naturally occurring Aib residues. 1 In the current study, systemic administration of COG1410 reduced functional disability and histological injury, a finding that is consistent with its palliative effects in other models of acute brain injury. 32
Thus, the current experimental paradigm offers the opportunity to study pharmacogenomic interactions between an apoE-based therapeutic and background humanized APOE genotype. In fact, we provide evidence that intravenous administration of apoE-mimetic peptide (COG1410) reduces inflammation and improves functional outcome regardless of the humanized APOE genetic background. This effect appears most robust in WT and APOETR4 mice, and when examined closely, administration of apoE-mimetic peptide results in neurological outcomes that are similar to their APOETR3 counterparts. These functional improvements are coupled with decreases in markers of microglial activation, inflammation, and cerebral edema. Together these data provide evidence that small mimetic peptides may represent a rational, targeted therapeutic strategy.
Several limitations to this study should be addressed. Although collagenase-induced ICH is a commonly used model, there are theoretical concerns that bacterial collagenase might induce an inflammatory response independent of that elicited by parenchymal blood. Although in this model it is impossible to tease out exact contributions to inflammation from exogenous (collagenase) versus endogenous (blood) substances, the anti-inflammatory properties of endogenous apoE and exogenous apoE-mimetic peptide are consistent throughout, and demonstration of the isoform-specificity of apoE is consistent with published data in the human condition, decreasing the likelihood that outcomes present in this preclinical model are related to inflammation secondary to bacterial collagenase alone. Second, although presence of the APOE4 allele was associated with significantly increased levels of IL-6 and eNOS RNA at 6 h after hemorrhage, an increase TNF-α was not wholly demonstrated. However, TNF-α elevation is an ultra-early event after ICH, and levels may have peaked prior to 6 h. 7 Finally, the reason(s) for the divergence in treatment effect of the mimetic peptide between the different genotypes is unclear. Although the apoE-mimetic peptide reduced inflammation and outcome in both APOE3TR and APOE4TR animals, it had a greater effect in the presence of APOE4. This might suggest that apoE4 is associated with a loss of adaptive anti-inflammatory function of endogenous apoE, and that this may be overcome with exogenous administration of an apoE-mimetic peptide. This pharmacogenomic interaction suggests that this therapeutic strategy might be of particular benefit to patients with an APOE4 allele who are at highest risk for poorer functional outcomes.
Summary
In summary, we provide data demonstrating that the presence of APOE4 is associated with enhanced neuroinflammatory responses and cerebral edema after ICH in our murine model. This is consistent with isoform-specific differences that have been demonstrated in a variety of both preclinical and clinical acute brain injury paradigms. The protective effect of the apoE3 isoform can be simulated by the administration of COG1410, an apoE-mimetic peptide, in our model of ICH. This therapeutic approach improves outcomes in both APOE3TR and APOE4TR animals, suggesting that it may be a viable, rational strategy for targeted pharmacogenomic therapy humans after ICH.
Acknowledgments
This study was possible through funding by NIH 1UL1 RR024128-01 (DTL), 2R44 AG 020473 (MPV) and grants from The Institute for the Study of Aging.
All authors have read and approved submission of the manuscript.
The material in the manuscript has not been published and is not being considered for publication elsewhere in whole or in part in any language.
No persons, outside of the authors, are mentioned in the acknowledgments.
Footnotes
Conflicts of Interest Disclosures: The following is a list of financial disclosures: Dr. Laskowitz serves as a consultant for Cognosci, Inc. Dr. Vitek is a principal in Cognosci, Inc.
Cognosci, Inc. did not provide any funding for this study and had no input into the study design, data collection, analysis, interpretation, or writing of reports.
References
- 1.Laskowitz DT, Vitek MP. Apolipoprotein e and neurological disease: Therapeutic potential and pharmacogenomic interactions. Pharmacogenomics. 2007;8:959–969. doi: 10.2217/14622416.8.8.959. [DOI] [PubMed] [Google Scholar]
- 2.Alberts MJ, Graffagnino C, McClenny C, DeLong D, Strittmatter W, Saunders AM, Roses AD. Apoe genotype and survival from intracerebral haemorrhage. Lancet. 1995;346:575. doi: 10.1016/s0140-6736(95)91411-0. [DOI] [PubMed] [Google Scholar]
- 3.Woo D, Kaushal R, Chakraborty R, Woo J, Haverbusch M, Sekar P, Kissela B, Pancioli A, Jauch E, Kleindorfer D, Flaherty M, Schneider A, Khatri P, Sauerbeck L, Khoury J, Deka R, Broderick J. Association of apolipoprotein e4 and haplotypes of the apolipoprotein e gene with lobar intracerebral hemorrhage. Stroke. 2005;36:1874–1879. doi: 10.1161/01.STR.0000177891.15082.b9. [DOI] [PubMed] [Google Scholar]
- 4.Tzourio C, Arima H, Harrap S, Anderson C, Godin O, Woodward M, Neal B, Bousser MG, Chalmers J, Cambien F, MacMahon S. Apoe genotype, ethnicity, and the risk of cerebral hemorrhage. Neurology. 2008;70:1322–1328. doi: 10.1212/01.wnl.0000308819.43401.87. [DOI] [PubMed] [Google Scholar]
- 5.Duan RS, Chen Z, Dou YC, Concha Quezada H, Nennesmo I, Adem A, Winblad B, Zhu J. Apolipoprotein e deficiency increased microglial activation/ccr3 expression and hippocampal damage in kainic acid exposed mice. Exp Neurol. 2006;202:373–380. doi: 10.1016/j.expneurol.2006.06.013. [DOI] [PubMed] [Google Scholar]
- 6.Lynch JR, Wang H, Mace B, Leinenweber S, Warner DS, Bennett ER, Vitek MP, McKenna S, Laskowitz DT. A novel therapeutic derived from apolipoprotein e reduces brain inflammation and improves outcome after closed head injury. Exp Neurol. 2005;192:109–116. doi: 10.1016/j.expneurol.2004.11.014. [DOI] [PubMed] [Google Scholar]
- 7.Lynch JR, Tang W, Wang H, Vitek MP, Bennett ER, Sullivan PM, Warner DS, Laskowitz DT. Apoe genotype and an apoe-mimetic peptide modify the systemic and central nervous system inflammatory response. J Biol Chem. 2003;278:48529–48533. doi: 10.1074/jbc.M306923200. [DOI] [PubMed] [Google Scholar]
- 8.Xue M, Del Bigio MR. Comparison of brain cell death and inflammatory reaction in three models of intracerebral hemorrhage in adult rats. J Stroke Cerebrovasc Dis. 2003;12:152–159. doi: 10.1016/S1052-3057(03)00036-3. [DOI] [PubMed] [Google Scholar]
- 9.McCarron MO, Hoffmann KL, DeLong DM, Gray L, Saunders AM, Alberts MJ. Intracerebral hemorrhage outcome: Apolipoprotein e genotype, hematoma, and edema volumes. Neurology. 1999;53:2176–2179. doi: 10.1212/wnl.53.9.2176. [DOI] [PubMed] [Google Scholar]
- 10.McCarron MO, Weir CJ, Muir KW, Hoffmann KL, Graffagnino C, Nicoll JA, Lees KR, Alberts MJ. Effect of apolipoprotein e genotype on in-hospital mortality following intracerebral haemorrhage. Acta Neurol Scand. 2003;107:106–109. doi: 10.1034/j.1600-0404.2003.01365.x. [DOI] [PubMed] [Google Scholar]
- 11.Linton MF, Gish R, Hubl ST, Butler E, Esquivel C, Bry WI, Boyles JK, Wardell MR, Young SG. Phenotypes of apolipoprotein b and apolipoprotein e after liver transplantation. J Clin Invest. 1991;88:270–281. doi: 10.1172/JCI115288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laskowitz DT, Thekdi AD, Thekdi SD, Han SK, Myers JK, Pizzo SV, Bennett ER. Downregulation of microglial activation by apolipoprotein e and apoe-mimetic peptides. Exp Neurol. 2001;167:74–85. doi: 10.1006/exnr.2001.7541. [DOI] [PubMed] [Google Scholar]
- 13.Herz J, Beffert U. Apolipoprotein e receptors: Linking brain development and alzheimer’s disease. Nat Rev Neurosci. 2000;1:51–58. doi: 10.1038/35036221. [DOI] [PubMed] [Google Scholar]
- 14.Hoe HS, Harris DC, Rebeck GW. Multiple pathways of apolipoprotein e signaling in primary neurons. J Neurochem. 2005;93:145–155. doi: 10.1111/j.1471-4159.2004.03007.x. [DOI] [PubMed] [Google Scholar]
- 15.Aono M, Bennett ER, Kim KS, Lynch JR, Myers J, Pearlstein RD, Warner DS, Laskowitz DT. Protective effect of apolipoprotein e-mimetic peptides on n-methyl-d-aspartate excitotoxicity in primary rat neuronal-glial cell cultures. Neuroscience. 2003;116:437–445. doi: 10.1016/s0306-4522(02)00709-1. [DOI] [PubMed] [Google Scholar]
- 16.Sullivan PM, Mezdour H, Aratani Y, Knouff C, Najib J, Reddick RL, Quarfordt SH, Maeda N. Targeted replacement of the mouse apolipoprotein e gene with the common human apoe3 allele enhances diet-induced hypercholesterolemia and atherosclerosis. J Biol Chem. 1997;272:17972–17980. doi: 10.1074/jbc.272.29.17972. [DOI] [PubMed] [Google Scholar]
- 17.Sullivan PM, Mace BE, Maeda N, Schmechel DE. Marked regional differences of brain human apolipoprotein e expression in targeted replacement mice. Neuroscience. 2004;124:725–733. doi: 10.1016/j.neuroscience.2003.10.011. [DOI] [PubMed] [Google Scholar]
- 18.Rosenberg GA, Estrada E, Wesley M, Kyner WT. Autoradiographic patterns of brain interstitial fluid flow after collagenase-induced haemorrhage in rat. Acta Neurochir Suppl (Wien) 1990;51:280–282. doi: 10.1007/978-3-7091-9115-6_95. [DOI] [PubMed] [Google Scholar]
- 19.Hamm RJ, Pike BR, O’Dell DM, Lyeth BG, Jenkins LW. The rotarod test: An evaluation of its effectiveness in assessing motor deficits following traumatic brain injury. J Neurotrauma. 1994;11:187–196. doi: 10.1089/neu.1994.11.187. [DOI] [PubMed] [Google Scholar]
- 20.Garcia JH, Wagner S, Liu KF, Hu XJ. Neurological deficit and extent of neuronal necrosis attributable to middle cerebral artery occlusion in rats. Statistical validation. Stroke. 1995;26:627–634. doi: 10.1161/01.str.26.4.627. discussion 635. [DOI] [PubMed] [Google Scholar]
- 21.Zhang X, Li H, Hu S, Zhang L, Liu C, Zhu C, Liu R, Li C. Brain edema after intracerebral hemorrhage in rats: The role of inflammation. Neurol India. 2006;54:402–407. doi: 10.4103/0028-3886.28115. [DOI] [PubMed] [Google Scholar]
- 22.Song EC, Chu K, Jeong SW, Jung KH, Kim SH, Kim M, Yoon BW. Hyperglycemia exacerbates brain edema and perihematomal cell death after intracerebral hemorrhage. Stroke. 2003;34:2215–2220. doi: 10.1161/01.STR.0000088060.83709.2C. [DOI] [PubMed] [Google Scholar]
- 23.Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative pcr and the 2(−delta delta c(t)) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- 24.Martinez-Gonzalez NA, Sudlow CL. Effects of apolipoprotein e genotype on outcome after ischaemic stroke, intracerebral haemorrhage and subarachnoid haemorrhage. J Neurol Neurosurg Psychiatry. 2006;77:1329–1335. doi: 10.1136/jnnp.2006.097543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Friedman G, Froom P, Sazbon L, Grinblatt I, Shochina M, Tsenter J, Babaey S, Yehuda B, Groswasser Z. Apolipoprotein e-epsilon4 genotype predicts a poor outcome in survivors of traumatic brain injury. Neurology. 1999;52:244–248. doi: 10.1212/wnl.52.2.244. [DOI] [PubMed] [Google Scholar]
- 26.James ML, Warner DS, Laskowitz DT. Preclinical models of intracerebral hemorrhage: A translational perspective. Neurocrit Care. 2007 doi: 10.1007/s12028-007-9030-2. [DOI] [PubMed] [Google Scholar]
- 27.Colton CA, Czapiga M, Snell-Callanan J, Chernyshev ON, Vitek MP. Apolipoprotein e acts to increase nitric oxide production in macrophages by stimulating arginine transport. Biochim Biophys Acta. 2001;1535:134–144. doi: 10.1016/s0925-4439(00)00092-2. [DOI] [PubMed] [Google Scholar]
- 28.Grunenfelder J, Umbehr M, Plass A, Bestmann L, Maly FE, Zund G, Turina M. Genetic polymorphisms of apolipoprotein e4 and tumor necrosis factor beta as predisposing factors for increased inflammatory cytokines after cardiopulmonary bypass. J Thorac Cardiovasc Surg. 2004;128:92–97. doi: 10.1016/j.jtcvs.2004.02.022. [DOI] [PubMed] [Google Scholar]
- 29.Moretti EW, Morris RW, Podgoreanu M, Schwinn DA, Newman MF, Bennett E, Moulin VG, Mba UU, Laskowitz DT. Apoe polymorphism is associated with risk of severe sepsis in surgical patients. Crit Care Med. 2005;33:2521–2526. doi: 10.1097/01.ccm.0000186368.96146.fb. [DOI] [PubMed] [Google Scholar]
- 30.Colton CA, Needham LK, Brown C, Cook D, Rasheed K, Burke JR, Strittmatter WJ, Schmechel DE, Vitek MP. Apoe genotype-specific differences in human and mouse macrophage nitric oxide production. J Neuroimmunol. 2004;147:62–67. doi: 10.1016/j.jneuroim.2003.10.015. [DOI] [PubMed] [Google Scholar]
- 31.Hoe HS, Pocivavsek A, Chakraborty G, Fu Z, Vicini S, Ehlers MD, Rebeck GW. Apolipoprotein e receptor 2 interactions with the n-methyl-d-aspartate receptor. J Biol Chem. 2006;281:3425–3431. doi: 10.1074/jbc.M509380200. [DOI] [PubMed] [Google Scholar]
- 32.Gao J, Wang H, Sheng H, Lynch JR, Warner DS, Durham L, Vitek MP, Laskowitz DT. A novel apoe-derived therapeutic reduces vasospasm and improves outcome in a murine model of subarachnoid hemorrhage. Neurocrit Care. 2006;4:25–31. doi: 10.1385/NCC:4:1:025. [DOI] [PubMed] [Google Scholar]