

SUMMARY
Voltage-gated potassium currents (Kv), primarily due to Kv2.1, are activated by glucose-stimulated pancreatic beta-cell depolarization, but the exact role(s) of this channel in regulating insulin secretion remains uncertain. Here we report that compared with controls the Kv2.1 null mouse has reduced fasting blood glucose levels and elevated serum insulin levels. Glucose tolerance is improved and insulin secretion enhanced compared to control animals, with similar results in isolated islets in vitro. Isolated Kv2.1−/− beta cells have residual Kv currents, decreased by 83% at +50 mV compared with control cells. The glucose-induced action potential duration is increased while the firing frequency is diminished, similar to the effect of specific toxins on control cells, but substantially different from the effect of the less-specific blocker tetraethylammonium (TEA). These results reveal the specific role of Kv2.1 in modulating glucose-stimulated action potentials of the beta cells, exposing additional important currents involved in regulating physiological insulin secretion.
INTRODUCTION
Glucose homeostasis is maintained through coordinated metabolic, hormonal and neural signals. These signals result in the tightly regulated release of insulin and glucagon. Glucose-induced ion fluxes in the pancreatic β-cell are critical for normal glucose homeostasis (Rorsman et al.,1994). Pancreatic β-cells metabolize glucose, generating an increase in the ATP to ADP ratio which closes the ATP sensitive potassium channel, KATP, resulting in membrane depolarization, calcium influx and insulin release (Ashcroft and Rorsman, 1989). Glucose-induced depolarization of the β-cell activates voltage gated channels including the delayed rectifier potassium channel Kv2.1. The resulting outward K+ current in turn controls the duration of depolarization and thus can affect insulin exocytosis (Smith et al., 1990; Dukes and Philipson, 1996; Roe et al., 1994; Macdonald et. al., 2001; Yan et. al., 2004; Herrington et al., 2005).
Kv2.1 is sensitive to a variety of pharmacological agents. These include highly specific agents such as the cysteine-knot spider venoms Hanatoxin (HaTx) and Stromatoxin (ScTx) and others that are less specific such as tetraethylammonium (TEA) and 4-amino-pyridine (4-AP) (Swartz and MacKinnon, 1995; Escoubas et al., 2002; Atwater et al., 1979; Bokvist et al., 1990; Kirsch et al., 1993). Our current understanding of the role of Kv channels in islet physiology has been generated primarily through pharmacological approaches using these agents. Kv channel blockade increases both the amplitude and duration of beta cell action potentials (MacDonald et al., 2002; Tamarina et al., 2005; Herrington et al., 2006). Augmentation of the beta cell action potential also leads to changes in glucose-induced calcium fluctuations leading to elevated islet insulin secretion (MacDonald et al., 2002; Tamarina et al., 2005; Herrington et al., 2006). These results have also been corroborated with a Kv-dominant negative construct that reduced Kv2.1 functional expression and enhanced glucose stimulated insulin secretion in an insulinoma cell line (MacDonald et al., 2001). However the role(s) of Kv2.1 in β-cell physiology in vivo have not been conclusively defined.
Here we investigated the physiological role of Kv2.1 in β-cells from a mouse model in which the Kv2.1 gene is disrupted. We found that the β-cells from these null animals have greatly reduced Kv currents, indicating that Kv2.1 is indeed the primary Kv channel of the β-cell. Action potentials are widened compared with controls while the frequency of action potentials is reduced. Kv2.1null mice also show significantly reduced fasting glucose levels with elevated insulin levels. These studies reveal the relationship between Kv channels, action potential regulation and insulin secretion. The lack of Kv2.1 exposed additional important β-cell currents and suggest that plasma membrane electrical activity regulates physiological insulin secretion.
RESULTS
To investigate the physiological role of Kv2.1 during glucose stimulated insulin secretion, a mouse model was utilized in which the Kv2.1 gene is disrupted at codon 346 (Fig. 1A). The targeting cassette removes 102 bases in S5 of the Kv2.1 coding gene sequence and adds nearly 7kB of extraneous sequence including a premature stop codon, 12 codons from the fusion with Kv2.1 (Fig. 1A). The disrupted gene has no significant affect on islet cell viability, assessed by caspase 3 cleavage positive cells per total islet cells (Fig. S1, n=7). Genomic DNA from homozygous targeted animals (Kv2.1−/−) contains the targeting sequences. The disrupted sequence is detected using the polymerase chain reaction to produce an amplicon with one primer inside the targeting sequence in combination with a Kv2.1 specific primer in the reverse orientation (3′ to 5′, Fig. 1B). Similarly, a Southern blot performed on EcoRV digested genomic DNA prepared with a Kv2.1 specific probe, using DNA from appropriately targeted ES cell lines, produces an additional 6.6kB fragment not present in controls (Fig. 1C). Kv2.1 protein was detected only in protein blots from wild-type tissue preparations using a Kv2.1 specific antibody which interacts with the C-terminus of the protein (Fig. 1D). Kv2.1−/− islet β-cell size and insulin staining as well as α-cell size and glucagon staining were equivalent to controls (Fig. S2, n=5 each).
Fig. 1.
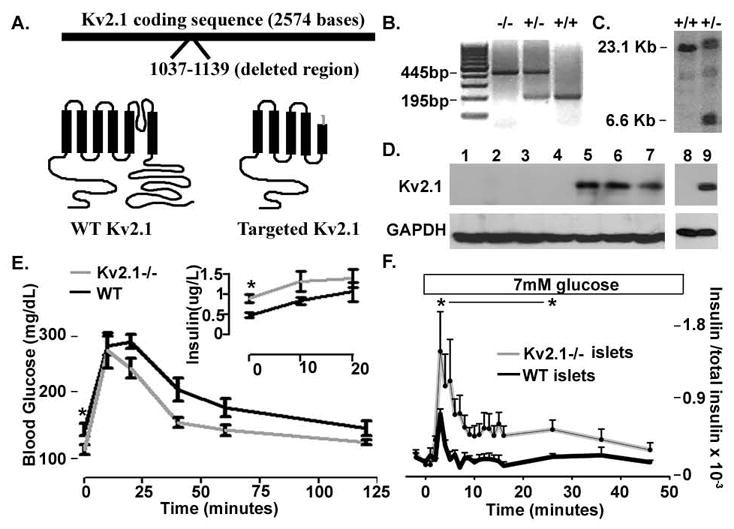
Kv2.1 knockout animals show aberrant glucose homeostasis. (A) Cartoon of the Kv2.1 targeted gene disruption and the resulting disrupted protein structure in grey versus WT in black. (B) PCR products from homozygous knockout, −/−, heterozygous, +/−, and control, +/+, animals with primers specific to the surrounding Kv2.1 gene disruption sequence and one primer specific to the targeting sequence. (C) Southern blot probed with a Kv2.1 specific probe run with genomic DNA from control,+/+, and targeted, +/−, embryonic stem cells cut with an endonuclease that removes part of the Kv2.1 sequence and the targeting cassette. (D) Western blot run with Kv2.1−/− brain extracts lanes 1–4, control brain extracts lanes 5–7, Kv2.1−/− islets lane 8, and control islets lane 9. (E) Glucose tolerance test on Kv2.1−/−, grey line, and control, black line, animals. Serum insulin levels from Kv2.1−/−, grey line, and control, black line, animals are shown in the inset. n= 10 each, * P<0.05. (F) Islet insulin secretion during the 7mM glucose treatment, boxed above, the islets are in 2mM glucose previous to stimulation, + SDEV (n=10 islet sets and 3 insulin assays each).
Kv2.1−/− animals were assessed for possible impairment in glucose homeostasis using an intraperitoneal glucose tolerance test (GTT). Kv2.1−/− animals had significantly decreased fasting blood glucose levels and showed improved glucose tolerance when compared to control animals (Fig. 1E, n=10 per group). This mild hypoglycemia likely results result from a significant elevation in the fasting serum insulin level in the Kv2.1−/− animals as compared to controls (Fig. 1E inset, n=10). Serum insulin levels are not significantly different between control and Kv2.1−/− animals at 10 and 20 minutes following a glucose load, attributable to the reduced fasting glucose levels of Kv2.1−/− animals(Fig. 1E inset, n=10). Accordingly Kv2.1 −/− animals have similar glucose levels to controls at 20 minutes post glucose although they show an accelerated return to baseline glucose levels (Fig. 1E, n=10).
To determine the role of islet insulin secretion on the defects seen in glucose homeostasis, insulin release from Kv2.1−/− and control islets was assessed. (Fig. 1F). Modest glucose stimulation (7mM)caused a significant elevation in the insulin peak that was sustained for over 20 minutes from the Kv2.1−/− islets compared to controls (Fig. 1F, n=10 sets of islets assayed in 3 independent groups). Since inhibition of Kv2.1 channel activity with tetraethylammonium (TEA) has been shown to increase glucose stimulated insulin secretion, insulin release from Kv2.1−/− and control islets stimulated with TEA and glucose were measured (Henquin, 1977). Quite unexpectedly, control and Kv2.1 null islets show augmented electrical activity with TEA stimulation, which results in equivalent insulin secretion from both groups of islets (Fig. S3, unpublished data). This indicates that channels other than Kv2.1, presumably permeable to potassium, are sensitive to TEA and play a critical role in repolarization following glucose stimulation.
Voltage gated potassium currents from Kv2.1−/− islet beta cells
Kv channels, other than Kv2.1, may participate in β-cell repolarization (Roe et al., 1996; Yan et al., 2004; MacDonald et al., 2001; Kukuljan et al., 1991). To confirm the presence of endogenous Kv channels, in addition to Kv2.1, Kv currents from islet β-cells devoid of Kv2.1 were recorded using the whole cell voltage clamp configuration. Kv2.1−/− β-cells do indeed show Kv currents, however these currents have significantly reduced amplitudes at voltage steps greater than −30 mV when compared to control β-cells. At 50mV the Kv2.1−/− β-cells have current amplitudes that are only 17% that of controls (recorded between 400–450 ms during a 500ms step to 50mV). Pharmacological inhibition of Kv2.1 with cysteine-knot containing spider venom peptides such as HaTx and ScTx-1 causes similar reductions in Kv currents from control β-cells, similar to that seen with the untreated Kv2.1−/− β-cells, whereas they have no significant effect on the Kv currents in Kv2.1−/− β-cells (Fig. 2E). Of the remaining Kv currents from Kv2.1−/− β-cells only 53% are sensitive to block by TEA (n=6 cells performed with a voltage step from −80 to 0mV treated with 20mM TEA) (Gopel et. al. 2000). Thus the channels responsible for the remaining Kv-currents of Kv2.1−/− β-cells are not from the Kv2 family and may provide some repolarizing influence on the β-cell during glucose-stimulated insulin secretion.
Fig. 2.
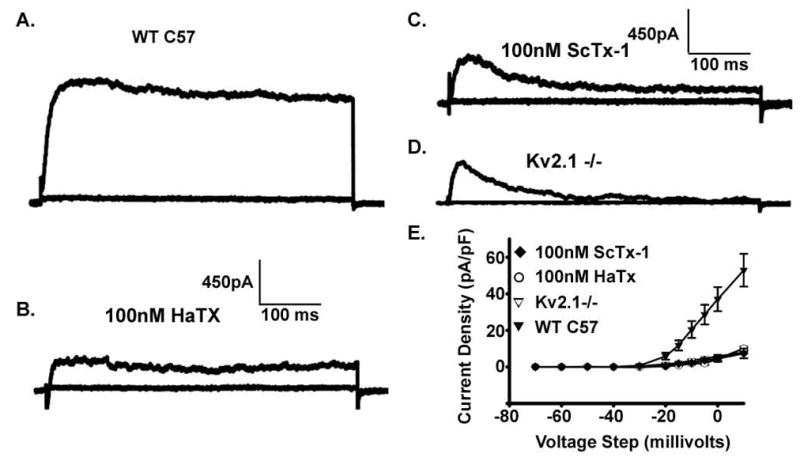
Kv2.1−/− β-cells have significantly reduced Kv currents. (A) Control β-cell Kv currents recorded in voltage clamp with voltage steps from −80 to +80 in 10mV increments. (B) Control β-cell Kv-currents 10 minutes post 100nM HaTx. (C) Control β-cell 10 minutes post 100nM ScTx-1. (E) Kv2.1−/− β-cell Kv-currents. (F) Current density vs Voltage plots for β-cells recorded in steps from −80mV to the indicated voltage +/− SEM’s (n>7 for each condition).
Kv2.1 regulation of the β-cell Action Potential
Kv2.1 channels have been implicated in the regulation of glucose stimulated action potential (AP) duration through their repolarizing influence primarily using pharmacological approaches (Atwater et al., 1979; MacDonald et al., 2002; Tamarina et al., 2005; Herrington et al., 2006). To test the role of Kv2.1 on glucose-stimulated APs, intact islet β-cell membrane potentials were recorded using the perforated patch current clamp configuration and stimulated with glucose. Glucose-stimulated action potentials recorded from Kv2.1−/− islets show a significantly increased duration (325.4 ms/AP versus 86.7 ms/AP for control islets, p<0.0001, n=7 each, Fig. S4) with a greatly reduced firing frequency (2/s versus 4.72/s for control islets, p<0.0001, n=7 each, Fig. S4) when compared to control islets at high concentrations, 14.4 mM, as well as at physiologically normal concentrations, 6.5mM, of glucose (Fig. 3). Interestingly ScTx-1 block of Kv2.1 causes the control glucose stimulated action potential duration to increase with an associated reduction in AP firing frequency, which resembles glucose treated Kv2.1−/− islet APs (Fig. 4). Islet calcium fluctuations recorded following depolarization, induced with tolbutamide to inhibit KATP, show a similar profile with significantly elevated amplitudes (1.9 fold increase in amplitude +/− 0.3, n=5) in response to 25nM ScTx-1. The changes in calcium dynamics of control islets in response to Kv2.1 inhibition likely represent an increased depolarizing influence of each AP resulting in activation of voltage dependent calcium channels and increased calcium influx (Fig. 4C and D). As expected, ScTx-1 has only minimal effects on Kv2.1−/− islet electrical activity and calcium fluctuations (1.2 fold increase in amplitude +/− 0.04, n=5), thus the difference in amplitudes between control and Kv2.1−/− islets were significantly different (Fig. 4, p<0.035).
Fig. 3.
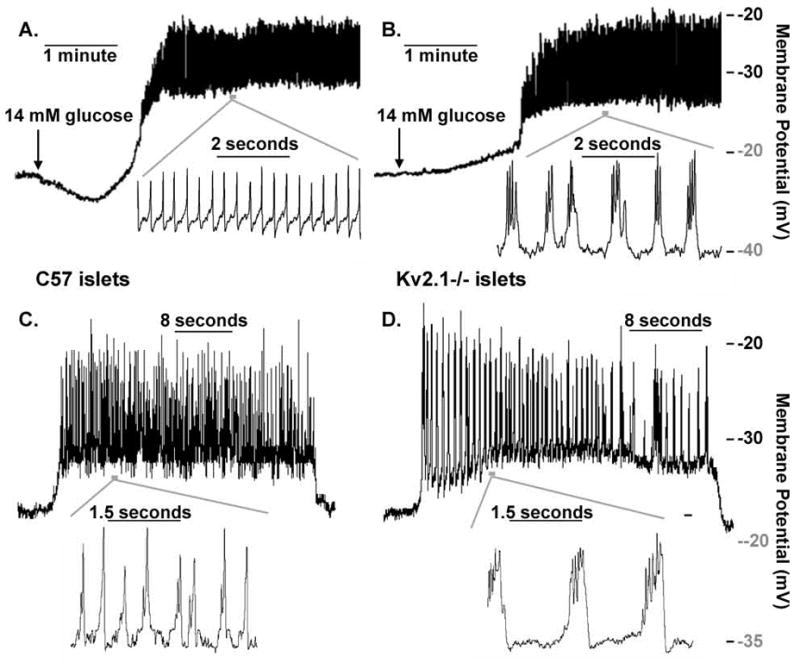
Kv2.1−/− islets have increased glucose induced action potential duration with decreased frequency. (A) Control islet electrical activity in response to 14mM glucose; inset shows the action potentials from the indicated segment of activity, horizontal grey bar. (B) Kv2.1−/− islet electrical activity in response to 14mM glucose; inset shows the action potentials from the indicated segment of activity, horizontal grey bar. (C) Control islet electrical activity in response to 6.5mM glucose; inset shows the action potentials from the indicated segment of activity, horizontal grey bar. (D) Kv2.1−/− islet electrical activity in response to 6.5mM glucose; inset shows the action potentials from the indicated segment of activity, horizontal grey bar.
Fig. 4.
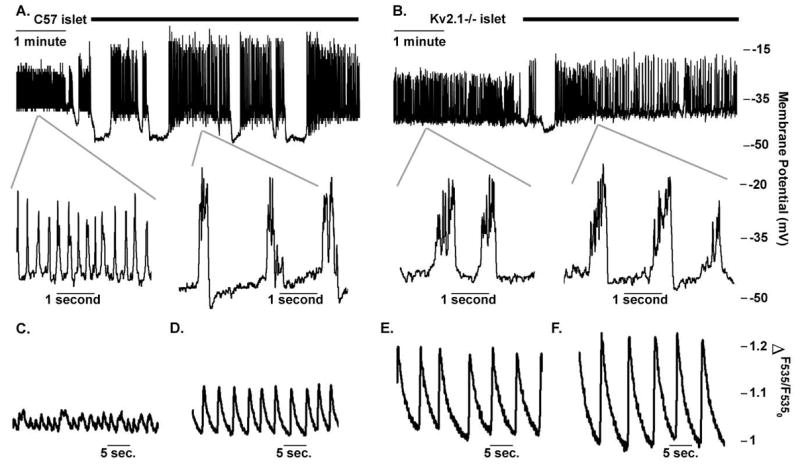
Control islets treated with ScTx-1 show similar bursting to untreated Kv2.1 islets. (A) Control islet electrical activity recorded in 14mM glucose and treated with 100nM ScTx-1, black bar. Insets show islet action potentials expanded during 14mM glucose alone or in combination with 100nM ScTx treatment. (B) Kv2.1 islet electrical activity recorded in 14mM glucose and treated with 100nM ScTx-1, black bar. Insets show islet action potentials expanded during 14mM glucose alone or in combination with 100nM ScTx treatment. (C–F) Representative fast acquisition calcium traces recorded with mouse islets loaded with Fluo-4 and incubated with 200uM Tolbutamide in 2mM glucose from control (C) and Kv2.1−/− (D) animals and from the same control (E) and Kv2.1−/− (F) islet four minutes post 25nM ScTx-1.
DISCUSSION
Pancreatic islet β-cells respond to elevations in glucose with an orchestrated cascade of ion channel inhibition and activation. (Ashcroft and Rorsman, 1989; Houamed et al., 2004). KATP channels are inhibited following glucose metabolism, allowing an inward cation current to depolarize the beta cell, activating voltage-dependent cation channels (Ashcroft and Rorsman, 1989). This activity results in membrane potential fluctuations, causing entry of calcium that facilitates granule-membrane interactions culminating in insulin secretion. Following glucose-induced depolarization rodent β-cells begin to repolarize through activation of voltage gated K+ channels, especially Kv2.1 (Smith et al., 1990; Roe et al., 1996; Yan et al., 2004; MacDonald et al., 2001; Kukuljan et al., 1991). Indeed human β-cells also express Kv2.1 protein resulting in Hannatoxin sensitive delayed rectifier currents that comprise 65% of the total Kv current recorded from these cells (Macdonald et. al., 2003; Tamarina et al., 2005; Herrington et al., 2005). The results presented here demonstrate the effects of removing Kv2.1 on electrical activity and insulin secretion using a mouse model with targeted ablation of the Kv2.1 gene. The results suggest that the most prominent effects of Kv2.1 are on the action potential itself. Kv2.1 null animals show improved glucose tolerance with a reduced fasting glucose, increased fasting serum insulin, and an accelerated return to baseline following a glucose challenge when compared to control animals. Electrical activity is also perturbed in the Kv2.1−/− islets and β-cells, as the remaining Kv currents show significantly reduced amplitudes at voltages above -30mV compared to controls. Thus there is no significant compensation for the loss of Kv2.1 current. The absence of Kv2.1 current results in increased glucose-stimulated action potential duration and, importantly, a decreased firing frequency from islets and individual β-cells. Interestingly, Kv2.1 null islets still respond to TEA (10–20 mM) with prolonged action potential duration, decreased firing frequency and a significant increase in action potential amplitude. Our studies on the Kv2.1 null islets therefore have elucidated specific modulatory roles for Kv2.1 in glucose stimulated electrical activity of the β-cell, and suggest that yet other channels are likely to underlie TEA-induced amplification of glucose stimulated insulin secretion.
The remaining Kv current observed in Kv2.1−/− β-cells is activated during glucose stimulated islet electrical activity and thus may help modulate insulin secretion. As the residual β-cell Kv-current is sensitive to TEA it may be partially responsible for the TEA amplification of glucose induced AP amplitude and resulting augmentation of insulin secretion. Of the TEA sensitive potassium channels expressed in pancreatic β-cells, other than Kv2.1, including Kv3.2, large conductance calcium activated potassium channels (BK), and intermediate conductance calcium activated potassium channels (IK), the current most closely resembles the biophysical properties of Kv3.2 currents (Bokvist et al., 1990; Kukuljan et al., 1991; Kirsch and Drewe, 1993; Roe et al., 1996; Philipson, 1999; Gopel et. al. 2000; Su et al., 2001; MacDonald et al., 2001; Yan et. al., 2004). However, the residual β-cell Kv current is insensitive to inhibitors specific to BK, IK, and Kv3.2 channels as well as to Kv1, Kv4, and voltage-gated subfamily H (eag-related) member 2 toxins. The molecular identity of the residual β-cell Kv-current remains undefined and might result from heteromeric assembly of Kv subunits. The activity of the remaining outward current is observed at voltages that an islet typically reaches during glucose-induced depolarization thus implicating an important role for this channel during insulin secretion.
Pharmacological inhibition of Kv2.1 potently amplifies glucose induced insulin secretion (MacDonald et al., 2002; Herrington et al., 2006; 2007). Interestingly, however, Kv2.1−/− islets stimulated with 14mM glucose have an initial insulin peak that is only slightly elevated compared to control islets with an accelerated rise to peak. This indicates that the inhibitors of Kv2.1 may not be completely specific, such as with TEA, or that the Kv2.1−/− mouse β-cell might be compensating for the loss of Kv2.1 with increased activity or expression of another channel. There is no compensation from a Kv channel as the amount of Kv current recorded from Kv2.1−/− β-cells is equivalent to control β-cells treated with Kv2.1 inhibitors. However, this does not rule out another type of potassium channel, such as a calcium activated potassium channel, functionally compensating for the Kv2.1 deficiency. The results suggest that under physiological concentrations of glucose, such as between meals, Kv2.1 exerts an important repolarizing role that can not be compensated for, and when absent results in significantly amplified insulin secretion.
Maintenance of the baseline blood glucose levels requires tightly regulated insulin release from the pancreatic islets. Baseline blood glucose between meals is modulated in part by small amounts of insulin secretion likely resulting from small bursts of islet electrical activity (Dean and Matthews, 1970; Beigelman et al., 1976). The data presented here suggest a novel role of Kv2.1 in regulating fasting blood sugar by determining the action potential duration and frequency, such as during the bursts of insulin secretion between meals. The reduced fasting glucose levels of Kv2.1−/− animals apparently result from longer action potential durations during the small bursts of electrical activity and resulting increased insulin secretion.
Voltage gated potassium channel mutations result in a variety of pathological conditions, such as Long QT syndrome (Shieh et al., 2000; Zhou et al., 1998). To date there have been no reports, however, of human Kv2.1 mutations, which may be due to a lack of insight into the expected phenotype due to lack of this channel, and heterozygous channel loss may have particularly subtle effects. Fasting hypoglycemia in humans has many etiologies, including potassium channel mutations, one of which could possibly result from mutations in the Kv2.1 gene (Ahn et al., 2007; Nestorowicz et al., 1996). These studies also suggest that the repolarization of the beta cell is far more complex than previously appreciated and involves channels in addition to Kv2.1. This includes both TEA-sensitive and insensitive current(s) present in the Kv2.1 null islets that govern AP amplitude. Our results show that the role of Kv2.1 is now clearly seen to lie in the regulation of the AP duration and frequency, so that there is increased fasting insulin secretion.
EXPERIMENTAL PROCEDURES
Mouse Kv2.1 genomic disruption
A targeting construct consisting of a 5′ fragment consisting of 2623 bp of genomic Kv2.1 sequence (Accession: M64228) through the fist base of codon 346 of the Kv2.1 gene sequence was inserted upstream of an IRES-LacZ- Neo cassette followed by 2457 bp of Kv2.1 genomic sequence from the 1st base of the380th codon of the Kv2.1 genomic sequence 3′. The targeting construct was electroporated into mouse embryonic stem cells resulting in homologous recombination with the Kv2.1 gene sequence disrupting the Kv2.1 gene sequence at codon 346. Southern blot analysis was performed to isolate correctly targeted ES cells. ES cell genomic DNA was degested with the restriction endonuclease EcoRV which cleaves a 23.1 Kb fragment from control C57 mice containing Kv2.1 sequence and also within the targeting cassette. Digested genomic DNA was run on an agarose gel transferred to a nitrocellulose membrane and probed with a P32 labeled Kv2.1 specific genomic probe located inside the EcoRV fragment. Correctly targeted ES cells show the 23.1 Kb fragment from the WT allele as well as a 6.6Kb fragment from the targeted allele. One of the correctly targeted ES cell clones was injected into C57Bl/6 blastocysts giving rise to chimeric offspring; the targeted allele was transmitted in crosses to C57Bl/6 females. This mouse was created by Deltagen Inc., San Mateo, California. Animal experimental protocols were all approved by the University of Chicago institutional animal care and use committee.
Western Blot Analysis
Protein extracts were prepared from mouse brain and HEK cells by extraction with SDS loading buffer (1% SDS, 30 mmol/l Tris HCl, pH 6.8, 5% β-mercaptoethanol, 5% glycerol, and 0.1% bromophenol blue) and heating at 70°C for 10 min. Proteins were prepared as a western blot on a polyvinylidine fluoride membrane (Whatman). After electrophoresis through an 14% denaturing polyacrylamide gel, Kv2.1antibody (Upstate Biotech,) was used to probe the membrane at 1/250 dilution in PBS, 0.1% Tween, and 3% powdered dried milk, followed by goat anti-rabbit horseradish peroxidase (HRP)-coupled secondary antibody (Santa-Cruz Biotechnology) at 1:5,000 in the same solution. The membranes were washed in PBS containing 0.1% Tween between and after antibody incubations; HRP was illuminated using Pico-signal (Pierce) and exposed on Kodak X-omat Blue film (Kodak).
Glucose and insulin homeostasis
We used 10 control and 10 Kv2.1−/− female animals for intraperitoneal glucose tolerance tests (Zhou et al., 2000). After a 4 h fast, animals received intraperitoneal glucose (2 mg/g body weight dextrose in PBS), and tail whole blood samples were taken at the indicated time intervals. Blood glucose was measured using a Freestyle blood glucose meter (Abbott, IL). Serum insulin was measured by enzyme-linked immunosorbent assay (ELISA) (Alpco diagnostics, NH).
Islet Immunofluorescent Staining
Pancreata cubes from Kv2.1−/− and C57 animals were frozen in OCT compound (Fisher Scientific, NH) and cut into 10uM sections on a cryotome (Lieca, IL) and fixed in 2% paraformaldehyde. Islet β- and α-cells were stained as described (Zhou et al., 2000), using insulin and glucagon antibodies at 1:300 in combination with fluorescently conjugated secondary antibodies, fluorescein isothiocyanate together with ToPro3 nuclear stain (Invitrogen). Apoptosis was assessed using an antibody specific to cleaved caspase 3 (Cell Signaling, MA) in combination with a fluorescein isothiocyanate conjugated secondary antibody together with TO-PRO-3 iodide nuclear stain (Invitrogen Corp., CA); on islets stuck to coverslips and incubated for 4 days as described below.
Mouse Islet and β-cell isolation
Islets were isolated from pancreata of 1–5-month-old C57BL/J6 wild-type mice (Jackson Laboratories) using collagenase digestion and Ficoll gradients as previously described (Philipson et al., 1994). Islets were dissociated in 0.005% trypsin, placed on glass coverslips, and cultured for 16 hr in RPMI-1640 medium supplemented with 10% FCS, concentrations of glucose specified in the figure legends, 100 IU/ml penicillin, and 100 μg/ml streptomycin. Cells and islets were maintained in a humidified incubator at 37 °C under an atmosphere of 95% air/5% CO2.
Hormone secretion measurements
Mouse islets were allowed to recover following isolation for four hours. For insulin measurements, 20 islets/animal (n=5 animals per group, Fig. S3; n=10 animals per group Figure 1F) were perifused with KRB solution containing 2mM glucose for 15 minutes followed by KRB with 7mM for Fig. 1F or with KRB with 14mM glucose for 30 minutes and return to 2mM glucose from 30 to 60 minutes in the presence of 15mM TEA between 20 and 35 minutes and 30mM KCl between 45 and 60 minutes for Fig. S3. Collected perfusate (1mL/minute) were analyzed for insulin content using an ELISA based detection kit (Alpco diagnostics, NH) and presented as ±SEM.
Electrophysiological Recordings
Whole cell rupture patch recording
Voltage activated currents were recorded using whole-cell ruptured patch clamps with an Axopatch 200B amplifier and pCLAMP software (Molecular Devices, Sunnyvale, CA). Patch electrodes (2 to 4 Megaohm) were loaded with intracellular solution containing (in mmol/L) KCl, 140; MgCl2.6 H2O, 1; EGTA, 10; Hepes, 10; MgATP, 5; pH 7.25 with KOH. Islets were perifused with a Krebs-Ringer buffer (KRB) containing (in mmol/L) NaCl, 119; CaCl2.[(H2O)6], 2; KCl, 4.7; Hepes, 10; MgS04, 1.2; KH2PO4, 1.2; glucose, 14.4; adjusted to pH 7.3 with NaOH. In some cases current was used to induce action potentials with an 80 pA 5ms step and the resulting resulting voltages representing a stimulated action potential were recorded. When indicated, cells were treated with 100nM ScTx-1 (Alomone Labs, Jerusalem Israel).
Islet perforated patch recording
Patch electrodes (2 to 4 μohm) were loaded with intracellular solution containing (in mmol/L) KCl, 140; MgCl2.6 H2O, 1; EGTA, 10; Hepes, 10; pH 7.25 with KOH containing the pore-forming antibiotic amphotericin B (Rae et al., 1991)(Sigma). Islets were perifused with a Krebs-Ringer buffer (KRB) containing (in mmol/L) NaCl, 119; CaCl2.[(H2O)6], 2; KCl, 4.7; Hepes, 10; MgS04, 1.2; KH2PO4, 1.2; glucose as indicated; adjusted to pH 7.3 with NaOH. Cells on the periphery of islets on glass coverslips were sealed in voltage clamp at −80 while the amphotericin allowed perforation and good access over a few minutes. After switching to current clamp cells that had a resting membrane voltage near −65 were assumed to be β-cells and the glucose containing solution (at 37C) was perfused into a heated chamber (at 37C) and TEA (15mM) or ScTx-1 (100nM) were added.
Slow Speed Intracellular Calcium Imaging
Mouse islets attached to the glass bottom Matek tissue culture dishes were loaded with 5uM Fura-2 AM loaded in KRH2 for 30 min. at 37°C. Loaded islets were placed in a temperature controller (TC-202, Medical Systems Corp.) mounted on the stage of an inverted microscope (Nikon TE2000, Nikon) for imaging and images were acquired at 5 second intervals. Experiments were performed with constant perfusion with KRH2 and treated with 200uM tolbutamide in combination with 5, 10, and 15 mM TEA.
High Speed Intracelluar Calcium Imaging
Mouse islets attached to the glass bottom of Matek tissue culture dishes were loaded with 5uM Fluo-4 AM loaded in KRH2 for 45 min. at 37°C. Loaded islets were placed in a temperature controller (TC-202, Medical Systems Corp.) mounted on the stage of an inverted microscope (Olympus IX70, Olympus) for imaging. Experiments were performed at 37°C in static conditions. Confocal imaging was performed with a custom-built system based on Yokogawa CSU10 spinning disk confocal unit. The islets were excited with the 488nm line of Ar-Kr laser (Series 43, Omnichrome), the emitted light was filtered with a 535/40 filter (Chroma) and recorded with Coolsnap HQ digital camera (Roper Scientific). The acquisition was controlled with MetaMorph software (Universal Imaging). In all experiments, to obtain data with sufficiently high time resolution we used 10fps stream acquisition mode. Data analysis was performed with MetaMorph and Microsoft Excel.
Supplementary Material
Fig. S1. Kv2.1−/− and control islets stain equivalently for caspase 3 cleavage. Kv2.1 (A) and Control (B) islets stained for cleaved caspase 3 (green) in combination with nuclei (red); these images are 3D reconstructions. ). In all images the white line corresponds to 20um.
Fig. S2. Kv2.1−/− and control pancreata stain equivalently for insulin and glucagon. Kv2.1−/− (A) and C57 (B) pancreatic sections stained for insulin (green) and nuclei (blue). Kv2.1 −/− (C) and C57 (D) pancreatic sections stained for glucagon (green) and nuclei (blue). In all images the white line corresponds to 15um.
Fig. S3. Kv2.1−/− islets respond to TEA with increased glucose induced action potential amplitude and duration. (A) Control mouse islet glucose induced (14mM) action potentials treated with 15mM TEA (black bar). (B) Kv2.1−/− islet glucose induced (14mM) action potentials treated with 15mM TEA (black bar). (C and D) Representative islet calcium fluctuations in response to treatment with 200uM tolbutamide in combination with the indicated TEA concentrations, boxes.
Fig. S4. Action potential duration is increased and frequency is decreased in the Kv2.1−/− islets compared to controls. (A) Average action potential duration during a 5 second duration 2 minutes post islet depolarization induced by 14mM glucose +/− SDEV, n=7, p<0.0001. (B) Average action potential frequency during a 5 second duration 2 minutes post islet depolarization induced by 14mM glucose +/− SDEV, n=7, p<0.0001.
Acknowledgments
We thank O Pongs, SA Goldstein, L.E. Fridlyand, J.P. Lopez, and N.A. Tamarina for helpful discussion and suggestions; F. Mendez, S. Eames for technical expertise on islet isolation, glucose tolerance tests and genotyping assistance. This work was supported by NIH grant DK48494 (L.H.P.), the University of Chicago DRTC DK 20595 and DJ was supported in part by a postdoctoral fellowship in beta cell research from Takeda Pharmaceuticals North America.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahn JE, Byun JH, Ko MS, Park SH, Lee MG. Case report: neuroendocrine carcinoma of the gallbladder causing hyperinsulinaemic hypoglycaemia. Clin Radiol. 2007;62:391–394. doi: 10.1016/j.crad.2006.11.016. [DOI] [PubMed] [Google Scholar]
- Ashcroft FM, Rorsman P. Electrophysiology of the pancreatic beta-cell. Prog Biophys Mol Biol. 1989;54:87–143. doi: 10.1016/0079-6107(89)90013-8. [DOI] [PubMed] [Google Scholar]
- Atwater I, Ribalet B, Rojas E. Mouse pancreatic beta-cells: tetraethylammonium blockage of the potassium permeability increase induced by depolarization. J Physiol. 1979;288:561–574. [PMC free article] [PubMed] [Google Scholar]
- Beigelman PM, Thomas LJ, Shu MJ, Bessman SP. Insulin from individual isolated islets of Langerhans 2. Effect of glucose in varying concentrations. J Physiol (Paris) 1976;72:721–728. [PubMed] [Google Scholar]
- Betsholtz C, Baumann A, Kenna S, Ashcroft FM, Ashcroft SJ, Berggren PO, Grupe A, Pongs O, Rorsman P, Sandblom J, et al. Expression of voltage-gated K+ channels in insulin-producing cells. Analysis by polymerase chain reaction. FEBS Lett. 1990;263:121–126. doi: 10.1016/0014-5793(90)80719-y. [DOI] [PubMed] [Google Scholar]
- Bokvist K, Rorsman P, Smith PA. Effects of external tetraethylammonium ions and quinine on delayed rectifying K+ channels in mouse pancreatic beta-cells. J Physiol. 1990;423:311–325. doi: 10.1113/jphysiol.1990.sp018024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dean PM, Matthews EK. Glucose-induced electrical activity in pancreatic islet cells. J Physiol. 1970;210:255–264. doi: 10.1113/jphysiol.1970.sp009207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dukes ID, Philipson LH. K+ channels: generating excitement in pancreatic beta-cells. Diabetes. 1996;45:845–853. doi: 10.2337/diab.45.7.845. [DOI] [PubMed] [Google Scholar]
- Escoubas P, Diochot S, Celerier ML, Nakajima T, Lazdunski M. Novel tarantula toxins for subtypes of voltage-dependent potassium channels in the Kv2 and Kv4 subfamilies. Mol Pharmacol. 2002;62:48–57. doi: 10.1124/mol.62.1.48. [DOI] [PubMed] [Google Scholar]
- Gopel SO, Kanno T, Barg S, Rorsman P. Patch-clamp characterisation of somatostatin-secreting -cells in intact mouse pancreatic islets. J Physiol. 2000;528:497–507. doi: 10.1111/j.1469-7793.2000.00497.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henquin JC. Tetraethylammonium potentiation of insulin release and inhibition of rubidium efflux in pancreatic islets. Biochem Biophys Res Commun. 1977;77:551–556. doi: 10.1016/s0006-291x(77)80014-4. [DOI] [PubMed] [Google Scholar]
- Herrington J, Sanchez M, Wunderler D, Yan L, Bugianesi RM, Dick IE, Clark SA, Brochu RM, Priest BT, Kohler MG, McManus OB. Biophysical and pharmacological properties of the voltage-gated potassium current of human pancreatic beta-cells. J Physiol. 2005;567:159–175. doi: 10.1113/jphysiol.2005.089375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrington J, Zhou YP, Bugianesi RM, Dulski PM, Feng Y, Warren VA, Smith MM, Kohler MG, Garsky VM, Sanchez M, et al. Blockers of the delayed-rectifier potassium current in pancreatic beta-cells enhance glucose-dependent insulin secretion. Diabetes. 2006;55:1034–1042. doi: 10.2337/diabetes.55.04.06.db05-0788. [DOI] [PubMed] [Google Scholar]
- Herrington J. Gating modifier peptides as probes of pancreatic beta-cell physiology. Toxicon. 2007;49:231–238. doi: 10.1016/j.toxicon.2006.09.012. [DOI] [PubMed] [Google Scholar]
- Houamed K, Fu J, Roe MW, Philipson LH. Electrophysiology of the pancreatic beta cell. In: LeRoith D, Taylor SI, Olefsky JM, editors. Diabetes Mellitus. Lippincott Williams and Wilkins; Philadelphia: 2004. pp. 51–68. [Google Scholar]
- Kirsch GE, Drewe JA. Gating-dependent mechanism of 4-aminopyridine block in two related potassium channels. J Gen Physiol. 1993;102:797–816. doi: 10.1085/jgp.102.5.797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kukuljan M, Goncalves AA, Atwater I. Charybdotoxin-sensitive K(Ca) channel is not involved in glucose-induced electrical activity in pancreatic beta-cells. J Membr Biol. 1991;119:187–195. doi: 10.1007/BF01871418. [DOI] [PubMed] [Google Scholar]
- MacDonald PE, Ha XF, Wang J, Smukler SR, Sun AM, Gaisano HY, Salapatek AM, Backx PH, Wheeler MB. Members of the Kv1 and Kv2 voltage-dependent K(+) channel families regulate insulin secretion. Mol Endocrinol. 2001;15:1423–1435. doi: 10.1210/mend.15.8.0685. [DOI] [PubMed] [Google Scholar]
- MacDonald PE, Sewing S, Wang J, Joseph JW, Smukler SR, Sakellaropoulos G, Wang J, Saleh MC, Chan CB, Tsushima RG, Salapatek AM, Wheeler MB. Inhibition of Kv2.1 voltage-dependent K+ channels in pancreatic beta-cells enhances glucose-dependent insulin secretion. J Biol Chem. 2002;277:44938–44945. doi: 10.1074/jbc.M205532200. [DOI] [PubMed] [Google Scholar]
- MacDonald PE, Wang G, Tsuk S, Dodo C, Kang Y, Tang L, Wheeler MB, Cattral MS, Lakey JR, Salapatek AM, Lotan I, Gaisano HY. Synaptosome-associated protein of 25 kilodaltons modulates Kv2.1 voltage-dependent K(+) channels in neuroendocrine islet beta-cells through an interaction with the channel N terminus. Mol Endocrinol. 2002;16:2452–2461. doi: 10.1210/me.2002-0058. [DOI] [PubMed] [Google Scholar]
- Nestorowicz A, Wilson BA, Schoor KP, Inoue H, Glaser B, Landau H, Stanley CA, Thornton PS, Clement JP, 4th, Bryan J, Aguilar-Bryan L, Permutt MA. Mutations in the sulonylurea receptor gene are associated with familial hyperinsulinism in Ashkenazi Jews. Hum Mol Genet. 1996;5:1813–1822. doi: 10.1093/hmg/5.11.1813. [DOI] [PubMed] [Google Scholar]
- Philipson LH, Rosenberg MP, Kuznetsov A, Lancaster ME, Worley JF, 3rd, Roe MW, Dukes ID. Delayed rectifier K+ channel overexpression in transgenic islets and beta-cells associated with impaired glucose responsiveness. J Biol Chem. 1994;269:27787–27790. [PubMed] [Google Scholar]
- Philipson LH. Beta-cell ion channels: keys to endodermal excitability. Horm Metab Res. 1999;31:455–461. doi: 10.1055/s-2007-978774. [DOI] [PubMed] [Google Scholar]
- Rae J, Cooper K, Gates P, Watsky M. Low access resistance perforated patch recordings using amphotericin B. J Neurosci Methods. 1991;37:15–26. doi: 10.1016/0165-0270(91)90017-t. [DOI] [PubMed] [Google Scholar]
- Roe MW, Worley JF, 3rd, Mittal AA, Kuznetsov A, DasGupta S, Mertz RJ, Witherspoon SM, 3rd, Blair N, Lancaster ME, McIntyre MS, Shehee WR, Dukes ID, Philipson LH. Expression and function of pancreatic beta-cell delayed rectifier K+ channels. Role in stimulus-secretion coupling. J Biol Chem. 1996;271:32241–32246. doi: 10.1074/jbc.271.50.32241. [DOI] [PubMed] [Google Scholar]
- Rorsman P, Trube G. Calcium and delayed potassium currents in mouse pancreatic beta-cells under voltage-clamp conditions. J Physiol. 1986;374:531–550. doi: 10.1113/jphysiol.1986.sp016096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rorsman P, Bokvist K, Ammala C, Eliasson L, Renstrom E, Gabel J. Ion channels, electrical activity and insulin secretion. Diabete Metab. 1994;20:138–145. [PubMed] [Google Scholar]
- Shieh CC, Coghlan M, Sullivan JP, Gopalakrishnan M. Potassium channels: molecular defects, diseases, and therapeutic opportunities. Pharmacol Rev. 2000;52:557–594. [PubMed] [Google Scholar]
- Smith PA, Bokvist K, Arkhammar P, Berggren PO, Rorsman P. Delayed rectifying and calcium-activated K+ channels and their significance for action potential repolarization in mouse pancreatic beta-cells. J Gen Physiol. 1990;95:1041–1059. doi: 10.1085/jgp.95.6.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su J, Yu H, Lenka N, Hescheler J, Ullrich S. The expression and regulation of depolarization-activated K+ channels in the insulin-secreting cell line INS-1. Pflugers Arch. 2001;442:49–56. doi: 10.1007/s004240000508. [DOI] [PubMed] [Google Scholar]
- Swartz KJ, MacKinnon R. An inhibitor of the Kv2.1 potassium channel isolated from the venom of a Chilean tarantula. Neuron. 1995;15:941–949. doi: 10.1016/0896-6273(95)90184-1. [DOI] [PubMed] [Google Scholar]
- Tamarina NA, Kuznetsov A, Fridlyand LE, Philipson LH. Delayed-rectifier (KV2.1) regulation of pancreatic beta-cell calcium responses to glucose: inhibitor specificity and modeling. Am J Physiol Endocrinol Metab. 2005;289:E578–E585. doi: 10.1152/ajpendo.00054.2005. [DOI] [PubMed] [Google Scholar]
- Yan L, Figueroa DJ, Austin CP, Liu Y, Bugianesi RM, Slaughter RS, Kaczorowski GJ, Kohler MG. Expression of voltage-gated potassium channels in human and rhesus pancreatic islets. Diabetes. 2004;53:597–607. doi: 10.2337/diabetes.53.3.597. [DOI] [PubMed] [Google Scholar]
- Zhou YP, Pena JC, Roe MW, Mittal A, Levisetti M, Baldwin AC, Pugh W, Ostrega D, Ahmed N, Bindokas VP, et al. Overexpression of Bcl-x(L) in beta-cells prevents cell death but impairs mitochondrial signal for insulin secretion. Am J Physiol Endocrinol Metab. 2000;278:E340–E351. doi: 10.1152/ajpendo.2000.278.2.E340. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Gong Q, Epstein ML, January CT. HERG channel dysfunction in human long QT syndrome: intracellular transport and functional defects. J Biol Chem. 1998;273:21061–21066. doi: 10.1074/jbc.273.33.21061. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Kv2.1−/− and control islets stain equivalently for caspase 3 cleavage. Kv2.1 (A) and Control (B) islets stained for cleaved caspase 3 (green) in combination with nuclei (red); these images are 3D reconstructions. ). In all images the white line corresponds to 20um.
Fig. S2. Kv2.1−/− and control pancreata stain equivalently for insulin and glucagon. Kv2.1−/− (A) and C57 (B) pancreatic sections stained for insulin (green) and nuclei (blue). Kv2.1 −/− (C) and C57 (D) pancreatic sections stained for glucagon (green) and nuclei (blue). In all images the white line corresponds to 15um.
Fig. S3. Kv2.1−/− islets respond to TEA with increased glucose induced action potential amplitude and duration. (A) Control mouse islet glucose induced (14mM) action potentials treated with 15mM TEA (black bar). (B) Kv2.1−/− islet glucose induced (14mM) action potentials treated with 15mM TEA (black bar). (C and D) Representative islet calcium fluctuations in response to treatment with 200uM tolbutamide in combination with the indicated TEA concentrations, boxes.
Fig. S4. Action potential duration is increased and frequency is decreased in the Kv2.1−/− islets compared to controls. (A) Average action potential duration during a 5 second duration 2 minutes post islet depolarization induced by 14mM glucose +/− SDEV, n=7, p<0.0001. (B) Average action potential frequency during a 5 second duration 2 minutes post islet depolarization induced by 14mM glucose +/− SDEV, n=7, p<0.0001.