

Abstract
Objective
Using a clinically relevant transduction strategy, we investigated to what extent hematopoietic stem cells in lineage-negative bone marrow (Linneg BM) could be genetically modified with a FV vector that expresses the DNA repair protein, O6-methylguanine DNA methyltransferase (MGMTP140K) and selected in vivo with submyeloablative versus myeloablative alkylator therapy.
Methods
Linneg BM was transduced at a low multiplicity-of-infection (MOI), with the FV vector, MD9-P140K, that co-expresses MGMTP140K and the enhanced green fluorescent protein, transplanted into C57BL/6 mice, and mice treated with submyeloablative or myeloablative alkylator therapy. The BM was analyzed for the presence of in vivo selected, MD9-P140K-transduced cells at 6 months post-transplantation and subsequently transplanted into secondary recipient animals.
Results
Following submyeloablative therapy, 55% of the mice expressed MGMTP140K in the BM. Proviral integration was observed in ∼50% of committed BM-derived progenitors and analysis of proviral insertion sites indicated up to 2 integrations per transduced progenitor colony. Transduced BM cells selected with submyeloablative therapy reconstituted secondary recipient mice for up to 6 months post-transplantation. In contrast, following delivery of myeloablative therapy to primary recipient mice, only 25% survived. Hematopoietic stem cells were transduced since BM cells from the surviving animals reconstituted secondary recipients with MGMTP140K positive cells for 5-6 months.
Conclusions
In vivo selection of MD9-P140K-transduced BM cells was more efficient following submyeloablative versus myeloablative therapy. These data indicate that a critical number of transduced-stem cells must be present to produce sufficient numbers of genetically modified progeny to protect against the acute toxicity associated with myeloablative therapy.
Keywords: gene therapy, hematopoietic stem cells, foamy virus vector, O6-methylguanine DNA methyltransferase (MGMT)
Introduction
Recent stem-cell gene therapy trials have demonstrated the feasibility of correcting genetically defective hematopoietic stem cells (HSC) by transfer of correct genetic sequences to HSC via gammaretroviral vectors [1-4]. Adverse events, however, directly related to gammaretroviral-mediated insertional mutagenesis, have been reported in a French trial for severe combined immunodeficiency (SCID)-X1 [5]. In addition, preclinical murine studies indicate that mouse HSC can occasionally be transformed by insertional mutagenesis [6, 7]. Another concern is that the extended ex vivo culture required for gammaretroviral transduction can lead to loss of HSC or as in the case of Fanconi anemia murine HSC, lead to genotoxic events resulting in MDS and AML months after transplantation [8].
To prevent adverse side effects related to viral vector transduction, alternative vector systems and transduction strategies requiring less ex vivo manipulation time and possessing a potentially safer proviral integration profile are needed. Retroviral vector systems based on the complex retroviridae lentivirus and foamy virus (FV) exhibit different integration patterns compared to the commonly used gammaretroviral vectors [9-11]. FV proviral integration was observed less often near promoter regions compared to oncoretroviral vectors [11] and less frequently in gene sequences compared to lentiviral vectors [11], suggesting that FV vectors may integrate in positions that modulate host gene expression to a lesser extent than oncoretroviral or lentiviral vectors. In addition, in contrast to all other retroviruses used for gene therapy, no known diseases have been associated with naturally occurring FV infections in cats, horses, chimpanzees, orangutans, and cows [12]. While there are several cases of health care providers in zoos who have accidentally contracted simian FV, none of these individuals have developed any symptoms or transmitted the virus to other humans despite the chronic persistent infection [13-16].
One feature of FV vectors that warrants consideration when designing transduction protocols, is that while these vectors efficiently enter resting cells, foamy viruses do not integrate into the host genome until cell division occurs [17, 18]. The FV virion contains full-length double stranded DNA (dsDNA) previously reverse transcribed from the viral RNA into dsDNA during the packaging process [12]. A substantial lag period of at least 2 weeks between exposure to FV and FV integration into the host genome can occur since the dsDNA intermediate is highly stable in the cytoplasm of the target cell [18]. Therefore in stem-cell transplant studies, promotion of cell division either in vitro or in vivo after transplantation may be required for efficient transduction of the transplanted stem-cell pool.
Viral vector systems derived from lentivirus or FV are currently under investigation in small and large animal models [9, 19-31]. Russell and colleagues demonstrated that murine stem cells residing in 5-fluorouracil (5FU)-treated bone marrow (BM) [24] and SCID-repopulating cells derived from umbilical cord blood and G-CSF-mobilized peripheral blood could be marked with FV vectors.[21, 22] We previously demonstrated similar levels of gene marking using FV or lentiviral vectors to transduce SCID-repopulating cells derived from human umbilical cord blood CD34+ cells [23]. In addition, FV-mediated gene transfer of canine hematopoietic long-term repopulating cells has been recently reported [32].
The objective of our study was to determine to what extent relatively quiescent HSC could be transduced overnight with a FV vector at low multiplicity-of-infection (MOI) and subsequently selected in vivo by alkylator treatment. We utilized the mutant form of O6-methylguanine DNA methyltransferase, MGMTP140K for in vivo selection of HSC [33, 34]. The FV backbone MD9 [35] which contains minimal cis-acting sequences and does not express any viral gene products was utilized to co-expresses MGMTP140K and the enhanced green fluorescent protein. Transplantation of FV-transduced cells into primary and secondary recipient mice demonstrated that FV-mediated gene transfer into long-term repopulating hematopoietic stem cells was feasible but in vivo selection was required to detect transduced progeny. In addition, following in vivo selection, expression of the MGMTP140K protein in the BM was stable for at least one year post-transplant.
Materials and Methods
Vector constructs
The MD9 vector is derived from the prototype (formerly human) FV and contains only minimal cis-acting sequences and does not express any viral gene products [35]. The MD9-P140K-IRES-EGFP (MD9-P140K) FV viral vector co-expresses the 6BG-resistant MGMT mutant, MGMTP140K, and EGFP (Fig 1A). The vector was constructed by subcloning a 5′-MGMTP140K-IRES-EGFP -3′cDNA fragment derived from the SF1-P140K-IRES-EGFP oncoretroviral vector downstream of the internal spleen focus-forming virus U3 promoter (SFFV U3) in the MD9 vector [36]. The GAG/POL packaging construct, pCgp-1, was a kind gift from Axel Rethwilm (Würzburg, Germany). The envelope plasmid, pczPFVenvEMO2, expresses the foamy virus wild-type envelope, EMO2, and was generously supplied by Dirk Lindemann (Universitat Wurzburg, Dresden, Germany). All recombinant DNA work was approved by the Indiana University Medical Center Institutional Biosafety committee and was performed under Biosafety Level-2.
Figure 1. MD9-P140K FV vector.
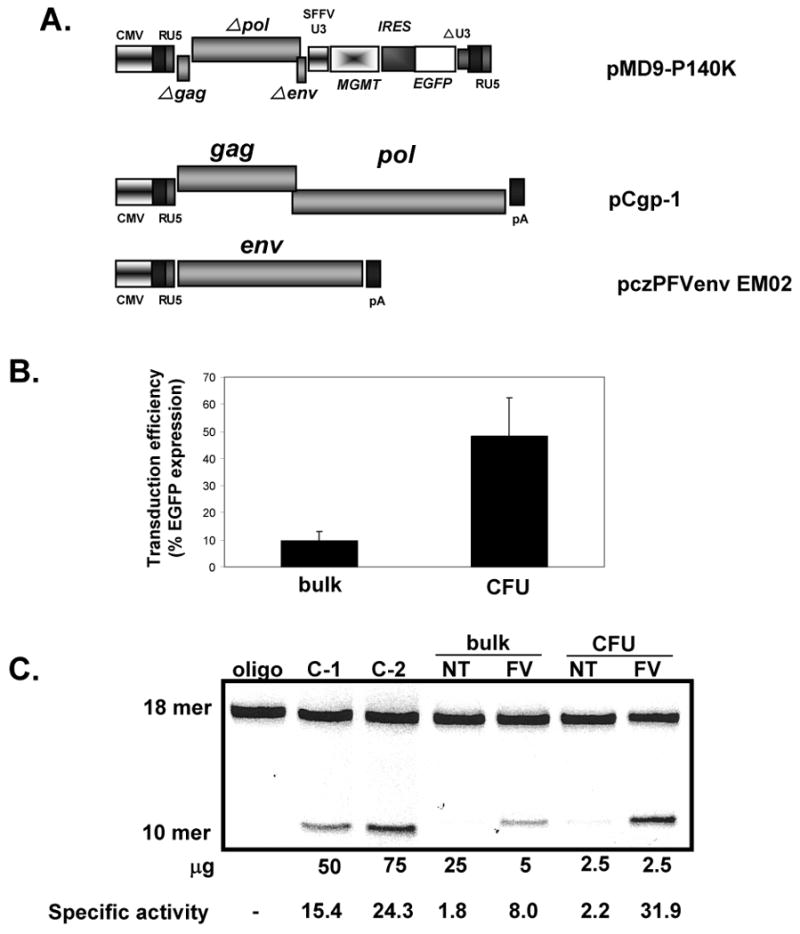
(A) FV expression (MD9-P140K), envelop (ENV: pczPFVenvEM02) and the GAG/POL transfer (GAG/POL: pCgp-1) plasmids. (B) EGFP expression in bulk and progenitor cells (CFU). (C) MGMT specific activity. Negative control (oligo) contains all reagents except lysate. Positive controls (C-1 and C-2) are lysates from an ovarian cancer cell line (OVCAR). Depending on the level of MGMT activity, different amounts of protein were run to avoid signal saturation. Specific activity = fmol O6 methylguanine removed per mg total protein.
Generation of MD9-P140K-containing supernatant
MD9-P140K supernatant was generated using the 293T cell line as previously described with some modification [23]. For transfection, 4 × 106 cells were plated on 10-cm tissue culture dishes and transfected at 75% confluency. Transfections were performed with 5 μg of each plasmid (MD9-P140K, pczPFVenvEMO2, and pCgp-1) in the presence of Polyfect (Qiagen Inc, Valencia, CA) according to the manufacturer's instructions. The following day, the media was replaced with fresh media containing 10 mM sodium butyrate; 8 hours later, the media was replaced with fresh media without sodium butyrate. MD9-P140K supernatants were harvested 24 hours later and passed through 0.45 μm filters (VWR International, West Chester, PA) and used fresh or stored at −80°C. Experiments 1, 3 and 4 used frozen supernatant and experiment 2 used fresh supernatant. Supernatants were concentrated and titered on HT1080 cells as previously described [23]. The titers of the viral supernatant were 1-2 × 107 infectious units/ml.
Mice
C57BL/6J mice purchased from Jackson Laboratories (Bar Harbor, ME) were used at 7-8 weeks of age for transplantation. Animals were housed in positive airflow ventilated racks and maintained in microisolators under specific pathogen-free conditions. Mice were placed on doxycycline-food pellets one week prior to split-dose total-body irradiation using a GammaCell 40 (Nordion International Inc., Ontario Canada) equipped with two opposing 137Cesium sources. A split-dose schema consisting of 700 cGy followed by 400 cGy 4 hours later was employed. All animal studies were approved by the Indiana University Animal Care and Use Committee. All transplanted animals were maintained for the duration of the experiment in a Biosafety Level-2 housing facility.
Isolation of Lineage negative (Linneg) BM cells
BM cells from 8- to 14-week-old C57BL/6J were collected from the femurs, tibias, and iliac crest. Mononuclear cells were stained using the mouse lineage panel (BD Biosciences, San Diego, CA) and Linneg BM cells were isolated via magnetic separation according to the manufacturer's instructions (Miltenyi Biotec, Bergisch Gladbach, Germany). In four independent experiments, flow cytometric analysis indicated that the purity was 85-94% Linneg cells.
Transduction protocols
Linneg BM cells were transduced at a density of 2.5×105/cm2 on nontissue culture plates (VWR) coated at 2 μg/cm2 RetroNectin™ (Takaraa Bio Inc., Otsu, Japan). [36, 37] The starting cell number per mouse was 4 × 105 Linneg cells per mouse. The transductions were perfomed in the presence of 100 ng/ml human granulocyte-colony stimulating factor, murine stem cell factor (SCF), and human thrombopoietin (Peprotech, Rocky Hill, NJ). At 10-14 hours post-transduction, cells were harvested using dissociation buffer (Invitrogen, Grand Island, NY) and plated as described below in colony-forming unit (CFU) assays to determine the transduction efficiency. An aliquot of cells was expanded in cytokines and analyzed for EGFP expression over the next week of culture. After 3 days, the level of EGFP expression did not change with increased culture time (data not shown).
Colony-forming unit (CFU) assays
Progenitor assays were performed immediately after the over-night transduction in complete methylcellulose medium (Methocult GF, M3434, Stem Cell Technologies Inc, Vancouver, BC) as previously described [36]. For assays using freshly transduced cells, the cells were plated at 1000 cells per ml; for assays using BM cells from transplanted mice, the cells were plated at 2.5 × 104 and 5 × 104 per ml. After 14 days of incubation, colony forming units-granulocyte-macrophage (CFU-GM), burst-forming units-erythroid (BFU-E), and colony forming units-mixed lineage (CFU-mix) were enumerated using the Axiovert 25 inverted fluorescence microscope. For in vitro experiments, direct visualization of EGFP fluorescence from colonies was used to determine transduction efficiency.
In vivo treatment with submyeloablative and myeloablative alkylator therapy
As described previously, cohorts of mice were randomly grouped and either vehicle-treated or treated with submyeloablative or myeloablative therapy at 4 weeks post-transplant [36]. Submyeloablative therapy consisted of 20 mg/kg 6BG followed by 5 mg/kg BCNU one hour later. Myeloablative therapy consisted of 30 mg/kg 6BG followed by 10 mg/kg BCNU one hour later and 15 mg/kg 6BG 7 hours later. Two cycles of therapy were given at weeks 4 and 6 post-transplantation.
Polymerase chain reaction (PCR) of proviral sequences from bone marrow-derived progenitor colonies from transplanted mice
Progenitor colonies were plucked from plates in which colonies were appropriately separated to prevent cross-contamination and placed in 200 μl Puregene lysis buffer and processed according to the manufacturer's instructions (Gentra Systems/ISIS, Wicklow, Ireland). Prior to final precipitation of the DNA, glycogen (10 μg/ml) was added to each DNA sample to increase recovery of the DNA. For each sample analyzed, one fourth of the sample was incubated with an EGFP-specific forward primer, 5′-TTTAAGCTTGCCACCATGGTGAGCAAGGGCGAG-3′ and the reverse 5′-TTTGGATCCTTACTTGTACAGCTCGTCCATGCC-3′ which amplifies a 468 bp fragment. Another aliquot was incubated with a β-globin-specific forward primer, 5′-CAATCCAGCTACCATTCTGC-3′ and the reverse primer, 5′-GAATCCAGATGCTCAAGGCC-3′ which amplifies a 344 bp fragment using previously published amplification conditions [38]. The negative control was DNA isolated from human erythroleukemia cells (HEL) and the positive control was DNA isolated from HEL cells containing one copy of the MFG-EGFP oncoretrovirus [39]. The amplified samples were run on 1% agarose gels and stained with ethidium bromide and photographed using the Kodak Digital Science camera (model TVL-312A).
Western analysis of MGMT expression in BM from primary and secondary reconstitution experiments
Protein lysates of BM cells were prepared using 1X Cell Lysis Buffer from Cell Signaling Technology, Danvers, MA, separated on 12% Tris-HCl polyacrylamide gels (Bio-Rad Laboratories, Inc., Hercules, CA), and proteins transferred to a nitrocellulose membrane with a 0.45 μm pore size (Bio-Rad). The membrane was then blocked with 5% milk containing 0.1% Tween 20 (Sigma-Aldrich Co., St. Louis, MO) and probed with an anti-MGMT antibody (clone MT3.1) (Chemicon International, Inc., Temecula, CA) overnight at 4 °C. The MT3.1 antibody is specific for human MGMT and does not recognize mouse MGMT. The next day, the membrane was washed and incubated with goat anti-mouse HRP-conjugated secondary antibody according to the manufacturer's instructions (Pierce Biotechnology, Inc., Rockford, IL). MGMT was detected with Supersignal West Femto Maximum Sensitivity Substrate (Pierce) and the blot exposed to x-ray film (Midwest Scientific, St. Louis, MO).
MGMT bioactivity assay
MGMT activity were determined as previously described by Kreklau et al [40]. Lysates were prepared from nontransduced and foamy viral-transduced bulk cultures at 3-days post-expansion and from forty pooled progenitor colonies derived from these samples. In vehicle- and drug-treated transplanted mice, lysates were prepared from the bone marrow and from forty pooled progenitor colonies derived from the bone marrow. Lysates were incubated with a fluorometric-5′-hexachloro-fluorescein phosphoramidite (HEX)-labeled oligonucleotide containing a single O6-methylguanine residue nested within a PvuII restriction site. Lysates were incubated with PvuII and PvuII-mediated cleavage occurred if O6-methylguanine residues were repaired by MGMT. To determine 6BG-resistant activity, some samples were run in parallel with a duplicate sample preincubated with 20 μM 6BG. Samples were run on a polyacrylamide gel and the amount of cleaved product was quantitated using the Hitachi FMBio II Fluorescence Imaging System (Hitachi Genetic Systems, South San Francisco, CA)
Ligation-mediated Polymerase Chain Reaction (LM-PCR)
For detection of FV integration sites, BM from four primary recipients transplanted with MD9-P140K-transduced BM and subsequently treated with submyeloablative therapy were plated in standard progenitor assay. From each of the mice, 20-30 progenitor colonies were picked and then subjected to LM-PCR as described previously [41] with minor variations. The restriction enzyme used here was Hae III (New England Biolabs, Frankfurt, Germany). The biotinylated primer 5′biotin-GTACAATCTAGGTGACCACTTTC-3′ (407) was used in a one step extension at 94°C, 15 sec; 58°C, 2 minutes; 72°C, 10 minutes; 2 cycles. The two internal primers for the nested PCR were 5′-TCTCATCCCAGGTACGTCTATGA-3′ (404) and AP2 as previously described[41]. The DNA from excised bands were cloned into pCR2.1 using the TOPO cloning kit (Invitrogen) and then sequenced on an ABI Gene Amp 3170 System. SeqMap (http://seqmap.compbio.iupui.edu/) was used to map the sequences against the mouse genome. This was independently confirmed by mapping the positions using the ENSEMBL website (http://ensemble.org) and mus musculus database release 42, Dec 2006 and the UCSC Genome Browser (http://genome.ucsc.edu).
Results
Transduction efficiency and MGMT activity in Linneg BM cells transduced with the MD9-P140K foamy virus vector
In order to test clinically relevant parameters, we transduced Linneg BM cells with recombinant FV MD9-P140K virions a MOI of 5-8 (Fig 1) in the presence of cytokines on the recombinant fibronectin fragment CH-296 for 10-14 hours. In 4 independent experiments, EGFP expression was utilized as the read-out for in vitro transduction efficiency and analyzed in bulk cultures via flow cytometry and in clonogenic progenitor assays using fluorescence microscopy (Fig 1B). The transduction efficiency of the bulk culture was 9.5% ± 3% at 3 days post-transduction, while the transduction efficiency of the clongenic cells as measured by the colony-forming unit (CFU) assay was 48% ± 14%. In the CFU assays, similar numbers of CFU were present in transduced and nontransduced cultures, indicating no gross toxicity occurred at these MOIs during the in vitro transduction (data not shown). MGMT activity assays performed in parallel correlated with these transduction levels (Fig 1C).
Detection of MD9-P140K-tranduced cells in primary and secondary recipient mice following submyeloablative treatment
MD9-P140K-transduced BM cells were transplanted into lethally irradiated recipient mice. One month post-transplant, mice were treated with vehicle or two cycles of a submyeloablative regimen (Table 1). Although >90% of the transplanted mice survived these treatment cycles, MGMT expression was not detectable in the BM of vehicle-treated mice at 6 months post-transplantation, indicating that the overall gene-transfer efficiency into repopulating HSC was below the detection level. In drug-treated mice, ∼55% of the mice expressed high levels of the MGMT protein in the BM (Table 1, expts 1 & 2 and Fig 2A) demonstrating that transduced repopulating HSC were present and in vivo selection was feasible. In addition, in vivo selection was observed at the progenitor level. MGMT bioactivity increased by more than 20 fold in pooled progenitor colonies derived from the bone marrow of drug-treated mice compared to progenitor colonies from vehicle-treated mice (Fig 2B). EGFP expression was not used as a read-out for gene transfer in the transplanted mice; in our experience, EGFP expression 3′ to an IRES element in FV vectors is not a reliable marker for determination of transduction efficiency in vivo (Hanenberg, Pollok, and Clapp, unpublished observations). EGFP PCR was used, however, to monitor transduction efficiency of the clonogenic progenitor cells. Amplification of proviral DNA from isolated progenitor colonies with EGFP-specific primers indicated that approximately 50% of the colonies contained integrated proviral DNA (Fig 3). As expected, analysis of proviral integration sites in progenitor colonies derived from drug-treated transplanted mice by LM-PCR analysis indicated a low number of proviral insertion sites. Analysis of 89 progenitor colonies from 4 mice revealed only 3 unique proviral integration sites in the Cugbp2, Ddx, and the Cnot61 genes (Table 2). To demonstrate that true long-term repopulating stem cells were initially transduced and selected in vivo, pooled BM from mice treated with the submyeloablative regimen were transplanted into secondary recipient mice and analyzed for engraftment of transduced cells 5-6 months later (Fig 4A). Subsequently, cohorts were treated twice with vehicle or submyeloablative therapy and 75% of the vehicle-treated mice and 67% of the drug-treated mice survived. The nonsurviving mice died within 2-4 months post transplant due to BM aplasia; no gross signs of leukemic cells were observed in any mouse in the peripheral blood, BM, or spleen. In surviving mice, MGMT expression in the BM was detected in the majority of vehicle and drug-treated secondary recipients (Fig 4A). In addition, 6BG-resistant MGMT activity was detected in the BM of drug-treated mice from both primary and secondary transplants confirming that MGMT expression was indeed derived from the MGMTP140K transgene introduced into repopulating stem cells with FV vectors (Fig 5).
Table 1.
Overview of primary transplantation experiments.
| Experiment | 6BG/BCNU dose | Vehicle-treated mice | 6BG/BCNU-treated mice | ||
|---|---|---|---|---|---|
| survival | MGMTP140K expression | survival | MGMTP140K expression | ||
| 1 | submyeloablativea | 3/4 | 0/4 | 6/6 | 3/6 |
| 2 | submyeloablative | 4/4 | 1/4 | 5/6 | 3/5 |
| 3 | myeloablativeb | 4/4 | 0/4 | 2/6 | 2/2 |
| 4 | myeloablative | 3/4 | 0/4 | 1/6 | 1/1 |
20 mg/kg 6BG followed one hour later by 5 mg/kg BCNU administered at 4 weeks and 6 weeks post-transplantation.
30 mg/kg 6BG followed one hour later by 10 mg/kg BNCU and 15 mg/kg 6BG 7 hours after initial 6BG bolus administered at 4 weeks and 6 weeks post-transplantation.
Figure 2. MGMTP140K expression in transplanted mice treated with vehicle or submyeloablative therapy.
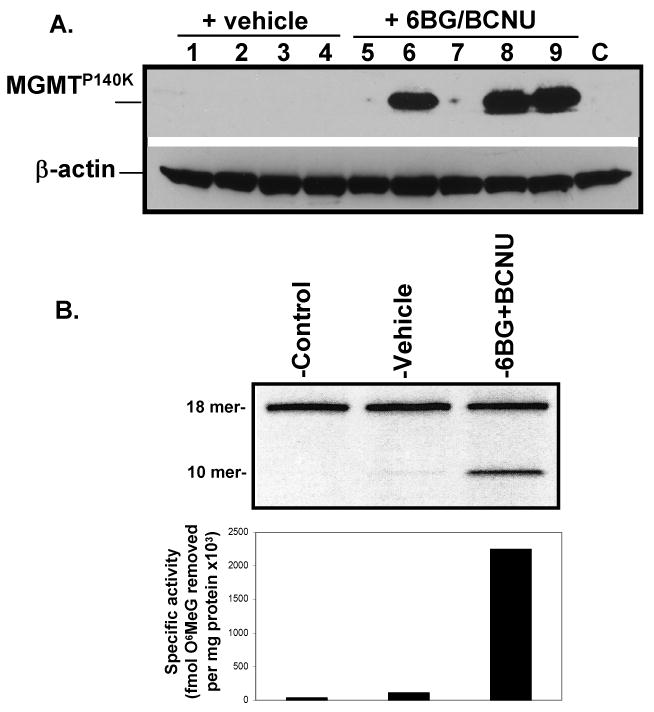
(A) Western analysis of BM from mice transplanted with MD9-P140K-transduced Linneg BM cells and selected in vivo. Data from experiment #2 are shown. (B) MGMT specific activity in pooled progenitor colonies derived from BM of control, vehicle- and drug-treated mice. Forty progenitor colonies were randomly plucked at 12-14 days after plating and analyzed for MGMT repair activity. These data are representative of individual mice from experiments 1 and 2. Specific activity = fmol O6 methylguanine removed per mg total protein.
Figure 3. Percentage of FV integration in CFU derived from drug-treated mice.
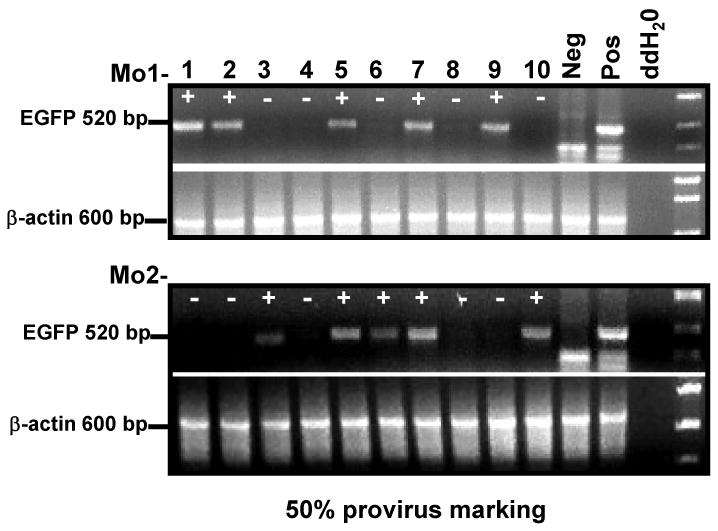
Progenitor colonies were analyzed for provirus integration by EGFP-specific PCR. Neg=HEL cells. Pos=HEL cell containing one copy of an EGFP oncoretrovirus per genome. [39] The marking frequency was determined by dividing the number of EGFP-positive colonies by the total number of colonies analyzed. EGFP PCR was performed on at least 40 colonies per mouse tested.
Table 2.
Proviral integration site analysis of progenitor colonies derived from in vivo selected MD9-P140K-transduced BM.
| Total # of mice analyzed* | Total # of colonies analyzed | Total # of specific integration sites | Location of proviral insertion-Chromosome/band | Specific insertion site-gene |
|---|---|---|---|---|
| 4 | 89 | 3 | 2, A1 12, A2 5, E3 |
Cugbp2 Ddx Cnot6l |
BM progenitors derived from transplanted mice treated with submyeloablative therapy.
Figure 4. MGMT expression in secondary transplants.
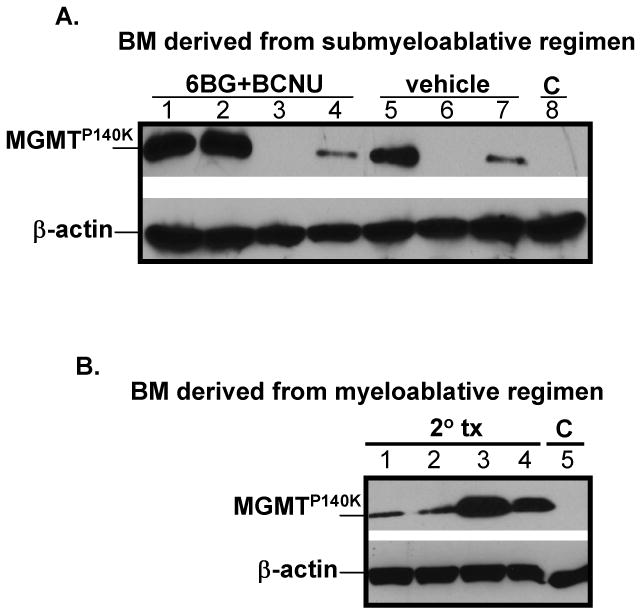
(A) Secondary recipients were transplanted with MD9-P140K-transduced BM cells from primary mice treated with submyeloablative therapy. These mice were subsequently treated with vehicle or submyeloablative therapy and MGMT expression determined. (B) BM of secondary recipient mice transplanted with MD9-P140K-transduced BM cells from primary mice treated with myeloablative doses of chemotherapy.
Figure 5. Total MGMT and 6BG-resistant activity in primary and secondary transplanted mice.
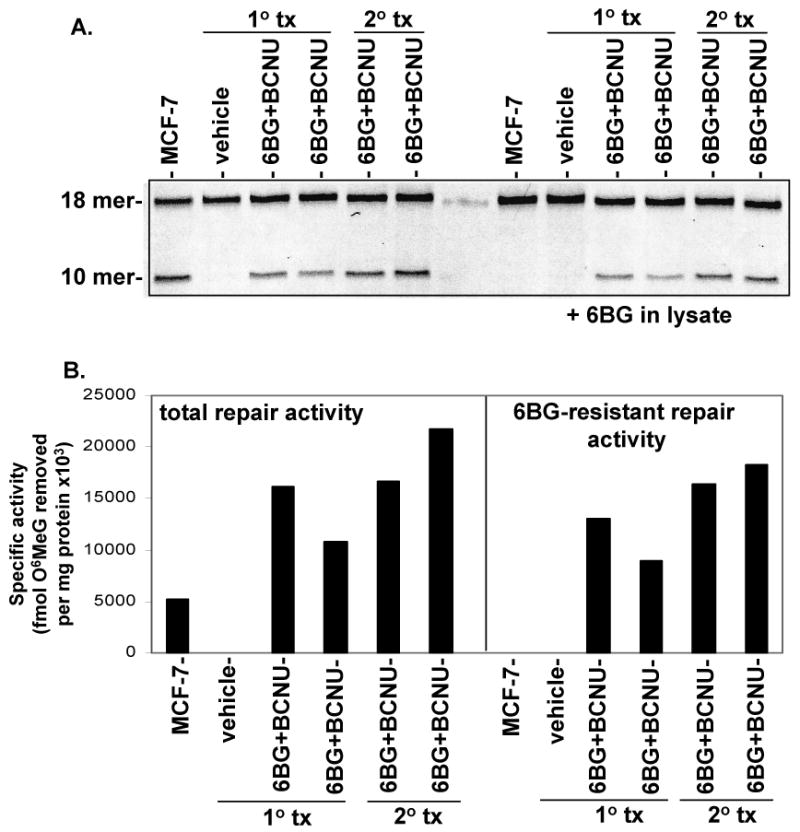
(A) BM from primary and secondary recipients (vehicle or treated with submyeloablative therapy) were analyzed for the presence of total MGMT and 6BG-resistant repair activity. (B) MCF-7 = breast cancer cell line that expresses wild-type MGMT activity. Depending on the level of MGMT activity, different amounts of protein were run to avoid signal saturation. Specific activity = fmol O6 methylguanine removed per mg total protein.
Detection of MD9-P140K-tranduced cells in primary and secondary recipient mice following myeloablative treatment
We next determined to what extent HSC transduced with the MD9-P140K vector at a low MOI could protect transplanted mice from a clinically relevant alkylator-based combinational regimen that can be potentially used for treatment of cancers with high levels of MGMT (Table 1) [42]. This regimen consists of a 6BG-double bolus combined with a single infusion of BCNU and results in ablation of the BM and subsequent morbidity unless MGMTP140K-expressing HSC are present. We and our collaborators previously used this myeloablative regimen to efficaciously kill human glioma cells in a mouse xenograft model [43]. Under these myeloablative conditions, 25% of the mice transplanted with MD9-P140K-transduced BM cells survived and all surviving mice expressed MGMTP140K. These data demonstrate that even under ablative conditions, an adequate number of MD9-P140K-transduced HSC still existed in some of the mice and afforded protection from chemotherapy-mediated lethality (Table 1, experiments 3 and 4). To demonstrate that transduced stem cells were indeed present following myeloablative therapy, BM cells from high-dose treated animals in experiment 3 were pooled and transplanted into secondary recipient mice. At 6 months post-transplantation, MGMT expression was detected in the BM of all transplanted mice indicating that long-term repopulating MGMTP140K-expressing stem cells had been transplanted into these mice (Fig 4B).
Discussion
The ability to deliver genetic sequences to HSC that encode therapeutic proteins in the absence of ex vivo cell cycle progression may provide a unique opportunity to transduce HSC without loss of long-term function [44-54]. Our objective was to test the ability of a FV vector that expresses a therapeutic protein, MGMTP140K, to transduce minimally manipulated murine BM at a low MOI due to the fact that FV can enter resting cells. In the past, transduction protocols using FV vectors have targeted murine BM isolated from 5-FU-treated donor mice. Therefore, more committed, mature cells are killed and cycling of the more primitive progenitor cells is induced [50, 52, 53]. In a murine transplant model utilizing FV viral vectors, Vassilopoulos et al employed a transduction protocol originally used for oncoretroviral transduction [24]. In this study, 5FU-treated BM was pre-stimulated with cytokines for 48 hours, transduced with FV vectors on retronectin-coated plates, and then maintained for an additional 48 hours in cytokines prior to transplantation. In this study, 7 out of 22 transplanted mice reconstituted with at least 50% donor cells and were analyzed in detail at 6 months post-transplantation. In 4 of the 7 mice, the average transduction level was 32% for BM-derived myeloid progenitor colonies. In the 3 other animals that lacked long-term reconstitution of FV-transduced cells, PCR analysis indicated this was due to engraftment of nontransduced stem cells as opposed to transgene silencing. Administration of 5FU, however, can compromise long-term engrafting cells and is not applicable to clinical gene therapy trials [49-54]. Therefore, in our study, we elected to use steady-state bone marrow as the target for FV-mediated gene transfer.
Consistent with the in vitro observations demonstrating that FV vectors require cell division for integration, Kiem et al recently demonstrated in a canine model that exposure of FV-transduced BM cells to G-CSF in vivo led to detection of FV-transduced long-term repopulating cells [32]. The marking of human SCID-repopulating cells with FV vectors without in vivo selection or administration of cytokines has been demonstrated by our group and others [21-23]. High levels of FV marking in human cells may be due in part to the rapid expansion of the SCID-repopulating cells in the BM following homing and engraftment in the bone marrow of NOD/SCID mice.
In our transduction strategy, transduced BM cells were exposed to cytokines during the overnight-transduction period to maintain cell viability. Even though a low MOI was employed, high levels of gene transfer in the committed progenitor cell populations were detected in the CFU assay which promotes rapid division of clonogenic cells. Toxicity to the progenitor pool was not evident since the absolute number of control nontransduced colonies versus transduced colonies were not significantly different in CFU assays (data not shown). Since foamy viral vectors require cycling of the target cells for integration into the target cell genome, the increased transduction into the progenitor compartment compared to the bulk cells that we observed, is most likely due to increased cycling of the progenitor population as these are the only cells that are assessed in the CFU assay. In addition, it is also possible that the cellular structure utilized as the foamy viral receptor is expressed at a higher level in the clonogenic population than the bulk cells in the lineage-negative bone marrow used in this study.
Once the transduced BM cells were transplanted into recipient mice, additional cytokines were not administered in vivo. The majority of hematopoietic stem cells residing in steady state BM do not routinely cycle and FV integration into the genome of the target cell will only occur following initiation of cell cycle progression. In our model, stem-cell division most likely occurs following homing and engraftment of the transduced HSC. The primary and secondary reconstitution experiments presented here demonstrate that a short transduction protocol using low MOI and minimal cytokine exposure was well suited to mediate FV vector integration into minimally manipulated long-term repopulating stem cells. Efficiency of in vivo selection, however, was dependent on the percentage of transduced HSC and the dose of alkylator therapy administered. In the submyeloablative setting, 6 of 11 mice expressed MGMTP140K following treatment. In the mice that did not express MGMTP140K, the level of transduction into the stem-cell population was presumably not sufficient to enable detectable in vivo selection in all animals since low MOIs were used for transduction. Due to the low gene transfer rate into long-term repopulating HSC and the acute toxicity of BCNU, MGMTP140K expression protected stem cell-derived progeny to a lesser extent in the myeloablative range. This in vivo model can now be used in the future to develop and test clinically relevant FV transduction protocols targeting minimally manipulated hematopoietic stem-cells. Using this murine model system, experiments are in progress to delineate the in vivo requirements for optimal donor chimerism and FV vector transduction efficiency into long-term repopulating cells.
Acknowledgments
This study was funded by the Biomedical Research Committee, Indiana University Medical Center (KEP): Hope Street Kids Foundation (KEP); KO8 HL75253 (WSG); Deutsche Forschungs-Gemeinschaft (SPP1230) and Elterninitiative Kinderkrebsklinik e.v (HH); the Riley Children's Foundation (WSG, HH, and KEP). The authors wish to thank Dr. Arthur R. Baluyut for his support and critical evaluation of this manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Cavazzana-Calvo M, Hacein-Bey S, de Saint Basile G, et al. Gene therapy of human severe combined immunodeficiency (SCID)-X1 disease. Science New York, NY. 2000;288:669–672. doi: 10.1126/science.288.5466.669. [DOI] [PubMed] [Google Scholar]
- 2.Chinen J, Davis J, De Ravin SS, et al. Gene therapy improves immune function in pre-adolescents with X-linked severe combined immunodeficiency. Blood. 2007 doi: 10.1182/blood-2006-11-058933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gaspar HB, Bjorkegren E, Parsley K, et al. Successful reconstitution of immunity in ADA-SCID by stem cell gene therapy following cessation of PEG-ADA and use of mild preconditioning. Mol Ther. 2006;14:505–513. doi: 10.1016/j.ymthe.2006.06.007. [DOI] [PubMed] [Google Scholar]
- 4.Ott MG, Schmidt M, Schwarzwaelder K, et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nature medicine. 2006;12:401–409. doi: 10.1038/nm1393. [DOI] [PubMed] [Google Scholar]
- 5.Hacein-Bey-Abina S, Von Kalle C, Schmidt M, et al. LMO2-associated clonal T cell proliferation in two patients after gene therapy for SCID-X1. Science New York, NY. 2003;302:415–419. doi: 10.1126/science.1088547. [DOI] [PubMed] [Google Scholar]
- 6.Baum C, von Kalle C, Staal FJ, et al. Chance or necessity? Insertional mutagenesis in gene therapy and its consequences. Mol Ther. 2004;9:5–13. doi: 10.1016/j.ymthe.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 7.Li Z, Dullmann J, Schiedlmeier B, et al. Murine leukemia induced by retroviral gene marking. Science New York, NY. 2002;296:497. doi: 10.1126/science.1068893. [DOI] [PubMed] [Google Scholar]
- 8.Li X, Le Beau MM, Ciccone S, et al. Ex vivo culture of Fancc-/- stem/progenitor cells predisposes cells to undergo apoptosis, and surviving stem/progenitor cells display cytogenetic abnormalities and an increased risk of malignancy. Blood. 2005;105:3465–3471. doi: 10.1182/blood-2004-06-2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Beard BC, Keyser KA, Trobridge GD, et al. Unique Integration Profiles in a Canine Model of Long-Term Repopulating Cells Transduced with Gammaretrovirus, Lentivirus, or Foamy Virus. Human gene therapy. 2007 doi: 10.1089/hum.2007.011. [DOI] [PubMed] [Google Scholar]
- 10.Nowrouzi A, Dittrich M, Klanke C, et al. Genome-wide mapping of foamy virus vector integrations into a human cell line. The Journal of general virology. 2006;87:1339–1347. doi: 10.1099/vir.0.81554-0. [DOI] [PubMed] [Google Scholar]
- 11.Trobridge GD, Miller DG, Jacobs MA, et al. Foamy virus vector integration sites in normal human cells. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:1498–1503. doi: 10.1073/pnas.0510046103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rethwilm A. Foamy virus vectors: an awaited alternative to gammaretro- and lentiviral vectors. Current opinions in Gene Therapy. doi: 10.2174/156652307781369092. In Press. [DOI] [PubMed] [Google Scholar]
- 13.Sandstrom PA, Phan KO, Switzer WM, et al. Simian foamy virus infection among zoo keepers. Lancet. 2000;355:551–552. doi: 10.1016/S0140-6736(99)05292-7. [DOI] [PubMed] [Google Scholar]
- 14.Schweizer M, Falcone V, Gange J, Turek R, Neumann-Haefelin D. Simian foamy virus isolated from an accidentally infected human individual. Journal of virology. 1997;71:4821–4824. doi: 10.1128/jvi.71.6.4821-4824.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Linial M. Why aren't foamy viruses pathogenic? Trends Microbiol. 2000;8:284–289. doi: 10.1016/s0966-842x(00)01763-7. [DOI] [PubMed] [Google Scholar]
- 16.Heneine W, Switzer WM, Sandstrom P, et al. Identification of a human population infected with simian foamy viruses. Nature medicine. 1998;4:403–407. doi: 10.1038/nm0498-403. [DOI] [PubMed] [Google Scholar]
- 17.Patton GS, Erlwein O, McClure MO. Cell-cycle dependence of foamy virus vectors. The Journal of general virology. 2004;85:2925–2930. doi: 10.1099/vir.0.80210-0. [DOI] [PubMed] [Google Scholar]
- 18.Trobridge G, Russell DW. Cell cycle requirements for transduction by foamy virus vectors compared to those of oncovirus and lentivirus vectors. Journal of virology. 2004;78:2327–2335. doi: 10.1128/JVI.78.5.2327-2335.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Guenechea G, Gan OI, Inamitsu T, et al. Transduction of human CD34+ CD38- bone marrow and cord blood-derived SCID-repopulating cells with third-generation lentiviral vectors. Mol Ther. 2000;1:566–573. doi: 10.1006/mthe.2000.0077. [DOI] [PubMed] [Google Scholar]
- 20.Horn PA, Keyser KA, Peterson LJ, et al. Efficient lentiviral gene transfer to canine repopulating cells using an overnight transduction protocol. Blood. 2004;103:3710–3716. doi: 10.1182/blood-2003-07-2414. [DOI] [PubMed] [Google Scholar]
- 21.Josephson NC, Trobridge G, Russell DW. Transduction of long-term and mobilized peripheral blood-derived NOD/SCID repopulating cells by foamy virus vectors. Human gene therapy. 2004;15:87–92. doi: 10.1089/10430340460732481. [DOI] [PubMed] [Google Scholar]
- 22.Josephson NC, Vassilopoulos G, Trobridge GD, et al. Transduction of human NOD/SCID-repopulating cells with both lymphoid and myeloid potential by foamy virus vectors. Proceedings of the National Academy of Sciences of the United States of America. 2002;99:8295–8300. doi: 10.1073/pnas.122131099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Leurs C, Jansen M, Pollok KE, et al. Comparison of Three Retroviral Vector Systems for Transduction of Nonobese Diabetic/Severe Combined Immunodeficiency Mice Repopulating Human CD34(+) Cord Blood Cells. Human gene therapy. 2003;14:509–519. doi: 10.1089/104303403764539305. [DOI] [PubMed] [Google Scholar]
- 24.Vassilopoulos G, Trobridge G, Josephson NC, Russell DW. Gene transfer into murine hematopoietic stem cells with helper-free foamy virus vectors. Blood. 2001;98:604–609. doi: 10.1182/blood.v98.3.604. [DOI] [PubMed] [Google Scholar]
- 25.Woods NB, Muessig A, Schmidt M, et al. Lentiviral vector transduction of NOD/SCID repopulating cells results in multiple vector integrations per transduced cell: risk of insertional mutagenesis. Blood. 2003;101:1284–1289. doi: 10.1182/blood-2002-07-2238. [DOI] [PubMed] [Google Scholar]
- 26.Miyoshi H, Smith KA, Mosier DE, Verma IM, Torbett BE. Transduction of human CD34+ cells that mediate long-term engraftment of NOD/SCID mice by HIV vectors. Science New York, NY. 1999;283:682–686. doi: 10.1126/science.283.5402.682. [DOI] [PubMed] [Google Scholar]
- 27.Mortellaro A, Hernandez RJ, Guerrini MM, et al. Ex vivo gene therapy with lentiviral vectors rescues adenosine deaminase (ADA)-deficient mice and corrects their immune and metabolic defects. Blood. 2006;108:2979–2988. doi: 10.1182/blood-2006-05-023507. [DOI] [PubMed] [Google Scholar]
- 28.Mostoslavsky G, Fabian AJ, Rooney S, Alt FW, Mulligan RC. Complete correction of murine Artemis immunodeficiency by lentiviral vector-mediated gene transfer. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:16406–16411. doi: 10.1073/pnas.0608130103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Scherr M, Battmer K, Blomer U, et al. Lentiviral gene transfer into peripheral blood-derived CD34+ NOD/SCID-repopulating cells. Blood. 2002;99:709–712. doi: 10.1182/blood.v99.2.709. [DOI] [PubMed] [Google Scholar]
- 30.Woods NB, Fahlman C, Mikkola H, et al. Lentiviral gene transfer into primary and secondary NOD/SCID repopulating cells. Blood. 2000;96:3725–3733. [PubMed] [Google Scholar]
- 31.Worsham DN, Schuesler T, von Kalle C, Pan D. In vivo gene transfer into adult stem cells in unconditioned mice by in situ delivery of a lentiviral vector. Mol Ther. 2006;14:514–524. doi: 10.1016/j.ymthe.2006.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kiem HP, Allen J, Trobridge G, et al. Foamy-virus-mediated gene transfer to canine repopulating cells. Blood. 2007;109:65–70. doi: 10.1182/blood-2006-04-016741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Milsom MD, Williams DA. Live and let die: in vivo selection of gene-modified hematopoietic stem cells via MGMT-mediated chemoprotection. DNA Repair (Amst) 2007;6:1210–1221. doi: 10.1016/j.dnarep.2007.03.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schambach A, Baum C. Vector design for expression of O(6)-methylguanine-DNA methyltransferase in hematopoietic cells. DNA Repair (Amst) 2007 doi: 10.1016/j.dnarep.2007.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heinkelein M, Dressler M, Jarmy G, et al. Improved primate foamy virus vectors and packaging constructs. Journal of virology. 2002;76:3774–3783. doi: 10.1128/JVI.76.8.3774-3783.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cai S, Hartwell JR, Cooper RJ, et al. In vivo effects of myeloablative alkylator therapy on survival and differentiation of MGMTP140K-transduced human G-CSF-mobilized peripheral blood cells. Mol Ther. 2006;13:1016–1026. doi: 10.1016/j.ymthe.2005.11.017. [DOI] [PubMed] [Google Scholar]
- 37.Hanenberg H, Xiao XL, Dilloo D, Hashino K, Kato I, Williams DA. Colocalization of retrovirus and target cells on specific fibronectin fragments increases genetic transduction of mammalian cells. Nature medicine. 1996;2:876–882. doi: 10.1038/nm0896-876. [DOI] [PubMed] [Google Scholar]
- 38.Bierhuizen MFA, Westerman Y, Visser TP, Dimjati W, Wognum AW, Wagemaker G. Enhanced green fluorescent protein as selectable marker of retroviral-mediated gene transfer in immature hematopoietic bone marrow cells. Blood. 1997;90:3304–3315. [PubMed] [Google Scholar]
- 39.Villella AD, Yao J, Getty RR, et al. Real-Time PCR: an Effective Tool for Measuring Transduction Efficiency in Human Hematopoeitic Progenitor Cells. Molecular Therapy. 2005;11:483–491. doi: 10.1016/j.ymthe.2004.10.017. [DOI] [PubMed] [Google Scholar]
- 40.Kreklau EL, Limp-Foster M, Liu N, Xu Y, Kelley MR, Erickson LC. A novel fluorometric oligonucleotide assay to measure O(6)-methylguanine DNA methyltransferase, methylpurine DNA glycosylase, 8-oxoguanine DNA glycosylase and abasic endonuclease activities: DNA repair status in human breast carcinoma cells overexpressing methylpurine DNA glycosylase. Nucleic Acids Res. 2001;29:2558–2566. doi: 10.1093/nar/29.12.2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Laufs S, Gentner B, Nagy KZ, et al. Retroviral vector integration occurs in preferred genomic targets of human bone marrow-repopulating cells. Blood. 2003;101:2191–2198. doi: 10.1182/blood-2002-02-0627. [DOI] [PubMed] [Google Scholar]
- 42.Quinn JA, Pluda J, Dolan ME, et al. Phase II trial of carmustine plus O(6)-benzylguanine for patients with nitrosourea-resistant recurrent or progressive malignant glioma. J Clin Oncol. 2002;20:2277–2283. doi: 10.1200/JCO.2002.09.084. [DOI] [PubMed] [Google Scholar]
- 43.Kreklau EL, Pollok KE, Bailey BJ, et al. Hematopoietic expression of O(6)-methylguanine DNA methyltransferase-P140K allows intensive treatment of human glioma xenografts with combination O(6)-benzylguanine and 1,3-bis-(2-chloroethyl)-1-nitrosourea. Mol Cancer Ther. 2003;2:1321–1329. [PubMed] [Google Scholar]
- 44.Baum C, Dullmann J, Li Z, et al. Side effects of retroviral gene transfer into hematopoietic stem cells. Blood. 2003;101:2099–2114. doi: 10.1182/blood-2002-07-2314. [DOI] [PubMed] [Google Scholar]
- 45.Goebel WS, Yoder MC, Pech NK, Dinauer MC. Donor chimerism and stem cell function in a murine congenic transplantation model after low-dose radiation conditioning: effects of a retroviral-mediated gene transfer protocol and implications for gene therapy. Experimental hematology. 2002;30:1324–1332. doi: 10.1016/s0301-472x(02)00927-x. [DOI] [PubMed] [Google Scholar]
- 46.Kittler EL, Peters SO, Crittenden RB, et al. Cytokine-facilitated transduction leads to low-level engraftment in nonablated hosts. Blood. 1997;90:865–872. [PubMed] [Google Scholar]
- 47.Qin S, Ward M, Raftopoulos H, et al. Competitive repopulation of retrovirally transduced haemopoietic stem cells. British journal of haematology. 1999;107:162–168. doi: 10.1046/j.1365-2141.1999.01664.x. [DOI] [PubMed] [Google Scholar]
- 48.Tisdale H, Sellers, Agricola, Metzger, Donahue, Dunbar Ex Vivo Expansion of Genetically Marked Rhesus Peripheral Blood Progenitor Cells results in Diminished Long-Term Repopulating Ability. Blood. 1998;92:1131–1141. [PubMed] [Google Scholar]
- 49.Austin TW, Salimi S, Veres G, et al. Long-term multilineage expression in peripheral blood from a Moloney murine leukemia virus vector after serial transplantation of transduced bone marrow cells. Blood. 2000;95:829–836. [PubMed] [Google Scholar]
- 50.Bodine DM, McDonagh KT, Seidel NE, Nienhuis AW. Survival and retrovirus infection of murine hematopoietic stem cells in vitro: effects of 5-FU and method of infection. Experimental hematology. 1991;19:206–212. [PubMed] [Google Scholar]
- 51.Dunbar CE, Seidel NE, Doren S, et al. Improved retroviral gene transfer into murine and Rhesus peripheral blood or bone marrow repopulating cells primed in vivo with stem cell factor and granulocyte colony-stimulating factor. Proceedings of the National Academy of Sciences of the United States of America. 1996;93:11871–11876. doi: 10.1073/pnas.93.21.11871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stewart FM, Crittenden RB, Lowry PA, Pearson-White S, Quesenberry PJ. Long-term engraftment of normal and post-5-fluorouracil murine marrow into normal nonmyeloablated mice. Blood. 1993;81:2566–2571. [PubMed] [Google Scholar]
- 53.Stewart FM, Temeles D, Lowry P, Thraves T, Grosh WW, Quesenberry PJ. Post-5-fluorouracil human marrow: stem cell characteristics and renewal properties after autologous marrow transplantation. Blood. 1993;81:2283–2289. [PubMed] [Google Scholar]
- 54.Wahlers A, Schwieger M, Li Z, et al. Influence of multiplicity of infection and protein stability on retroviral vector-mediated gene expression in hematopoietic cells. Gene therapy. 2001;8:477–486. doi: 10.1038/sj.gt.3301426. [DOI] [PubMed] [Google Scholar]