

Abstract
Proliferation of vascular endothelial cells (EC) and smooth muscle cells (SMC) is a critical event in angiogenesis and atherosclerosis. We previously showed that the C5b-9 assembly during complement activation induces cell cycle in human aortic EC (AEC) and SMC. C5b-9 can induce the expression of Response Gene to Complement (RGC)-32 and over expression of this gene leads to cell cycle activation. Therefore, the present study was carried out to test the requirement of endogenous RGC-32 for the cell cycle activation induced by C5b-9 by knocking-down its expression using siRNA. We identified two RGC-32 siRNAs that can markedly reduce the expression of RGC-32 mRNA in AEC. RGC-32 silencing in these cells abolished DNA synthesis induced by C5b-9 and serum growth factors, indicating the requirement of RGC-32 activity for S-phase entry. RGC-32 siRNA knockdown also significantly reduced the C5b-9 induced CDC2 activation and Akt phosphorylation. CDC2 does not play a role in G1/S transition in HeLa cells stably overexpressing RGC-32. RGC-32 was found to physically associate with Akt and was phosphorylated by Akt in vitro. Mutation of RGC-32 protein at Ser 45 and Ser 47 prevented Akt mediated phosphorylation. In addition, RGC-32 was found to regulate the release of growth factors from AEC. All these data together suggest that cell cycle induction by C5b-9 in AEC is RGC-32 dependent and this is in part through regulation of Akt and growth factor release.
Keywords: RGC-32, C5b-9, complement activation, endothelial cells, Akt
Introduction
Proliferation of EC is a critical process involved in angiogenesis and atherogenesis (Herrmann et al., 2006). Through cell-cell contact inhibition, normal vascular EC are in a quiescent non-proliferative state (Bazzoni and Dejana, 2004). However, rapid proliferative burst occurs in EC when they are exposed to injuries that include iatrogenic factors such as balloon angioplasty (Cunningham and Gotlieb, 2005) and various pathophysiologic events such as modified LDL and angiogenic or atherogenic factors (Libby et al., 2006). Proliferation and migration of EC can also be induced by complement activation through insertion of the C5b-9 complex into the EC membrane (Benzaquen et al., 1994; Fosbrink et al., 2006).
Assembly of C5b-9 causes cell death by forming multiple pores in target cell membranes. However, nucleated cells survive when cells are exposed to limited numbers of the C5b-9 complex by eliminating membrane-inserted terminal complement complexes C5b-7, C5b-8 and C5b-9 through endocytosis and/or membrane shedding (Carney et al., 1985; Scolding et al., 1989). Membrane-inserted C5b-9 complex leads to the activation and progression of cell cycle by activating signaling pathways of Gi/Go family of G-proteins (Niculescu et al., 1994), ERK (Niculescu et al., 1997) and phosphatidylinositol 3-kinase (PI3K) pathways (Hila et al., 2001; Niculescu et al., 1999). Cell cycle activation by C5b-9 is associated with expression of c-JUN and c-FOS proto-oncogenes (Rus et al., 1996a) and increased CDK4, CDK2 and CDC2 activities (Fosbrink et al., 2006; Niculescu et al., 1999; Rus et al., 1996a). In our attempt to better understand the molecular mechanisms responsible for cell cycle activation, we screened for the expression of cell cycle genes induced by C5b-9 by differential display (Badea et al., 1998) and identified a novel gene, Response Gene to Complement (RGC)-32. Over expression of RGC-32 induced DNA synthesis and activated cell cycle (Badea et al., 2002; Badea et al., 1998). RGC-32 was also found to be physically associated with CDC2 and to increase CDC2 activity in vitro (Badea et al., 2002). CDC2-cyclin B1 phosphorylated RGC-32 in vitro, and mutation of RGC-32 at Thr-91 prevented this CDC2-mediated phosphorylation and the CDC2 kinase enhancing activity of RGC-32. These findings indicate that the C5b-9 induced cell cycle may involve CDC2 activation through RGC-32. Thus, RGC-32 appears to be a cell cycle regulatory factor, which functions as an activator and substrate of CDC2 (Badea et al., 2002). We found that RGC-32 is over expressed in human malignant tumors (Fosbrink et al., 2005), suggesting that an over expression of RGC-32 may participate in cell cycle deregulation required for tumor growth. RGC-32 mRNA expression was up regulated in malignant tumors of colon, kidney, stomach (Fosbrink et al., 2005) and ovary (Donninger et al., 2004) or during oncogenic transformation induced by ras (Sagiv et al.). RGC-32 is also co expressed with other genes involved in cell proliferation during the treatment with steroids (James et al., 2005) and estrogens (Fujimoto et al., 2004). These studies collectively indicate that an increased expression of RGC-32 is associated with cell proliferation in vivo (Vlaicu et al., 2008).
In this study, we investigated the role of endogenous RGC-32 in cell cycle activation induced by C5b-9 in primary human AEC by knocking down RGC-32 expression using specific siRNA. RGC-32 silencing abolished the ability of C5b-9 to induce cell cycle and CDC2 activation. In addition, RGC-32 silencing also significantly inhibited C5b-9-mediated release of growth factors as well as Akt phosphorylation. Our data indicate that endogenous RGC-32 plays a critical role in regulating cell cycle activation induced by C5b-9, in part by regulating phosphorylation of cell cycle-related substrates and release of growth factors.
Materials and Methods
Primary Aortic Endothelial Cell Culture
Human AEC (Lonza, Walkersville, MD) were cultured in endothelial cell basal medium (EBM) supplemented with 2% fetal bovine serum, 10 ng/ml human epidermal growth factor, 2 ng/ml human fibroblast growth factor, 5 μg/ml vascular endothelial growth factor, 0.1 μg/ml of hydrocortisone, and 0.5 μg/ml of heparin. AEC were used after 3-5 passages and were starved for 18 h in EBM without serum and growth supplements prior treatment with complement components.
Activation of serum complement and terminal complement complex assembly
Pooled normal human sera (NHS) from healthy adult donors were used as a source of serum complement. To assemble serum C5b-9, AEC were sensitized with anti-human HLA class A, B, C monoclonal IgG (Dako, Carpenteria, CA), then exposed with dilutions of NHS. Control cells were stimulated with antibody to HLA and K76 COONa (K76) -treated NHS. As previously described, K76 (Otsuka Pharmaceuticals Co, Tokyo, Japan) prevents C5b-9 assembly in serum by binding to and inactivating C5 (Niculescu et al., 1997; Rus et al., 1996a). We also assembled C5b-9 using purified components (Advances Researches Technologies, San Diego, CA) by treating cells sequentially with C5b6, C7, and C8/C9, as described previously (Rus et al., 1996a). The concentrations of complement proteins used in this study were sublytic for AEC as measured by the release of cytoplasmic lactate dehydrogenase, as an indicator of cell death (Carney et al., 1990).
siRNA target sequences and transfection
Control siRNA (siCTR) was synthesized by Qiagen Inc, (Valencia CA): 5′ – AATTCTCCGA ACGTGTCACGT – 3′. The following Si RGC-32 sequences were synthesized by Dharmacon Inc., Chicago IL: siRGC-32-1: 5′ – AAGTGCAGATTCACTTTATAG – 3′ and siRGC-32-2: 5′ – AAAATCAGCTACTAGAA TCTG – 3′. AEC were transfected with siRNA duplexes at a final concentration of 25 nM of siRNA using TransIT siQuest reagent (Mirus, Madison, WI) for 6 h and then transfected again for another 24 h.
Real-time PCR
RNA was isolated using RNeasy (Qiagen, Santa Clarita, CA) according to the manufacturer's protocol. One μg of RNA was reversed transcribed to obtain cDNA. RGC-32 forward and reverse primers and hybridization probe sequences were provided by TIB Mol Biol (Adelphia, NJ). Real-time PCR was performed according to the manufacturer's protocol using a LightCycler FastStart DNA MasterHybProbe (Roche) and Real-Time Light Cycler System (Roche Biochemicals). The fold change in target gene samples, after normalization with the housekeeping gene (18S), was calculated as described (Livak and Schmittgen, 2001).
[3H]-Thymidine incorporation
DNA synthesis was performed as previously described (Niculescu et al., 1997). Cultured cells were synchronized in G0/G1-phase by starvation in EBM. After stimulation, 1 μCi/ml of [3H]-thymidine (Perkin Elmer, Boston MA) was added to cells. The plate was incubated at 37° C, 5% CO2 for 18 h. Cells were lysed, precipitated by adding 20% trichloroacetic acid and the precipitated DNA was filtered through Whatman GF/A filter paper (Whatman, Maidstone, UK). The filter paper was air-dried and radioactivity was counted by liquid scintillation.
Cell cycle kinase assays
Immunoprecipitation kinase assay were performed as previously described (Niculescu et al., 1999). Cells were lysed by adding RIPA lysis buffer and CDC2 or CDK2 were immunoprecipitated using rabbit IgG anti-CDC2 (Santa Cruz Biotechnol) in the presence of protein A/G (Santa Cruz Biotech.). Immunoprecipitated CDC2 were incubated in reaction buffer (25 mM Tris-HCl, pH 7.4, 10 mM MgCl2) containing 5 μg of histone H1 (Roche, Indianapolis, IN) and 1 μCi of [γ-32P] ATP/sample (Perkin Elmer). Phosphorylated histone H1 bands were analyzed by 10% SDS-PAGE and autoradiography.
Western blot
20-30 μg of protein were separated by SDS-PAGE, and then transferred to a nitrocellulose membrane. The membrane was incubated with primary antibody in blocking buffer at 4° C overnight. Then the membrane was washed and incubated HRP-conjugated goat anti-rabbit IgG (Santa Cruz Biotech) for 1 h at RT. The bands were visualized by ECL and autoradiography.
Angiogenic factor protein array
To determine the effect of silencing RGC-32 on C5b-9 induced growth factor release, Human Angiogenesis Antibody Array 1.1 (RayBiotech Inc., Norcross, GA) was used to test the expression of the most important angiogenic factors. AEC were transfected with RGC-32 siRNA or control siRNA then cultured in serum and growth factor free media for 18 h. They were treated with C5b-9 or C5b6 assembled from purified components for 1 h. The conditioned media was incubated with the membrane array overnight at 4° C. The array was processed following manufacturer recommendations.
Binding of Akt to GST-RGC-32
Binding of Akt from AEC lysates to GST-RGC-32 fusion protein was determined using the Profound pull-down GST protein-protein interaction kit (Pierce). The proteins binding to GST-RGC-32 in the presence of glutathione-Sepharose beads were eluted using gluathione and analyzed by Western blotting using anti-Akt (Upstate Biotechnology, Lake Pacid, NY) and anti-GST (Calbiochem, San Diego, CA) antibodies.
Phosphorylation of RGC-32 by Akt and the target site analysis by site-directed mutagenesis
Serine 45 and 47 of RGC-32, possible targets for Akt phosphorylation were mutated using the QuickChange site-directed mutagenesis kit (Stratagene, LaJolla, CA). The desired mutation was confirmed by DNA sequencing at the University of Maryland Biopolymer Facility. Mutated and wild type GST-RGC-32 proteins were extracted from bacterial lysate and purified by affinity chromatography using GST purification module (GE HealthCare, Piscataway, NJ) (Badea et al., 2002). The recombinant proteins were used for in vitro phosphorylation assays. Purified and mutated GST-RGC-32, Akt (NEBLabs, Beverly, MA) or CDC2/cyclin B1 (NEBLabs), in the presence of 1 μCi[γ32P]-ATP/sample were incubated for 30 min at 37°C. Protein phosphorylation was then analyzed by 10% SDS-PAGE followed by autoradiography.
Construction of an inducible RGC-32 expressing cell line
In order to control RGC-32 expression, an inducible cell line was created using the T-Rex system (Invitrogen, Carlsbad, CA). Briefly, HeLa cells were stably transfected with pCDNA6 which encodes the tetracycline repressor (TR). HeLa cells expressing the TR were then stably transfected with pCDNA4 vector which encodes for RGC-32 controlled under the tet operator. Stable cells were obtained in the presence of Blasticidin (3μg/ml) and Zeocin (100μg/ml). Stable clones were induced to express RGC-32 in the presence of tetracycline (2μg/ml). Two clones, with the highest level of inducibility, were chosen for subsequent studies. In some experiments cells were arrested in M phase using nocodazole 40 ng/ml for 12h. Mitotic HeLa cells were isolated by selective detachment after nocodazole block then replated in DMEM with 10% FBS in the presence of tetracycline.
Results
RGC-32 is required for cell cycle activation by C5b-9
To test the requirement of RGC-32 in cell cycle activation, we designed and synthesized two siRNAs targeting the RGC-32 mRNA, one siRNA (siRGC-32-1) is targeting the open reading frame of RGC-32 towards the 5′ end and one (siRGC-32-2) targeting the 3′UTR. AEC were transfected with siRNA and then tested for their effectiveness in blocking the endogenous expression of RGC-32 mRNA using real time-PCR. Both siRNAs (siRGC-32-1 and 2) effectively decreased the mRNA expression by 91% and 87% respectively when compared to cells transfected with control siRNA (siCTR) as determined by real-time PCR (Fig. 1 A). We then examined the ability of siRNA to block the cell cycle activation induced by C5b-9 using [3H] thymidine uptake. Cells were transfected with siRGC-32 or siCTR. Cells were then cultured in serum and growth factor-deprived medium for 18 h to synchronize in G0/G1-phase of the cell cycle. They were treated with serum C5b-9 or C5D (Fig. 1B). In control cells transfected with siCTR, serum C5b-9 induced a 14.5-fold increase in [3H] thymidine incorporation over the unstimulated cells. The thymidine uptake caused by serum and serum growth factors, as shown by C5D, was a 5.4-fold increase over the unstimulated cells. We found that both siRGC-32-1 and -2 were very effective in blocking the thymidine incorporation induced by C5b-9 (Fig. 1B). Since RGC-32 is also induced by growth factors, (Fosbrink, 2003) we examined the requirement of RGC-32 in growth factor induced cell cycle activation using endothelial growth media (EGM). AEC were transfected with siRNA, cultured in serum-free medium for 18 h, then stimulated with EGM (Fig. 1C). EGM increased thymidine incorporation 7.8-fold in the control siRNA transfected cell level over unstimulated cells. Both RGC-32 siRNAs effectively reduced thymidine incorporation induced by EGM (Fig. 1C). These data clearly indicate that RGC-32 is required for cell cycle activation induced by C5b-9 and by other growth factors.
Figure 1. RGC-32 is required for C5b-9 and growth factors induced cell cycle.
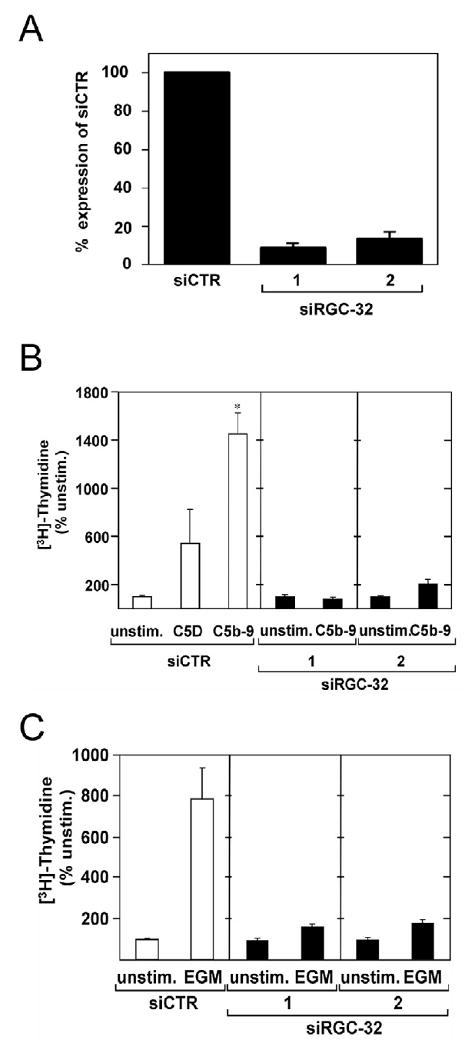
A. AEC were transfected with siRGC-32-1, siRGC-32–2, or control siRNA (siCTR) at a final concentration of 25 nM using Mirus SiQuest transfection reagent. After 48 h, total RNA was extracted, and analyzed for RGC-32 and Actin mRNA expression by real time - PCR. Both siRGC-32 significantly reduced RGC-32 mRNA expression.
B. AEC were stimulated with serum C5b-9 (C5b-9) or C5D serum for 24 h in the presence of 1 μCi [3H]-thymidine. The level of [3H]-thymidine incorporation was used to assess cell cycle activation. C5b-9 significantly increased [3H]-thymidine incorporation compared to C5D (*p<0.001). Both siRGC-32-1 and −2 abolished thymidine incorporation induced by C5b-9.
C. AEC were transfected with siRGC-32-1, siRGC-32-2, or control siCTR for 48 h. Cells were starved in serum- and growth factor-free medium for 18 h to synchronize in G0/G1-phase of the cell cycle. Cells were then stimulated with endothelial growth media (EGM) for 24 h, followed by [3H]-thymidine incorporation. Both RGC-32 siRNAs abolished EGM-induced [3H]-thymidine incorporation. Results are expressed as % of unstimulated cell level (Unstim).
RGC-32 is required for the activation of CDC2
RGC-32 forms a complex with CDC2 as shown by co-immunoprecipitation both in vivo and in vitro (Badea et al., 2002; Badea et al., 1998). In addition, RGC-32 enhances CDC2 kinase activity in vitro and this activity is inhibited by the CDK inhibitor p27 (Badea et al., 2002). Since RGC-32 regulates CDC2 function in vitro, we tested whether silencing RGC-32 also affects CDC2 activation induced by C5b-9. First, the time course of C5b-9 to induce CDC2 activation was determined by growing AEC in serum- and growth factor-free medium for 18 h then exposed to serum C5b-9. CDC2 activity was assayed by an in vitro kinase assay using histone H1 as a substrate in the presence of [γ-32P] ATP (Fig. 2A). A significantly increased CDC2 activity was noted after 24 h of stimulation (Fig. 2A). We then determined the CDC2 activity in RGC-32 siRNA transfected EC following stimulation with serum C5b-9 or C5D for 24 h (Fig. 2B). In cells transfected with siCTR, C5b-9 increased CDC2 activity by 5.8-fold as compared with 3.4-fold in cells stimulated with C5D. In the presence of siRGC-32-1 or -2, both C5b-9 and C5D induced CDC2 activation was abolished (Fig. 2C). The level of CDC2 activity was decreased by RGC-32 siRNA even though CDC2 proteins were not changed (Fig. 2B).
Figure 2. RGC-32 is required for the C5b-9 induced CDC2 activation.
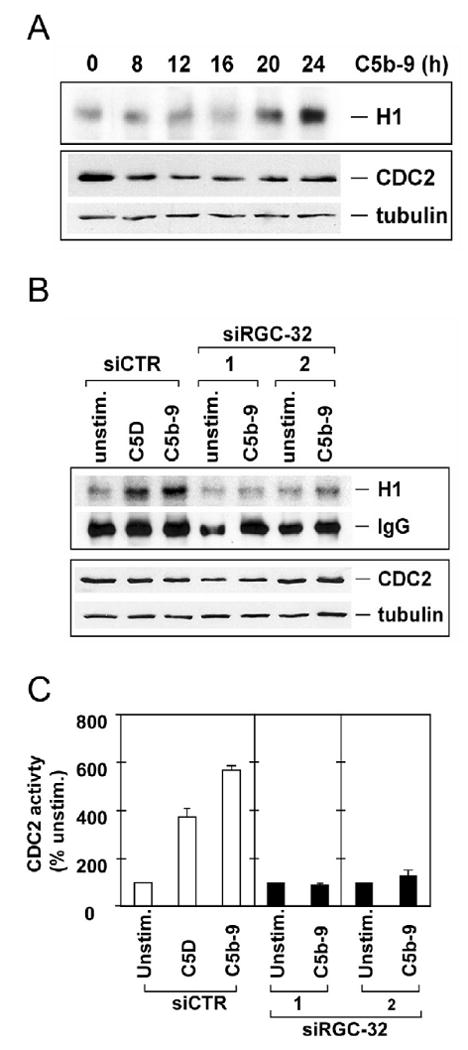
A. AEC were cultured in serum- and growth factor-free medium for 18 h, then exposed to serum C5b-9 for the times indicated. CDC2 was then immunoprecipitated and the activity was assayed using histone H1 as a substrate in the presence of γ32P-ATP (upper panel – H1) followed by SDS-PAGE and transfer to nitrocellulose membrane. Autoradiography revealed phosphorylated histone H1 bands, representing CDC2 activity. The CDC2 and tubulin protein levels were determined by western blot using anti-CDC2 IgG (lower panel). C5b-9 increased CDC2 activity in a time-dependent manner without any significant changes in the CDC2 and tubulin protein level.
B. siRGC-32-1, siRGC-32-2, or control siCTR were complexed with Mirus siQuest transfection reagent, and added to EC, at a final concentration of 25 nM. After 48 h, cells were starved in serum- and growth-factor-free medium for 18 h and then stimulated with serum C5b-9 (C5b-9) or C5D for 24 h. Cells were lysed and CDC2 activity was assayed (upper panel – H1). The IgG levels were used as loading control. The same blot was probed with anti-CDC2 IgG and tubulin by western blotting.
C. The CDC2 activity was quantitated by densitometric scanning of phosphorylated histone H1 bands and by normalizing the values to IgG. CDC2 was expressed as % of unstimulated cell activity (n=3). Significantly higher levels of CDC2 activity were induced by C5b-9, when compared to the level of unstimulated- or C5D-stimulated cells. The CDC2 activity induced by C5b-9 was abolished in cells transfected with both RGC-32 siRNAs.
Enhanced CDK2 activity by RGC-32 overexpression in HeLa cells
To investigate the role of cell cycle kinases in cell cycle induced by RGC-32 we used Hela cells stably overexpressing RGC-32. Forty eight clones were screened and two clones with the highest level of RGC-32 expression in the presence of tetracycline are shown in Fig. 3 A, B. To test if RGC-32 overexpression may affect CDK2 activity, HeLa overexpressing RGC-32 were arrested in M phase by nocodazole block in the presence of tetracycline and growth media containing 10% FBS. CDK2 activity was monitored by in vitro kinase assay. At 4 h after nocodazole block (post-M), there was a significant increase in CDK2 activity in HeLa expressing RGC-32 than in HeLa without RGC-32 expression (Fig. 3C). The enhanced CDK2 activity continued up to 12 h post-nocodazole when some of the cells were already in S-phase (data not shown). CDC6 is an endogenous substrate of CDK2 involved in the early steps of DNA replication. When CDC6 is phosphorylated by CDK2, it is stabilized by protecting it from proteolysis. Induction of RGC-32 expression led to increased CDC6 protein levels at 4 h and levels stabilized at 12 h (Fig. 3C). Our data suggests that RGC-32 overexpression may increase CDK2 activation to accelerate S-phase entry. In contrast CDC2 activity which is significantly higher in mitotic cells decreased significantly after 8h (Fig. 4A), suggesting that CDC2 does not play a role in G1/S transition. As expected, p27 expression was down regulated at an earlier time point in HeLa overexpressing RGC-32 when compared with control cells (Fig. 4 B).
Figure 3. Effect of RGC-32 overexpression on CDK2 and CDC6 activity.
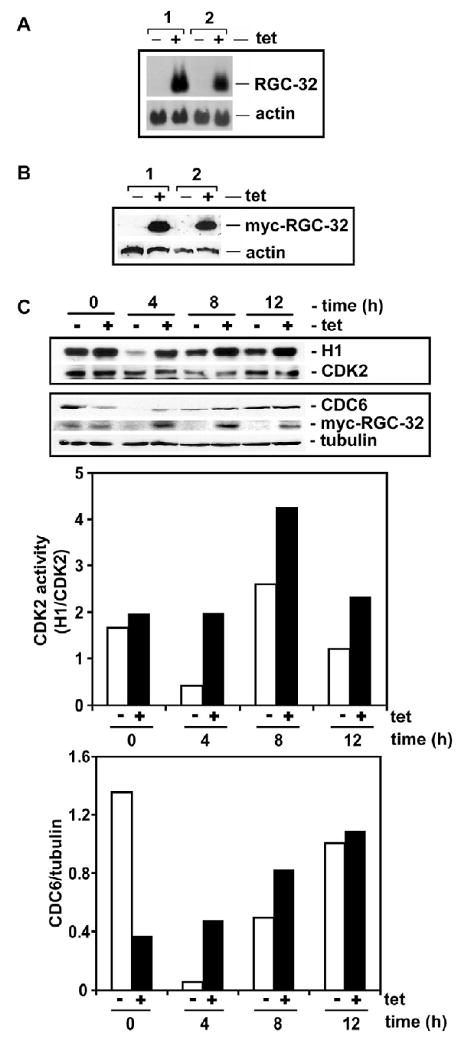
A, B. HeLa cell line stably overexpressing RGC-32. Two clones were examined for tetracycline induction of RGC-32 expression by northern blotting (A) and western blotting (B). For western blotting we used an antibody that recognizes the Myc-tag that is fused to the C-terminal of RGC-32. RGC-32 expression is induced by exposure to tetracycline (2 μg/ml).
C. RGC-32 accelerates and enhances CDK2 and CDC6 activity. HeLa cells overexpressing RGC-32 were arrested in M-phase by nocodazole block and released into growth media in the presence (+) or absence (-) of 2 μg/ml of tetracycline (tet). At the times indicated, cells were collected and lysed. 0 h post-M represents cells that were released from nocodazole blocked then immediately lysed. For CDK2 kinase assay (upper panel) immunoprecipitated CDK2 was incubated with [γ-32P] ATP and histone H1. Phosphorylated histone H1 (upper panel - H1) was detected by autoradiography. The same blot was used to detect CDK2 by western blotting (upper panel – CDK2). Whole lysates were used to detect CDC6, myc-RGC-32 and tubulin expression. To quantitate CDK2 activity and CDC6 densitometric scanning was performed.
Figure 4. Effect of RGC-32 overexpression on CDC2 activity and p27 expression.
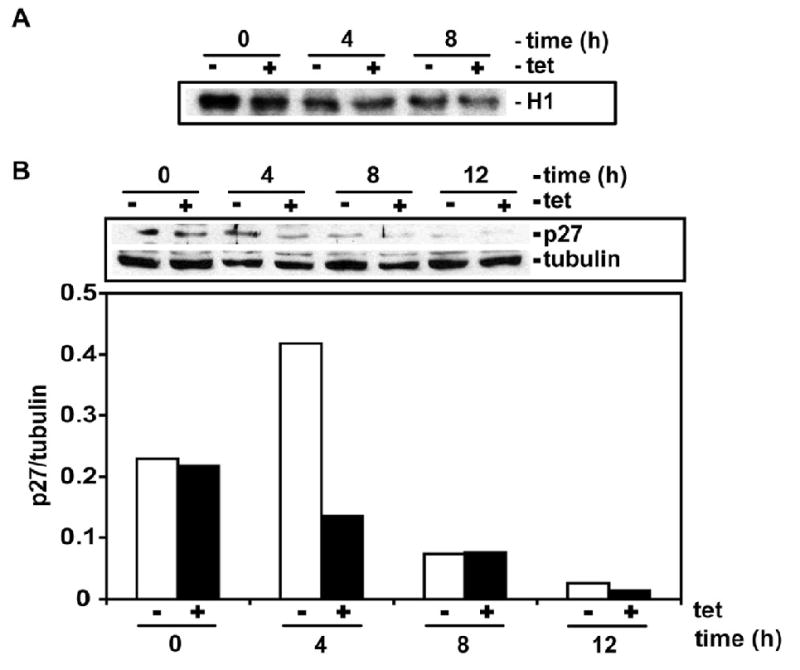
HeLa RGC-32 overexpressing were arrested in M-phase by nocodazole block and released into growth media in the presence (+) or absence (-) of 2 μg/ml of tetracycline (tet). At the times indicated, cells were collected and lysed. 0 h post-M represents cells that were released from nocodazole blocked then immediately lysed. For CDC2 kinase assay (upper panel) immunoprecipitated CDC2 was incubated with [γ-32P] ATP and histone H1. Phosphorylated histone H1 (upper panel - H1) was detected by autoradiography. Whole lysates was used to detect p27 and tubulin expression.
RGC-32 regulates Akt phosphorylation
We have previously shown that the growth factor release induced by C5b-9 is Akt dependent, possibly through phosphorylation of Akt via PI3K pathway activation (Fosbrink et al., 2006); therefore, we tested whether RGC-32 also binds and regulates Akt. A possible interaction of RGC-32 with Akt was first examined in vitro, by GST pull down assay. Akt was recovered with RGC-32 in GST pull down assay (Fig. 5A). As expected, GST alone was unable to pull down Akt (Fig. 5B). We also found that RGC-32 co-immunoprecipitate in vivo with Akt (Fig. 5C).
Figure 5. RGC-32 interacts and regulates Akt.
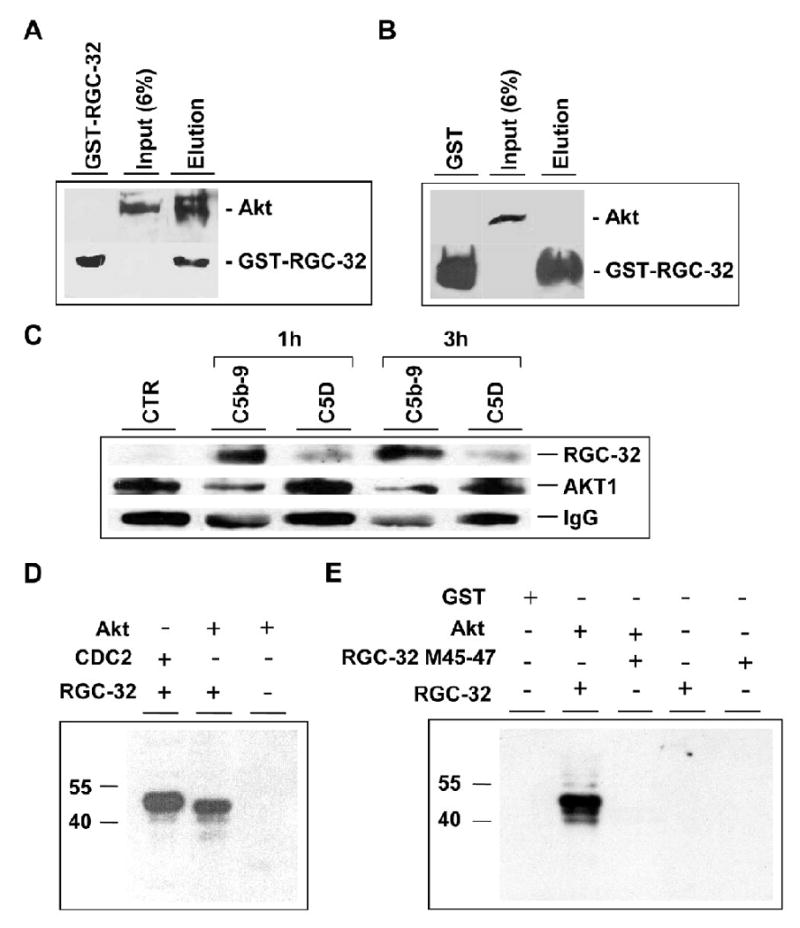
A, B. Formation of RGC-32-Akt complexes. GST-RGC-32 fusion protein (A) or GST (negative control) (B) interaction with Akt in AEC lysates was analyzed using the Profound pull-down GST protein-protein interaction kit (Pierce). The proteins binding to GST-RGC-32 or GST in the presence of glutathione-Sepharose beads were eluted and analyzed by Western blotting using anti-Akt antibodies. A. Akt was eluted together with GST-RGC-32 (A; lane Elution). Akt input is also shown (A; Input 6% lane). B. Akt was not recovered with GST alone (B; lane GST). Akt input is shown (B; Input 6% lane).
C. Coimmunoprecipitation of RGC-32 with Akt in vivo. AEC treated with C5b-9 or C5b6 (C5D) for 1 and 3 h were lysed and immunoprecipitated with anti-Akt IgG in the presence of protein A/G-agarose. Protein eluted from the beads separated in 10% SDS-PAGE and then immunoblotted with anti-RGC-32 and anti-Akt. IgG was used for normalization. CTR., unstimulated cells.
D, E. Phosphorylation of RGC-32 by Akt in vitro. Phosphorylation of RGC-32 by Akt was examined by in vitro kinase assay. Recombinant RGC-32-GST (1 μM) or GST (1 μM) were incubated with Akt (1μM) in the presence of 1 μCi of [γ-32P] ATP/sample as described in Material and Methods section. The phosphorylation of GST-RGC-32 was assessed by SDS-PAGE and then autoradiography. GST-RGC-32 (revealed as a 40 kD band) but not GST was phosphorylated by Akt (D). Phosphorylation of GST- RGC-32 by CDC2/cyclin B1 was used as a positive control (Badea et al., 2002). Mutation of Serine 45 and Seine 47 in RGC-32 molecule resulted in the abolition of GST-RGC-32 phosphorylation by Akt (E).
We then explored whether RGC-32 serves as a substrate for Akt using purified proteins. As shown in Fig. 5D, Akt phosphorylated GST-RGC-32 in vitro, and CDC2 was also able to phosphorylate RGC-32. GST alone was not phosphorylated by Akt (data not shown). We searched for an Akt kinase motif in human RGC-32 using a software program available at http://scansite.mit.edu that identifies likely sequence motifs. Using this program we identified a putative Akt kinase motif at position Ser-45 and Ser-47. To test whether Ser-45 and Ser-47 are phosphorylated by Akt, the sites were mutated to alanine, and the corresponding GST fusion protein was expressed in Escherichia coli. The mutated proteins were purified and used to examine the role in Akt phosphorylation of RGC-32. As shown in Fig. 5E, Akt failed to phosphorylate RGC-32 S45A-S47A mutant. These data indicate that Ser 45 and Ser 47 may be the RGC-32 phosphorylation sites for Akt kinase.
We then tested the effect of RGC-32 knockdown on Akt phosphorylation induced by C5b-9 (Fig. 6). C5b-9 was able to induce Akt phosphorylation in AEC treated with siCTR but not in cells transfected with siRGC-32, indicating an involvement of RGC-32 in regulating Akt phosphorylation at Ser-473.
Figure 6. RGC-32 is required for C5b-9-induced Akt phosphorylation.
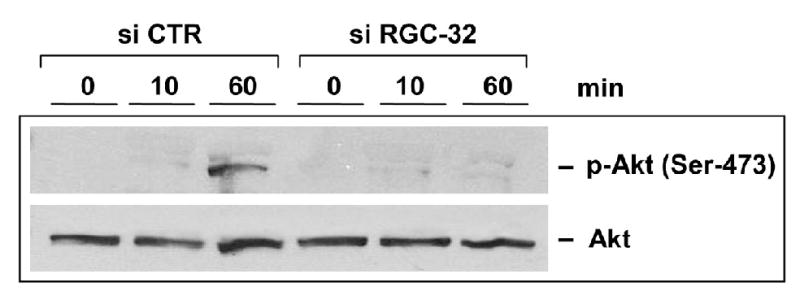
AEC were transfected with RGC-32 siRNA or control CTR siRNA, starved in serum- and growth factor-free media for 18 h, then stimulated with serum C5b-9, as described earlier, for 10 and 60 min. Cells were lysed and Akt phosphorylation was assessed by western blot, using anti-Akt IgG specific for Akt phosphorylated at Ser 473. RGC-32 knockdown results in inhibition of Akt phosphorylation by C5b-9.
RGC-32 regulates the release of growth factors by C5b-9
C5b-9 is known to induce the release of multiple growth factors from EC, which are required for cell proliferation (Benzaquen et al., 1994; Fosbrink et al., 2006). Since C5b-9 induced cell cycle activation is RGC-32-dependent, we tested the effect of RGC-32 knockdown on growth factors release by C5b-9. EC were transfected with RGC-32 siRNA and stimulated with purified C5b-9 or control C5b6. Then the conditioned media was used to probe a protein antibody array containing antibodies to various growth and angiogenic factors. Silencing of RGC-32 expression in AEC caused an altered growth factor release profile in response to C5b-9 compared to control siRNA transfected cells (Fig. 7A). RGC-32 siRNA knockdown caused increased levels of ENA-78, IL-8, TIMP-1, VEGF-D, while the levels of Leptin, PlGF and RANTES decreased. From these growth factors we have chosen to validate the effect on PlGF using ELISA (Fig. 7B). A similar decrease in PlGF levels was found after RGC-32 knockdown to that seen with the protein array.
Figure 7. RGC-32 is required for the release of angiogenic factors by C5b-9 and mediation of PIGF release.
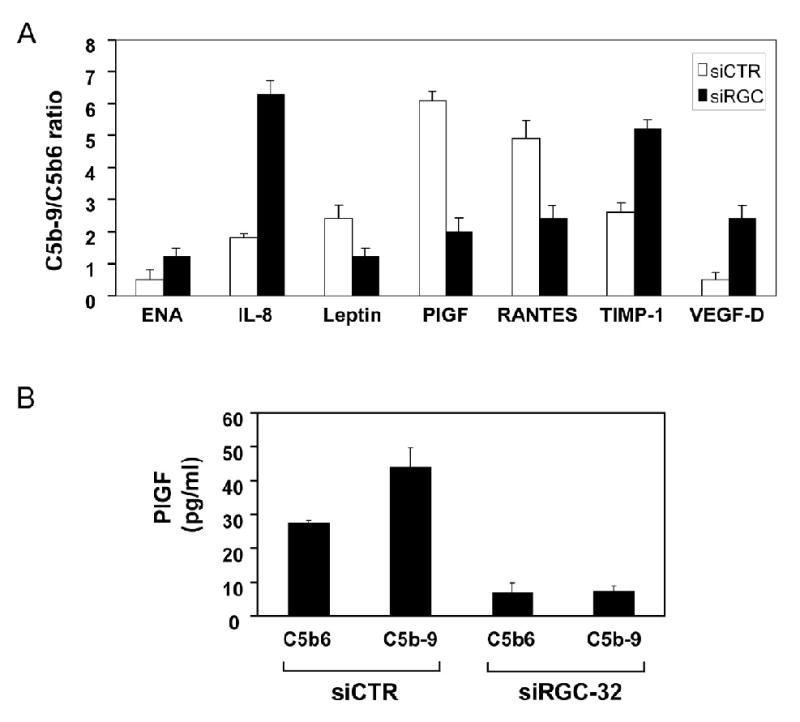
A. AEC were transfected with siRGC-32-1, or control siCTR, then starved in serum- and growth factor-free media for 18 h. Cells were then stimulated with C5b6 or C5b-9 assembled from purified components for 1 h. The conditioned media were subjected to an angiogenic factor antibody protein array. The array was quantitated by densitometric scanning and C5b-9/C5b6 ratios were calculated. Only those factors exhibiting a 1.5-fold difference in C5b-9/C5b6 ratio are shown.
B. AEC transfected with siRGC-32 or control siCTR and cultured in serum and growth factor free media, as described, were stimulated with C5b6 or C5b-9 assembled from purified components for 1 h. The conditioned media were tested for the presence of PlGF by ELISA. PlGF release was inhibited in cells transfected with siRGC-32.
Discussion
We have shown previously that over expression of RGC-32 induced cell cycle activation and progression through S and G2/M phases (Badea et al., 2002; Badea et al., 1998) suggesting an important role of RGC-32 in cell cycle activation. This observation led us to investigate whether endogenous RGC-32 is required for cell cycle activation induced by C5b-9 or other growth factors. To assess the requirement for endogenous RGC-32 in activating cell cycle by C5b-9 and growth factors, we used siRNA to silence RGC-32 expression.
We were able to show for the first time that RGC-32 silencing effectively abolished C5b-9 and growth factors induced cell proliferation. These findings suggest that endogenous RGC-32 is an essential mediator of cell cycle activation induced by growth factors. Our data also show that RGC-32 silencing abolished C5b-9 induced CDC2 activation and suggesting that at least in part the effect on the cell cycle is mediated by down regulation of CDC2 activation. Since RGC-32 binds to and up regulates CDC2 activity we can speculate that RGC-32 is an essential regulator of CDC2 activation. CDC2 is known to be required during mitosis (Sherr and Roberts, 1999) and accumulating evidence indicates that CDC2 also affects the G1/S transition and S-phase. In human T-lymphocytes, CDC2 is expressed at G1/S transition (Furukawa et al., 1990) and CDC2 phosphorylation is also required for DNA replication (Dutta and Stillman, 1992). CDC2-cyclin B1 complex, when expressed in nuclei, can phosphorylate S-phase substrates and promote S-phase entry (Moore et al., 2003). In CDK2 knockout mice, cyclin E-CDC2 complex was found to induce S-phase entry (25). Despite these evidences, in an inducible tet system we were not able to show that CDC2 plays a role in RGC-32 induced G1/S transition. These data support the model that CDC2 activation requires RGC-32 during mitosis and that both might be required for C5b-9 induced cell cycle activation. In addition CDK2 seems to play an important role in G1/S transition both in AEC and in stable overexpression of RGC-32 in HeLa cells.
C5b-9, like many other growth factors, achieves its pleotrophic biological activities by activating multiple signaling pathways in target cells (Niculescu and Rus, 2001). Cell proliferation induced by C5b-9 is dependent on the activation of PI3K/Akt/FOXO1 signaling pathway (Fosbrink et al., 2006). This signaling pathway also regulates the C5b-9 induced release of growth factors, such as FGF, PDGF (Benzaquen et al., 1994), IL-8 (Rus et al., 1996b) and PlGF (Fosbrink et al., 2006). To assess if RGC-32 plays a role in growth factor release, we analyzed their release in the AEC supernatants in response to C5b-9. Interestingly, RGC-32 silencing caused an altered expression profile of growth factors in AEC by decreasing the release of Leptin, PlGF and RANTES, and increasing the release of ENA-78, IL-8, TIMP-1 and VEGF-D. These results clearly indicate that RGC-32 is involved in regulation of C5b-9 induced growth factors release. The fact that RGC-32 can affect PlGF and Leptin release may be significant since PlGF and Leptin induce cell proliferation, migration and neovascularization (Sierra-Honigmann et al., 1998; Ziche et al., 1997). In addition, local delivery of PlGF promotes neointima formation in hypercholesterolemic rabbits and macrophage accumulation in atherosclerotic lesions in ApoE-/- mice (Khurana et al., 2005). Moreover, RANTES can bind to the surface of activated endothelium, where it triggers the firm arrest and transmigration of monocytes under flow conditions, which may be instrumental for its involvement in atherogenic processes, such as neointima formation after arterial injury (Schober et al., 2002).
Since the release of many growth factors is Akt dependent, we have tested if the silencing of RGC-32 had any effect on Akt phosphorylation. It is reasonable to speculate that the role-played by RGC-32 in growth factor release is most probably mediated through binding and regulation of Akt phosphorylation. Since Akt and the release of growth factors play important roles in cell cycle activation induced by C5b-9, the inhibition of growth factors by RGC-32 knockdown might be responsible for the inhibition of cell cycle activation.
Our data thus indicate that endogenous RGC-32 plays a critical role in regulating cell cycle activation induced by C5b-9, in part by regulating phosphorylation of multiple cell cycle and signal transduction-related substrates. Further exploration of role played by RGC-32 in vivo is necessary to fully understand its role in cell cycle activation and proliferation.
Acknowledgments
We thank Dr. Moon L. Shin for editing the manuscript. This work was supported, in part, by a Veterans Administration Merit Award (HR), the Veterans Administration Maryland Health Care System, Multiple Sclerosis Center of Excellence, Baltimore, MD (HR) and by the US Public Health Grant, RO1 NS42011 (HR). M. Fosbrink was supported in part by an NIH training grant ES 07263.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Badea T, Niculescu F, Soane L, Fosbrink M, Sorana H, Rus V, Shin ML, Rus H. RGC-32 increases p34CDC2 kinase activity and entry of aortic smooth muscle cells into S-phase. J Biol Chem. 2002;277:502–8. doi: 10.1074/jbc.M109354200. [DOI] [PubMed] [Google Scholar]
- Badea TC, Niculescu FI, Soane L, Shin ML, Rus H. Molecular cloning and characterization of RGC-32, a novel gene induced by complement activation in oligodendrocytes. J Biol Chem. 1998;273:26977–81. doi: 10.1074/jbc.273.41.26977. [DOI] [PubMed] [Google Scholar]
- Bazzoni G, Dejana E. Endothelial cell-to-cell junctions: Molecular organization and role in vascular homeostasis. Physiol Rev. 2004;84:869–901. doi: 10.1152/physrev.00035.2003. [DOI] [PubMed] [Google Scholar]
- Benzaquen LR, Nicholson-Weller A, Halperin JA. Terminal complement proteins C5b-9 release basic fibroblast growth factor and platelet-derived growth factor from endothelial cells. J Exp Med. 1994;179:985–92. doi: 10.1084/jem.179.3.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carney DF, Koski CL, Shin ML. Elimination of terminal complement intermediates from the plasma membrane of nucleated cells: the rate of disappearance differs for cells carrying C5b-7 or C5b-8 or a mixture of C5b-8 with a limited number of C5b-9. J Immunol. 1985;134:1804–9. [PubMed] [Google Scholar]
- Carney DF, Lang TJ, Shin ML. Multiple signal messengers generated by terminal complement complexes and their role in terminal complement complex elimination. J Immunol. 1990;145:623–9. [PubMed] [Google Scholar]
- Cunningham KS, Gotlieb AI. The role of shear stress in the pathogenesis of atherosclerosis. Lab Invest. 2005;85:9–23. doi: 10.1038/labinvest.3700215. [DOI] [PubMed] [Google Scholar]
- Donninger H, Bonome T, Radonovich M, Pise-Masison CA, Brady J, Shih JH, Barrett JC, Birrer MJ. Whole genome expression profiling of advance stage papillary serous ovarian cancer reveals activated pathways. Oncogene. 2004;23:8065–77. doi: 10.1038/sj.onc.1207959. [DOI] [PubMed] [Google Scholar]
- Dutta A, Stillman B. cdc2 family kinases phosphorylate a human cell DNA replication factor, RPA, and activate DNA replication. Embo J. 1992;11:2189–99. doi: 10.1002/j.1460-2075.1992.tb05278.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fosbrink M, Badea T, Shin ML, Rus H. Regulation of RGC-32 mRNA expression by terminal complement complex C5b-9 and growth factors. Faseb J. 2003;17:323. [Google Scholar]
- Fosbrink M, Cudrici C, Niculescu F, Badea TC, David S, Shamsuddin A, Shin ML, Rus Overexpression of RGC-32 in colon cancer and other tumors. Exp Mol Pathol. 2005;78:116–122. doi: 10.1016/j.yexmp.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Fosbrink M, Niculescu F, Rus V, Shin ML, Rus H. C5b-9-induced endothelial cell proliferation and migration are dependent on Akt inactivation of forkhead transcription factor FOXO1. J Biol Chem. 2006;281:19009–18. doi: 10.1074/jbc.M602055200. [DOI] [PubMed] [Google Scholar]
- Fujimoto N, Igarashi K, Kanno J, Honda H, Inoue T. Identification of estrogen-responsive genes in the GH3 cell line by cDNA microarray analysis. J Steroid Biochem Mol Biol. 2004;91:121–9. doi: 10.1016/j.jsbmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- Furukawa Y, Piwnica-Worms H, Ernst TJ, Kanakura Y, Griffin JD. cdc2 gene expression at the G1 to S transition in human T lymphocytes. Science. 1990;250:805–8. doi: 10.1126/science.2237430. [DOI] [PubMed] [Google Scholar]
- Herrmann J, Lerman LO, Mukhopadhyay D, Napoli C, Lerman A. Angiogenesis in atherogenesis. Arterioscler Thromb Vasc Biol. 2006;26:1948–57. doi: 10.1161/01.ATV.0000233387.90257.9b. [DOI] [PubMed] [Google Scholar]
- Hila S, Soane L, Koski CL. Sublytic C5b-9-stimulated Schwann cell survival through PI 3-kinase-mediated phosphorylation of BAD. Glia. 36:58–67. doi: 10.1002/glia.1095. [DOI] [PubMed] [Google Scholar]
- James ER, Fresco VM, Robertson LL. Glucocorticoid-induced changes in the global gene expression of lens epithelial cells. J Ocul Pharmacol Ther. 2005;21:11–27. doi: 10.1089/jop.2005.21.11. [DOI] [PubMed] [Google Scholar]
- Khurana R, Moons L, Shafi S, Luttun A, Collen D, Martin JF, Carmeliet P, Zachary IC. Placental growth factor promotes atherosclerotic intimal thickening and macrophage accumulation. Circulation. 2005;111:2828–2836. doi: 10.1161/CIRCULATIONAHA.104.495887. [DOI] [PubMed] [Google Scholar]
- Libby P, Aikawa M, Jain MK. Vascular endothelium and atherosclerosis. Handb Exp Pharmacol. 2006:285–306. doi: 10.1007/3-540-36028-x_9. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001;25:402–8. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Moore JD, Kirk JA, Hunt T. Unmasking the S-phase-promoting potential of cyclin B1. Science. 2003;300:987–90. doi: 10.1126/science.1081418. [DOI] [PubMed] [Google Scholar]
- Niculescu F, Badea T, Rus H. Sublytic C5b-9 induces proliferation of human aortic smooth muscle cells: role of mitogen activated protein kinase and phosphatidylinositol 3-kinase. Atherosclerosis. 1999;142:47–56. doi: 10.1016/s0021-9150(98)00185-3. [DOI] [PubMed] [Google Scholar]
- Niculescu F, Rus H. Mechanisms of signal transduction activated by sublytic assembly of terminal complement complexes on nucleated cells. Immunol Res. 2001;24:191–9. doi: 10.1385/ir:24:2:191. [DOI] [PubMed] [Google Scholar]
- Niculescu F, Rus H, Shin ML. Receptor-independent activation of guanine nucleotide-binding regulatory proteins by terminal complement complexes. J Biol Chem. 1994;269:4417–23. [PubMed] [Google Scholar]
- Niculescu F, Rus H, van Biesen T, Shin ML. Activation of Ras and mitogen-activated protein kinase pathway by terminal complement complexes is G protein dependent. J Immunol. 1997;158:4405–12. [PubMed] [Google Scholar]
- Rus HG, Niculescu F, Shin ML. Sublytic complement attack induces cell cycle in oligodendrocytes. J Immunol. 1996a;156:4892–900. [PubMed] [Google Scholar]
- Rus HG, Vlaicu R, Niculescu F. Interleukin-6 and interleukin-8 protein and gene expression in human arterial atherosclerotic wall. Atherosclerosis. 1996b;127:263–71. doi: 10.1016/s0021-9150(96)05968-0. [DOI] [PubMed] [Google Scholar]
- Sagiv E, Memeo L, Karin A, Kazanov D, Jacob-Hirsch J, Mansukhani M, Rechavi G, Hibshoosh H, Arber N. CD24 is a new oncogene, early at the multistep process of colorectal cancer carcinogenesis. Gastroenterology. 2006;131:630–9. doi: 10.1053/j.gastro.2006.04.028. [DOI] [PubMed] [Google Scholar]
- Schober A, Manka D, von Hundelshausen P, Huo Y, Hanrath P, Sarembock IJ, Ley K, Weber C. Deposition of platelet RANTES triggering monocyte recruitment requires P-selectin and is involved in neointima formation after arterial injury. Circulation. 2002;106:1523–9. doi: 10.1161/01.cir.0000028590.02477.6f. [DOI] [PubMed] [Google Scholar]
- Scolding NJ, Morgan BP, Houston WA, Linington C, Campbell AK, Compston DA. Vesicular removal by oligodendrocytes of membrane attack complexes formed by activated complement. Nature. 1989;339:620–2. doi: 10.1038/339620a0. [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–12. doi: 10.1101/gad.13.12.1501. [DOI] [PubMed] [Google Scholar]
- Sierra-Honigmann MR, Nath AK, Murakami C, Garcia-Cardena G, Papapetropoulos A, Sessa WC, Madge LA, Schechner JS, Schwabb MB, Polverini PJ, Flores-Riveros JR. Biological action of leptin as an angiogenic factor. Science. 1998;281:1683–6. doi: 10.1126/science.281.5383.1683. [DOI] [PubMed] [Google Scholar]
- Vlaicu SI, Cudrici C, Ito T, Fosbrink M, Tegla CA, Rus V, Mircea PA, Rus H. Role of response gene to complement 32 in diseases. Arch Immunol Ther Exp. 2008;56:115–22. doi: 10.1007/s00005-008-0016-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziche M, Maglione D, Ribatti D, Morbidelli L, Lago CT, Battisti M, Paoletti I, Barra A, Tucci M, Parise G, Vincenti V, Granger HJ, Viglietto G, Persico MG. Placenta growth factor-1 is chemotactic, mitogenic, and angiogenic. Lab Invest. 1997;76:517–31. [PubMed] [Google Scholar]