

Abstract
The design and synthesis of α-helix peptidomimetics using inverse electron demand Diels-Alder reactions is described. The potency of the resulting pyridazine-based library to disrupt the Bak/Bcl-XL interaction was tested using an in vitro Fluorescence Polarization Assay.
Protein-protein interactions are involved in the regulation of a wide variety of biological processes. Since the sequencing of the human genome, some research groups have put forward the challenge of developing a small molecule inhibitor for every protein-protein interaction.1 This is unlikely for many protein–protein complexes: large surface areas with ∼1600 Å2 or 170 atoms are involved,2,3 and their relatively flat shapes do not offer purchase for small molecules. Two exceptions are possible. Allosteric sites, particularly those deep within the core of a protein can modify protein-protein interactions such as those well known in the association of hemoglobin subunits.4a A second favorable situation arises when one of the interacting surfaces features a deep, narrow invagination. An appropriate small molecule can be more or less surrounded in this environment, with the consequence of high binding affinity through the molecular recognition elements on offer. The natural complement for the cleft may be a strand or loop structure,4b but α-helices are often involved.4c Moreover, only a few side chains of the helices typically occupy the binding site on complexation, and we are concerned with these cases here.
The helices present their i, i+3/i+4 and i+7 residues to make crucial contacts with the target protein that constitute the majority of binding energy (Figure 1a).5 These residues project from one face of the helix with well-known distance and angular relationships as shown in Figure 1a.
Figure 1.
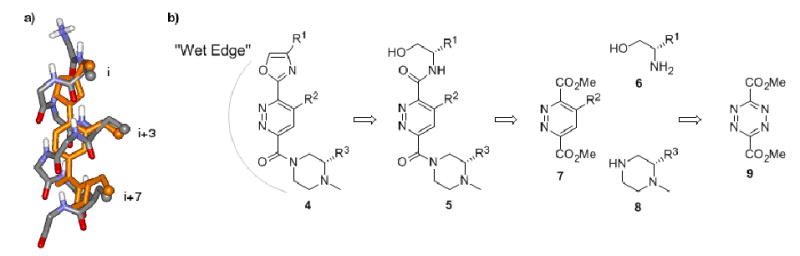
a) Overlay of energy minimized 4 with a generic α-helix (Minimization carried out using Macromodel (AMBER)); b) General retrosynthetic approach of the target molecule 4.
The development of non-peptide based scaffolds capable of displaying functionality in a fashion imitating the relevant binding residues of an α-helix was pioneered by Hamilton.6-9 Many of these compounds have shown relatively high affinity for α-helix binding sites, as well as in vitro, and in vivo activity. Terphenyl derivatives functionalized at the 3, 2′, and 2″ positions such as 1 can achieve a staggered conformation where the substituents are displayed in a way that closely resembles the i, i+3, and i+7 residues of an α-helix (Scheme 1). A series of these molecules were synthesized in a modular fashion by Hamilton and co-workers, allowing for the incorporation of multiple components.10
Scheme 1.

Examples of alpha-helix mimetics: terphenyl 1,10 oligoamide foldamer 2,11 terephtalamide 3.12
Inspired by the success of the Hamilton terphenyl scaffolds, we set out to devise synthetic methods for access to structurally similar molecules that feature more hydrophilic components via an easier synthetic route. Our major alteration was the incorporation of a pyridazine heterocycle as the center ring of our scaffolds. These heterocycles are readily accessible through an inverse electron demand Diels-Alder reaction between an electron deficient 1,2,4,5-tetrazine and an electron rich dienophile. This type of chemistry has been extensively studied over the past two decades by the Boger and Snyder groups.13-16
An overlay of an α-helix with a general depiction of one of our target molecules (4, Figure 1b) is shown in Figure 1a. The i, i+3 and i+7 residues of the α-helix as well as their counterparts on the synthetic scaffold are highlighted as small spheres. It is clear that the presentation of functionality by scaffold 4 is close to that of a natural α-helix. The new scaffolds are intended to present both a hydrophobic surface for recognition and a “wet edge” that is rich in hydrogen bond donors and acceptors. This was intended to enhance solubility and to ensure that during complexation with its target, the wet edge remains directed toward the solvent; in other words, no entropic penalty (or advantage) should occur by solvation at the wet edge as it is not altered during docking of the helix mimetic. We also hoped that these molecules would exhibit an increased solubility in water through protonation of the basic piperazine ring at physiological pH. These scaffolds may be thought of as synthetic counterparts of amphiphilic α-helices.
The general retrosynthetic approach to these molecules is laid out in Figure 1b. The major disconnections from the final oxazole-pyridazine-piperazine compound 4 are made at the amide bonds to give a pyridazine diester (7), an amino alcohol (6) and a piperazine (8). This synthesis is modular, as each piece can be synthesized separately and attached in sequence. Many amino-alcohols are commercially available bearing either natural amino acid side chains or some of the common homologs. The central pyridazine ring is readily available from the inverse electron demand Diels-Alder reaction of known dimethyl-1,2,4,5-tetrazine-dicarboxylate 913 and a suitable dienophile. Any piperazine (bearing a variety of protecting groups) can be easily prepared from the corresponding dipeptide. This synthesis also allows for the attachment of the piperazine module (or amino-alcohol) to either position of the pyridazine ring.
A small variety of mono-protected 2-substituted piperazines are commercially available. These compounds present standard, hydrophobic side chains (i.e. benzyl, isobutyl, etc.) and are available as the 2-Boc or 4-benzyl derivatives. As an alternative, these structures can be prepared containing any of the desired amino acid residues following the procedure of Hartman et al.17
Since some of our desired piperazines were not commercially available, we synthesized a series of 2-N-methyl piperazines containing a variety of substituents.18 A representative example is shown in Scheme 2, (S)-2-sec-butyl-1-methylpiperazine. Coupling of amino acid derivative 10 and Boc-protected glycine using standard coupling reagents provided the dipeptide 11 in good yield. Removal of the Boc group with TFA followed by thermal cyclization in the presence of a hydrogen bonding solvent19 gave the crystalline diketopiperazine 12. Reduction with lithium aluminum hydride proceeded smoothly with mild heating to give the enantiomerically pure piperazine 13.
Scheme 2.
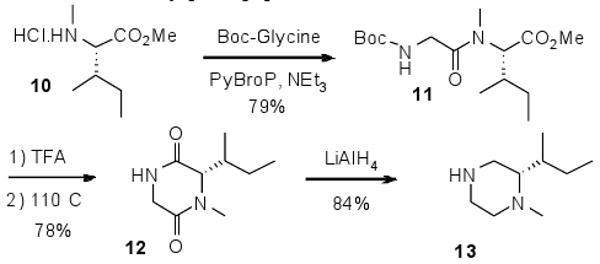
Synthesis of N-methyl-piperazine derivative 13.
For construction of the central pyridazine ring, the electron deficient 3,6-dimethyl-1,2,4,5-tetrazine dicarboxylate 9 was synthesized following the procedure of Boger and co-workers.13 Tetrazine 9 was reacted with 4-methylpentyne 14 to give pyridazine 15 in good yield (Scheme 3). The two heterocycles can then be connected by preparation of the aluminum amide20-22 of piperazine 13. Exposure of this intermediate to a solution of pyridazine diester 15 with gentle heating (41°C) results in smooth conversion to the mono-amide 16.23 It is clear from the 1H NMR spectrum of product 16 that only one methyl ester is present, as the two esters in the starting material are not magnetically equivalent. An HMBC NMR spectrum of the mono-ester 16 shows coupling between the protons of the piperazine ring and the pyridazine aryl proton to the same carbonyl carbon; the carbonyl at the less hindered 6-position.
Scheme 3.

Modular synthesis of the oxazole-pyridazine-piperazine scaffold 21 bearing hydrophobic substituents.
Exposure of mono-ester 16 to protected-valinol in the presence of AlMe3 also resulted in the aminolysis of the remaining methyl ester function, however the yield of this reaction was quite low (20%). To increase the yield, the Curtius coupling procedure was used.24 The methyl ester was transformed into the acyl hydrazide 17, diazotized to give the acyl-azide 18 and displaced with (S)-valinol to give the di-amide 19 in 64% overall yield.
To increase the rigidity of the final α-helix mimetics molecule and decrease loss of entropy upon binding, we pursued methods to close the β-hydroxy amide to an oxazole. Our first attempt to form an oxazole ring from the β-hydroxy amide moiety required oxidation of the alcohol to the aldehyde 20, and this proceeded as expected using the Dess-Martin periodinane. Exposure of the amide-aldehyde to PPh3/I2/DMAP,25 was not effective in the conversion of this function to an oxazole. A slight modification of the reaction conditions to PPh3, 2,6-di-tert-butyl pyridine, dibromo-tetrachloroethane and DBU,26 resulted in the transformation of amide-aldehyde 20 to the oxazole 21 in good yield.
We were also interested in preparing substituted pyridazines where the groups at the 3- and 6- positions are equivalent. As discussed above, while AlMe3 is a very reactive reagent, the yields obtained for aminolysis at the 3-position of substituted pyridazine rings are quite low. Magnesium chloride is another effective Lewis Acid capable of carrying out the aminolysis of pyridazine methyl esters.27 Exposure of iso-butyl substituted diester 15 to an excess of MgCl2 and (S)-valinol proceeds smoothly to give the diamide 22 in 63% yield (Scheme 4). Oxidation to the dialdehyde followed by oxazole formation as described above gives the dioxazole-pyridazine compound 23. While these bis-oxazole compounds do not display functionality in exactly the same orientation as the oxazole-pyridazine-piperazine scaffold 4, their synthesis is straightforward and is a potential theme for preparation of a small library for structure-activity correlations.
Scheme 4.

Modular synthesis of the bis-oxazole scaffold.
The synthetic sequences described above were applied to combine a series of piperazines, piperidines, pyridazines, and amino alcohols bearing hydrophobic groups to prepare a library of twenty four compounds shown in Figure 2b.
Figure 2.
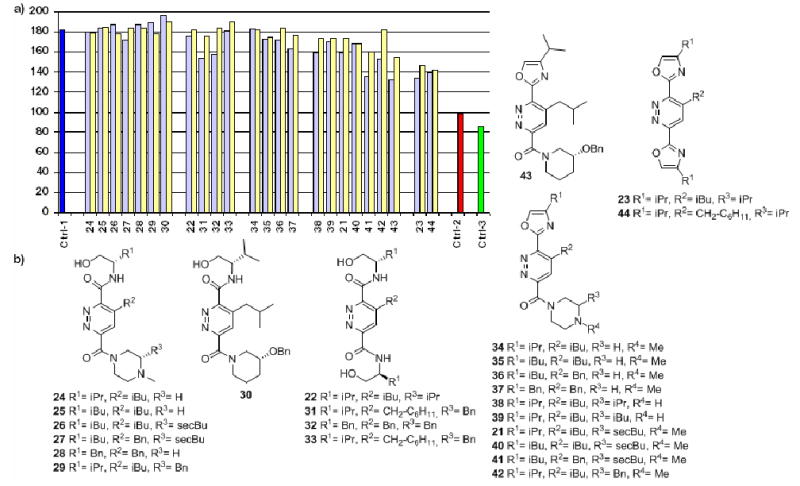
a) FPA for the disruption of the Bak-BH3 peptide and Bcl-XL interaction; pale yellow bars: 10 μM of assayed compound, pale blue bars: 50 μM of assayed compound. Ctrl-1 (dark blue bar): measured polarization for Bak-BH3 and Bcl-XL alone, Ctrl-2 (red bar): terphenyl 1 positive control, Ctrl-3 (green bar): measured polarization for Bak-BH3 peptide alone; b) Library tested in the FPA.
To evaluate the activity of our α-helix mimetics we tested their ability to disrupt the Bak/Bcl-XL interaction. Bak and Bcl-XL are two proteins involved in apoptosis and thus have become targets as new therapeutic approaches for the treatment of cancer and other diseases.9 It is known that when the domain of interaction of Bak (termed “BH3”) interacts with Bcl-XL, it adopts an α-helix conformation that lies in a hydrophobic cleft at the surface of Bcl-XL. Key contacts are made between the hydrophobic residues Val74, Leu78, Ile81 and Ile85 of Bak and hydrophobic residues lining the pocket of Bcl-XL.5b
The binding affinity of our molecules for Bcl-XL was determined by a fluorescence polarization assay (FPA)28 using fluorescently labeled Bak-BH3 peptide.29 The polarization amount (mP) is plotted on the y-axis in the graph in Figure 2. It provides a rough estimate of the affinity for the small molecules 22-44 for Bcl-XL. The dark blue bar in the column on the far left of the graph represents the amount of polarization observed when the fluorescein-labeled Bak peptide is bound to Bcl-XL. The green bar in the column on the far right is the fluorescein-labeled Bak peptide alone in solution. Terphenyl 130 has been previously shown to bind to Bcl-XL with nanomolar affinity10 and acts as a positive control (red bar, far right). Values from ca. 100-140 mP represents a “target area” for polarization values. Since this was a preliminary screening assay, we sought compounds having high enough affinity for Bcl-XL to generate a polarization value within this range.
The left part of Figure 2a lists the results from compounds shown in Figure 2b that appear to have little to no affinity for Bcl-XL. This group of compounds is composed of molecules containing β-hydroxyamides (compounds 22 and 24 to 33) and/or unsubstituted piperazines (compounds 34 to 37). It is not surprising that this series of molecules have low affinity for Bcl-XL. Those compounds presenting three R-groups also contain β-hydroxy amide functions that possess six freely rotating bonds each. The entropic cost for “freezing” out these rotations on receptor binding is high. However, the compounds containing the more rigid oxazole function in place of the β-hydroxy amide (three freely rotating bonds) only present two R-groups. It is likely these structures are not involved in enough interactions with the receptor to induce binding. Also, most of the compounds in this set contain basic piperazine functions that are protonated under the assay conditions. While we included this feature in these molecules for ease of synthesis and to increase water-solubility, the cationic ammonium group may be causing unfavorable interactions on binding to the receptor. It may be involved in repulsive electrostatic contacts with residues near the hydrophobic cleft on Bcl-XL or the large enthalpic costs for desolvation of this ammonium center upon binding. The second set of data shown in Figure 2a contains compounds belonging to the oxazole-pyridazine-piperazine and bis-oxazole scaffold classes. Of this group, four compounds approximate our desired mP values: 23, 41, 43 and 44. Three of these compounds (23, 43 and 44) are neutral while the fourth (41) presents a hydrophobic phenyl ring. These compounds represent the strongest binders for Bcl-XL of the small library depicted in Figure 2b, however they do not approach the affinity demonstrated by terphenyl 1 (red bar on the far right).
Some insight can be gleaned from these results that suggest a strategy for second-generation structures. We see that neutral compounds have a higher affinity for Bcl-XL than those containing a positive charge, except in the case of 41. The cationic ammonium may cause unfavorable electrostatic interactions while in the hydrophobic binding pocket and/or the penalty for desolvation of this ionic center is too high. Compound 1 possesses two carboxylate functions thought to be involved in favorable electrostatic interactions with Bcl-XL along the upper ridge of the binding site.10 The extra favorable interaction(s) experienced by compound 1 may be enough to bring its binding affinity into the nanomolar range.
Acknowledgments
We are grateful to the Skaggs Institute for Research, the ARCS Foundation for a Fellowship to S.M.B. and the National Institutes of Health (CA 113318) for support. Both Kemia, Inc. and Novartis provided support. We thank Prof. A. D. Hamilton for a gift of terphenyl 1 and Prof. D. L. Boger and Prof. E. Roberts for advice and helpful discussions. We are pleased to acknowledge Drs. D.-H. Huang and L. Pasternack for assistance with NMR experiments.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Schreiber SL. Bioorg Med Chem. 1998;6:1127. doi: 10.1016/s0968-0896(98)00126-6. [DOI] [PubMed] [Google Scholar]
- 2.Jones S, Thornton JM. Proc Natl Acad Sci. 1996;93:13. doi: 10.1073/pnas.93.1.13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bogan AA, Thorn KS. J Mol Biol. 1998;280:1. doi: 10.1006/jmbi.1998.1843. [DOI] [PubMed] [Google Scholar]
- 4.a) Dickerson RE, Geis I. Hemoglobin: Structure, Function, Evolution, and Pathology. Benjamin Cummings; 1983. pp. 48–58. [Google Scholar]; b) Davis CN, Mann E, Behrens MM, Gaidarova S, Rebek M, Rebek J, Jr, Bartfai T. Proc Natl Acad Sci USA. 2006;103:2953. doi: 10.1073/pnas.0510802103. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Tsai CJ, Xu D, Nussinov R. Protein Sci. 1997;6:1793. doi: 10.1002/pro.5560060901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.a) Fairlie DP, West ML, Wong AK. Curr Med Chem. 1998;5:29. [PubMed] [Google Scholar]; b) Sattler M, Liang H, Nettesheim D, Meadows RP, Harlan JE, Eberstadt M, Yoon HS, Shuker SB, Chang BS, Minn AJ, Thompson CB, Fesik SW. Science. 1997;275:983. doi: 10.1126/science.275.5302.983. [DOI] [PubMed] [Google Scholar]
- 6.a) Davis JM, Tsou LK, Hamilton AD. Chem Soc Rev. 2007;36:326. doi: 10.1039/b608043j. [DOI] [PubMed] [Google Scholar]; b) Fletcher S, Hamilton AD. J R Soc Interface. 2006;3:215. doi: 10.1098/rsif.2006.0115. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Yin H, Hamilton AD. Angew Chem Int Ed. 2005;44:4130. doi: 10.1002/anie.200461786. [DOI] [PubMed] [Google Scholar]
- 7.Antuch W, Menon S, Chen QZ, Lu Y, Sakamuri S, Beck B, Schauer-Vukasinovic V, Agarwal S, Hess S, Dömling A. Bioorg Med Chem Let. 2006;16:1740. doi: 10.1016/j.bmcl.2005.11.102. [DOI] [PubMed] [Google Scholar]
- 8.a) Arkin MR, Wells JA. Nature Rev Drug Disc. 2004;3:301. doi: 10.1038/nrd1343. [DOI] [PubMed] [Google Scholar]; b) Babine RE, Bender SL. Chem Rev. 1997;97:1359. doi: 10.1021/cr960370z. [DOI] [PubMed] [Google Scholar]
- 9.Walensky LD. Cell Death and Differentiation. 2006;1 doi: 10.1038/sj.cdd.4401992. [DOI] [PubMed] [Google Scholar]
- 10.Yin H, Lee Gi, Sedey KA, Kutzki O, Park HS, Orner BP, Ernst JT, Wang HG, Sebti SM, Hamilton AD. J Am Chem Soc. 2005;127:10191. doi: 10.1021/ja050122x. [DOI] [PubMed] [Google Scholar]
- 11.Ernst JT, Becerril J, Park HS, Yin H, Hamilton AD. Angew Chem Int Ed. 2003;42:535. doi: 10.1002/anie.200390154. [DOI] [PubMed] [Google Scholar]
- 12.Yin H, Lee Gi, Sedey KA, Rodriguez JM, Wang HG, Sebti SM, Hamilton AD. J Am Chem Soc. 2005;127:5463. doi: 10.1021/ja0446404. [DOI] [PubMed] [Google Scholar]
- 13.Boger DL, Panek JS, Patel M. Org Synth. 1992;70:79. [Google Scholar]
- 14.Spencer GH, Cross PC, Wiberg KB. J Chem Phys. 1939;35:1919. [Google Scholar]
- 15.Sauer J, et al. Chem Ber. 1965;98:1435. [Google Scholar]
- 16.Boger DL, Patel M. J Org Chem. 1987;53:1405. [Google Scholar]
- 17.Neville AJ, Ciccarone TM, Dinsmore CJ, Gomez RP, Williams TM, Hartman GD. 5,856,326. US Patent. 1999
- 18.Jung ME, Rohloff JC. J Org Chem. 1985;50:4909. [Google Scholar]
- 19.Nitecki DE, Halpern B, Westley JW. J Am Chem Soc. 1968;33:864. doi: 10.1021/jo01266a091. [DOI] [PubMed] [Google Scholar]
- 20.Basha A, Lipton M, Weinreb SM. Tetrahedron Lett. 1977;48:4171. [Google Scholar]
- 21.Sidler DR, Lovelace TC, McNamara JM, Reider PJ. J Org Chem. 1994;59:1231. [Google Scholar]
- 22.Levin JI, Turos E, Weinreb SM. Synth Comm. 1982;12:989. [Google Scholar]
- 23.Wan ZK, Woo GHC, Snyder JK. Tetrahedron. 2001;57:5497. [Google Scholar]
- 24.Meienhofer J. In: The peptides, Analysis, Synthesis, Biology. Gross E, Meienhofer J, editors. Vol. 1. Academic Press, Inc.; 1979. pp. 197–228. [Google Scholar]
- 25.Ceide SC, Trembleau L, Haberhauer G, Somogyi L, Lu X, Bartfai T, Rebek J., Jr Proc Natl Acad Sci. 2004;101:16727. doi: 10.1073/pnas.0407543101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wipf P, Lim S. J Am Chem Soc. 1995;117:558. [Google Scholar]
- 27.González-Gómez JC, Santana L, Uriarte E. Tetrahedron. 2003;59:8171. [Google Scholar]
- 28.Zhang H, Nimmer P, Rosenberg SH, Ng SC, Joseph M. Anal Biochem. 2002;307:70. doi: 10.1016/s0003-2697(02)00028-3. [DOI] [PubMed] [Google Scholar]
- 29.Assays for Bcl-XL were performed using as the binding partner the 5-carboxyfluorescein-labeled 25-mer peptide tracer Flu-Bak-BH3 (FITC-Ahx-PSSTMGQVGRQLAIIGDDINRRYDS) derived from the Bak BH3 domain. The assays were performed in a 96-well format, in 20 mM potassium phosphate buffer, pH 7.4, containing 150 mM NaCl. The final concentration of DMSO in all assays was 1%. The reaction was carried out in a 100 microL volume and the resulting polarization signal was measured at kex = 485 nm/kem = 535 nm using an Analyst TM AD Assay Detection System (LJL Biosystem, Sunnyvale, CA) after 20 min incubation of the reaction mixture at room temperature.
- 30.Compound 1 was generously provided by Prof. A. D. Hamilton.