

Abstract

Anomeric O-alkylation/arylation is applied to the synthesis of 2-deoxy-β-glycosides. Treatment of lactols with NaH in dioxane followed by the addition of electrophiles leads to the formation of 2-deoxy-β-glycosides in high yield and high selectivity. The high β-selectivity observed here demonstrates a powerful stereoelectronic effect for the stereoselective formation of acetals under kinetic control.
2-Deoxy-β-glycosides are present in biologically active natural products such as the lomaiviticins, olivomycin A, OSW-1, and durhamycin. The stereoselective preparation of 2-deoxy-β-glycosides is difficult1 because substituents at C2 often serve as directing groups during the glycosylation event. The synthesis challenge posed by 2-deoxy-β-glycosides coupled with their presence in nature has inspired a variety of approaches aimed at accessing these important glycosides. The most common methods involve the use of a heteroatom substituent at C2 of the glycosyl donor followed by its reductive removal after glycosylation.2 Other methods include the use of α-glycosyl phosphites,3 displacement of α-glycosylhalides,4 palladium catalyzed glycosylation reactions,5 utilization of alkoxy-substituted anomeric radicals, 6 and the use of glycosyl imidates as glycosyl donors under oxidative conditions. 7
We became interested in the synthesis of 2-deoxy-β-glycosides since they are present in the lomaiviticins, molecules we are targeting for synthesis (Figure 1). In particular, our recent synthesis of the central ring system of the lomaiviticins involves incorporation of the C4 and C4′ carbinols by SN2 displacement of allylic sulfones with methoxide anions.8 This raised the possiblity that the 2-deoxy-β-glycosides at C4 and C4′ might be incorporated via an anomeric O-alkylation using glycosyl-1- alkoxides.
Figure 1.
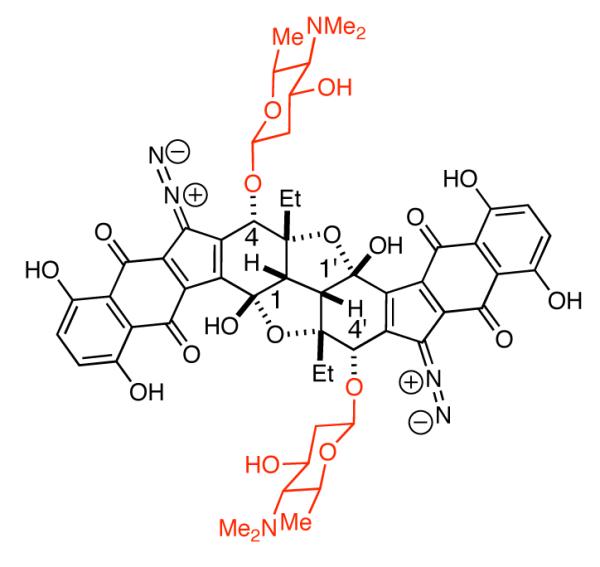
Lomaiviticin B
Anomeric O-alkylation with glycosyl-1-alkoxides generally affords high levels of β-glycosides.9 An explanation for this selectivity involves rapid equilibrium between axial and equatorial alkoxides with the enhanced nucleophilicity of the equatorial alkoxide leading to selective generation of β-glycosides (Scheme 1). It has been proposed that the enhanced nucleophilicity of the equatorial alkoxide (II), compared to the axial alkoxide (I), is due to increased electron-electron repulsion resulting from the alkoxide of II being gauche to both electron lone pairs of the ring oxygen compared to a single gauche interaction in I. This phenomenon has been referred to as the kinetic anomeric effect,9 or the β-effect, and may be similar to the α-effect, which is observed in molecules where the nucleophilic atom is directly attached to another heteroatom. To date, most examples of anomeric O-alkylation have been performed with substituents in the C2 position.10 This has made it difficult to assess the contribution of the β-effect versus steric (or electronic) influences of C2 substitutents in the selective formation of β-glycosides by anomeric O-alkylation. In this paper we report the first examples of anomeric O-alkylation/arylation in the absence of a C2 substituent, stereoselectively forming 2-deoxy-β-glycosides.
Scheme 1.
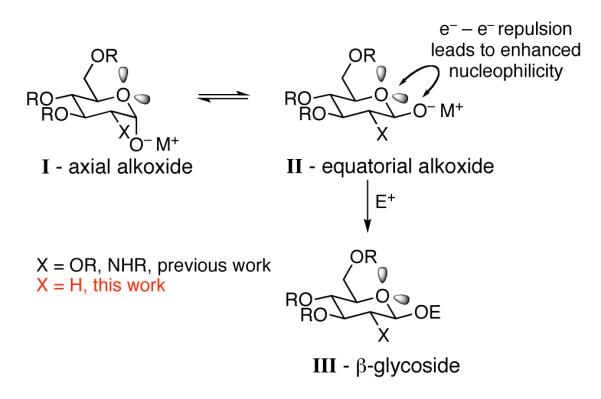
Kinetic Anomeric Effect
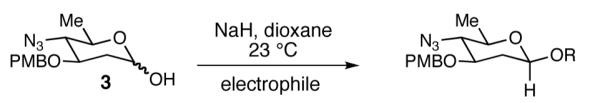
Before beginning our investigation into anomeric O-alkylation/arylation, a synthesis of a protected form of the N,N-dimethylpyrrolosamine sugar of lomaiviticin B was accomplished (Scheme 2). Beginning from the known glycal 1,11 triflation was followed by displacement with NaN3.12 The azido-glycal 2 was hydrated in a 2-step procedure involving addition of AcOH to 2 catalyzed by triphenylphosphine hydrogen bromide13 and cleavage of the resulting anomeric acetate with Me2NH. Lactol 3 was isolated as a 4:1 (α:β) mixture of anomers .
Scheme 2.
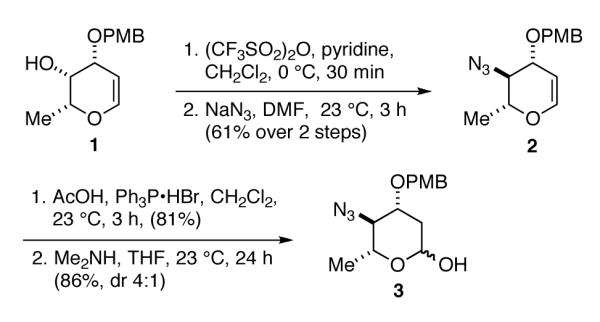
Synthesis of protected N,N-dimethylpyrrolosamine
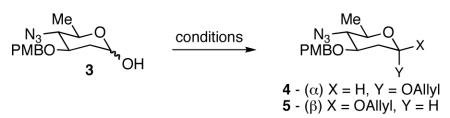
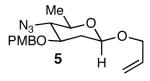
Our next goal was to establish general reaction conditions to access 2-deoxy-β-glycosides directly from an anomeric mixture of lactols (Table 1). During the course of our optimization studies we found that treatment of lactol 3 with KHMDS in THF at −78 °C followed by the addition of allyl bromide led to the exclusive formation of the α-anomer (Entry 1).14 Changing the base to NaH and raising the reaction temperature to 0 °C led to a 1:1 mixture of α and β anomers. The addition of LiBr, an additive known to enhance β-selectivity in anomeric O-alkylation15 provided only minor improvements in selectivity (Entry 3). Gratifyingly, we found that changing the solvent from DMF to dioxane and increasing the reaction temperature to 23 °C led to a dramatic increase in selectivity. These conditions afforded the 2-deoxy-β-glycoside 5 exclusively in 91% yield.
Table 1.
Optimization of Reaction Conditionsa
 | ||
|---|---|---|
| Entry | conditions | α:βb |
| 1 | KHMDS, THF, −78 °C, allyl bromide | 95:5 |
| 2 | NaH, DMF, allyl bromide, 0 °C | 1:1 |
| 3 | NaH, DMF, LiBr, allyl bromide, 0 °C | 1:1.5 |
| 4 | NaH, dioxane, allyl bromide, 23 °C | 5:95 |
Performed at 0.1 M.
As determined by 1H NMR (600 MHz)
With optimal reaction conditions in hand, the scope of this reaction was evaluated (Table 2). We considered nucleophilic aromatic substitution as a valuable application of this methodology due to its potential use in the synthesis of the aureolic acid family of natural products. Towards that end, when lactol 3 was treated with NaH in dioxane followed by 1-bromo-2,4-dinitrobenzene, the 2-deoxy-β-glycoside product 8 was isolated as an 18:1 mixture of anomers favoring the β-anomer. Primary triflates such as 916 proved to be suitable electrophiles for anomeric O-alkylation, as disaccharide 10 was isolated in 90% yield exclusively as the β diastereomer. Following the successful anomeric O-alkylation of a C6 triflate, we set out to determine if secondary triflates were competent electrophiles for this reaction. Despite previous reports by Schmidt of anomeric O-alkylations on similar systems,17 triflates 11 and 12 failed to undergo displacement (Entries 4 & 5). In each case, addition of the alkoxide took place at sulfur resulting in detriflation of 11 and 12.
Table 2.
Electrophile Scopea
 | |||
|---|---|---|---|
| Entry | electrophile | 2-deoxy-β-glycoside | yieldb (β:α)c |
| 1 |  |
 |
91% (20:1) |
| 2 |  |
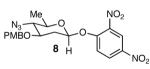 |
87% (18:1) |
| 3 | 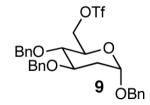 |
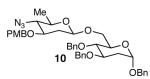 |
90% (20:1) |
| 4 | 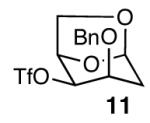 |
--- | n/a |
| 5 | 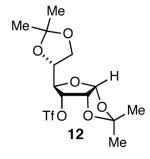 |
--- | n/a |
Reactions performed at 0.1 M.
Isolated yield after chromatographic purification.
As determined by 1H NMR (600 MHz)
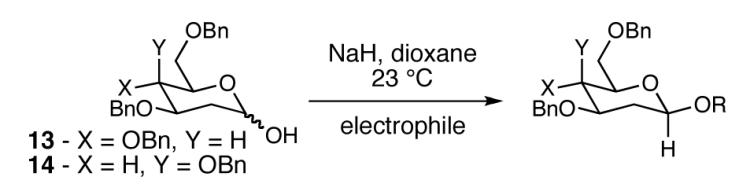
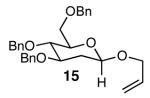
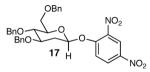
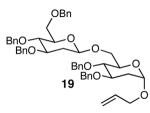
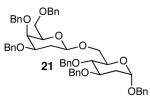
The methodology proved to be general with respect to the starting lactol component (Table 3). Lactol 13, derived from glucose, was allylated in high yield and provided exclusively the β-anomer (15). Nucleophilic aromatic substitution with 16 took place in high yield, albeit with slightly diminished stereoselectivity. The displacement of primary triflate 18 furnished 2-deoxy-β-glycoside 19 in 75% yield.18 The lactol derived from galactose (14) also provided 2-deoxy-β-glycosides in high yields and with high levels of sterocontrol (Entries 4 and 5). Galactose and mannose derived C6 triflates were found to be suitable electrophiles for this reaction (Entries 6 and 7). The ability to utilize lactols and C6 triflates with varying substitution patterns is a key feature of this methodology. This compares favorably to traditional glycosylation reactions where subtle changes to either the glycosyl donor or acceptor can have a dramatic impact on the stereoselectivity of the reaction.19
Table 3.
Lactol and Electrophile Scopea
 | ||||
|---|---|---|---|---|
| Entry | LACTOL | electrophile | 2-deoxy-β-glycoside | yieldb (β:α)c |
| 1 | 13 |  |
 |
70% (20:1) |
| 2 | 13 | 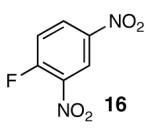 |
 |
85% (8:1) |
| 3 | 13 | 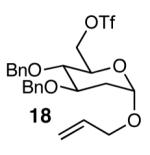 |
 |
75% (20:1) |
| 4 | 14 | 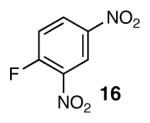 |
 |
90% (16:1) |
| 5 | 14 | 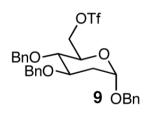 |
 |
80% (20:1) |
| 6 | 13 | 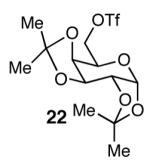 |
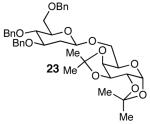 |
61% (20:1) |
| 7 | 13 | 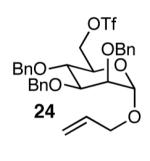 |
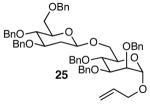 |
77% (20:1) |
Reactions performed at 0.1 M.
Isolated yield after chromatographic purification.
As determined by 1H NMR (600 MHz)
We next devised a competition experiment between lactol 3 and trans-4-tert-butylcyclohexanol20 to determine whether the increased reactivity of equatorial alkoxides derived from lactols (II, Scheme 1) can be attributed to steric effects or the aforementioned β-effect. An equimolar mixture of lactol 3 and trans-4-tert-butylcyclohexanol (26) was dissolved in dioxane and treated with 2 equivalents of NaH (Scheme 3). After 10 minutes, 1 equivalent of allyl bromide was added to the reaction mixture. After workup, 1H NMR of the crude reaction mixture showed only 2 products: 2-deoxy-β-glycoside 5 and unreacted trans-4-tert-butylcyclohexanol 26. The mixture was purified by flash column chromatography yielding 81% of 5 and quantitative recovery of trans-4-tert-butylcyclohexanol. This experiment is the best support to date that the high β-selectivity in anomeric O-alkylations/arylations is due to the increased nucleophilicity of the β-configured anomeric alkoxides.
Scheme 3.
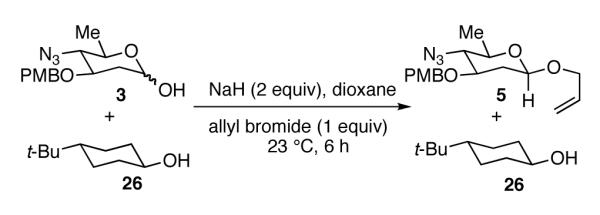
Competition Experiment
In conclusion, we report that anomeric O-alkylation/arylation can be used to form 2-deoxy-β-glycosides with high stereoselectivity. The high β-selectivity afforded with 2-deoxy-1-lactols supports the theory that the β-effect, rather than the substituent at C2, is the stereocontrol element in anomeric O-alkylations/arylations. This conclusion has been obscured in previous reports concerning anomeric O-alkylations/arylations since they all involved substrates with C2 substituents which may have influenced the stereoselectivity. The competition experiment reported here demonstrates that the nucleophilicity of β-configured anomeric alkoxides is enhanced over similar cyclohexyl alkoxides, presumably due to the proximity of the alkoxide and the lone pair electrons of the ring oxygen. These results suggest that the β-effect is a powerful stereoelectronic effect that may be useful in designing other stereoselective reactions.
Supplementary Material
Acknowledgment
Financial support for this project was provided by the NIH (CA125240), Merck Research Laboratories, Novartis, and AstraZeneca.
Footnotes
Supporting Information Available: Experimental procedures and spectral data for all new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Toshima K, Tatsuta K. Chem. Rev. 1993;93:1503. [Google Scholar]
- (2) (a).Roush WR, Bennett CE. J. Am. Chem. Soc. 1999;121:3541. [Google Scholar]; (b) Blanchard N, Roush WR. Org. Lett. 2003;5:81. doi: 10.1021/ol027257h. [DOI] [PubMed] [Google Scholar]; (c) Thiem J, Gerken M. J. Org. Chem. 1985;50:954. [Google Scholar]; (d) Ito Y, Ogawa T. Tetrahedron Lett. 1987;28:2723. [Google Scholar]; (e) Grewal G, Kaila N, Franck RW. J. Org. Chem. 1992;57:2084. [Google Scholar]; (f) Preuss R, Schmidt RR. Synthesis. 1988:694. [Google Scholar]; (g) Franck RW, Marzabadi CH. J. Org. Chem. 1998;63:2197. [Google Scholar]; (h) Nicolaou KC, Ladduwahetty T, Randall JL, Chucholowski A. J. Am. Chem. Soc. 1986;108:2466. doi: 10.1021/ja00269a066. [DOI] [PubMed] [Google Scholar]; (i) Perez M, Beau J-M. Tetrahedron Lett. 1989;30:75. [Google Scholar]; (j) Gervay J, Danishefsky S. J. Org. Chem. 1991;56:5448. [Google Scholar]; (k) Tavecchia P, Trumtel M, Veyrieres A, Sinay P. Tetrahedron Lett. 1989;30:2533. [Google Scholar]; (l) Wiesner K, Tsai TYR, Jin H. Helv. Chim. Acta. 1985;68:300. [Google Scholar]
- (3) (a).Pongdee R, Wu B, Sulikowski GA. Org. Lett. 2001;3:3523. doi: 10.1021/ol016593f. [DOI] [PubMed] [Google Scholar]; (b) Hashimoto S, Yanagiya Y, Honda T, Ikegami S. Chem. Lett. 1992:1511. [Google Scholar]
- (4) (a).Binkley RW, Koholic DJ. J. Org. Chem. 1989;54:3577. [Google Scholar]; (b) Toshima K, Misawa M, Ohta K, Tatsuta K, Kinoshita M. Tetrahedron Lett. 1989;30:6417. [Google Scholar]
- (5).Zhou M, O’Doherty GA. J. Org. Chem. 2007;72:2485. doi: 10.1021/jo062534+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6) (a).Crich D, Ritchie TJ. J. Chem. Soc. Perkin Trans. I. 1990:945. [Google Scholar]; (b) Kahne D, Yang D, Lim JJ, Miller R, Paguaga E. J. Am. Chem. Soc. 1988;110:8716. [Google Scholar]
- (7).Tanaka H, Yoshizawa A, Takahashi T. Angew. Chem. Int. Ed. 2007;46:2505. doi: 10.1002/anie.200604031. [DOI] [PubMed] [Google Scholar]
- (8).Krygowski ES, Murphy-Benenato K, Shair MD. Angew. Chem. Int. Ed. 2008;47:1680. doi: 10.1002/anie.200704830. [DOI] [PubMed] [Google Scholar]
- (9) (a).Schmidt RR. Angew. Chem. Int. Ed. 1986;25:212. [Google Scholar]; (b) Klotz W, Schmidt RR. Liebigs Ann. Chem. 1993:683. [Google Scholar]; (c) Schmidt RR, Michel J. Tetrahedron Lett. 1984;25:821. [Google Scholar]; (d) Box VGS. Heterocycles. 1982;19:1939. [Google Scholar]
- (10).Schmidt RR, Esswein A. Angew. Chem. Int. Ed. 1988;27:1178. This alkylation affords the α-glycoside.
- (11).Halcomb RL, Wittman MD, Olson SH, Danishefsky S. J. Am. Chem. Soc. 1991;113:5080. [Google Scholar]
- (12).Werz DB, Seeberger PH. Angew. Chem. Int. Ed. 2005;44:6315. doi: 10.1002/anie.200502615. [DOI] [PubMed] [Google Scholar]
- (13).Bolitt V, Mioskowski C, Lee S-G, Falck JR. J. Org. Chem. 1990;55:5812. [Google Scholar]
- (14).Xiong X, Ovens C, Pilling AW, Ward JW, Dixon DJ. Org. Lett. 2008;10:565. doi: 10.1021/ol702693m. [DOI] [PubMed] [Google Scholar]
- (15).Vauzeilles B, Dausse B, Palmier S, Beau J-M. Tetrahedron Lett. 2001;42:7567. [Google Scholar]
- (16).Yadav JS, Reddy BVS, Reddy KB, Satyanarayana M. Tetrahedron Lett. 2002;43:7009. [Google Scholar]
- (17).Tsvetkov YE, Klotz W, Schmidt RR. Liebigs Ann. Chem. 1992:371. [Google Scholar]
- (18).This methodology has been applied in an iterative manner to access polysaccharides. See Supporting Information.
- (19) (a).Durham TB, Roush WR. Org. Lett. 2003;5:1871. doi: 10.1021/ol034393t. [DOI] [PubMed] [Google Scholar]; (b) Hashimoto S, Yanagiya Y, Honda T, Ikegami S. Chem. Lett. 1992:1511. [Google Scholar]
- (20).Spiniello M, White JM. Org. Biomol. Chem. 2003;17:3094. doi: 10.1039/b303453d. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.