

Abstract
Heme-oxygenase-1 (HO-1), a stress-inducible protein, is an important cytoprotective agent in various models of ischemia/reperfusion (I/R) injury. However, the role of downstream mediators involved in HO-1 induced cytoprotection is not clear. In the current study we investigated the role of biliverdin reductase, an enzyme involved in the conversion of HO-1 derived biliverdin into bilirubin and the PI3K/Akt pathway in mediating the cytoprotective effects of HO-1 against hypoxia and reoxygenation (H/R) injury in vitro and in vivo. H9c2 cardiomyocytes were transfected with a plasmid expressing HO-1 or LacZ and exposed to 24 hours of hypoxia followed by 12 hours of reoxygenation. At the end of reoxygenation, reactive oxygen species generation was determined using CM-H2DCFDA dye and apoptosis was assessed by TUNEL, caspase activity and Bad phosphorylation. p85 and Akt phosphorylation were determined using cell based ELISA and phospho-specific antibodies, respectively. HO-1 overexpression increased phosphorylation of the regulatory subunit of the PI3K (p85α) and downstream effector Akt in H9c2 cells, leading to decreased ROS and apoptosis. Furthermore, cardiac specific HO-1 expression increased basal phosphorylated Akt levels and decreased infarct size in response to LAD ligation and release induced I/R injury. Conversely, PI3K inhibition reversed the effects of HO-1 on Akt phosphorylation, cell death and infarct size. In addition, knockdown of biliverdin reductase (BVR) expression with siRNA attenuated HO-1 induced Akt phosphorylation and increased H/R-induced apoptosis of H9c2 cells. Co-immunoprecipitation revealed protein-protein interaction between BVR and the phosphorylated p85 subunit of the PI3 kinase. Taken together, these results suggest that the enzyme biliverdin reductase plays an important role in mediating cytoprotective effects of HO-1. This effect is mediated, at least in part, via activation of the PI3K/Akt pathway.
Introduction
Oxidative stress is a well established cause of myocardial damage associated with ischemia and reperfusion (I/R) injury [1]. In response to reactive oxygen species (ROS), several stress proteins and anti-oxidant enzymes are upregulated as a cytoprotective response to limit cellular damage [2]. Heme oxygenase-1 (HO-1) is an inducible protein whose expression is increased several fold in response to a variety of cellular stresses and stimuli including ischemia [3] hypoxia [4], oxidative stress [5] and inflammatory cytokines [6], suggesting an important role for this enzyme in tissue protection. Several studies have shown that HO-1 over-expression protects against ischemia/reperfusion injury in a variety of tissues including the myocardium [7-11]. Conversely, reduced HO-1 levels increase susceptibility to injury in a variety of stress conditions. For example, HO-1 null mice show impaired ventricular function [12] and increased incidence of right ventricular infarcts with mural thrombi in response to chronic pulmonary hypoxia [13].
We reported previously that constitutive [11] or hypoxia induced [10] over-expression of HO-1 markedly reduces infarct size after ischemia/reperfusion injury. This was associated with reduced oxidative stress and decreased expression of pro-inflammatory and apoptotic mediators [14]. Another mechanism by which HO-1 may protect cells or tissues against oxidative injury is by generating bilirubin from catabolism of heme. Several studies have demonstrated that exogenous bilirubin can protect against hypoxia induced injury in cultured cells and in isolated perfused hearts subjected to I/R injury [15-17]. Furthermore, a strong correlation has been reported between elevated plasma bilirubin levels and lower risk of cardiovascular diseases [18, 19]. However, the signaling mechanism(s) underlying the cytoprotective effects of HO-1 are not clear. Bilirubin is produced via reduction of heme-derived biliverdin by biliverdin reductase (BVR) [15]. In addition to its role in biliverdin metabolism, BVR has recently been reported to function as a protein kinase with serine/threonine/tyrosine kinase activity [20]. In this regard, BVR shares similarities with insulin receptor kinases in their ability to regulate the PI3 kinase pathway. The PI3K/Akt cascade is an important survival pathway in the heart. Akt overexpression leads to increased survival and improved myocardial function following I/R injury [21-24] in a manner similar to HO-1. Recent evidence suggests that these two enzyme systems interact both at the post-transcriptional and post-translational levels [25, 26], and may function in a co-dependent function to confer cytoprotection [25]. However, it is not known whether PI3K/Akt pathway is required for the cytoprotective effects of HO-1 in myocardial I/R injury.
In this study we investigated the role of the PI3K/Akt in the cardioprotective effects of HO-1 against ischemia/hypoxia and reperfusion injury. Our results indicate that PI3K/Akt activity is essential for HO-1 induced protection against H/R induced injury. Furthermore, the mechanism of PI3K/activation by HO-1 is dependent on biliverdin reductase activity and its metabolic by-product bilirubin.
Materials and Methods
Reagents
All chemicals were purchased from Sigma (Sigma-Aldrich, St. Louis, MO, USA) with the exception of MTT reagent and TUNEL (Roche, USA), Hoecsht 33342 and CM-H2DCFDA (Invitrogen, Carlsbad, CA, USA) and LY294002 (Cell Signaling Technologies, Beverly, MA, USA). HO-1 and BVR siRNAs were purchased from Ambion. LY294002 and CM-H2DCFDA were initially dissolved in DMSO and diluted in PBS with a final DMSO concentration of < 0.01%. All the antibodies were from Cell Signaling except for HO-1 and BVR which were from Stressgen.
Constructs
HO-1 and LacZ constructs were generated using PCR amplification. BVR construct was a kind gift from Dr. Mahim Maines, University of Rochester, Minnesota. BVR was amplified from this construct and ligated into a pCMV-3Tag (#240200) (Stratagene, CA) expression vector in BAMH1/Xho1 restriction sites to generate a FLAG tagged BVR expression vector.
In-Vivo Gene Delivery and I/R injury
Adult male HO-1 transgenic mice (HO-1 Tg) and wild type (WT) littermates were subjected to I/R injury as described previously [11]. For the gene delivery studies, adult male C57Bl6 mice were purchased from Charles River Laboratories and maintained on a 12:12-h light-dark cycle at an ambient temperature of 24°C and 60% humidity. Food and water were provided ad libitum. Intramyocardial gene delivery and I/R injury were performed 5 weeks in advance of I/R injury using adeno-associated virus (AAV) to constitutively express HO-1 or LacZ as described previously [11]. For the PI3 kinase inhibition experiments, mice transduced with AAV-HO-1 or AAV-LacZ were administered LY294002 i.p. at a dose of 100 mg/kg (≈2 mg/mouse) one hour prior to the ischemic injury. All surgical and experimental procedures were approved by the Duke University Committee on Animal Welfare.
Cell Culture and hypoxia/reoxygenation
H9c2 cells were purchased from ATCC (Manassus, VA, USA), seeded at constant density (10,000/cm2) and grown to 80% in DMEM containing 10% fetal bovine serum and antibiotics. Cells were washed with HBSS and rendered quiescent in serum free DMEM for 24 hours prior to experiments. Embryonic fibroblasts (MEFs) from HO-1 null mice were a kind gift from Dr. Shaw-Feng Yet, Harvard Medical School, Boston, MA. For the hypoxia/reoxygenation studies, cells were transfected with plasmids expressing HO-1 or LacZ. Forty eight hours after transfection, cells were subjected to twenty four hours of hypoxia (< 0.5% O2) using pre-conditioned hypoxic medium followed by twelve hours of reoxygenation. Hypoxic medium was changed to fresh medium upon initiation of reoxygenation.
siRNA transfection
siRNA transfections were performed using TRANS-IT-TKO reagent (Mirus Corporation) prior to transfer to hypoxia chamber. Sequences of siRNA oligonucleotides were as follows: HO-1—(5′→ 3′) Sense: GGAACUUUCAGAAGGGUCAtt; Antisense: UGACCCUUCUGAAAGUUCCtc; BVR (5′→ 3′) Sense; GGAUAUAUUUGUUCAGAAGtt; Antisense; CUUCUGAACAAAUAUAUCCtg.
Quantification of reactive oxygen species generation
Reactive oxygen species was determined using CM-H2DCFDA dye. Following transfections, cells were subjected to 24 hours of hypoxia followed by 24 hours of reoxygenation. One hour before end of reoxygenation, CM-H2DCFDA dye (5μM) was loaded to cells in 12-well culture slides and kept at 37°C in 5% CO2:21% O2. Wells were washed once with HBSS and incubated with Hoechst 33342 [10μg/ml] for 5 min. Fluorescence was viewed with a Leica DRB inverted microscope at 100× magnification. Flow cytometry was used in quantification of ROS generation.
Co-immunoprecipitation and Western Immunoblot Analysis
Cells were harvested and lysed in Tris HCL buffer containing protease inhibitors, and protein concentration was determined by Bradford assay. Mouse heart tissues were harvested following reperfusion and homogenized in tris-HCl buffer. For co-immunoprecipitation experiments, protein concentration was measured using Bradford assay and 100ug of cell lysate was subjected to co-immunoprecipitation using ProFound co-i.p. kit from Pierce Technologies according to manufacturer's instructions. For western blots, cells or tissues were lysed in the same lysis buffer and protein (10-20ug) was loaded on acrylamide gels, transferred to polyvinylidiene difluoride membrane, and probed with phospho-Akt (ser473), phospho-Bad (Ser136) total Akt (Cell Signaling, Danvers, Massachusetts), or HO-1 (Stressgen, Canada) antibodies as described previously [14].
Determination of p85 phosphorylation
Phosphorylated levels of p85α were determined using a cell based ELISA according to manufacturer's instructions (Activ Motif, Carlsbad California, USA). Briefly, H9c2 cells were cultured in 96 well plates. Twenty four hours after seeding, cells were transfected with either HO-1 or lacZ expression plasmid. Forty eight hours after transfection, cells were washed with PBS and fixed with 4% formaldehyde. Cells were incubated with primary antibody that recognizes either phosphorylated PI3K p85 or total PI3K p85. After overnight incubation, cells were washed and incubated with secondary HRP conjugated antibody. p85α phosphorylation was quantified at 450 nm using a spectrophotometer. The readings were normalized to relative cells number in each well.
Bilirubin Quantitation
Bilirubin levels were measured in medium and in whole cell lysates using a colorimetric bilirubin determination assay (Bioassays, Inc, CA) according to manufacturer's instructions. The color change was measured at 530nm wavelength and the following formula was used for determination of bilirubin levels: Bilirubin= ODsample- ODBlank/ODcalibrator-ODwater X 10 mg/dl.
TUNEL staining
Detection of apoptotic cells was determined using TUNEL assay using a fluorescence kit from Roche according to manufacturer's protocols. Caspase 3 activity was determined using a colorimetric assay from Sigma.
Statistical Analysis
All results are expressed as mean ± standard error of the mean (SEM). One way ANOVA was used to compare differences in caspase activity assay, apoptotic index, infarct size and bilirubin levels. P<0.05 was considered to be statistically significant.
Results
HO-1 protects against hypoxia/reoxygenation induced oxidative stress
The effect of HO-1 over-expresssion in protection from H/R-induced injury of H9c2 cells in shown in Figure 1. Exposure of cells of 24 hr of hypoxia and reoxygenation led to significant increase in ROS generation in the LacZ transfected control cells compared to nearly undetectable levels in the HO-1 transfected cells (Figure 1 A, B), indicating that HO-1 reduces H/R induced oxidative stress. To establish whether endogenous HO-1 activity is essential for protection against H/R-induced oxidative stress, we used embryonic fibroblasts from wild type and HO-1 null mice. Exposure of the cells to H/R led to a significant increase in ROS generation compared to fibroblasts from wild-type animals (Figure 1C). We then assessed the effect of H/R on apoptosis in the HO-1 and LacZ-transfected H9C2 cardiomyocytes. H/R led to a significant increase in the number of TUNEL positive nuclei in the LacZ-transfected cells while there was no increase in apoptotic nuclei in the HO-1 transfected cells (Figure 1D, E).
Figure 1. Effect of HO-1 over-expression on oxidative stress in vitro.
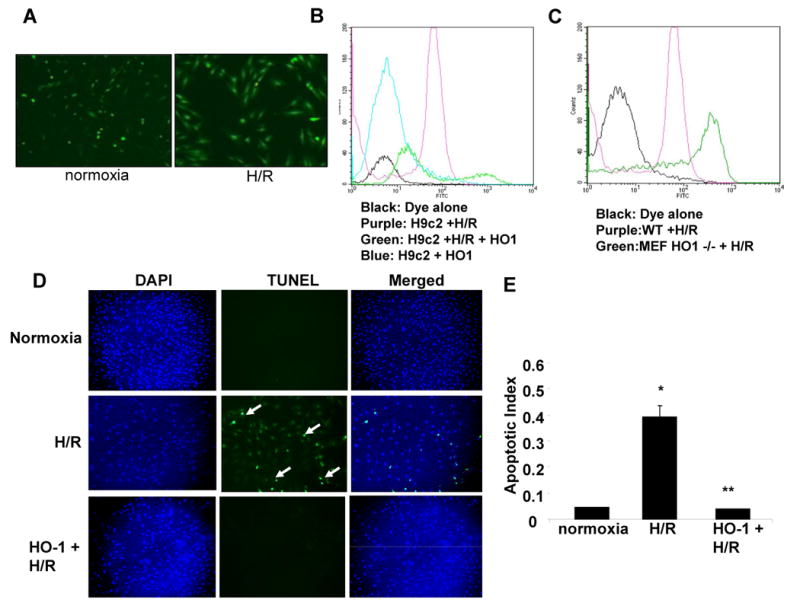
A. CM-H2DCFDA fluorescence after hypoxia and reoxygenation in H9c2 cells. (B-C). Quantification of CM-H2DCFDA fluorescence by flow cytometry in non-transfected and HO-1 transfected H9c2 cells (B) and in mouse embryonic fibroblasts (C) from WT and HO-1 null mice exposed to hypoxia and reoxygenation. D. Fluorescent TUNEL staining in H9c2 cells transfected with either HO-1 or control plasmid under normoxia or H/R. E. Quantification of apoptosis in H9c2 under normoxia or H/R. Values are mean ± SEM. *, p < 0.05 vs. normoxia; **, p < 0.05 vs.H/R.
Effect of HO-1 over-expression on Akt activation in vitro and in vivo
We investigated whether HO-1 may exert its cytoprotective effects by activating the PI3K-Akt pathway. The effect of HO-1 on PI3K and Akt activity is shown in Figure 2. HO-1 overexpression led to approximately four fold increase in basal levels of phosphorylated Akt (ser 473) in H9c2 cells compared to the LacZ-transfected cells, without any change in total Akt (Figure 2A). Interestingly, Akt phosphorylation was increased in the LacZ-transfected cells but not in the HO-1 transfected cells following hypoxia and reoxygenation (H/R) (Figure 2A and 2B), suggesting that the elevated basal level of Akt activity in the HO-1-transfected may be sufficient to withstand the stress imposed by H/R. In agreement with the results in the H9c2 cells, basal levels of phosphorylated Akt were elevated in myocardial homogenates form cardiac-specific over-expressing HO-1 transgenic mice compared to the wild type control littermates (Figure 2C, D). The elevated levels of phosphorylated Akt in the HO-1 transgenic animals were accompanied by increased phosphorylation of its downstream target, glycogen synthase kinase 3-β (GSK3-β) (Figure 2E) further confirming that Akt activity is increased in the myocardium of HO-1 transgenic animals.
Figure 2. Effect of exogenous HO-1 over-expression on Akt phosphorylation in vitro and in vivo.
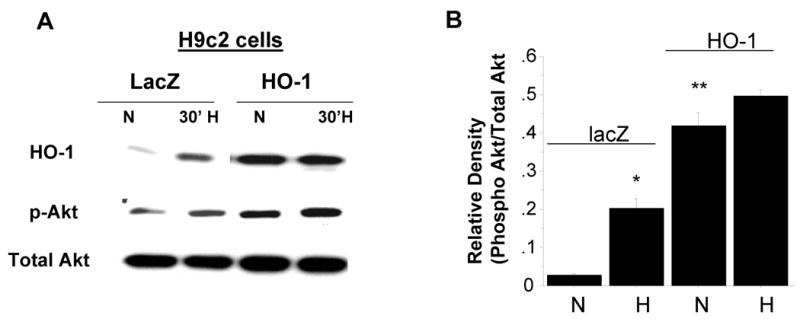
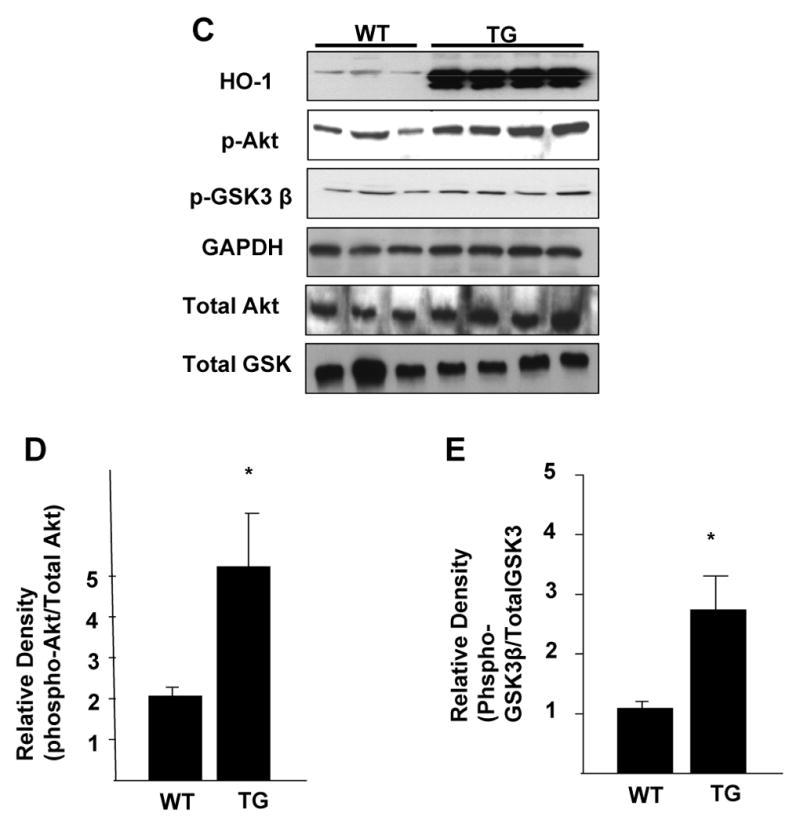
A-B. Representative blot of phosphorylated Akt levels in H9c2 cells transfected with either lacZ or HO-1 expression plasmid in normoxic conditions and after exposure to 30 minutes of hypoxia (< 0.5% O2). B. Densitometric data analysis of the data in panel A (* vs. normoxia lacZ; ** vs. normoxia lacZ; n=3 independent experiments). C-E. Representative blots of HO-1, p-Akt, and p-GSK3β (C) and densitometric analysis of p-Akt (D) and p-GSK3β (E) in myocardial tissue homogenates from wild type and HO-1 transgenic mice (*TG vs. WT, p<.05, n = 3-4/group) using specific antibodies.
We determined further whether Akt activity is dependent on HO-1 expression, using siRNA to knockdown endogenous HO-1 expression in H9c2 cells exposed to H/R. Transfection efficiency was 50-60% as determined with FITC-labeled oligonucleotide (data not shown). We tested three different sets of siRNA oligonucleotides to knockdown expression of HO-1. As shown in Figure 3A, HO-1 protein expression was decreased by >50% with oligonucleotide set #2, and this set was used in subsequent experiments. Inhibtion of HO-1 with siRNA decreased phosphorylated Akt levels both in normoxic conditions and in response to H/R (Figure 3B) by approximately 41% and 32%, respectively (Supplementary Figure 1). The knockdown of HO-1 protein was accompanied by increase in apoptosis in the H9C2 exposed to H/R, as indicated by increased capase-3 activity (Figure 3C) and increased number of TUNEL-positive cells (Figure 3D).
Figure 3. Effect of HO-1 knockdown on Akt phosphorylation levels and apoptosis in vitro.
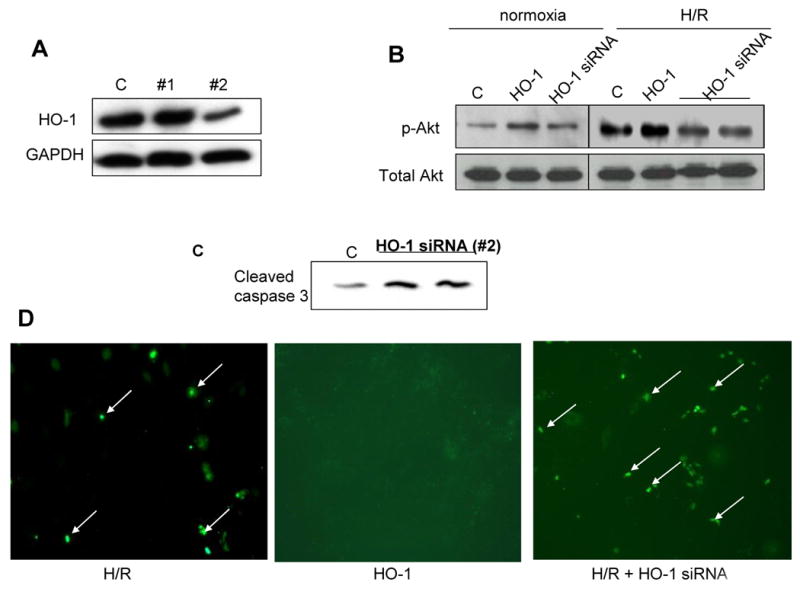
A. Evaluation of the effectiveness of different siRNA oligonucleotides in knockdown of HO-1 protein expression; B. Representative blot showing the effect of HO-1 knockdown on p-Akt (Ser 473) in normoxic conditions and in response to H/R in H9c2 cells; C. Representative blot showing the effect on HO-1 knockdown on cleaved caspase 3 levels in H9c2 cells;. D. Representative fluorescent TUNEL showing the effect of HO-1 protein knockdown on apoptosis of H9c2 cells in response to hypoxia and reoxygenation. Knockdown of endogenous HO-1 protein markedly increases H/R-induced apoptosis, whereas exogenous HO-1 overexpression markedly reduces apoptosis compared to the control cells.
Role of Biliverdin Reductase in HO-1 induced Akt activation and cellular protection
Having established the role of HO-1 in enhancing PI3K-Akt activity, we then determined whether this effect is mediated by downstream mechanisms involved in catabolism of biliverdin, one of the catalytic by-products of heme degradation by HO-1. Biliverdin is converted to bilirubin by biliverdin reductase (BVR). Recently, BVR was reported to have serine/threonine kinase activity [20]. Thus, we investigated whether BVR is involved in HO-1 mediated phosphorylation of Akt. Initially we determined the effects of HO-1 on BVR protein levels. As shown in Figure 4A and Supplementary Figure 2, HO-1 overexpression increased BVR levels in H9c2 cells resulted in expected increase in HO-1 levels. To further determine the role of BVR in HO-1 induced Akt activation, we transfected H9c2 cells either with HO-1 expression alone or with simultaneous inhibition of BVR with 200nM siRNA. We confirmed dose-dependent knockdown of BVR using a specific siRNA oligonucleotide (Figure 4B). Importantly, our data show that BVR knockdown with 300nM siRNA reduced the stimulatory effect of HO-1 on Akt phosphorylation by > 50% in H9c2 (Figure 4C and Supplementary Figure 2), thus indicating that HO-1-dependent activation of Akt is, at least in part, mediated by BVR. In contrast there was no effect of scrambled siRNA (300nM) co-transfection on p-Akt levels. Furthermore, treatment of H9c2 cells with 500 nM bilirubin, the final product in the catabolism of biliverdin by BVR, dose dependently increased the phosphorylation of Akt (Figure 4D) and its downstream target protein Bad (Figure 4E), a pro-apoptotic member of the Blc-2 family that is negatively regulated by phosphorylation. Interestingly, the effect of bilirubin on Akt and Bad phosphorylation was attenuated by pre-treatment with BVR siRNA. To determine if bilirubin-induced Akt and Bad phosphorylation was mediated by BVR, we transfected H9c2 cells with BVR siRNA (300nM) prior to treatment with bilirubin. As shown in Figure 4E and Supplementary Figure 2, the effect of bilirubin on Akt and Bad phosphorylation was attenuated by BVR siRNA, indicating that bilirubin-induced Akt phosphorylation is mediated, at least in part, by BVR.
Figure 4. Effect of Biliverdin Reductase inhibition on HO-1 and bilirubin induced Akt phosphorylation and cytoprotection in vitro.
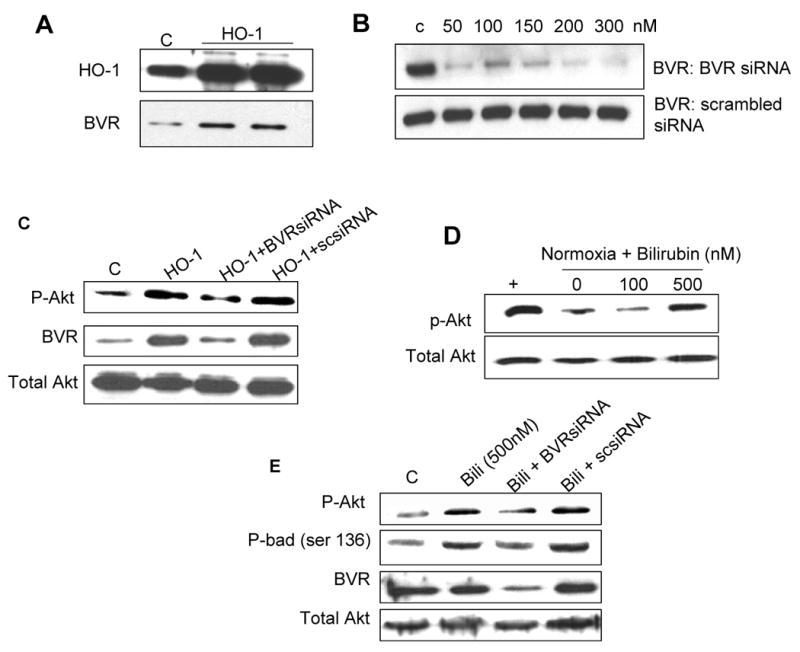
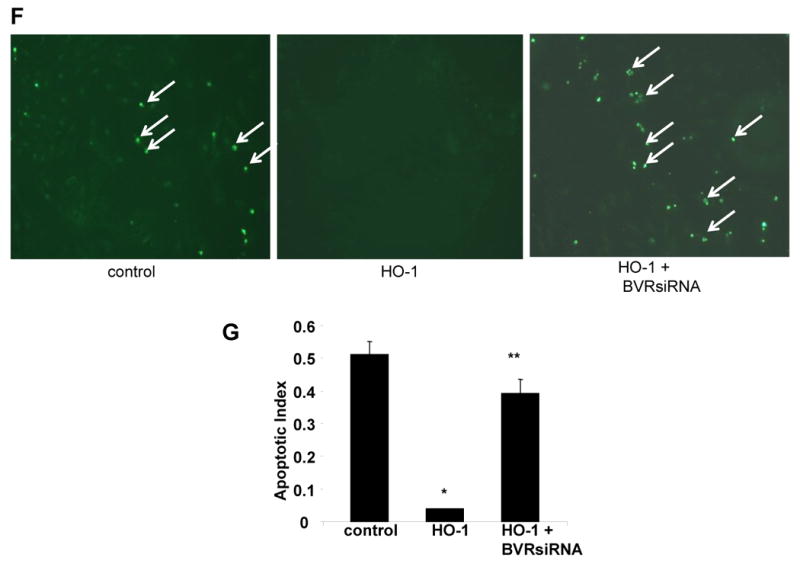
A. Western blot analyses of total cell lysates from H9c2 cells transfected with HO-1-expressing plasmid B. Optimization of BVR knockdown using BVR siRNA C. Representative blot of p-Akt and BVR expression in H9c2 cells transfected with either HO-1 plasmid alone or jointly with BVR siRNA D. Representative blot showing the effect of exogenous bilirubin on p-Akt expression in H9c2 cells maintained in normoxic conditions. E. Effect of BVR knockdown on bilirubin-induced changes in p-Akt, p-Bad (ser136), biliverdin and total Akt. in lysates from H9c2 cells. F. Representative fluorescent TUNEL staining in H9c2 cells transfected with either HO-1 or HO-1 plus BVR siRNA and exposed to hypoxia and reoxygenation G. Effect of BVR knockdown on apoptosis of H9c2 cells exposed to H/R. BVR protein knockdown reduced the anti-apoptotic effect of HO-1 overexpression in response to H/R by > 70%. (Values are mean ± SEM. p<.05, * HO-1 vs. control; p<.05, ** HO-1 + BVR siRNA vs. HO-1; n = 3 independent experiments.). “+” is the positive control generated by over-expression of HO-1 in H9c2 cells.
We further determine the functional consequence of biliverdin reductase inhibition on cell viability in response to H/R. Inhibition of BVR using siRNA markedly attenuated the anti-apoptotic effect of HO-1 in H9c2 cells after exposure to H/R (Figure 4F). Compared, to control cells, exogenous HO-1 overexpression reduced the number of TUNEL positive nuclei by approximately 50%, and pre-treatment with BVR siRNA reduced the anti-apoptotic effect of HO-1 by 80% (Figure 4G), suggesting that the cytoprotective effects of HO-1 against H/R injury in H9c2 myocytes is mediated, at least in part, by BVR and subsequent activation of the PI3K-Akt survival pathway.
Effect of biliverdin reductase on bilirubin concentration
In order to determine the effect of BVR and exogenous bilirubin on total bilirubin levels, we measured bilirubin concentration in both cell lysates and the medium of cells treated with BVR siRNA alone or together with the addition of exogenous bilirubin. As shown in Figure 5, treatment with BVR siRNA reduced endogenous bilirubin levels in whole cell lysates by > 50%. (Figure 5A). However, the inhibition of BVR did not have any effect on the concentration of bilirubin after addition of exogenous bilirubin (Figure 5A). Bilirubin levels were undetectable in the medium from untreated (control) cells (Figure 5B), indicating negligible secretion of bilirubin. As expected, addition of exogenous bilirubin increased the concentration of bilirubin in the media in a dose-dependent fashion (Figure 5B). However, treatment of cells with BVR siRNA led to decreased concentration of bilirubin in the media even after addition of 500 nM exogenous bilirubin (Figure 5B).
Figure 5. Effect of Biliverdin Reductase inhibition on bilirubin levels in vitro.
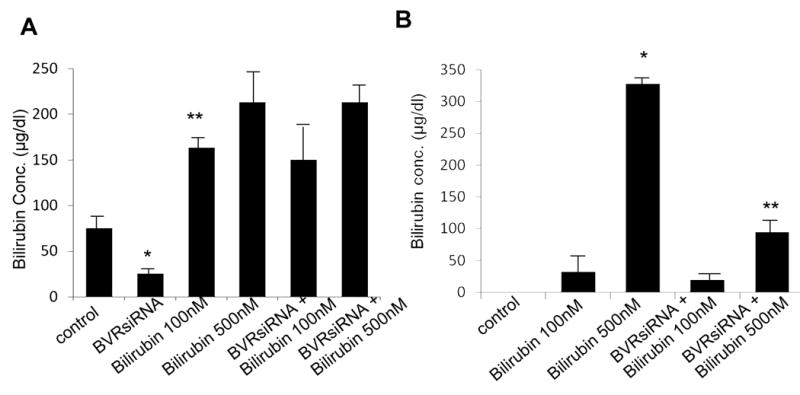
A-B. Measurement of bilirubin concentration in whole cell lysates (A) or medium (B) of H9c2 cells treated with or without BVR siRNA. Addition of exogenous bilirubin increases bilirubin concentration in a dose dependent fashion both in the cytosol and media. BVR siRNA decreases endogenous bilirubin concentration, but not exogenous bilirubin levels in the cytosol (A, * vs. control; ** vs. BVR siRNA; B, * vs. control; ** vs. bilirubin 500nM; *, **, p < 0.05, n=2 independent experiments performed in duplicates).
Effect of PI3 kinase inhibition on HO-1 induced protection in vitro and in vivo
We determined the role of PI3 kinase in HO-1 mediated cellular protection against H/R injury in vivo and in vitro. Under normoxic conditions, pharmacological inhibition of PI3K with LY 294002 had no effect on caspase 3 activity (Figure 6A). Hypoxia and reoxygenation increased caspase activity approximately 4 fold. HO-1 overexpression decreased H/R-induced caspase activity to the levels seen in normoxic conditions (Figure 6A). However, inhibition of PI3K abrogated the inhibitory effect of HO-1 on caspase activity (Figure 6A), suggesting a dependence on PI3K-Akt for HO-1 mediated cytoprotection. In contrast treatment of control H9c2 cells with LY294002 did not significantly alter caspase activity in control cells during hypoxia/reoxygenation (Figure 6A). We determined whether HO-1 induced protection against ischemia and reperfusion injury in vivo is also mediated via the PI3K-Akt pathway in mice treated by intramyocardial injection of AAV-HO-1 or AAV-LacZ. For evaluation of the role of PI3K-Akt, a subset of animals from each group was administered LY294002 one hour before the injury. Animals treated with AAV-HO-1 had approximately 68% reduction in infarct size as compared to LacZ treated animals (infarct size, 7.6 ± 2 vs. 25 ± 1.5, p < 0.05) (Figure 6B, C). Administration of the inhibitor reversed this protective effect of HO-1 (HO-1 + LY 294002, 18 ± 2 vs. HO-1, 7.6 ± 2) indicating dependence on PI3K pathway.
Figure 6. Effect of PI3K inhibition on HO-1 induced cytoprotection in vitro and in vivo.
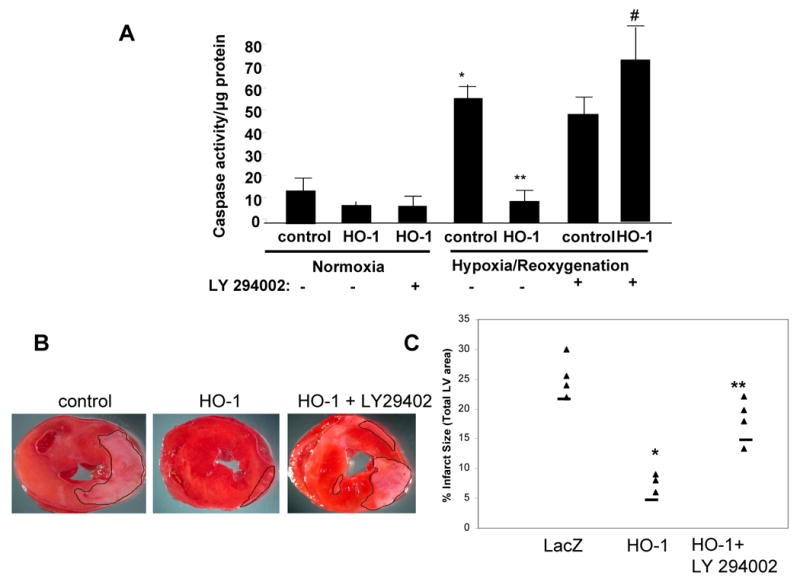
A. Caspase 3 activity in lysates from control and HO-1 transfected H9c2 exposed to normoxia or hypoxia/reoxygenation in the presence or absence of PI3K inhibition with LY 294002. Inhibition of PI3K had no effect on caspase activity in normoxic conditions. However, LY 294002 markedly increased caspase activity in both control and HO-1-transfected cells in response to H/R (* vs. control normoxia; ** vs. control H/R; # vs. HO-1 H/R. p<.05. n=3 independent experiments performed in duplicates). B. Representative bi-ventricular sections showing the effect of PI3K inhibition with LY 294002 on anterior wall infarct in mice pre-treated with intramyocardial injection of AAV-LacZ or AAV-HO-1. C. Infarct size in mouse hearts transduced with either AAV-HO-1 or AAV-LacZ followed by administration of LY294002 before the I/R injury (* vs. lacZ; ** vs. HO-1, p < 0.05; n = 4-5 mice/group)
Effect of HO-1 expression on physical interaction between PI3 kinase and BVR
As shown above, PI3 kinase pathway plays an important role in mediating the protective effect of HO-1 and BVR on cellular apoptosis Since Akt activity is regulated upstream by PI3 kinase, which is activated by phosphorylation of its p85 regulatory subunit [27], we determined whether p85 activity is affected by HO-1 levels. As shown in Figure 7A, the phosphorylated levels of p85α were increased in H9C2 cells transfected with HO-1 plasmid relative to control cells transfected with LacZ, despite comparable levels of total p85, indicating that HO-1 enhances Akt activity, at least in part, by increasing the activity of its upstream activator p85. Furthermore, since our data shows that BVR mediates the effect of HO-1 on Akt phosphorylation, we sought to determine if there is any physical interaction between BVR and the p85 subunit of PI3 kinase. As shown in Figure 7B, phosphorylated p85 co-immunoprecipitated with BVR in homogenates from the left ventricles of wild type mice. This interaction was further increased in LV homogenates from HO-1 transgenic mice. To further confirm the specificity of this interaction we over-expressed FLAG-BVR in H9c2 cells. As shown in Figure 7C, BVR transfected cells demonstrated increased binding with the phosphorylated p85 subunit of the PI3 kinase. Taken together, these data suggest that BVR may mediate the anti-apoptotic effects of HO-1 by binding to and modulating the activity of the PI3K/Akt pathway.
Figure 7. Effect of HO-1 on PI3kinase p85 regulatory subunit phosphorylation and physical interaction between BVR and p85 in vitro and in vivo.
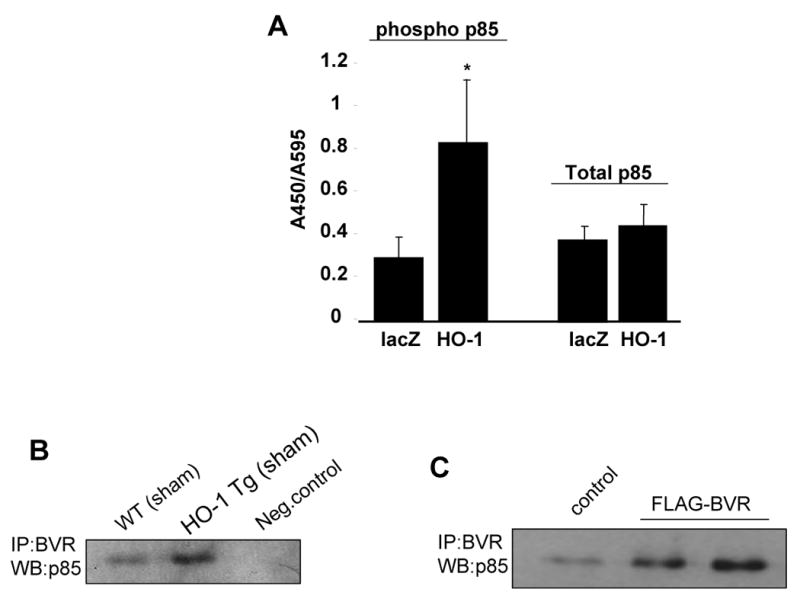
A. Effect of HO-1 over-expression on basal p85 phosphorylation levels H9c2 cells using a cell-based ELISA assay. (Values are mean ± sem. p <0.05, *HO-1 vs. LacZ. n = 3 independent experiments performed in triplicates). B. Representative blot showing effect of HO-1 over-expression on physical interaction between BVR and phosphorylated p85 as determined by co-immunoprecipitation experiments in different groups as indicated. IP was conducted with the BVR antibody, samples were run on a SDS gel and transferred to PVDF membrane and probed with phosphor p85 antibody. Negative control lane: cell lysates incubated with unconjugated beads C. Interaction between BVR and p85 by overexpression of FLAG-BVR construct in H9c2 cells followed by co-immunoprecipitation using BVR antibody followed by western blot using p85 phosphorylated antibody.
Discussion
Myocardial ischemia and reperfusion injury triggers a series of cytoprotective mechanisms aimed at counteracting the cellular damage induced by inflammatory mediators and reactive oxygen species [1]. Heme oxygenase-1 plays a central role in myocardial protection from I/R injury. The enzyme is induced several fold by hypoxia and reoxygenation [28, 29], and genetic mouse models of HO-1 deficiency have greater propensity towards ischemia and hyperoxia induced tissue injury [12, 13]. Moreover, pre-emptive delivery of HO-1 gene markedly reduces infarct size following acute I/R [10, 11], suggesting that enhanced basal level of HO-1 preconditions the myocardium [12, 30] and renders it resistant to subsequent episodes of I/R injury [14]. Thus, strategies aimed at increasing basal HO-1 levels may yield therapeutic potential in protection against I/R induced myocardial injury and failure. However, the mechanism(s) underlying the cardioprotective actions of HO-1 have not been fully elucidated. The cytoprotective properties of HO-1 have traditionally been attributed to the by-products of heme degradation, namely bilirubin and carbon monoxide (CO). Indeed, within a narrow therapeutic range, these catalytic by-products exert powerful antioxidant [15, 31], anti-inflammatory [32] and anti-apoptotic effects [33-35], leading to reduced infarct size [16, 17, 36-37]. However, emerging evidence, suggests that HO-1 may also exert cytoprotective effects, independent of heme breakdown [38] by interacting with survival signaling pathways such as PI3K-Akt and p38. We reported recently that HO-1 functions co-dependently with Akt to confer protection from pro-oxidant-induced injury in human aortic smooth muscle cells [25]. Moreover, HO-1 and biliverdin reductase function as phosphoproteins [20, 26] and may participate in phosphorylation and activation of PI3K kinase. Here we show that the cardioprotective effect of HO-1 against I/R injury is critically dependent on Akt activity both in vitro and in vivo. Furthermore, HO-1-dependent activation of Akt is mediated, at least in part, by biliverdin reductase and is mimicked by bilirubin. Thus, these findings reveal a novel mechanism of HO-1-induced cardioprotection that appears to be mediated by phosphorylation and activation of PI3K/p85 subunit and its downstream target Akt by BVR or HO-1.
The mechanisms by which HO-1 activates Akt are not fully understood. HO-1 appears to interact with Akt both at transcriptional [25] the post-translational level [26]. Akt has been reported to induce HO-1 transcriptionally by promoting the activation and nuclear translocation of Nrf2, a key transcription factor involved in regulation of HO-1 gene expression [25, 39, 40], suggesting that Akt-mediated induction of HO-1 may represent an essential protective response to cellular stress. In addition, Akt phosphorylates HO-1 at serine 188 in vitro and vivo, leading to a modest increase in HO-1 activity [26]. It remains to be established whether or not HO-1 directly activates PI3K/Akt. Our results show that HO-1 overexpression increases phosphorylation of PI3K/p85 subunit without alteration in total p85 levels. However, the possibility that HO-1 may also function as a protein kinase capable of phosphorylating PI3K/Akt is an intriguing prospect that merits further investigation. In this regard, HO-1 was recently reported to inhibit apoptosis of Caco-2 cells via activation of Akt [41]. Interestingly, the effects of HO-1 on Akt activity and cell survival in this colon cancer cell line were mimicked by exogenous bilirubin, analogous to our current findings in H9c2 cells exposed to hypoxia and reoxygenation. Others have reported that CO also protects against I/R injury by increasing Akt activity [35, 36]. Thus Akt and HO-1 may function cooperatively in cellular protection by reciprocally enhancing each other activity through direct transcriptional and post-translational events, and indirectly via the by-products of heme degradation, bilirubin and CO.
Another possible mechanism by which HO-1 may increase Akt activity is via BVR, the downstream enzyme in the heme degradation pathway involved in the reduction of biliverdin to bilirubin [15, 42]. Our results indicate that HO-1 increases BVR levels. Moreover, BVR knockdown attenuates Akt phosphorylation and increases apoptosis in HO-1-overexpressing H9c2 myocytes exposed to H/R. Furthermore, we have also demonstrated that BVR binds to the phosphorylated p85 regulatory subunit of the PI3K complex. Taken together these data suggest that BVR may serve an intermediary role in the regulation of Akt activity by HO-1. In addition it has been reported that BVR exhibit serine-threonine protein kinase activity [20]. Specifically, BVR has been suggested to act as a kinase for insulin receptor substrate (IRS) [20]. Since the activation of Akt is initiated by docking of PI3 kinase to phosphorylated IRS via its p85α subunit [43], it is plausible that BVR, by activating IRS phosphorylation, initiates PI3 kinase docking and subsequent activation of the Akt pathway. Additionally, BVR also has a sequence motif similar to IRS [20] which is required for SH2 binding proteins such as p85, suggesting that the enzyme, itself, may serve as a docking site for PI3 kinase and subsequent activation of the pathway. Thus, BVR could potentially modulate Akt activity at multiple levels in the PI3K/Akt signaling cascade. However, the structural basis for BVR being a protein kinase is inconclusive and no evidence of any appropriate motifs has yet been provided in literature reporting the structure of BVR [44] [45].
Alternatively, BVR may inhibit Akt dephosphorylation by reducing oxidative stress [46]. BVR may also exert anti-apoptotic effects independent of Akt activity by recycling and maintaining appropriate levels of bilirubin [47], and by promoting metabolic stability and directly stimulating suppressors of apoptosis [48]. The exact role that bilirubin may play in the regulation of the interplay between BVR and p85 is not clear. Our results show that exogenous bilirubin increases basal BVR levels, suggesting that bilirubin and BVR may operate in a positive feedback fashion to maintain each other's levels in the cell. Indeed Baranano et al [47] showed that BVR and bilirubin constitute a catalytic cycle, whereby bilirubin is oxidized back to biliverdin, providing in this fashion the substrate for continuous regeneration of bilirubin by BVR. Furthermore, our results demonstrate that BVR inhibition decreases basal levels of endogenous bilirubin, while having no effect of the levels of exogenously added bilirubin in cytosolic homogenates. Interestingly, inhibition of BVR reduces the amount of bilirubin in the media after addition of 500 nM exogenous bilirubin. One possible explanation for this phenomenon is that inhibition of intracellular bilirubin by BVR knockdown causes uptake/diffusion of bilirubin from the medium thus reducing the concentration of extracellular bilirubin. Alternatively, inhibition of endogenous bilirubin production by BVR siRNA may reduce the amount of bilirubin that may diffuse into the media. However, we believe that the former explanation is a more likely mechanism. These scenarios are currently being investigated.
A plausible working model for the mechanism of HO-1-induced cytoprotection is shown in Figure 8. Accordingly, we propose that hypoxia and reoxygenation leads to the production of reactive oxygen species, possible via activation of membrane bound NADPH oxidase and as result of mitochondrial oxidative metabolism. Increased ROS generation may dephosphorylate Akt, leading to its inactivation. The decrease in Akt activity reduces cellular resistance to stress, culminating in mitochondrial permeability transition pore opening and activation of the mitochondrial death pathway. HO-1 activity may counteract the effect of oxidative stress directly by inhibiting ROS generation and mitochondrial apoptosis pathway activated by cytochrome c via the anti-oxidant actions of bilirubin. The decrease in ROS load by bilirubin may also indirectly lead to preservation of Akt activity be inhibiting its dephosphorylation. In addition HO-1 and biliverdin reductase may directly enhance Akt activity by phosphorylating p85 and/or Akt itself leading to inhibition of pro-apoptotic moieties such as Bad and GSK-3β, and preservation of mitochondrial function. The end result is inhibition of the intrinsic apoptotic pathway, resulting in improved cell survival in the presence of cellular stress such as triggered by hypoxia and reoxygenation.
Figure 8. Model describing the potential interactions between the HO-1-BVR enzyme axis and the PI3K-Akt signaling pathway in cardioprotection from I/R injury.
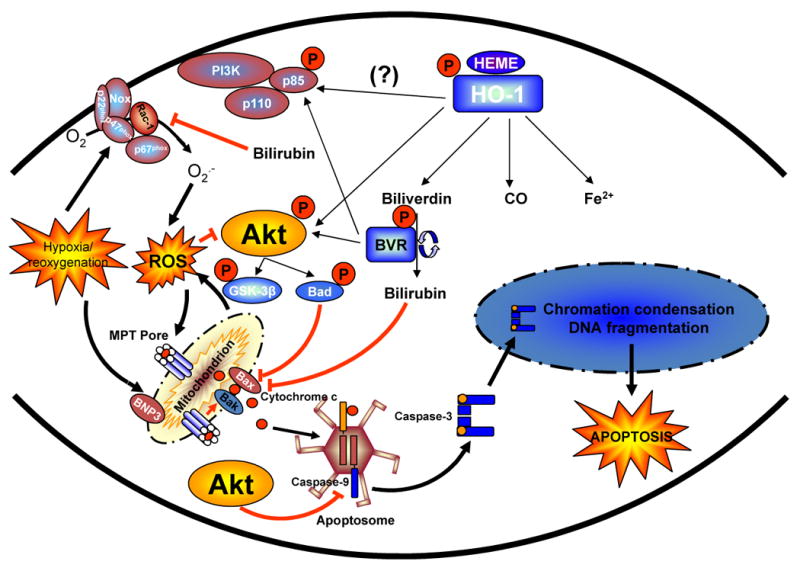
Increased basal HO-1 may confer cytoprotection against I/R injury by increasing Akt activity directly via phosphorylation of p85 and/or Akt by BVR and/or HO-1 itself. In addition the increased bilirubin production by BVR buffers intracellular ROS production, leading to decreased dephosphorylation of Akt. The enhanced Akt activity preserves metabolic stability and mitochondrial membrane potential, resulting in inhibition of the intrinsic apoptotic pathway mediated by cytochrome c and caspase-9. The end result is enhanced cell survival in the presence of cellular stress such as triggered by hypoxia and reoxygenation.
In conclusion, we provide evidence of a novel mechanism of HO-1-mediated cardioprotection, whereby inhibition of I/R-induced injury is critically dependent on activation of the PI3K-Akt signaling pathway and subsequent inhibition of downstream mitochondrial apoptotic pathways. The mechanism of Akt activation by HO-1 appears to be mediated at the post-translational level by multiple mechanisms, and possibly include direct phosphorylation events on p85 and Akt by HO-1 or BVR or both, and inhibition of Akt dephosphorylation by bilirubin-dependent buffering of ROS. Unequivocal confirmation of this novel paradigm of HO-1-mediated remains to be established. Nevertheless, the current findings provide a basis for the development of novel strategies to enhance the therapeutic potential of HO-1 in myocardial protection.
Supplementary Material
Figure 1. Densitometric analysis of data shown in figure 3B. HO-1 overexpression in H9c2 cells leads to significantly higher levels of phospho-Akt which were attenuated in the presence of HO-1 siRNA both under normal as well as hypoxia/reoxygenation conditions (* vs. control; ** vs. HO-1; # vs. HO-1 normoxia; ## vs. HO-1 hypoxia/reoxygenation. P<0.05, n=3 independent experiments performed in duplicate)
Figure 2A-C. Densitmetric analysis of data shown in figure 4, panels A, C and E. A. Effect of HO-1 overexpression on BVR protein levels. B. HO-1 overexpression in H9c2 cells leads to significantly higher levels of phospho-Akt which was significantly attenuated with co-transfection of BVRsiRNA. There was no effect on phospho-Akt levels when cells were co-transfected with HO-1 along with scrambled siRNA. (* vs. control; ** vs. HO-1; p<0.05, n=3 independent experiments performed in duplicate) C. Similarly, bilirubin treatment (500nM) of H9c2 cells leads to high levels of phospho-Akt which were completely attenuated in the presence of BVR siRNA. Scrambled siRNA transfection had no effect on the phospho-Akt levels. (* vs. control; ** vs. bilirubin; p<0.05, n=3 independent experiments performed in duplicate)
Acknowledgments
This work was supported by an NIH grant (HL 696901) to Drs. Pachori and Dzau, and by grants from the Heart and Stroke Foundation of Ontario (HSFO, NA 5779) and CIHR (MOP 79506) to Dr. Melo. Dr. Melo is Canada Research Chair in Molecular Cardiology. We thank Dr. Shaw-Feng Yet (Harvard Medical School) for providing the MEFs from the HO-1 null mice.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Carden DL, Granger DN. Pathophysiology of ischaemia-reperfusion injury. J Pathol. 2000;190(3):255–266. doi: 10.1002/(SICI)1096-9896(200002)190:3<255::AID-PATH526>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- 2.Ryter SW, Choi AM. Heme oxygenase-1: redox regulation of a stress protein in lung and cell culture models. Antioxid Redox Signal. 2005;7(12):80–91. doi: 10.1089/ars.2005.7.80. [DOI] [PubMed] [Google Scholar]
- 3.Stuhlmeier KM. Activation and regulation of Hsp32 and Hsp70. Eur J Biochem. 2000;267(4):1161–1167. doi: 10.1046/j.1432-1327.2000.01112.x. [DOI] [PubMed] [Google Scholar]
- 4.Borger DR, Essig DA. Induction of HSP 32 gene in hypoxic cardiomyocytes is attenuated by treatment with N-acetyl-L-cysteine. Am J Physiol. 1998;274(3 Pt 2):H965–973. doi: 10.1152/ajpheart.1998.274.3.H965. [DOI] [PubMed] [Google Scholar]
- 5.Ishii T, Itoh K, Takahashi S, Sato H, Yanagawa T, Katoh Y, Bannai S, Yamamoto M. Transcription factor Nrf2 coordinately regulates a group of oxidative stress-inducible genes in macrophages. J Biol Chem. 2000;275(21):16023–16029. doi: 10.1074/jbc.275.21.16023. [DOI] [PubMed] [Google Scholar]
- 6.Otterbein LE, Choi AM. Heme oxygenase: colors of defense against cellular stress. Am J Physiol Lung Cell Mol Physiol. 2000;279(6):L1029–1037. doi: 10.1152/ajplung.2000.279.6.L1029. [DOI] [PubMed] [Google Scholar]
- 7.Tulis DA, Durante W, Liu X, Evans AJ, Peyton KJ, Schafer AI. Adenovirus-mediated heme oxygenase-1 gene delivery inhibits injury-induced vascular neointima formation. Circulation. 2001;104(22):2710–2715. doi: 10.1161/hc4701.099585. [DOI] [PubMed] [Google Scholar]
- 8.Otterbein LE, Kolls JK, Mantell LL, Cook JL, Alam J, Choi AM. Exogenous administration of heme oxygenase-1 by gene transfer provides protection against hyperoxia-induced lung injury. J Clin Invest. 1999;103(7):1047–1054. doi: 10.1172/JCI5342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sabaawy HE, Zhang F, Nguyen X, ElHosseiny A, Nasjletti A, Schwartzman M, Dennery P, Kappas A, Abraham NG. Human heme oxygenase-1 gene transfer lowers blood pressure and promotes growth in spontaneously hypertensive rats. Hypertension. 2001;38(2):210–215. doi: 10.1161/01.hyp.38.2.210. [DOI] [PubMed] [Google Scholar]
- 10.Pachori AS, Melo LG, Hart ML, Noiseux N, Zhang L, Morello F, Solomon SD, Stahl GL, Pratt RE, Dzau VJ. Hypoxia-regulated therapeutic gene as a preemptive treatment strategy against ischemia/reperfusion tissue injury. Proc Natl Acad Sci U S A. 2004;101(33):12282–12287. doi: 10.1073/pnas.0404616101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Melo LG, Agrawal R, Zhang L, Rezvani M, Mangi AA, Ehsan A, Griese DP, Dell'Acqua G, Mann MJ, Oyama J, Yet SF, Layne MD, Perrella MA, Dzau VJ. Gene therapy strategy for long-term myocardial protection using adeno-associated virus-mediated delivery of heme oxygenase gene. Circulation. 2002;105(5):602–607. doi: 10.1161/hc0502.103363. [DOI] [PubMed] [Google Scholar]
- 12.Yoshida T, Maulik N, Ho YS, Alam J, Das DK. H(mox-1) constitutes an adaptive response to effect antioxidant cardioprotection: A study with transgenic mice heterozygous for targeted disruption of the Heme oxygenase-1 gene. Circulation. 2001;103(12):1695–1701. doi: 10.1161/01.cir.103.12.1695. [DOI] [PubMed] [Google Scholar]
- 13.Yet SF, Perrella MA, Layne MD, Hsieh CM, Maemura K, Kobzik L, Wiesel P, Christou H, Kourembanas S, Lee ME. Hypoxia induces severe right ventricular dilatation and infarction in heme oxygenase-1 null mice. J Clin Invest. 1999;103(8):R23–29. doi: 10.1172/JCI6163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pachori AS, Melo LG, Zhang L, Solomon SD, Dzau VJ. Chronic recurrent myocardial ischemic injury is significantly attenuated by pre-emptive adeno-associated virus heme oxygenase-1 gene delivery. J Am Coll Cardiol. 2006;47(3):635–643. doi: 10.1016/j.jacc.2005.09.038. [DOI] [PubMed] [Google Scholar]
- 15.Sedlak TW, Snyder SH. Bilirubin benefits: cellular protection by a biliverdin reductase antioxidant cycle. Pediatrics. 2004;113(6):1776–1782. doi: 10.1542/peds.113.6.1776. [DOI] [PubMed] [Google Scholar]
- 16.Clark JE, Foresti R, Green CJ, Motterlini R. Dynamics of haem oxygenase-1 expression and bilirubin production in cellular protection against oxidative stress. Biochem J. 2000;348(Pt 3):615–619. [PMC free article] [PubMed] [Google Scholar]
- 17.Clark JE, Foresti R, Sarathchandra P, Kaur H, Green CJ, Motterlini R. Heme oxygenase-1-derived bilirubin ameliorates postischemic myocardial dysfunction. Am J Physiol Heart Circ Physiol. 2000;278(2):H643–651. doi: 10.1152/ajpheart.2000.278.2.H643. [DOI] [PubMed] [Google Scholar]
- 18.Mayer M. Association of serum bilirubin concentration with risk of coronary artery disease. Clin Chem. 2000;46(11):1723–1727. [PubMed] [Google Scholar]
- 19.Lin JP, O'Donnell CJ, Schwaiger JP, Cupples LA, Lingenhel A, Hunt SC, Yang S, Kronenberg F. Association between the UGT1A1*28 allele, bilirubin levels, and coronary heart disease in the Framingham Heart Study. Circulation. 2006;114(14):1476–1481. doi: 10.1161/CIRCULATIONAHA.106.633206. [DOI] [PubMed] [Google Scholar]
- 20.Lerner-Marmarosh N, Shen J, Torno MD, Kravets A, Hu Z, Maines MD. Human biliverdin reductase: a member of the insulin receptor substrate family with serine/threonine/tyrosine kinase activity. Proc Natl Acad Sci U S A. 2005;102(20):7109–7114. doi: 10.1073/pnas.0502173102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matsui T, Li L, del Monte F, Fukui Y, Franke TF, Hajjar RJ, Rosenzweig A. Adenoviral gene transfer of activated phosphatidylinositol 3′-kinase and Akt inhibits apoptosis of hypoxic cardiomyocytes in vitro. Circulation. 1999;100(23):2373–2379. doi: 10.1161/01.cir.100.23.2373. [DOI] [PubMed] [Google Scholar]
- 22.Matsui T, Tao J, del Monte F, Lee KH, Li L, Picard M, Force TL, Franke TF, Hajjar RJ, Rosenzweig A. Akt activation preserves cardiac function and prevents injury after transient cardiac ischemia in vivo. Circulation. 2001;104(3):330–335. doi: 10.1161/01.cir.104.3.330. [DOI] [PubMed] [Google Scholar]
- 23.Fujio Y, Nguyen T, Wencker D, Kitsis RN, Walsh K. Akt promotes survival of cardiomyocytes in vitro and protects against ischemia-reperfusion injury in mouse heart. Circulation. 2000;101(6):660–667. doi: 10.1161/01.cir.101.6.660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matsui T, Rosenzweig A. Convergent signal transduction pathways controlling cardiomyocyte survival and function: the role of PI 3-kinase and Akt. J Mol Cell Cardiol. 2005;38(1):63–71. doi: 10.1016/j.yjmcc.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 25.Brunt KR, Fenrich KK, Kiani G, Tse MY, Pang SC, Ward CA, Melo LG. Protection of human vascular smooth muscle cells from H2O2-induced apoptosis through functional codependence between HO-1 and AKT. Arterioscler Thromb Vasc Biol. 2006;26(9):2027–2034. doi: 10.1161/01.ATV.0000236204.37119.8d. [DOI] [PubMed] [Google Scholar]
- 26.Salinas M, Wang J, Rosa de Sagarra M, Martin D, Rojo AI, Martin-Perez J, Ortiz de Montellano PR, Cuadrado A. Protein kinase Akt/PKB phosphorylates heme oxygenase-1 in vitro and in vivo. FEBS Lett. 2004;578(12):90–94. doi: 10.1016/j.febslet.2004.10.077. [DOI] [PubMed] [Google Scholar]
- 27.Cantley LC. The phosphoinositide 3-kinase pathway. Science. 2002;296(5573):1655–1657. doi: 10.1126/science.296.5573.1655. [DOI] [PubMed] [Google Scholar]
- 28.Sharma HS, Maulik N, Gho BC, Das DK, Verdouw PD. Coordinated expression of heme oxygenase-1 and ubiquitin in the porcine heart subjected to ischemia and reperfusion. Mol Cell Biochem. 1996;157(12):111–116. doi: 10.1007/BF00227888. [DOI] [PubMed] [Google Scholar]
- 29.Maulik N, Sharma HS, Das DK. Induction of the haem oxygenase gene expression during the reperfusion of ischemic rat myocardium. J Mol Cell Cardiol. 1996;28(6):1261–1270. doi: 10.1006/jmcc.1996.0116. [DOI] [PubMed] [Google Scholar]
- 30.Yet SF, Tian R, Layne MD, Wang ZY, Maemura K, Solovyeva M, Ith B, Melo LG, Zhang L, Ingwall JS, Dzau VJ, Lee ME, Perrella MA. Cardiac-specific expression of heme oxygenase-1 protects against ischemia and reperfusion injury in transgenic mice. Circ Res. 2001;89(2):168–173. doi: 10.1161/hh1401.093314. [DOI] [PubMed] [Google Scholar]
- 31.Stocker R, Yamamoto Y, McDonagh AF, Glazer AN, Ames BN. Bilirubin is an antioxidant of possible physiological importance. Science. 1987;235:1043–1046. doi: 10.1126/science.3029864. [DOI] [PubMed] [Google Scholar]
- 32.Otterbein LE, Bach FH, Alam J, Soares M, Lu HT, Wysk M, Davis RJ, Flavell RA, Choi AMK. Carbon monoxide has anti-inflammatory effects involving the mitogen-activated protein kinase pathway. Nat Med. 2000;6(4):422–428. doi: 10.1038/74680. [DOI] [PubMed] [Google Scholar]
- 33.Wang X, Wang Y, Kim HP, Nakahira K, Ryter SW, Choi AM. Carbon monoxide protects against hyperoxia-induced endothelial cell apoptosis by inhibiting reactive oxygen species formation. J Biol Chem. 2006 doi: 10.1074/jbc.M607610200. [DOI] [PubMed] [Google Scholar]
- 34.Ryter SW, Alam J, Choi AM. Heme oxygenase-1/carbon monoxide: from basic science to therapeutic applications. Physiol Rev. 2006;86(2):583–650. doi: 10.1152/physrev.00011.2005. [DOI] [PubMed] [Google Scholar]
- 35.Zhang X, Shan P, Alam J, Fu XY, Lee PJ. Carbon monoxide differentially modulates STAT-1 and STAT-3 and inhibits apoptosis via a phosphotidylinositol 3-kinase/Akt and p38 kinase-dependent STAT-3 pathway during anoxia-reoxygenation injury. J Biol Chem. 2005;280(10):8714–8721. doi: 10.1074/jbc.M408092200. [DOI] [PubMed] [Google Scholar]
- 36.Fujimoto H, Ohno M, Ayabe S, Kabayashi H, Ishizaka N, Kimura H, Yoshida K, Nagai R. Carbon monoxide protects against cardiac ischemia-reprfusion injury in vivo via MAPK and Akt-eNOS pathway. Arteriscler Thromb Vasc Biol. 2004;24:1848–1853. doi: 10.1161/01.ATV.0000142364.85911.0e. [DOI] [PubMed] [Google Scholar]
- 37.Akamatsu Y, Haga M, Tyagi S, Yamashita K, Graça-Souza AV, Ollinger R, Czismadia E, May GA, Ifedigbo E, Otterbein LE, Bach FH, Soares MP. Heme oxygenase-1 derived carbon monoxide protects hearts from transplant associated ischemia reperfusion injury. FASEB J. 2004;18:771–772. doi: 10.1096/fj.03-0921fje. [DOI] [PubMed] [Google Scholar]
- 38.Hori R, Kashiba M, Toma T, Yachie A, Goda N, Makino A, Sowjima A, Nagasawa T, Nakabayashi K, Suematsu M. Gene transfection of H25A mutant heme oxygenase-1 protects cells against hydroperoxide -induced cytoxicity. J Biol Chem. 2002;277:10713–10718. doi: 10.1074/jbc.M107749200. [DOI] [PubMed] [Google Scholar]
- 39.Martin D, Rojo AI, Salinas M, Diaz R, Gallardo G, Alam J, Galarreya CMR, Cuadrado A. Regulation of heme oxygenase-1 expression through the phosphatydilinositol 3-kinase/Akt pathway and the Nrf2 transcription factor in responsew to the antioxidant phytochemical carnosol. J Biol Chem. 2004;279(10):8919–8929. doi: 10.1074/jbc.M309660200. [DOI] [PubMed] [Google Scholar]
- 40.Kang KW, Lee SJ, Park JW, Kim SG. Phosphatidylinositol 3-kinase regulates nuclear translocation of NF-E2 related factor 2 through actin rearanngement in response to oxidative stress. Mol Pharmacol. 2002;62(5):1001–1010. doi: 10.1124/mol.62.5.1001. [DOI] [PubMed] [Google Scholar]
- 41.Busserolles J, Megias J, Terencio MC, Alcaraz MJ. Heme oxygenase-1 inhibits apoptosis in Caco-2 cells via activation of Akt pathway. Int J Biochem Cell Biol. 2006;38:1510–1517. doi: 10.1016/j.biocel.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 42.Kutty RK, Maines MD. Purification and characterization of biliverdin reductase from rat liver. J Biol Chem. 1981;256(8):3956–3962. [PubMed] [Google Scholar]
- 43.Backer JM, Myers MG, Jr, Shoelson SE, Chin DJ, Sun XJ, Miralpeix M, Hu P, Margolis B, Skolnik EY, Schlessinger J, et al. Phosphatidylinositol 3′-kinase is activated by association with IRS-1 during insulin stimulation. Embo J. 1992;11(9):3469–3479. doi: 10.1002/j.1460-2075.1992.tb05426.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pereira P, Macedo-Ribeiro S, Parraga A, Perez-Luque R, et al. Structure of human biliverdin IXβ reductase, an early fetal bilirubin IXβ producing enzyme. Nature Structural Biology. 2001;8(3):215–220. doi: 10.1038/84948. [DOI] [PubMed] [Google Scholar]
- 45.Kikuchi A, Park S, Miyatake H, et al. Crystal structure of rat biliverdin reductase. Nature Structural Biology. 2001;8(3):221–225. doi: 10.1038/84955. [DOI] [PubMed] [Google Scholar]
- 46.Hiroaki M, Yoshito I, Hajime N, Junji Y, Koji S, Takahito K. Glutaredoxin exerts an antiapoptotic effect by regulating the redox state of Akt. J Biol Chem. 2003;278:50226–50233. doi: 10.1074/jbc.M310171200. [DOI] [PubMed] [Google Scholar]
- 47.Baranano DE, Rao M, Ferris D, Snyder SH. Biliverdin reductase: A major physiological cytoprotectant. Proc Natl Acad Sci. 2002;99(25):16093–16098. doi: 10.1073/pnas.252626999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maines MD. New insights into biliverdin reductase functions: Linking heme metabolism to cell signaling. Physiology. 2005;20:382–389. doi: 10.1152/physiol.00029.2005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure 1. Densitometric analysis of data shown in figure 3B. HO-1 overexpression in H9c2 cells leads to significantly higher levels of phospho-Akt which were attenuated in the presence of HO-1 siRNA both under normal as well as hypoxia/reoxygenation conditions (* vs. control; ** vs. HO-1; # vs. HO-1 normoxia; ## vs. HO-1 hypoxia/reoxygenation. P<0.05, n=3 independent experiments performed in duplicate)
Figure 2A-C. Densitmetric analysis of data shown in figure 4, panels A, C and E. A. Effect of HO-1 overexpression on BVR protein levels. B. HO-1 overexpression in H9c2 cells leads to significantly higher levels of phospho-Akt which was significantly attenuated with co-transfection of BVRsiRNA. There was no effect on phospho-Akt levels when cells were co-transfected with HO-1 along with scrambled siRNA. (* vs. control; ** vs. HO-1; p<0.05, n=3 independent experiments performed in duplicate) C. Similarly, bilirubin treatment (500nM) of H9c2 cells leads to high levels of phospho-Akt which were completely attenuated in the presence of BVR siRNA. Scrambled siRNA transfection had no effect on the phospho-Akt levels. (* vs. control; ** vs. bilirubin; p<0.05, n=3 independent experiments performed in duplicate)