

Abstract
A new, stereoselective synthesis of substituted tetrahydrofurans via Pd-catalyzed reactions of aryl and vinyl bromides with γ-hydroxy terminal alkenes is described. This transformation affords trans-2,5- and trans-2,3-disubstituted tetrahydrofurans with up to >20:1 dr. This methodology also provides access to bicyclic and spirocyclic tetrahydrofuran derivatives in good yield with 10-20:1 dr. The scope and limitations of these transformations are discussed in detail, as are the effect of substrate sterics and electronics on yield and stereoselectivity. A proposed mechanism of these transformations is presented along with a model that rationalizes the stereochemical outcome of the reactions.
Introduction
A wide variety of interesting biologically active molecules contain substituted tetrahydrofuran subunits.1 For example, the annonaceous acetogenins are a large family of natural products with a diverse range of biological activities including antitumor, antihelmic, antimalarial, antimicrobial, and antiprotozoal.2 Substituted tetrahydrofurans are also found in polyether antibiotics,3 lignans,4 and C-glycosides.5
Due to the medicinal importance of compounds bearing tetrahydrofuran units, there has been considerable interest in the development in efficient, stereoselective methods for the preparation of the tetrahydrofuran ring.1-6 However, few methods have been described that allow for formation of a tetrahydrofuran ring from an acyclic precursor with concomitant formation of both a new stereogenic center and a new carbon-carbon bond at the C1′-position.7 These methods are effective for the preparation of tetrahydrofurans bearing ester functionality at C1′, but often exhibit only modest stereocontrol (ca. 1-3:1) for the preparation of 2,3- and 2,5-disubstituted products, and do not allow for the installation of aryl, vinyl, or alkyl chains at C1′.7
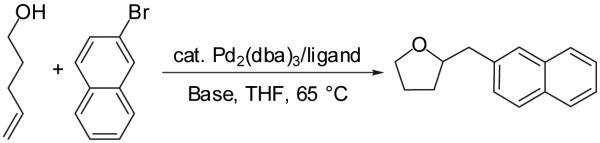
We recently reported a new method for the stereoselective synthesis of tetrahydrofurans from aryl or vinyl bromides and γ-hydroxyalkenes.8,9,10,11,12,13 These reactions lead to the formation of both a C-O and a C-C bond along with up to two new stereocenters in a single step. Transformations of γ-hydroxyalkenes bearing substituents at the 1- or 3-position proceed with good to excellent levels of diastereoselectivity (up to >20:1 dr). However, reactions of substrates that are substituted at the 2-position afford products with low dr (ca. 2:1). In this article we describe the development, scope, and limitations of the synthesis of substituted tetrahydrofurans from 4-penten-1-ol derivatives. We also describe unexpected electronic effects that influence the chemical yield of these transformations, and present further studies on the effect of substituent size on the diastereoselectivity of these transformations. Finally, we propose a refined model to explain the stereochemical outcome of these transformations.
Results
Development and Optimization of Reaction Conditions
Larock has previously demonstrated that the Pd-catalyzed reaction of 4-penten-1-ol with aryl halides in the presence of a weak base (NaHCO3) affords 5-arylpentanal products (eq 1), which are believed to derive from Heck-type olefination of the aryl halide followed by reversible β-hydride elimination.14 We felt that the synthesis of 2-benzyltetrahydrofurans from a similar combination of substrates (eq 2) could be achieved through two key modifications of the Larock conditions: 1) use of a stronger base, which would lead to deprotonation of the alcohol substrate and provide access to intermediate palladium alkoxides,15 and 2) use of a palladium/phosphine catalyst that would promote tetrahydrofuran formation via alkene insertion followed by reductive elimination. We reasoned that the reactivity of the intermediate palladium alkoxide complex could be controlled by varying the steric and electronic properties of the phosphine ligands.
 |
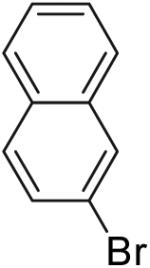
In our preliminary experiments, we examined the reaction of 4-penten-1-ol with 2-bromonaphthalene (eq 3) using a catalyst comprised of Pd2(dba)3 and P(o-tol)3 and found that the desired tetrahydrofuran product was formed in 20% isolated yield when NaOtBu was employed as base.16 The main side product observed in this transformation was naphthalene, which derives from Pd-catalyzed reduction of the aryl bromide. Previous studies have shown that aryl halides undergo Pd-catalyzed reduction in the presence of an alcohol and a base by a mechanism involving β-hydride elimination from a Pd(Ar)(OR) intermediate.17 Chelating bis(phosphine) ligands are known to decrease the rate of β-hydride elimination reactions,18 and we were pleased to find that use of dpe-phos19 as ligand increased the yield of the desired product to 45%. This yield was further improved to 76% by adding two equivalents of both the aryl bromide and NaOtBu to the reaction mixture. Further experiments demonstrated that use of the dppb ligand provided results comparable to those obtained with dpe-phos. However, most other ligands and bases that were examined provided unsatisfactory results (Table 1). A brief survey of reaction media showed that use of toluene as solvent provided similar results to those obtained in THF. However, reactions conducted in polar solvents such as DMF and acetonitrile gave little or no desired products.
Table 1.
Optimization Studies(a)
| ||||||
|---|---|---|---|---|---|---|
| Entry | Ligand | Base | GC Yield (%) | Isolated Yield (%) | ||
| 1 | P(o-tol)3 | KHCO3 | 0(g),(h) | |||
| 2 | P(o-tol)3 | NaOAc | 0(g),(h) | |||
| 3 | P(o-tol)3 | NaHCO3 | 0(g),(h) | |||
| 4 | P(o-tol)3 | K2CO3 | 0(g),(h) | |||
| 5 | P(o-tol)3 | Et3N | 0(g),(h) | |||
| 6 | P(o-tol)3 | Na2CO3 | 0(g),(h) | |||
| 7 | P(o-tol)3 | LiOtBu | 5 | |||
| 8 | P(o-tol)3 | KOtBu | 11 | |||
| 9 | P(o-tol)3 | NaOtBu | 17 | 20 | ||
| 10 | PPh3 | NaOtBu | 0(h) | |||
| 11 | P(2-furyl)3 | NaOtBu | 0(g),(h) | |||
| 12 | tBu2P(o-biphenyl) | NaOtBu | 0 | |||
| 13 | Cy2P(o-biphenyl) | NaOtBu | <3 | |||
| 14(b) | Pd[P(tBu)3]2 | NaOtBu | 0(h) | |||
| 15(c) | Pd[P(Cy)3]2 | NaOtBu | 0(h) | |||
| 16 | dppe | NaOtBu | <1 | |||
| 17 | (±)-BINAP | NaOtBu | 0(g),(h) | |||
| 18 | dppf | NaOtBu | 8 | |||
| 19 | dppb | NaOtBu | 52 | |||
| 20 | xantphos | NaOtBu | 40 | |||
| 21 | dpe-phos | NaOtBu | 55 | 45 | ||
| 22(d) | dpe-phos | NaOtBu | 79 | |||
| 23(e) | dpe-phos | NaOtBu | 80 | |||
| 24(f) | dpe-phos | NaOtBu | 93 | 76 | ||
Conditions: 1.0 equiv alcohol, 1.1 equiv ArBr, 1.2 equiv NaOtBu, 1 mol % Pd2(dba)3, 4 mol % ligand (monodentate ligands) or 2 mol % ligand (bidentate ligands), solvent (0.25 M), 65 °C.
Pd[P(tBu)3]2 used in place of Pd2(dba)3/ligand.
Pd[P(Cy)3]2 used in place of Pd2(dba)3/ligand.
2.0 equiv base used.
2.0 equiv ArBr used.
2.0 equiv ArBr and 2.0 equiv base used.
Less than 10% of the starting material was consumed.
Naphthalene was the sole product detected by GC analysis of the crude reaction mixture.
Synthesis of 2-Monosubstituted and 2,2-Symmetrically Disubstituted Tetrahydrofurans
Transformations of 4-penten-1-ol (1) and the analogous symmetrically substituted tertiary alcohol derivatives 3 and 4 proceed in moderate to good yield with a variety of electron-neutral aryl bromides (Table 2).20 This method is amenable to the synthesis of spirocyclic ring systems (entries 11-12), and ortho-substitution on the aryl bromide is tolerated (entries 9 and 12). High regioselectivity (>20:1) for 2-benzyltetrahydrofuran formation was observed in all reactions examined in these studies.21
Table 2.
| entry | alcohol | aryl bromide | product | yield(b) |
|---|---|---|---|---|
| 1 | 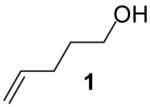 |
 |
76% | |
| 2 | 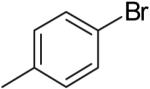 |
65% | ||
| 3 | 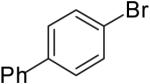 |
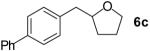 |
70% | |
| 4 | 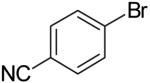 |
40% | ||
| 5 | 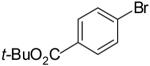 |
36% | ||
| 6 | 32% | |||
| 7 | 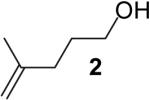 |
 |
19% | |
| 8 | 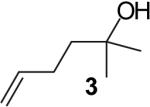 |
 |
80% | |
| 9 |  |
 |
60% | |
| 10 | 81% | |||
| 11 |  |
 |
70% | |
| 12 |  |
60% | ||
| 13 |  |
 |
No Reaction |
Conditions: 1.0 equiv alcohol, 2.0 equiv ArBr, 2.0 equiv NaOtBu, 1 mol % Pd2(dba)3, 2 mol % dpe-phos, THF (0.13-0.25 M), 65 °C.
Yields represent average isolated yields for two or more experiments.
The main side products in these reactions are arenes derived from reduction of the aryl halide,22 and/or ethers derived from O-arylation/vinylation of the alcohol substrate.23,24 These side reactions are more problematic with primary alcohol substrates. For example, a significantly lower yield was obtained in the reaction of β-bromostyrene with 4-penten-1-ol (32%, entry 6) than in the analogous reaction with 3 (81%, entry 10) due to competing O-vinylation of the primary alcohol. The O-arylation reactions are most problematic with electron-deficient aryl bromides such as 4-bromobenzonitrile (entry 4) and t-butyl-4-bromobenzoate (entry 5).
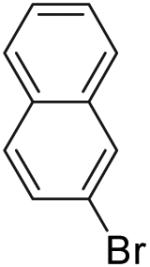
In contrast, reactions of electron-neutral aryl halides proceed efficiently with both primary and tertiary alcohols. For example, the reaction of 2-bromonaphthalene with 4-penten-1-ol proceeded in 76% isolated yield (entry 1) and treatment of 2-bromonaphthalene with 2-methyl-4-hexen-2-ol (3) afforded an 80% isolated yield of the tetrahydrofuran product 8a (entry 8).
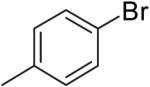
The steric properties of the alcohol substrate also have a large effect on the overall reactivity in these transformations. Substrates 2 and 5, that are substituted with a methyl group at the internal olefinic carbon are less reactive than the unsubstituted analogs 1 and 3. As shown in Table 2, the reaction of 1 with 4-bromotoluene proceeded in 65% yield at 65 °C, whereas the reaction of primary alcohol 2 with 4-bromotoluene afforded a modest 19% yield of tetrahydrofuran 7. The tertiary alcohol 5 failed to react with 4-bromotoluene even when heated to 140 °C in xylenes.
Stereoselective Synthesis of 2,5-Disubstituted and 2,5,5-Trisubstituted Tetrahydrofurans
As shown in Table 3, the reactions of aryl bromides with several different 4-penten-1-ol derivatives bearing C-1 substituents were examined. In all transformations, a single tetrahydrofuran product was formed with >20:1 diastereoselectivity favoring the trans-stereoisomer.25,26 The size of the R-group did not have an impact on the stereoselectivity of the transformations. However, the steric bulk of the R-group had a pronounced effect on chemical yield. For substrates with alkyl substituents (R = Me, Et, t-Bu) the yield of tetrahydrofuran products decreased and larger amounts of ketone side products were formed as the size of the alkyl group increased. For example, the reaction of 1-bromo-4-t-butylbenzene with 5-hexen-2-ol (11a, entry 1) afforded a 43% yield of the desired product (12a), whereas the analogous reaction of 6-hepten-3-ol (11b) proceeded in 37% yield (entry 2) and the transformation of 2,2-dimethyl-6-hepten-3-ol (11c) did not provide a tetrahydrofuran product. Reactions of 1-phenyl-4-penten-1-ol with aryl bromides (11e, Table 3 entries 5-9 and Table 5 entry 3) typically afforded yields of 60-69%, although lower yields (29-37%) were obtained when vinyl bromides were employed as the coupling partners due to competing O-vinylation of the alcohol (entries 8 and 9). The nature of the aryl/vinyl bromide did not have a noticeable effect on the stereoselectivity of these transformations. An alcohol substrate bearing a strongly electron withdrawing ester substituent at the 1-position (11d) afforded only products of Heck arylation of the alkene moiety; no tetrahydrofuran products were observed (entry 4).24
Table 3.
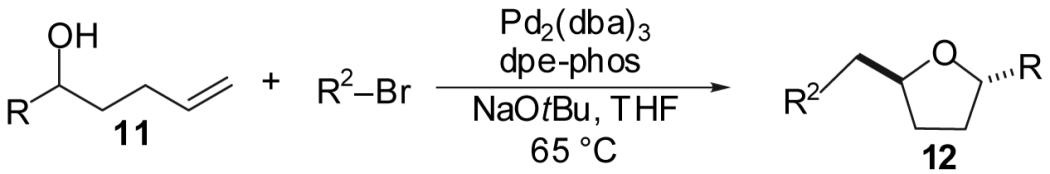 | ||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | R | R2 | Product | Yield(b) | dr |
| 1 | 11a | Me |  |
12a | 43%(c) | >20:1 |
| 2 | 11b | Et | 12b | 37%(c) | >20:1 | |
| 3 | 11c | t-Bu | No Reaction | --- | ||
| 4 | 11d | CO2Et | 0%(d) | --- | ||
| 5 | 11e | Ph | 12c | 62% | >20:1 | |
| 6 | 11e | Ph |  |
12d | 60% | >20:1 |
| 7 | 11e | Ph |  |
12e | 61% | >20:1 |
| 8 | 11e | Ph | 12f | 37% | >20:1 | |
| 9 | 11e | Ph | 12g | 29% | >20:1 | |
Conditions: 1.0 equiv alcohol, 2.0 equiv ArBr, 2.0 equiv NaOtBu, 1 mol % Pd2(dba)3, 2 mol % dpe-phos, THF (0.13-0.25 M), 65 °C.
Yields represent average isolated yields for two or more experiments.
A reductive workup was employed to facilitate removal of a ketone side product. See supporting information for complete details.
Heck arylation was observed.
Table 5.
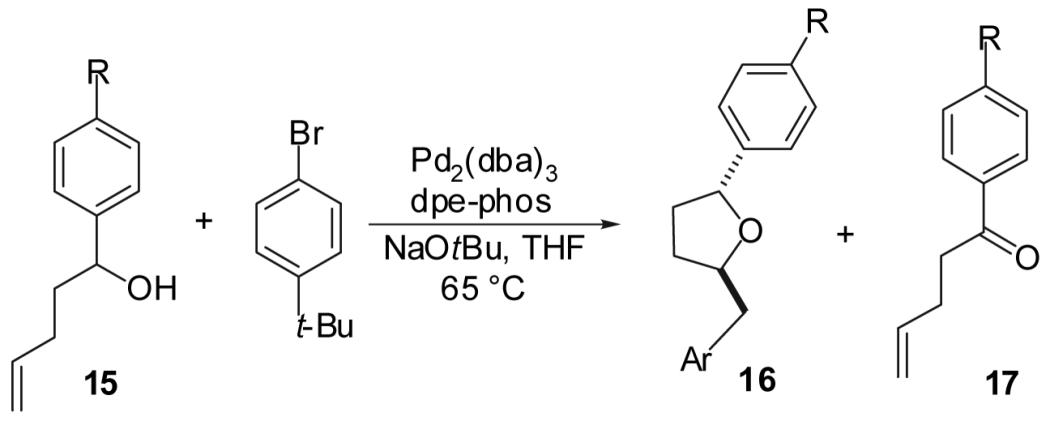 | ||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | R | Product | Yieldb | dr | 16:17c |
| 1 | 15a | CN | 16a | 76% | >20:1 | >20:1 |
| 2 | 15b | CF3 | 16b | 64% | >20:1 | >20:1 |
| 3 | 11e | H | 16c | 69% | >20:1 | >20:1 |
| 4 | 15c | OMe | 16d | 58% | >20:1 | 5:1 |
| 5 | 15d | N(Me)2 | 16e | 35% | >20:1 | 3:1 |
(a) Conditions: 1.0 equiv alcohol, 2.0 equiv ArBr, 2.0 equiv NaOtBu, 1 mol % Pd2(dba)3, 2 mol % dpe-phos, THF (0.13-0.25 M), 65 °C.
(b) Yields represent average isolated yields for two or more experiments.
(c) The formation of t-butylbenzene in amounts comparable to the amount of 17 observed in these reactions was detected by GC analysis.
Curiously, reactions of 1-alkoxymethyl substituted substrates provided significantly higher yields than were obtained in transformations of substrates bearing a 1-methyl or 1-ethyl group. To further probe this effect, substrates bearing different alcohol protecting groups were treated with 1-bromo-4-t-butylbenzene under the optimized reaction conditions (Table 4). These reactions afforded the desired tetrahydrofurans with high diastereoselectivities and isolated yields of 68-81%. No clear correlation between protecting group size and chemical yield was observed, suggesting that the high yields obtained with these substrates is not due to a chelation effect. As observed in reactions of primary alcohol substrates, a low yield was obtained with a vinyl bromide coupling partner (entry 5) due to competing O-vinylation of the substrate.
Table 4.
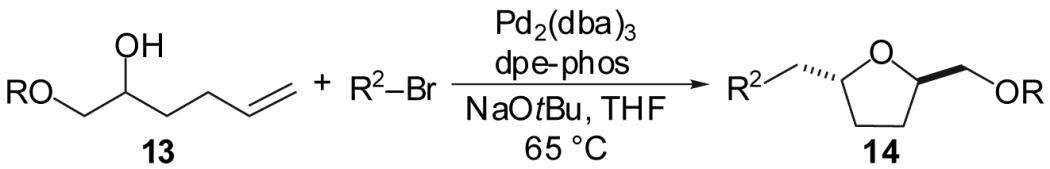 | ||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | R | R2 | Product | Yield(b) | dr |
| 1 | 13a | Me |  |
14a | 72% | >20:1 |
| 2 | 13b | TBS | 14b | 68% | >20:1 | |
| 3 | 13c | BHT | 14c | 81% | >20:1 | |
| 4 | 13b | TBS |  |
14d | 52% | >20:1 |
| 5 | 13b | TBS | 14e | 38% | >20:1 | |
Conditions: 1.0 equiv alcohol, 2.0 equiv ArBr, 2.0 equiv NaOtBu, 1 mol % Pd2(dba)3, 2 mol % dpe-phos, THF (0.13-0.25 M), 65 °C.
Yields represent average isolated yields for two or more experiments.
In the absence of chelation, the relatively high yields obtained in reactions of 1-alkoxymethyl substituted alcohols could potentially derive from the electron-withdrawing nature of the alkoxy substituent. To further explore the effects of electronics on these transformations, a series of 1-aryl-4-penten-1-ols bearing various substituents in the para-position of the aryl group were prepared and treated with 1-bromo-4-t-butylbenzene in the presence of NaOtBu and the Pd2(dba)3/dpe-phos catalyst. As shown in Table 5, as the electron-donating ability of the p-substituent increased, the yield of the desired product 16 decreased and the amount of oxidized side product 17 increased.24 These results suggest that inductive effects have a larger impact on yield than chelation effects in reactions of 1-alkoxymethyl substituted substrates.
The stereoselectivity of transformations involving tertiary alcohol substrates bearing two different groups at the 1-position was also briefly examined. As shown in Table 6, high diastereoselectivities are obtained in reactions involving electron-rich or -neutral aryl/vinyl halides provided that the two substituents (R and R1) are sufficiently different in size. For example, the Pd-catalyzed reaction of 3-methyl-6-hetpen-3-ol (18a, R = Et, R1 = Me) with 1-bromo-4-t-butylbenzene provided a 59% yield of the tetrahydrofuran product 19a as a 3:1 mixture of diastereomers (entry 1). In contrast, the analogous transformation of 1-phenyl-5-hexen-2-ol (18b, R = Ph, R1 = Me) afforded the desired product in 77% yield with >20:1 diastereoselectivity (entry 2). The use of β-bromostyrene as a coupling partner with 18b provided a slightly lower chemical yield (58%) of the tetrahydrofuran product (entry 5), although diastereoselectivity was high (>20:1). In contrast, the reaction of 18b with the electron-deficient t-butyl-4-bromobenzoate proceeded in excellent yield (70%) but modest (4:1) diastereoselectivity (entry 4). Resubjection of each isolated diastereomer of 19d to the reaction conditions demonstrated that the diastereomers do not interconvert, and the stereoselectivity is kinetically controlled.
Table 6.
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Substrate | R | R1 | R2 | Product | Yield(b) | dr |
| 1 | 18a | Et | Me | 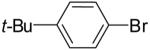 |
19a | 59% | 3:1 |
| 2 | 18b | Ph | Me | 19b | 77% | 20:1 | |
| 3 | 18b | Ph | Me | 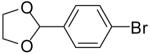 |
19c | 84% | >20:1 |
| 4 | 18b | Ph | Me | 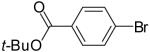 |
19d | 70% | 4:1 |
| 5 | 18b | Ph | Me | 19e | 58% | >20:1 | |
Conditions: 1.0 equiv alcohol, 2.0 equiv ArBr, 2.0 equiv NaOtBu, 1 mol % Pd2(dba)3, 2 mol % dpe-phos, THF (0.13-0.25 M), 65 °C.
Yields represent average isolated yields for two or more experiments.
Stereoselective Synthesis of 2,3-Disubstituted Tetrahydrofurans
As shown in Table 7, transformations of 4-penten-1-ol derivatives bearing a substituent at the 3-position afford trans-2,3-disubstituted tetrahydrofuran products with good to excellent levels of diastereocontrol. In contrast to reactions of substrates bearing substituents at the 1-position, the stereoselectivity in these reactions was dependent both on the size of the 3-substituent, and the size of the substituents at the 1-position. For example, the reaction of 3-methyl-4-penten-1-ol (20a) with 4-bromobiphenyl provided the desired product 21a in 78% yield with modest (3:1) diastereoselectivity (entry 1). In contrast, the reaction of the analogous tertiary alcohol substrate 20e proceeded in similar (78%) yield but with 8:1 diastereoselectivity (entry 6). The stereoselectivity of these transformations improved as the size of the 3-subsituent was increased from methyl (3:1) to t-butyl (>20:1). Additionally, the size of the aryl bromide also had a small influence on the diastereoselectivity in these transformations. The reaction of 3-ethyl-4-penten-1-ol (20b) with a para-substituted aryl bromide afforded 21b 66% yield with 6:1 dr (entry 2), whereas treatment of this substrate with an ortho-substituted aryl bromide afforded a 57% yield of the tetrahydrofuran product 21c as an 8:1 mixture of diastereomers (entry 3).
Table 7.
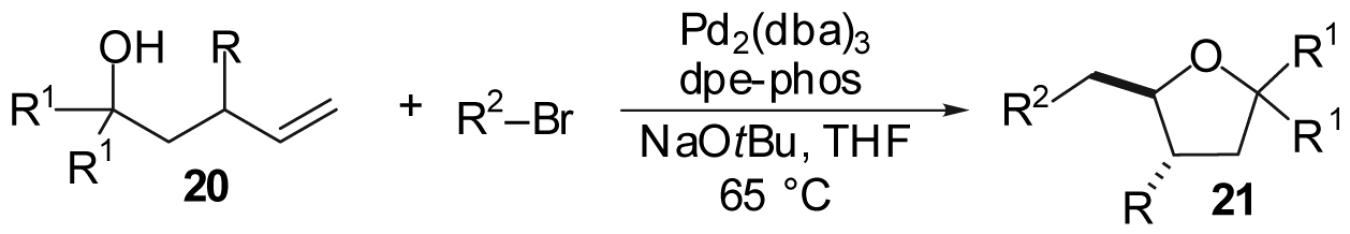 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Substrate | R | R1 | R2 | Product | Yield(b) | dr |
| 1 | 20a | Me | H | 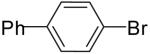 |
21a | 78% | 3:1 |
| 2 | 20b | Et | H | 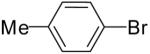 |
21b | 66% | 6:1 |
| 3 | 20b | Et | H | 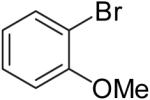 |
21c | 57% | 8:1 |
| 4 | 20c | Ph | H | 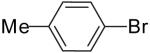 |
21d | 51% | 18:1 |
| 5 | 20d | t-Bu | H | 21e | 55% | <20:1 | |
| 6 | 20e | Me | Me |  |
21f | 78% | 8:1 |
Conditions: 1.0 equiv alcohol, 2.0 equiv ArBr, 2.0 equiv NaOtBu, 1 mol % Pd2(dba)3, 2 mol % dpe-phos, THF (0.13-0.25 M), 65 °C.
Yields represent average isolated yields for two or more experiments.
Synthesis of 2,4-Disubstituted Tetrahydrofurans
In contrast to reactions that afford 2,5- and 2,3-disubstituted tetrahydrofurans, transformations that produce 2,4-disubstituted tetrahydrofurans proceeded with generally low (1-2:1) diastereoselectivity (Table 8). For example, treatment of 2-methyl-4-penten-1-ol (22a) with 4-bromobiphenyl under standard reaction conditions afforded a 1.5:1 mixture of diastereomeric 2,4-disubstituted tetrahydrofuran products in 70% yield (entry 1); a slight preference for the cis-diastereomer was observed. Increasing the size of the 2-substituent from methyl to tert-butyl did not lead to significant improvements in selectivity. Despite the low diastereoselectivities of these transformations, the tetrahydrofuran products were obtained in good to excellent yields. It is noteworthy that the reaction of substrate 22e (R = Me, R1 = Me) with 4-bromobiphenyl afforded an 88% yield of the desired product (entry 7). This result sharply contrasts to the analogous reaction of 2 (Table 2, entry 7, R = H, R1 = Me) which proceed in low yield. The high yield obtained in the reaction of 22e may derive from a Thorpe-Ingold conformational effect induced by the C-3 substituent,27 which could potentially increase the rate of the desired cyclization reaction relative to the rate of competing alcohol oxidation/aryl bromide reduction side reactions.
Table 8.
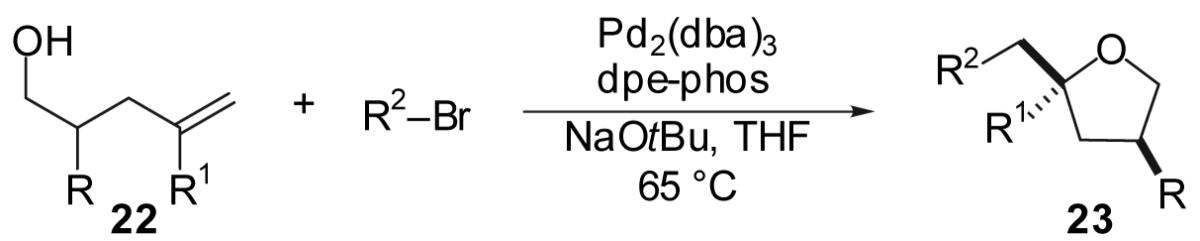 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Substrate | R | R1 | R2 | Product | Yield(b) | dr |
| 1 | 22a | Me | H |  |
23a | 70% | 1.5:1 |
| 2 | 22b | Et | H | 23b | 88% | 1.5:1 | |
| 3 | 22c | Ph | H |  |
23c | 84% | 2:1 |
| 4 | 22d | t-Bu | H |  |
23d | 88% | 1.5:1 |
| 5 | 22d | t-Bu | H | 23e | 77% | 2:1 | |
| 6 | 22d | t-Bu | H | 23f | 66% | 2:1 | |
| 7 | 22e | Me | Me |  |
23g | 88% | 1:1 |
Conditions: 1.0 equiv alcohol, 2.0 equiv ArBr, 2.0 equiv NaOtBu, 1 mol % Pd2(dba)3, 2 mol % dpe-phos, THF (0.13-0.25 M), 65 °C.
Yields represent average isolated yields for two or more experiments.
Synthesis of Fused Bicyclic Oxygen Heterocycles
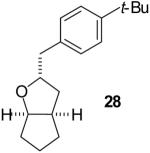
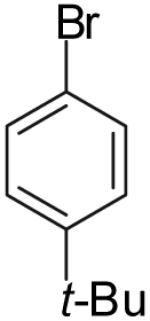
The utility of this transformation for the synthesis of fused bicyclic oxygen heterocycles from 2-allyl-1-cycloalkanols was also briefly explored (Table 9). The stereochemistry of the starting material has a pronounced effect on both the chemical yield and diastereoselectivity of these reactions. For example, the Pd-catalyzed reaction of cis-2-allylcyclohexanol (24a) with 4-bromobiphenyl afforded bicyclic product 26a in 60% yield as a 10:1 mixture of diastereomers. In contrast, the analogous reaction of trans-2-allylcyclohexanol (24b) provided a 70% yield of 26b with >20:1 diastereoselectivity (entry 2). Transformation of cis-2-allylcyclopentanol (25a) to cis-fused product 28 proceeded in modest yield with excellent (>20:1) dr (entry 4). However, the preparation of the analogous trans fused product from trans-2-allylcyclopentanol (25b) was not achieved; the sole product detected was 2-allylcyclopentanone (entry 5).
Table 9.
| Entry | Alcohol | R2-Br | Product | Yield(b) | dr |
|---|---|---|---|---|---|
| 1 | 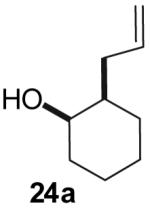 |
 |
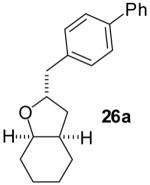 |
60% | 10:1 |
| 2 | 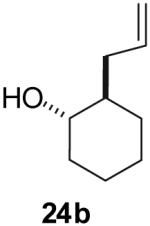 |
 |
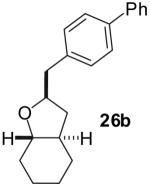 |
70% | >20:1 |
| 3 |  |
 |
40% | >20:1 | |
| 4 |  |
 |
 |
53% | >20:1 |
| 5(a)(b) | 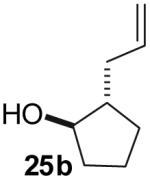 |
 |
0%(c) | -- |
Conditions: 1.0 equiv alcohol, 2.0 equiv ArBr, 2.0 equiv NaOtBu, 1 mol % Pd2(dba)3, 2 mol % dpe-phos, THF (0.13-0.25 M), 65 °C.
Yields represent average isolated yields for two or more experiments.
Oxidation of the alcohol substrate to 2-allylcyclopentanone was observed.
Discussion
Synthetic Scope and Limitations
The palladium-catalyzed reaction of γ-hydroxy terminal alkenes with aryl bromides allows for the synthesis of a variety of substituted tetrahydrofuran products under relatively mild reaction conditions. This transformation is effective for the preparation of 2,5-, 2,4-, and 2,3-disubstituted tetrahydrofurans, as well as spirocyclic and fused bicyclic heterocycles. A number of common functional groups are well tolerated, including nitriles, acetals, tert-butyl esters, silyl ethers, and N,N-dimethylanilines. In all reactions examined, high regioselectivites for 2-benzyltetrahydrofuran derivatives are observed.
The highest yields in these reactions are obtained with electron-neutral or electron-rich aryl bromides, and in many cases 3° alcohols afford higher yields of products than 1° and 2° alcohols. The main side products formed in these reactions are ethers that result from O-arylation or O-vinylation of the alcohol, and arenes and aldehdyes or ketones, which derive from oxidation of the alcohol substrate with concomitant reduction of the aryl bromide. These side reactions are more problematic with 1° and 2° alcohol substrates, and larger amounts of O-arylated products are observed when electron-deficient aryl bromides are employed as coupling partners.
The yields of products in these transformations are dependent on the steric properties of both the alcohol and the aryl bromide coupling partner. In general, reactions of alcohol substrates bearing large substituents at the 1- or 3-positions provide lower yields than the corresponding reactions of less hindered substrates; competing oxidation/reduction processes are more problematic with substrates that are sterically encumbered. Bulky, ortho-substituted aryl bromides are also less efficiently transformed than meta- or para-substituted derivatives. The nature of the aryl or vinyl bromide has little impact on the diastereoselectivity of these transformations when electron-rich or -neutral aryl halides are employed. In contrast, use of an electron-deficient aryl bromide led to a significant decrease in diastereoselectivity in the preparation of a 2,5,5-trisubstituted tetrahydrofuran product (Table 6, entry 4).
The electronic properties of the alcohol affect the chemical yield of these transformations, but do not appear to have a significant impact on stereocontrol. Higher yields of tetrahydrofuran products are obtained in reactions of substrates bearing weak electron-withdrawing groups (e.g. p-C6H4CN) at the 1-position than in corresponding reactions of substrates bearing weak electron-donating groups (e.g. p-C6H4OMe) at the 1-position. The more electron-rich substrates provide increased amounts of oxidation/reduction side products. However, strong C-1 electron-withdrawing groups (e.g. CO2Et) inhibit the reaction and result in the formation of Heck-olefination products.
Mechanism and Stereochemistry
A plausible catalytic cycle for the conversion of γ-hydroxyalkenes and aryl bromides to 2-benzyltetrahydrofuran derivatives is shown below (Figure 1).8 Oxidative addition of the aryl bromide to a LnPd(0) complex would provide 29, which could be transformed into palladium(aryl)(alkoxide) intermediate 30 upon reaction with the alcohol substrate and NaOtBu.17 Intermediate 30 could undergo intramolecular insertion of the alkene into the Pd-O bond28 to afford 31, which would undergo C-C bond-forming reductive elimination29 to provide the 2-benzyltetrahydrofuran with concomitant regeneration of the Pd(0) catalyst. This proposed mechanism is supported by the fact that reactions of internal alkene substrates afford products derived from syn-addition of the oxygen and the aryl group across the carbon-carbon double bond.8,30,31,32 This mechanistic proposal is also consistent with the observation of oxidation/reduction side products, which could derive from β-hydride elimination from 30, and with the formation of O-arylated side products, which would derive from C-O bond-forming reductive elimination of 30.17
Figure 1. Proposed Catalytic Cycle.

The decreased yields observed with increasing steric hindrance around the alkene or the alcohol are likely due to a decrease in the rate of alkene insertion as the size of substituents near the reacting sites increases, which leads to increased amounts of side products from competing β-hydride elimination. The electronic effects observed in reactions of 1-substituted 4-penten-1-ols (Table 5) are likely due, in part, to changes in the rate of β-hydride elimination, which should be facilitated by electron-donating groups at C1 and inhibited by electron withdrawing groups.33 It is also possible that weak electron-withdrawing groups at C-1 may increase the rate of the desired transformation by increasing the acidity of the OH proton, which would facilitate deprotonation of the alcohol substrate, or by decreasing the strength of the Pd-O bond, which may facilitate the alkene insertion.34 The fact that the Pd-catalyzed reaction of 11d with 1-bromo-4-t-butylbenzene provides only Heck-type products (Table 3, entry 4) suggests that if the alkoxide is not sufficiently nucleophilic, the Heck arylation is faster than formation of the requisite Pd-alkoxide intermediate.
The precise mechanistic details of the alkene insertion into the Pd-O bond have not yet been elucidated. However, the most likely pathways involve either direct insertion of the alkene via five-coordinate intermediate 32 (Figure 2, path A) to afford 31,35 or insertion through four coordinate intermediate 33 that is formed by an associative ligand substitution process (Figure 2, path B) that also proceeds through five-coordinate complex 32.36 Dissociative ligand substitution processes (Figure 3, Path C) are extremely rare in reactions of d-8, 16-electron palladium (II) complexes;37,38 the two known examples both involve complexes bearing large and exceptionally labile monodentate ligands. Thus, 34 is unlikely to be an intermediate along the pathway from 30 to 31.
Figure 2. Proposed Insertion Mechanism.

Figure 3. Stereochemistry of 2,5-Disubstituted Tetrahydrofuran Products.

The palladium-catalyzed conversion of 1-substituted γ-hydroxy alkenes to trans-2,5-disubstituted tetrahydrofurans (Tables 3-5) proceeds with generally excellent levels of diastereoselectivity (dr ≥ 20:1), and the size of the 1-substituent does not appear to have a significant impact on the stereoselectivity of these transformations. In contrast, although 3-substituted γ-hydroxyalkenes are efficiently transformed to trans-2,3-disubstituted tetrahydrofurans (Table 7), the diastereomeric ratio of the products is highly dependent on the size of the 3-substituent and is also affected by the presence of disubstitution at the 1-position of the substrate. To explain the stereochemical outcome of these transformations, we suggest that the stereochemistry determining step of the reaction is the insertion of the alkene into the Pd-O bond of intermediate 30 (Figure 2).32 As shown below (Figure 3), we propose that the conversion of a 1-substituted γ-hydroxyalkene (11) to a trans-2,5-disubstituted tetrahydrofuran (12) proceeds via conformer 35 in which the R-substituent is oriented in a pseudoequatorial position to minimize nonbonding interactions with the C-3 hydrogen substituent and the aryl group or phosphine ligand bound to the Pd-complex.39 The combination of these two interactions would disfavor reaction through conformer 36 in which the R-group is oriented in a pseudoaxial position regardless of whether the insertion proceeds directly through five-coordinate intermediate 32 to 31 (Figure 2, Path A) or through the associative substitution mechanism (30→32→33→31) shown in Figure 2, Path B. The alkene insertion via conformer 35 would afford intermediate 31a, which would provide the observed trans-2,5-disubstituted product 12 upon C-C bond-forming reductive elimination.
We propose that the conversion of a 3-substituted γ-hydroxyalkene (20) to a trans-2,3-disubstituted tetrahydrofuran (21) likely proceeds through a similar mechanism via conformer 37 in which the R-group is placed in a pseudoequatorial orientation (Figure 4). However, in contrast to the cyclization of a 1-substituted substrate, the only unfavorable interaction that is present in conformer 38 is the developing 1,3-diaxial interaction between the C-3 substituent and the axial C-1 hydrogen. The C-3 carbon is extended out of the coordination plane of the square planar metal complex, which leads to little or no interaction between the C-3 substituent and the metal-bound aryl group (or phosphine ligand). Thus, an axial orientation of the C-3 substituent leads to a smaller difference in energy between conformers 37 and 38 (Figure 4) than exists between conformers 35 and 36 (Figure 3), and observed diastereoselectivities are more dependent on the size of the R-group in reactions of 3-substituted alcohol substrates than in transformations of alcohols bearing a 1-substituent. Further evidence for the importance of the developing 1,3-diaxial interaction is provided by the observation that stereoselectivity is higher (dr = 8:1) in the transformation of a 3-substituted alcohol bearing a gem-dimethyl group at C-1 (Table 7, 20e) than the analogous reaction of the corresponding primary alcohol substrate (Table 7, 20a, dr = 3:1).
Figure 4. Stereochemistry of 2,3-Disubstituted Tetrahydrofuran Products.

In contrast to the reactions of 1- and 3-substituted γ-hydroxyalkenes, transformations of substrates bearing substituents at the 2-position proceed with low levels (1.5-2:1) of diastereoselectivity. The reasons for this low stereoselectivity are not entirely clear, but it is possible that the energy difference between the pseudoequatorial and the pseudoaxial orientation of the substituent is relatively low. The only potential 1,3-diaxial interaction would occur between the 2-substituent and the substituent at the internal position of the alkene, which would be much smaller than a typical 1,3-diaxial interaction due to the 120° bond angle between the groups on the sp2-hybridized carbon.
The origins of the decreased diastereoselectivity observed in the reaction of 18b with the electron-deficient aryl bromide t-butyl-4-bromobenzoate are not clear. It is possible that reactions of electron-deficient arenes proceed through a different mechanism than that outlined in Figure 1.40
Conclusion
In conclusion, the palladium-catalyzed reaction of aryl bromides with γ-hydroxyalkenes is a new and useful method for the stereoselective construction of a wide variety of substituted tetrahydrofuran derivatives from readily available precursors. This transformation allows for the installation of a tetrahydrofuran moiety on a broad range of arenes, and has a number of potential applications towards the synthesis of interesting biologically active compounds. Further studies on transformations of substrates bearing internal alkenes and applications of this methodology to the synthesis of complex molecules are currently underway.
Experimental
General Procedure for the Palladium Catalyzed Synthesis of Tetrahydrofurans
An oven or flame-dried Schlenk tube was cooled under a stream of argon or nitrogen and charged with Pd2(dba)3 (1 mol % complex, 2 mol % Pd), dpe-phos (2 mol %), NaOtBu (2.0 equiv), and the aryl bromide (2.0 equiv). The tube was purged with argon or nitrogen and the alcohol substrate (1.0 equiv), and THF (4 mL/mmol aryl bromide) were added. The reaction mixture was heated to 65°C with stirring until the alcohol substrate was consumed as judged by capillary GC analysis. The reaction mixture was cooled to rt and saturated aqueous NH4Cl (2 mL) and ethyl acetate (10 mL) were added. The layers were separated and the aqueous layer was extracted with ethyl acetate (2 × 10 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered, and concentrated in vacuo. The crude product was then purified by flash chromatography on silica gel.
2-Naphthalen-2-ylmethyltetrahydrofuran (6a)
Reaction of 43 mg (0.5 mmol) of 4-penten-1-ol with 2-bromonaphthalene (208 mg, 1.0 mmol) following the general procedure afforded 80 mg (76 %) of the title compound as a pale yellow oil. 1H NMR (500 MHz, CDCl3) δ 7.83-7.79(m, 3 H), 7.70 (s, 1 H), 7.52-7.40 (m, 3 H), 4.22-4.17 (m, 1 H), 3.96-3.92 (m, 1 H), 3.80-3.76 (m, 1 H), 3.13-3.09 (dd, J = 6.0, 13.5 Hz, 1 H), 2.96-2.92 (dd, J = 6.5, 13.5 Hz, 1H), 1.99-1.83 (m, 3 H), 1.67-1.62 (m, 1 H); 13C NMR (125 MHz, CDCl3) δ 136.8, 133.8, 132.4, 128.1, 128.09, 127.9, 127.8, 127.76, 126.1, 125.5, 80.2, 68.3, 42.3, 31.3, 25.9; IR (film) 1062 cm-1. Anal calcd for C15H16O: C, 84.87; H, 7.60. Found: C, 84.70; H, 7.51.
Supplementary Material
Acknowledgment
The authors thank the University of Michigan for financial support of this work. JPW acknowledges the Camille and Henry Dreyfus Foundation for a New Faculty Award, and Research Corporation for an Innovation Award. ARH thanks the University of Michigan Department of Chemistry for a Summer Undergraduate Research Fellowship. The authors also thank Amgen, 3M, and Eli Lilly for additional unrestricted support.
References and Notes
- 1.Hou X-L, Yang Z, Wong HNC. Prog. Heterocycl. Chem. 2002;14:139–179. [Google Scholar]
- 2.(a) Alali FQ, Liu X-X, McLaughlin JL. J. Nat. Prod. 1999;62:504–540. doi: 10.1021/np980406d. [DOI] [PubMed] [Google Scholar]; (b) Figadere B. Acc. Chem. Res. 1995;28:359–365. [Google Scholar]; (c) Zafra-Polo MC, Figadere B, Gallardo T, Tormo JR, Cortes D. Phytochemistry. 1998;48:1087–1117. [Google Scholar]
- 3.(a) Faul MM, Huff BE. Chem. Rev. 2000;100:2407–2474. doi: 10.1021/cr940210s. [DOI] [PubMed] [Google Scholar]; (b) Westley JW, editor. Polyether Antibiotics: Naturally Occurring Acid Ionophores. 1-2. Marcel Dekker; New York: 1982. [Google Scholar]
- 4.Ward RS. Nat. Prod. Rep. 1999;16:75–96. [Google Scholar]
- 5.(a) Nicotra F. Top. Curr. Chem. 1997;187:55–83. [Google Scholar]; (b) Postema MHD. Tetrahedron. 1992;48:8545–8599. [Google Scholar]
- 6.(a) Elliott MC. J. Chem. Soc., Perkin Trans. 1. 2002:2301–2323. [Google Scholar]; (b) Harmange JC, Figadere B. Tetrahedron: Asymmetry. 1993;4:1711–1754. [Google Scholar]
- 7.(a) Semmelhack MF, Bodurow C. J. Am. Chem. Soc. 1984;106:1496–1498. [Google Scholar]; (b) Semmelhack MF, Kim C, Zhang N, Bodurow C, Sanner M, Dobler W, Meier M. Pure Appl. Chem. 1990;62:2035–2040. [Google Scholar]; (c) Semmelhack MF, Epa WR. Tetrahedron Lett. 1993;34:7205–7208. [Google Scholar]; (d) Semmelhack MF, Shanmugam P. Tetrahedron Lett. 2000;41:3567–3571. [Google Scholar]; (e) Hayes PY, Kitching W. J. Am. Chem. Soc. 2002;124:9718–9719. doi: 10.1021/ja026728s. [DOI] [PubMed] [Google Scholar]; (f) Gracza T, Jäger V. Synlett. 1992:191–193. [Google Scholar]; (g) Gracza T, Hasenöhrl T, Stahl U, Jäger V. Synthesis. 1991:1108–1118. [Google Scholar]; (h) Mikami K, Shimizu M. Tetrahedron. 1996;52:7287–7296. [Google Scholar]
- 8.A portion of this work has been previously communicated. See:Wolfe JP, Rossi MA. J. Am. Chem. Soc. 2004;126:1620–1621. doi: 10.1021/ja0394838.
- 9.For related transformations of γ-aminoalkenes that afford nitrogen heterocycles see:Ney JE, Wolfe JP. Angew. Chem. Int. Ed. 2004;43:3605–3608. doi: 10.1002/anie.200460060.Lira R, Wolfe JP. J. Am. Chem. Soc. 2004;126:13906–13907. doi: 10.1021/ja0460920.
- 10.For related transformations of γ-alkylidene malonates that afford carbocycles see:Balme G, Bouyssi D, Lomberget T, Monteiro N. Synthesis. 2003:2115–2134.Balme G, Bouyssi D, Faure R, Gore J, Van Hemelryck B. Tetrahedron. 1992;48:3891–3902.
- 11.For related transformations of γ-hydroxy allenes see:Kang S-K, Baik T-G, Kulak AN. Synlett. 1999:324–326.Walkup RD, Guan L, Mosher MD, Kim SW, Kim YS. Synlett. 1993:88–90.
- 12.For related transformations of γ-hydroxy alkynes, see:Luo FT, Schreuder I, Wang RT. J. Org. Chem. 1992;57:2213–2215.
- 13.For related heteroannulations of internal alkynes with o-haloanilines, o-halophenols, and related compounds see:Larock RC, Yum EK, Doty MJ, Sham KKC. J. Org. Chem. 1995;60:3270–3271.
- 14.Small amounts of 5-aryl-4-penten-1-ol products were also observed. See:Larock RC, Leung W-Y, Stoltz-Dunn S. Tetrahedron Lett. 1989;30:6629–6632.
- 15.Bryndza HE, Tam W. Chem. Rev. 1988;88:1163–1188. [Google Scholar]
- 16.Trost has noted the formation of a tetrahydrofuran side product in the Heck arylation of an enyne bearing a secondary OH group and suggested a mechanism of intermolecular carbopalladation to account for its formation. See:Trost BM, Pfrengle W, Urabe H, Dumas J. J. Am. Chem. Soc. 1992;114:1923–1924.
- 17.Widenhoefer RA, Buchwald SL. J. Am. Chem. Soc. 1998;120:6504–6411.and references cited therein.Mann G, Hartwig JF. J. Am. Chem. Soc. 1996;118:13109–13110.
- 18.Jensen DR, Schultz MJ, Mueller JA, Sigman MS. Angew. Chem. Int. Ed. 2003;42:3810–3813. doi: 10.1002/anie.200351997.Steinhoff BA, Stahl SS. Org. Lett. 2002;4:4179–4181. doi: 10.1021/ol026988e.and references cited therein.
- 19.Ligand abbreviations: dppe = 1,2-bis(diphenylphosphino)ethane; BINAP = 2,2′-bis(diphenylphosphino)-1,1′-binaphthyl; dppf = 1,1′-bis(diphenylphosphino)ferrocene; dppb = 1,1′-bis(diphenylphosphino)butane; xantphos = 9,9-dimethyl-4,5-bis(diphenylphosphino)xanthene; dpe-phos = bis(2-diphenylphosphinophenyl)ether. All ligands are readily available from commercial sources.
- 20.Most reactions proceeded to completion in ca. 2 h as judged by GC analysis of crude reaction mixtures.
- 21.No evidence for the formation of regioisomeric products in reactions of terminal alkene substrates was obtained in analysis of crude reaction mixtures by 1H NMR and GC.
- 22.It is likely that oxidation of the alcohol accompanies reduction of the aryl bromide. However, the aldehyde side products that would be generated in this process were not detected, and are likely unstable under the reaction conditions. See reference 17.
- 23.Only trace amounts of side products derived from Heck arylation of the olefin were detected.
- 24.Side products were characterized by1H and 13C NMR spectroscopy and MS. See supporting information.
- 25.Stereochemistry of tetrahydrofuran products was established by nOe experiments or by comparison of NMR spectra to related compounds of known configuration. See Supporting Information for complete details.
- 26.Diastereomeric ratios were determined by 1H NMR and/or GC analysis of crude reaction mixtures.
- 27.(a) Allinger NL, Zalkow V. J. Org. Chem. 1960;25:701–704. [Google Scholar]; (b) Jager J, Graafland T, Schenk H, Kirby AJ, Engberts JBFN. J. Am. Chem. Soc. 1984;106:139–143. [Google Scholar]
- 28.(a) Bryndza HE. Organometallics. 1985;4:406–408. [Google Scholar]; (b) Bryndza HE, Calabrese JC, Wreford SS. Organometallics. 1984;3:1603–1604. [Google Scholar]; (c) Hayashi T, Yamasaki K, Mimura M, Uozumi Y. J. Am. Chem. Soc. 2004;126:3036–3037. doi: 10.1021/ja031946m. [DOI] [PubMed] [Google Scholar]
- 29.Milstein D, Stille JK. J. Am. Chem. Soc. 1979;101:4981–4991. [Google Scholar]
- 30.Tetrafluoroethylene undergoes selective insertion into the Pt-O bond of (dppe)Pt(Me)(OMe). See references 28a-b.
- 31.Related transformations of γ-aminoalkenes to pyrrolidines have been shown to proceed through an analogous mechanism. Experimental evidence in the pyrrolidine-forming reactions supports the idea that alkene insertion occurs into the Pd-heteroatom bond rather than the Pd-carbon bond. See reference 9a.
- 32.Although we currently favor this mechanism, an alternative mechanism involving alkene insertion into the Pd-C bond followed by C-O bond-forming reductive elimination cannot be unambiguously ruled out in all instances. This mechanistic pathway would also likely proceed via a cyclic, chelated intermediate such as 32, or 35. High 1,4 asymmetric induction would be unlikely if insertion into the Pd-C bond occurred from an acyclic, unchelated intermediate. For further details, see reference 8.
- 33.There is a significant buildup of positive charge on the C1 carbon in the transition state leading to β-elimination. Thus, substrates bearing electron-donating C1 substituents are more prone to β-elimination than substrates with electron-withdrawing substituents. See:Mueller JA, Sigman MS. J. Am. Chem. Soc. 2003;125:7005–7013. doi: 10.1021/ja034262n.
- 34.For a discussion on the effect of Pt-O bond strength on rates of CO insertion, see:Dockter DW, Fanwick PE, Kubiak CP. J. Am. Chem. Soc. 1996;118:4846–4852.
- 35.The insertion of tetrafluoroethylene into the Pt-O bond of (dppe)Pt(Me)(OMe) has been shown to occur through a five-coordinate intermediate. See reference 28a.
- 36.For a discussion of mechanistic issues in related alkene insertions into Pd-C bonds, see:Ashimori A, Bachand B, Calter MA, Govek SP, Overman LE, Poon DJ. J. Am. Chem. Soc. 1998;120:6488–6499.Oestreich M, Sempere-Culler F, Machotta AB. Angew. Chem. Int. Ed. 2005;44:149–152. doi: 10.1002/anie.200460921.Samsel EG, Norton JR. J. Am. Chem. Soc. 1984;106:5505–5512.
- 37.(a) Tobe ML. In: Comprehensive Coordination Chemistry. Wilkinson G, Gilard RD, McCleverty JA, editors. Vol. 1. Pergamon; Oxford: 1987. pp. 281–329. [Google Scholar]; (b) Cross RJ. Adv. Inorg. Chem. 1989;34:219–291. [Google Scholar]
- 38.The very rare examples of dissociative ligand substitution at d-8, 16-electron Pd(II) complexes involve the extremely bulky, monodentate, and unusually labile ligands tris(2,4,6-trifluoromethylphenyl)phosphine and P(o-tolyl)3. See:Bartolome C, Espinet P, Martin-Alvarez JM, Villafane F. Eur. J. Inorg. Chem. 2004:2326–2337.Louie J, Hartwig JF. J. Am. Chem. Soc. 1995;117:11598–11599.
- 39.The insertion of alkenes into Pd-C bonds in intramolecular Heck reactions occurs through a conformation in which the alkene π-bond and the Pd-C bond are eclipsed. The analogous insertion into the Pd-O bond via an eclipsed conformation is depicted in Figures 3-4. See:Link JT. Org. React. 2002;60:157–534.
- 40.Diastereoselectivities in related Pd(II)-catalyzed carbonylations of 1-substituted 4-penten-1-ols that proceed via a Wacker-type mechanism are typically in the range of 1-3:1. See references 7a-d.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.