

Summary
Monoterpene indole alkaloids from Catharanthus roseus (Madagascar periwinkle), such as the anticancer agents vinblastine and vincristine, have important pharmacological activities. Metabolic engineering of alkaloid biosynthesis can provide an efficient and environmentally friendly route to analogs of these synthetically challenging and pharmaceutically valuable natural products. However, the narrow substrate scope of strictosidine synthase, the enzyme at the entry point of the pathway, limits a pathway engineering approach. We demonstrate that with a new expression system and screening method it is possible to rapidly identify strictosidine synthase variants that accept tryptamine analogs not turned over by the wild-type enzyme. The variants are used in stereoselective synthesis of β-carboline analogs and assessed for biosynthetic competence within the terpene indole alkaloid pathway. These results present an opportunity to explore metabolic engineering of “unnatural” product production in the plant periwinkle.
Introduction
Monoterpene indole alkaloids (MIAs) from Catharanthus roseus (Madagascar periwinkle) are structurally complex natural products with well-established pharmacognosy [1, 2]. The MIA biosynthetic pathway produces the anti-hypertensive compound ajmalicine 1, the topoisomerase II inhibitor serpentine 2 and the anticancer agents vinblastine 3 and vincristine 4 (Figure 1) [1, 3, 4]. Interestingly, derivatives of vinblastine such as Vinflunine (4′-deoxy-20′,20′-difluoro-C′-norvincaleukoblastine) 5 show excellent anti-cancer activity in the clinic, indicating that small changes to the alkaloid scaffold can improve or modulate bioactivity (Figure 1) [5–7]. However, the current supply of MIA analogs is primarily dependent on semi-synthesis from isolated pathway intermediates that are produced in low yields.
Figure 1.

Structures of selected medicinal MIAs produced or derived from Madagascar periwinkle (C. roseus). Ajmalicine 1 acts as an antihypertensive agent, serpentine 2 is a topoisomerase II inhibitor, vinblastine 3 and vincristine 4 are anticancer agents and Vinflunine 5 is a modified vinblastine analog with promising bioactivity.
Metabolic engineering of a biosynthetic pathway can provide an efficient and environmentally friendly route to novel natural products or natural product derivatives. We previously demonstrated that the MIA biosynthetic pathway in C. roseus root cultures and seedlings can utilize non-natural substrates to synthesize an array of unnatural MIA analogs [8]. However, the low substrate promiscuity of the enzyme at the entry point in the pathway, strictosidine synthase (STS), limits the scope of precursor directed biosynthesis efforts. STS catalyzes the stereoselective Pictet-Spengler reaction between tryptamine 6a and secologanin 7 to yield the tetrahydro-β-carboline (3S)-strictosidine 8a as a single diastereomer (Figure 2) [9]. Deglucosylation of 8a by the second enzyme of the pathway, strictosidine glucosidase (SG), produces the aglucone 9 that is in turn enzymatically converted into the structurally diverse group of MIAs produced in C. roseus (Figure 2) [1, 2].
Figure 2.

STS-catalyzed formation of 8a via a Pictet-Spengler reaction between 6a and 7. The reaction proceeds via an iminium intermediate, which undergoes electrophilic aromatic substitution and cyclization to give 8a. Deglucosylation by a dedicated β-glucosidase forms an aglucone 9 that rearranges into dehydrogeissochizine 10 and cathenamine 11. The deglucosylated products are then enzymatically converted into the MIA structures observed in periwinkle. However, aglucone 9 can be intercepted by amine nucleophiles to form colored adducts.
Reengineering STS substrate specificity is key for biosynthetic production of many MIA analogs. We recently reported that a secologanin analog already accepted by wild-type STS is selectively turned over by an STS mutant (Asp177Ala) [10]. The observed switch in selectivity could be rationalized based on the crystal structure of STS from Rauvolfia serpentina in complex with secologanin (PDB ID: 2FPC) [11]. The efficiency of rational redesign of enzyme active sites can often be improved by using saturation mutagenesis protocols. However, the ineffectiveness of current methods to assess STS activity limits this “random” rational approach. Therefore, to rapidly obtain STS mutants with expanded tryptamine specificity, we developed a streamlined expression system and colorimetric assay for the Pictet-Spengler reaction. Herein, we describe the discovery of two strictosidine synthase mutants using this discovery strategy that have broadened substrate specificity. We highlight the potential applications of these mutants in biosynthesis of unnatural alkaloid analogs.
Results and Discussion
Development of a colorimetric assay for STS activity
The time consuming high-performance liquid chromatography (HPLC) assay used to measure catalytic activity limits rapid reengineering of STS substrate specificity. There are no reports of a high throughput assay for Pictet-Spengler activity. However, previous literature reports noted that a yellow pigment forms after deglycosylation of 8a by SG [12, 13]. Inspired by these results, we developed an assay for visual detection of STS activity that could be adapted to medium- or high-throughput screening.
In vitro deglycosylation by SG yields an equilibrium mixture of isomers of 4,21-dehydrogeissoschizine 10 and cathenamine 11 (Figure 2) [14]. Previous literature suggested that the yellow chromophore was a low molecular weight degradation product of 10 (dihydroflavopereirine) [15]. However, liquid chromatography coupled to high-resolution mass spectrometry (LC-HRMS) of yellow-colored assay mixtures produced by us indicated formation of compounds with m/z 493, 491 and 489 from the initial deglycosylation products 10 and 11 (m/z 351). The compound corresponding to m/z 493 disappeared after overnight incubation to form compounds with m/z 491 and 489 exclusively.
It has been reported that deglycosylated 8a can form amine adducts in the presence of excess ammonium sulfate, and that these adducts can further rearrange to form a dihydropyridine ring, which can easily air-oxidize to an aromatic pyridine ring [16]. HRMS suggested that the compounds in our assay corresponding to m/z 493, 491 and 489 have molecular formulae consistent with tryptamine adducts of 10 in different oxidation states (Supplemental Data). Purification of the compounds corresponding to m/z 491 and 489 and analysis by 1D- and 2D-NMR suggested that the tryptamine adducts formed in the assay undergo rearrangement and air oxidation, similar to what is reported in reference 16 (Figure 2) (Supplemental Data). The UV-VIS spectrum of the compound corresponding to m/z 489 showed absorption maxima at 314 nm and 427 nm; m/z 491 had one unique absorption maximum at 470 nm (data not shown).
If this assay is to be used for STS substrate reengineering, SG must accept analogs of 8a and the resulting deglycosylated adducts must form the pigment. To determine the scope of the assay, we synthesized a variety of strictosidine analogs (maleic acid buffer, 10 mM, pH 2) to obtain a diastereomeric mixture of strictosidine analogs that were then subjected to the assay conditions. Pigment formation in the presence of excess tryptamine 8a or tryptamine analog yielded strictosidine 8a or strictosidine analogs having substituents on the indole ring or on the side-chain (data not shown). Careful analysis showed that the purified strictosidine analog 8m (m/z 561, 531+30) formed compounds corresponding to the expected aglucone analogs 9m and 10m after deglycosylation by SG (m/z 381, 351+30). Only after addition of excess tryptamine analog 6m to the deglycosylated products did the color form and the expected masses of the analogous tryptamine adducts appear (m/z 553, 551, and 549, M+60). Control assays lacking tryptamine or a tryptamine analog did not form the putative adducts and were not colored (Supplemental Data). Because of the slow rate of chromophore formation relative to strictosidine formation, this assay cannot be used as a quantitative coupled assay of STS activity. Nevertheless, this assay appeared to be a robust method for qualitative detection of STS activity.
Expression system for extra-cellular STS activity
To facilitate screening for STS activity, we constructed a plasmid that allows export of STS to the media upon expression in S. cerevisiae. Although the STS gene from C. roseus contains a putative native signal peptide (residues 1–26), expression in yeast did not result in significant protein export in S. cerevisiae. Replacement of the nucleotides encoding the first 26 residues with an N-terminal yeast mating-factor alpha signal sequence [17] efficiently directed the export of STS to the culture media (Figure 3A, Lane 2). A C-terminal FLAG-peptide was also introduced to allow purification by affinity chromatography (Figure 3A, Lanes 3–4) [18].
Figure 3.
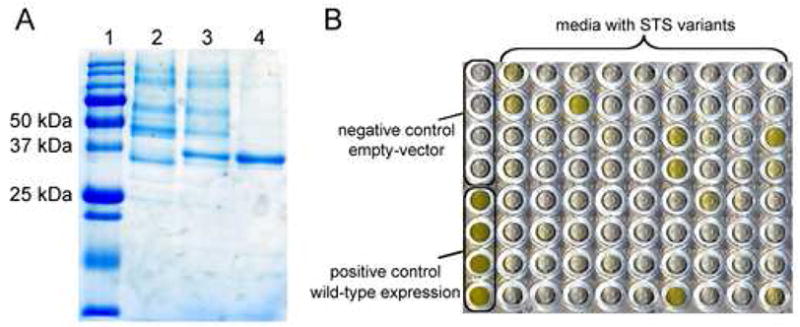
Visualization of expression and purification of C. roseus STS and verification of mutagenesis and screening methods. (A) Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) of STS samples. STS has a calculated molecular weight of ~37 kDa. Lane 1: Relative mass marker. Lane 2: STS in media without FLAG-tag. Lane 3: STS in media with FLAG-tag. Lane 4: Anti-FLAG antibody purified STS from supernatant. FLAG-tag: Asp-Tyr-Lys-Asp4-Lys. (B) Representative screen of STS activity after randomization of a residue distant from the active site (Tyr167). All wells contain the same assay mixture (2 mM 6a, 1 mM 7, SG and 25 mM sodium phosphate buffer at pH 7.0). The first column contains no-insert and wild-type-insert controls (rows 1–4 and 5–8, respectively). Columns 2–10 (all rows) contain media from mutagenesis libraries.
Characterization of purified wild-type STS expressed in yeast by a quantitative HPLC assay afforded kinetic constants (Vmax = 21 U mg−1, KM = 1.4 μM; turnover number assuming all enzyme is active: kcat = 78 min−1) (Table 1). These values are consistent with constants reported for STS purified from C. roseus (average of several isoforms: Vmax = 14 U mg−1, KM = 9 μM) [19], and from heterologously expressed STS from the medicinal plant Rauvolfia serpentina (kcat = 78 min−1, KM = 6 μM, 88% sequence similarity to C. roseus STS) [11]. Mutagenesis studies of R. serpentina STS suggested that Glu309 is important for catalysis (Glu309 corresponds to Glu315 in the C. roseus protein sequence, Figure 4A) [11]. The >1000-fold reduced turnover number of the analogous mutant of C. roseus STS (Glu315Ala, Vmax = 0.012 U mg−1, KM = 0.44 mM) confirms this conclusion. When subjected to the colorimetric assay conditions, Glu315Ala produced a faint yellow pigment (data not shown), suggesting that this screen is sufficiently sensitive to detect weakly active mutants.
Table 1.
Kinetic constants for STS variants varying tryptamine 6a or analog of 6a.a
| Variant, substrate | Vmax [U mg−1]b | KM [mM] | Vmax/KM [U mg−1 mM−1]b |
|---|---|---|---|
| Wild-type, 6a | 21 | 0.0014 | 15,000 |
| Glu315Ala, 6a | 0.012 | 0.44 | 0.027 |
| Val214Met, 6a | 3.1 | 0.42 | 7.5 |
| Val214Met, 6lc | 0.051 | 0.37 | 0.14 |
| Phe232Leu, 6a | 0.24 | 0.62 | 0.39 |
| Phe232Leu, 6mc | 0.23 | 0.94 | 0.24 |
Kinetic assays were performed in 100 mM sodium phosphate buffer at pH 7.0 using an internal standard (naphthyl acetic acid) and samples were analyzed by HPLC for formation of product. The error-limit for kinetic assays is estimated at 20%. V and V/K parameters are obtained by non-linear regression analysis using the Northrop equation and KM is obtained from the Michaelis-Menten equation;
1 U = formation of 1 μmol product (8a or analog) per minute;
Compounds 6l and 6m are not accepted by wild-type STS or Glu315Ala STS.
Figure 4.

Tryptamine binding site and tryptamine substrate scope of STS. (A) Tryptamine binding site of STS from R. serpentina (PDB ID: 2FPB, residues are numbered according to the C. roseus peptide sequence). Side-chains interacting with tryptamine are in stick-representation. The protein main-chain is represented in cartoon form. (B) Tryptamine analogs with indole ring substitutions that are accepted by wild-type STS [21].
Validation of mutagenesis and screening strategy
The crystal structure of R. serpentina STS in complex with tryptamine (PDB ID: 2FPB) was used to evaluate residues involved in substrate binding and catalysis [11]. The relatively high B-factor of 6a in the crystal structure (3-fold higher than the remainder of the protein) suggests that 6a may adopt a relatively flexible orientation in the active site [11]. Moreover, the known substrate specificity of STS cannot always be rationalized from the structural data; for example, it is not clear why 5-hydroxytryptamine 6f is weakly turned over while 5-methyltryptamine 6l is not. To expand our rational redesign approach, we used saturation mutagenesis in combination with our newly developed screen to probe all twenty naturally occurring amino acids at selected residues in the tryptamine binding pocket.
Codons were randomized using primers containing NNK/MNN degenerate codons (32 sequence possibilities). To validate the mutagenesis strategy, expression system and screening method, a saturation mutagenesis library of a tyrosine residue distant from the active site (Tyr167Ala greatly reduces STS catalytic activity, unpublished data) was constructed. The mutated gene and the linearized expression vector were transformed into the yeast expression host (S. cerevisiae BJ5465) and homologous recombination resulted in the production of the desired STS variant library. Colonies were inoculated (88 per mutated site, an oversampling that gives 94% certainty that the library is complete [20]) into expression media in deep-well 96-well plates. The culture media was then transferred to 96-well microtiter plates containing the assay components 6a, 7 and SG. A positive control from a yeast strain expressing wild-type and a negative (empty-vector) control were included in the screen (Figure 3B, column 1). Five clones that led to the formation of a strong yellow pigment were sequenced and each contained the wild-type gene. Cultures lacking pigment yielded genes with a variety of mutations at tyrosine 167. This test assay with the natural substrate 6a indicated that the screen was robust enough to use to search for STS variants with altered substrate specificity.
Selection of substrates for screening and residues for mutagenesis
With the exception of Glu315, which is crucial for the catalysis of the Pictet-Spengler reaction, the tryptamine binding site consists primarily of hydrophobic and aromatic residues, including Trp153, Tyr155, Val171, Val182, Val214 and Phe232 (Figure 4A). Therefore, the pocket may simply serve to foster tryptamine binding to the enzyme in a productive orientation that promotes iminium formation with the aldehyde substrate and cyclization to form the tetrahydro-β-carboline. We hypothesized that mutagenesis of residues in the tryptamine binding pocket will allow productive binding and catalysis of substrates not accepted by the wild-type enzyme.
We selected substrates for screening based on previous work on STS substrate specificity (Figure 4B). Tryptamine analogs with a methyl substituent at the 4- or 7-position (6c, 6j) are accepted by the wild-type enzyme (11- and 27-fold increased KM and 5- and 3-fold decreased kcat with 6c and 6j, respectively) [21]. Substitutions at the 5- and 6-positions are less tolerated: although 5-hydroxytryptamine 6f and 6-methoxytryptamine 6h are weakly turned over by the wild-type enzyme, 5-methyltryptamine 6l is not a substrate. We selected 6l as a model compound to assay mutants with increased tolerance to 5 indole substitutions. The 2′-position of the tryptamine side-chain is also highly sensitive towards substitution. A methyl group (racemic starting material) is only weakly tolerated at the 2′ position [21]. We determined that (2′R)-tryptophanol 6m, (2′S)-tryptophanol 6p, (2′R)-tryptophan 6q, (2′S)-tryptophan 6r and (2′R)-tryptophan methyl ester 6s are not accepted by the wild type enzyme. Compounds 6m and 6p were selected for screening.
The intriguing limitation at the 5-position may be due to the proximity of Val171 or Val214 (closest distances: 4.4 Å and 3.2 Å, respectively). The 2′-position is close to the reactive amine and large substituents at this position may interfere with the catalytic machinery. In addition, Phe232 could prevent reaction with tryptamine analogs that have substitutions at the 2′-proR hydrogen atom (closest distance: 3.8 Å), while Glu315 could prevent the proS-substitutions (3.3 Å).
We anticipated that saturation mutagenesis of residues Val171 or Val214 could help turnover of 6l and mutagenesis of Phe232 or Glu315 would allow reaction with 6m and 6p (Figure 5). These four residues formed the basis for our first efforts to alter the tryptamine substrate specificity for STS.
Figure 5.

Expanded substrate scope: tryptamine analogs assayed with STS variants used in this study.
Discovery of active mutants
The Val171, Val214, Phe232 or Glu315 libraries were expressed in 96-well plates. After protein expression, the media containing the enzyme was transferred to microtiter plates for screening using an assay solution containing the appropriate analogs of 6a, as well as 7 and SG. Color formation was detected visually after 24 h incubation (Supplemental Data). The Val171 and Glu315 libraries did not produce positive hits toward 6l, 6m or 6p (Figure 5). However, several members of the Val214 library showed activity towards 6l as evidenced by the colorimetric assay. Interestingly, sequencing of three active mutants revealed a Val214Met mutation. The methionine substitution eliminates the side-chain β-branching that could hinder reaction with 6l in wild-type STS. Since no mutants containing smaller amino acids at Val214 were isolated from the screen, we speculate that a longer side-chain is key for orienting the tryptamine analog optimally for catalysis. (A different R. serpentina STS variant that accepts 6l was also found by J. Stöckigt and co-workers, personal communication.) Further evaluation of this mutant by LC-MS and HPLC showed that the Val214Met mutant also accepts 5-chlorotryptamine 6n and 5-bromotryptamine 6o. Halogenated precursors are useful because the distinctive isotopic signature allows facile detection of metabolites. Furthermore, aryl chlorides and bromides can be used to derivatize pathway products via aromatic cross-coupling reactions [22]. There is also some evidence that bromination at this position in synthetic vinblastine analogs has resulted in compounds displaying promising anti-tumor activity [23].
The colorimetric assay also indicated that 6m was accepted by members of the Phe232 library (Figure 5). Sequencing of six active clones showed a substitution at residue 232 to leucine (four of the six sequenced cultures) or methionine (two of the six sequenced cultures). The 2′-proR hydrogen atom projects towards Phe232 (closest distance: 3.4 Å) and mutagenesis of Phe232 to the smaller leucine or methionine residue may help accommodate the hydroxymethylene substituent. Initial experiments using HPLC indicated that Phe232Leu was slightly more active under the assay conditions, so the Phe232Leu mutant was chosen for further characterization. Notably, 6p, 6r and other tryptamine analogs with a substitution at the 2′-proS hydrogen are directed towards the catalytic Glu315 residue. Continued redesign of mutants that accept these substituents would likely be unwarranted as Glu315 is required for catalysis. Turnover of L-tryptophan 6r by STS could impact the cellular supply of tryptophan and was likely subject to strong negative selection during the evolution of STS.
Kinetic analysis of STS mutants
Larger-scale (500 mL) expression of the enzyme mutants in yeast followed by purification of the concentrated media using anti-FLAG antibody resin afforded Val214Met and Phe232Leu in apparent homogeneity as indicated by SDS-PAGE (data not shown). Steady-state kinetic analysis of Val214Met and Phe232Leu was performed with both the natural substrate 6a and the substrate analogs 6l and 6m (Table 1). The mutants each have reduced catalytic capacity compared to the wild-type enzyme (Vmax,wt/Vmax,mutant) using 6a (7-fold and 90-fold, respectively). In addition to the 90-fold reduced Vmax, Phe232Leu shows a >400-fold increased Michaelis constant (Table 1). This may be attributed to loss of the aromatic residue, since phenylalanine may help orient the substrate correctly for catalysis or stabilize high-energy intermediates in the reaction pathway. A comparison of the catalytic efficiency (Vmax/KM) of the mutants using 6a or the respective analogs of 6a suggested that both mutants are selective for tryptamine in the presence of the analog (50-fold and 1.5-fold for Val214Met and Phe232Leu, respectively, Table 1). Lowering the amount of 6a in cell culture by inhibiting tryptophan decarboxylase - the enzyme directly responsible for tryptamine biosynthesis - may be necessary to suppress formation of native MIA analogs.
Preparative enzymatic synthesis of 8l-n and characterization of products
We used the Val214Met and Phe232Leu mutants in the stereoselective preparative (mmol-scale) synthesis of (3S)-strictosidine analogs 8l-n. Although several chemical strategies for Pictet-Spengler reactions exist, formation of 8a in acidic aqueous media gave a low-yielding diastereomeric mixture of products (3R and 3S), as well as numerous side-products (data not shown). Expression of the STS mutants in E. coli afforded crude cell extracts containing either Val214Met or Phe232Leu. Despite the reduced Vmax of the mutants relative to wild type enzyme (Table 1), the extracts were conveniently used to synthesize 8l-n on a mmol-scale. Gratifyingly, side-products were minimal and the expected (3S)-diasteromer was formed exclusively (d.r. >99.5%). Preparative HPLC purification was used to separate the products from the unreacted starting materials. NMR and high-resolution mass spectrometry (HRMS) validated the structures of the predicted strictosidine analogs (Experimental Procedures).
Biosynthetic competence of strictosidine analogs
We envision that Val214Met and Phe232Leu will be useful for metabolic pathway engineering of alkaloid biosynthesis. Previous reports demonstrate that MIA pathways accept and process tryptamine and secologanin analogs to form unnatural alkaloids [8, 10]. However, it is important to validate whether the strictosidine analogs that are produced by STS mutants are in fact accepted by the remainder of the pathway.
Decades of research on C. roseus plants and differentiated cell cultures have yielded a wealth of structural and biochemical information of the alkaloid profile, thereby facilitating analysis of the complex alkaloid production in C. roseus [1]. LC-MS analysis of hairy root cultures supplemented with the natural substrate tryptamine 6a showed, as expected, formation of the heteroyohimbine alkaloids ajmalicine 1 (m/z 353), serpentine 2 (m/z 349), and yohimbine 12(m/z 355) as well as the more complex alkaloid akuammacine 13 (m/z 323) [8].
To assess the metabolic fate of tryptamine analogs that are not accepted by the wild-type enzyme, the corresponding strictosidine analogs have to be preformed and then fed to the hairy root cell cultures. Reports in the literature indicated that the relatively large strictosidine biosynthetic intermediate could be taken up by plant cell culture [24]. C. roseus hairy root cultures were supplemented with preformed 8m (product of Phe232Leu) and 8o (product of Val214Met) (Figure 5) at a final concentration of 0.5 mM, and the alkaloid content was extracted from the media and the roots using established protocols [25]. Analysis of the media and the extracted cell cultures by LC-MS suggested the presence of compounds with masses corresponding to MIA analogs (with M+30 for alkaloids derived from 8m and M+78/80 for alkaloids derived from 8o, Figure 6 and Supplemental Data). These compounds were not observed in control cultures that contained only the natural substrate tryptamine 6a. Compounds derived from 8o could be identified by the characteristic isotopic distribution expected for brominated molecules (79Br/81Br), providing further support that these new metabolites are derived from the halogenated strictosidine precursor (Figure 6). Previous directed biosynthesis experiments indicated that the MIA pathway tolerates substituents on the indole ring [8], so it was not surprising to see evidence for incorporation of 8o into products with molecular masses corresponding to analogs of the known MIAs 1, 2 (Figure 6A), 12 and 13 (Supplemental Data).
Figure 6.
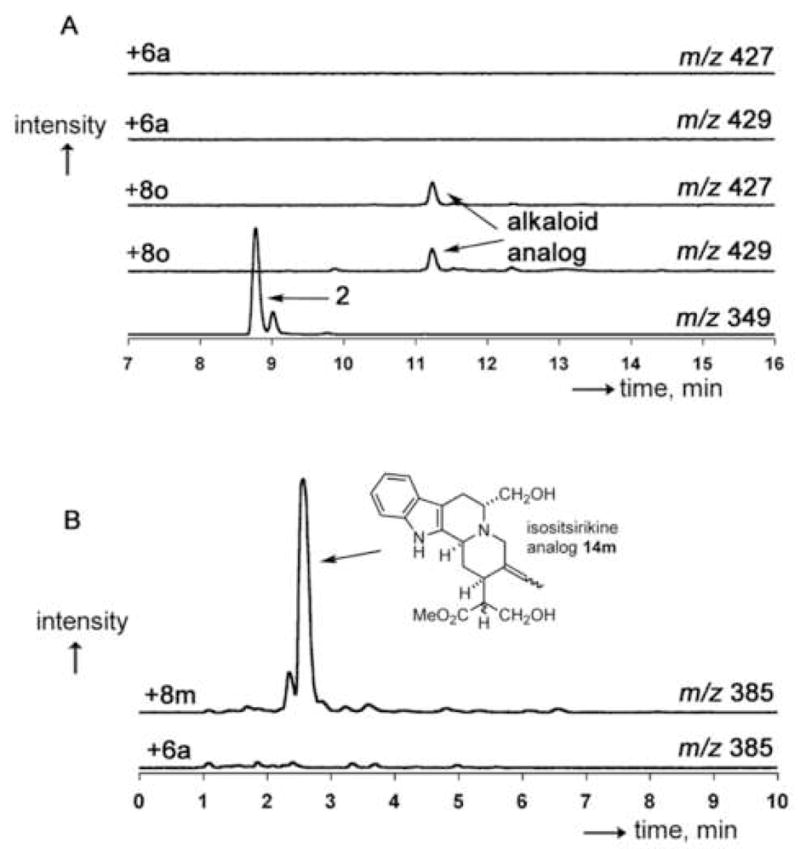
LC-traces of selected alkaloid analogs formed by feeding strictosidine analogs to two-week-old C. roseus hairy root cultures. (A) Formation of putative serpentine or serpentine isomer analog upon feeding of 8o to periwinkle. The two top traces show that no compounds corresponding to m/z 427 and 429 exist in the culture supplemented with tryptamine 6a. The bottom trace shows the elution of an authentic standard of serpentine 2. The remaining two traces show the formation of an alkaloid analog displaying the expected isotopic pattern that could be a brominated MIA analog. (B) Formation of isositsirikine 14m in a feeding study of 8m. The top trace shows the expected mass of the alkaloid analog; the bottom trace shows that no naturally occuring alkaloid of m/z 385 co-elutes with 14m.
For this study, we isolated and characterized an alkaloid analog derived from 8m. A metabolite derived from 8m was chosen because while tryptamine analogs with substituents on the indole ring have been previously shown by rigorous structural characterization to be widely incorporated into the MIA pathway, the effect of a tryptamine side-chain substituent has not yet been explored. Furthermore, while alkaloids derived from 8o contain a unique isotopic signature from the bromine atom, there is no distinctive isotopic signal for alkaloids derived from 8m. The major product derived from 8m was therefore purified and characterized by NMR spectroscopy to validate the formation of a terpene indole alkaloid analog.
Initial analysis of cultures supplemented with 8m by LC-MS suggested the formation of an analog of a yohimbine-type alkaloid (m/z 385, 353+30, 14m) and an analog of ajmalicine 1 or an isomer of 1 (m/z 383, 353+30 1m, Supplemental Data). Additionally, an unknown analog with m/z 426 (396+30) 15m was formed (Supplemental Data). The absence of m/z 379 in the media and extracts suggests that the serpentine analog 2m cannot form from 8m. This could be due to the absence of a (5-pro-R)-hydrogen atom in the substrate, or the intracellular localization of the biosynthetic intermediates may be altered. A compound corresponding to the analog of the more complex alkaloid 13 was also not observed, suggesting that the (unknown) enzyme forming akuammacine 13 does not accept 8m as a precursor. Isolation, cloning and reengineering of these enzymes will be key for studies relating to the biosynthesis of these analogs. Interestingly, while the cell culture media did not appear to contain any naturally occurring MIAs, analogs 14m and 15m were present in the media. Although relatively little is known about export mechanisms in plant cell culture, it is intriguing to speculate that the plant cell cultures exported these products into the media [26].
Compounds 14m and 15m, found in both the media and in root extracts, appeared to be the major unnatural products. Preparative HPLC purification afforded 14m (2 mg, 3% yield) and 15m (1 mg, 1%) from 500 mL of plant cell culture. Compound 15m proved intractable to structural assignment as it was prone to decomposition and consisted of several isomers that could not be separated. The observations of NMR signals for indole aromatic protons strongly suggested assignment as an indole alkaloid (δ 7.41 (d, 1H, J 7.7, H-9), 7.34 (d, 1H, J 8.2, H-12), 7.14 (t, 1H, J 7.6, H-10), 7.04 (t, 1H, J 7.5, H-11)), but no natural alkaloid corresponding to either the proposed parent ion m/z of 396 or the observed m/z 426 has been reported from C. roseus. However, NMR analysis revealed that 14m is an analog of the MIA isositsirikine (Figure 6) [27, 28]. This compound was shown to have an unsubstituted indole (7.5-7.1 ppm, 4H), an intact hydroxymethylene group (4.0 and 3.8 ppm, 2H; compare to 4.0 and 3.8 ppm for 8m) and the methyl ester (s, 3.8 ppm, 3H). Also, the spectrum revealed an ethylidene group containing a trisubstituted alkene, since one olefinic hydrogen atom appears as a quartet (6.0 ppm, J = 6.8 Hz) coupled to three hydrogen atoms that appear as a doublet (1.9 ppm, J = 6.8 Hz). There are no reports on isolation of isositsirikine from either roots or hairy root cultures of C. roseus, suggesting that unusual biosynthetic precursors may form unexpected products [1].
Large scale synthesis of the strictosidine analogs (~600 Da) with the expensive secologanin substrate 7 limits the scope and rigor of these feeding studies. We anticipate that C. roseus cell lines expressing these STS mutants (using previously reported transformation methods [29]) will have the capacity to synthesize the strictosidine analogs from the more readily available tryptamine starting materials. More detailed analysis of the resulting alkaloid profiles by NMR spectroscopy would therefore be greatly facilitated. Construction of these transgenic cell lines is currently underway.
Significance
Modulating the substrate specificity of strictosidine synthase (STS) is a critical step towards indole alkaloid pathway engineering to yield non-natural alkaloids. We developed a streamlined method to expand the range of substrate analogs accepted by STS. The expression system and assay described here was used to identify two STS mutants that yield strictosidine analogs that cannot be produced by the wild-type enzyme. We anticipate that this assay can be applied to identify additional mutants with alternate substrate specificity or optimized selectivity. The assay may also facilitate the identification of functional homologs of STS and the introduction of STS activity in structurally related enzymes. The Val214Met mutant allows formation of halogenated indole alkaloids, which may be derivatized further by cross-coupling strategies. Inspection of the isotopic pattern of crude cell lysates suggests that a brominated strictosidine analog can be incorporated into several alkaloid metabolites of C. roseus. STS mutant Phe232Leu allows formation of a strictosidine analog with a modification at an aliphatic carbon. Feeding studies of this strictosidine analog showed that minor modifications to the side-chain of tryptamine can cause major perturbations of the alkaloid profile of C. roseus. These results represent a starting-point for investigations into how the STS variants can interface with the later steps of the biosynthetic machinery. Future research will seek to understand how these enzyme mutants function in the context of transgenic Madagascar periwinkle cell lines.
Experimental Procedures
General methods and analytical techniques
Secologanin was obtained by methanol-extraction of Lonicera tatarica as described previously [21]. Tryptamine and tryptamine analogs were purchased in highest available purity and used without further purification. Protein was obtained as described below and the protein concentration was determined by measuring the absorbance at 280 nm (ε = 50,300 M−1 cm−1, www.expasy.ch/tools/protparam.html). NMR spectra were acquired on Varian Inova 500 MHz or Bruker 600 MHz spectrometers.
LC-MS analysis was performed on a Waters LC-MS system. Compounds were separated by an Acquity Ultra-Performance LC (UPLC) BEH C18-column (1.7 μm, 2.1 × 100 mm) equipped in tandem with an MS technologies Micromass LCT Premier with an electrospray ionization (ESI) source and time-of-flight (TOF) detector. The chromatography solvent system consisted of acetonitrile/water(0.1% formic acid). The capillary and sample cone voltages were 2000 V and 30 V, respectively. The desolvation and source temperature were 350 °C and 100 °C, respectively. The cone and desolvation gas flow-rates were 20 L/h and 700 L/h, respectively. High-resolution mass spectra (HRMS) were obtained in accurate mass mode (W-mode) with reserpine (m/z 609.2812, as 1 nM solution, 40 μL/min) as reference. The HPLC-based assay was carried out on a Beckman-Coulter System Gold system equipped with a 125 solvent module, 125 autosampler and a 168 detector. Data was processed (integrated) using 32Karat (v 7). A solvent system composed of acetonitrile/water (0.1% TFA) was used with a reverse-phase column (HiBar RT 250-4, RP-select B, 5 μm).
Construction of expression vectors
The wild-type strictosidine synthase synthetic gene (with codon usage optimized for expression in yeast) was amplified with PCR primers that introduce sites for Xho I and Hind III (underlined) and the FLAG-tag epitope (italics): 5′-CCG AAG CTT TCA CCA ATC TTG AAG AAG-3′, 5′-C TCC TAC GTT TCC TCC GGA TCT GAC TAC AAG GAT GAC GAC AAC AAG TAG CTC GAG CGG. The primers were designed to eliminate the first 26 residues corresponding to a putative native N-terminal signal sequence. The PCR fragment was subcloned into the pGEM-T Easy Vector (Promega, Madison, WI) and then excised and ligated into pGAL-MF (Dualsystems Biotech AG) to obtain pSTSMF-FLAG. The hexa histidine-tagged protein expressed in E. coli was obtained by PCR amplification of the STS gene in pSTSMF-FLAG using primers that introduce sites for Nco I and Xho I: 5′-CAT GCC ATG GGC TCA CCA ATC TTG AAG AAG ATC-3′, 5′-CCG CTC GAG GGA GGA AAC GTA GGA GTT TCC C-3′ and was then ligated into pET-28a(+) to obtain pSTSMF-HIS.
Library generation and expression
Primers were designed to overlap with the plasmid sequence to allow homologous recombination in vivo. Degenerate codons (NNK/MNN) reduced the library size by two-fold as compared to complete randomization (NNN/NNN). Two PCR reactions using the pSTSMF-FLAG as template afforded gene halves ending at the mutagenized codon and the plasmid and overlapping regions (>20 bp) with each other and with the plasmid. Digestion of the parental DNA and agarose gel elecrophoresis purification was followed by a splicing-overlap extension PCR reaction to assemble the mutated gene. Homologous recombination of the mutated gene and digested pGAL-MF vector created the saturation mutagenesis libraries. See Supplemental Data for more information.
Yeast colonies were inoculated in 300 μL SCMM-U (S. cerevisiae minimal media, composition per liter: 6.7 g yeast extract without amino acids and 1.92 g yeast supplemental media without uracil) supplemented with 2% glucose and incubated overnight at 30 °C in 96-well square-bottom deep-well plates (2 mL volume capacity per well) with shaking at 225 rpm. An aliquot of the overnight culture (100 μL) was then used to inoculate 900 μL of SCMM-U supplemented with 0.5% glucose and 2% galactose and incubated for an additional 72–96 h at 20 °C. Each well contained one glass bead (5 mm) to facilitate aeration and all plates were protected from contamination during the incubation by sterile breathable sealing film. The plates were centrifuged (10 °C, 3000g, 10 min) and the supernatant was used directly for screening. We verified that the supernatant could be stored with cells at 4 °C for at least four weeks without noticeable loss of activity. 96-well plates can be recultured in SCMM-U media or supplemented with 25% glycerol and stored at −80 °C for future use.
Kinetic analysis of STS variants
Reaction conditions were modified to obtain less than 15% conversion for all data points used in steady-state kinetic analysis. All enzymatic reactions (0.100 mL) contained secologanin (4.0 mM) in sodium phosphate buffer (100 mM, pH 7.0) with internal standard (0.075 mM naphthyl acetic acid) and one of several tryptamine or tryptamine analog concentrations (always added as a 10-fold dilution from stock solution). Tryptamine or tryptamine analog concentrations were at least two-fold above and below the estimated KM value. The HPLC detection-limit did not allow substrate concentrations lower than 1 μM. Formation of strictosidine was monitored at 280 nm using the experimentally determined extinction coefficient of 5,140 μM−1. Wild-type reactions were quenched with two equivalents of sodium hydroxide due to the fast reaction between tryptamine and secologanin. This quench resulted in the quantitative formation of strictosamide (C26H31N2O8 calculated: 499.2091, found: 499.2080) from strictosidine and the extinction coefficient of the strictosamide product was experimentally determined to be 12,000 μM−1. Plotted experimental data were fitted to the Michaelis-Menten and Northrop equations by non-linear regression analysis using Origin (v 7.0552) (OriginLab Corp., Northampton, MA) to directly afford Vmax, Vmax/KM and KM. All kinetic assays were repeated three times to ensure reproducibility. Representative raw data using the Val214Met mutant and 5-methyltryptamine are shown in the Supplemental Data.
Feeding study and alkaloid isolation
Hairy root cell cultures of C. roseus were inoculated in half-strength Gamborg media and grown for three weeks. Enzymatic synthesis and purification afforded 8m (ESI-MS C28H36N2O10: calculated m/z 561.2448 [M+H]+, found m/z 561.2448 [M+H]+) and chemical synthesis and purification afforded 8o (ESI-MS C27H34N2O979Br: calculated m/z 609.1448 [M+H]+, found m/z 609.1456 [M+H]+; ESI-MS C27H34N2O981Br: calculated m/z 611.1427 [M+H]+, found m/z 611.1453 [M+H]+). Each strictosidine analog was dissolved in water to a final concentration of 0.5 mM, the solution was filter-sterilized and then added to the hairy root culture. After one week, the media was separated from the hairy roots and the roots washed with water three times. The media and roots were extracted according to standard procedures (Supplemental Data) [25, 28]. The extracted alkaloids were analyzed by LC-MS and the two major species were isolated using preparative HPLC and analyzed by NMR.
NMR characterization of enzymatially generated strictosidine analogs
(5R)-hydroxymethylstrictosidine 8m
1H NMR (methanol-d4, 500 MHz) δ7.80 (s, 1H, H-17), 7.46 (td, 1H, 3J10 7.8, 4J11 1.0, H-9), 7.33 (td, 1H, 3J11 8.2, 4J10 0.84, H-12), 7.15 (ddd, 1H, 3J10 7.1, 3J12 8.2, 4J9 1.1, H-11), 7.05 (ddd, 1H, 3J11 7.1, 3J9 8.2, 4J12 0.92, H-10), 5.84 (ddd, 1H 3J18trans 17.4, 3J18cis 10.6, 3J20 7.6, H-19), 5.80 (d, 1H, 3J20 8.8, H-21), 5.37 (ddd, 1H, 3J19 17.4, 2J18cis 1.3, 4J20 1.0, H-18trans), 5.28 (ddd, 1H, 3J19 10.6, 2J18trans 1.3, 4J20 1.0, H-18cis), 4.78 (d, 1H, 3J2′ 7.9, GlcH-1′), 4.67 (dd, 1H, 3J14proR 10.9, 3J14proS 4.0, H-3), 4.03 (dd, 1H, 3J23 10.7, 3J5a 3.4, H-23), 3.94 (dd, 1H, 2J6′b 11.8, 3J5′ 2.1, GlcH-6′a), 3.84-3.75 (m, 2H, H-5a, H-23), 3.78 (s, 3H, H-24), 3.62 (dd, 1H, 2J6′a 11.9, 3J5′ 6.9, GlcH-6′b), 3.40 (tapp, 1H, 3J2′,4′ 9.1, GlcH-3′), 3.36-3.32 (m, 1H, GlcH-5′), 3.25-3.19 (m, 2H, GlcH-2′,4′), 3.12 (dd, 1H, 2J6b 16.3, 3J5a 4.8, H-6a), 3.04 (td, 1H, 3J14proS 11.2, 3J20,14proR 4.6, H-15), 2.89 (dd, 1H, 2J6a = 16.4, 3J5a 8.9, H-6b), 2.77 (ddd, 1H, 3J15 5.0, 3J19 7.8, 3J21 8.6, H-20), 2.32 (ddd, 1H, 3J15 4.4, 3J3 11.1, 2J14proS 15.2, H-14proR), 2.24 (ddd, 1H, 3J3 4.2, 3J15 11.4, 2J14proR 15.2, H-14proS); 13C NMR (methanol-d4, 125 MHz) δ 171.28, 156.93, 138.45, 135.51, 130.46, 127.50, 123.79, 120.76, 120.08, 119.24, 112.48, 108.84, 106.25, 100.56, 97.54, 78.82, 78.08, 74.78, 71.82, 63.02, 62.03, 54.10, 52.76, 51.16, 45.34, 35.38, 32.63, 21.89; ESI-MS (C28H37N2O10+): calculated m/z 561.2448 [M+H]+, found m/z 561.2448 [M+H]+.
10-methylstrictosidine 8l
1H NMR (methanol-d4, 500 MHz) δ 7.81 (s, 1H, H-17), 7.26 (s, 1H, H-9), 7.19 (d, 1H, 3J11 8.3, H-12), 6.98 (dd, 1H, 4J9 1.3, 3J12 8.4, H-11), 5.85 (ddd, 1H, 3J20 7.6, 3J18cis 10.6, 3J18trans 17.5, H-19), 5.84 (d, 1H, 3J20 9.1, H-21), 5.35 (dd, 1H, 2J18cis 1.3, 3J19 17.4, H-18trans), 5.28 (dd, 1H, 2J18trans 1.1, 3J19 10.6, H-18cis), 4.80 (d, 1H, 3J2′ 7.9, GlcH-1′), 4.64 (br-d, 1H, 3J14proR,14proS 10.6, H-3), 3.98 (dd, 1H, 3J5′ 2.1, 2J6′b 11.8, GlcH-6′a), 3.80 (s, 3H, H-23), 3.73 (ddt, 1H, 3J5′ 5.0, 2J6′a 12.5, GlcH-6′b), 3.49-3.42 (m, 1H, H-5a), 3.40 (tapp, 1H, 3J2′, 4′ 9.1, GlcH-3′), 3.39-3.35 (m, 1H, H-5b), 3.25-3.19 (m, 2H, GlcH-2′, GlcH-4′), 3.10-2.97 (m, 3H, H-6a, H-6b, H-15), 2.75 (ddd, 1H, 3J15 5.1, 3J19 7.8, 3J21 8.7, H-20), 2.40 (s, 3H, H-23), 3.36-2.16 (m, 2H, H-14proS, H-14proR); 13C NMR (methanol-d4, 125 MHz) δ 171.43, 157.05, 136.67, 135.62, 130.30, 130.03, 127.82, 125.24, 119.90, 118.91, 112.10, 109.08, 106.79, 100.56, 97.43, 78.96, 78.135, 74.80, 71.88, 63.15, 53.19, 52.74, 45.52, 42.80, 34.92, 32.75, 21.71, 19.71; ESI-MS (C28H37N2O9+): calculated m/z 545.2499 [M+H]+, found m/z 545.2523 [M+H]+.
10-chlorostrictosidine 8n
1H NMR (methanol-d4, 500 MHz) δ 7.81 (s, 1H), 7.47 (dd, 1H, 5J12 0.6, 4J11 2.1, H-9), 7.29 (dd, 1H, 5J9 0.6, 3J12 8.7, H-12), 7.11 (dd, 1H, 4J9 2.0, 3J12 8.7, H-11), 5.85 (ddd, 1H, 3J20 7.7, 3J18trans 10.7, 3J18trans 17.4, H-19), 5.84 (d, 1H, 3J20 9.1, H-21), 5.35 (dtapp, 1H, 2J18cis 1.4, 4J20 1.4, 3J19 17.4, H-18trans), 5.28 (dtapp, 1H, 2J18trans 1.3, 4J20 1.3, 3J19 10.7, H-18cis), 4.80 (d, 1H, 3J2′ 7.9, H-1′), 4.68 (br-d, 1H, 3J14proS,R 10.4, H-3), 3.98 (dd, 1H, 3J5′ 2.0, 2J6′b 11.8, H-6′a), 3.80 (s, 3H, H-23), 3.74 (td, 1H, 3J6ax,6eq 5.2, 2J5ax 12.5, H-5eq), 3.64 (dd, 1H, 3J5 7.1, 2J6′a 11.8, H-6′b), 3.47 (ddd, 1H, 3J6eq 5.6, 3J6ax 8.5, 2J5eq 12.5, H-5ax), 3.40 (tapp, 1H, 3J2′,4′ 9.1, H-3′), 3.39-3.34 (m, 1H, H-5′), 3.23 (dd, 1H, 3J3′ 8.9, 3J5′ 9.9, H-4′), 3.22 (dd, 1H, 3J1′ 7.9, 3J3′ 9.3, H-2′), 3.13-3.05 (m, 2H, H-6ax, H-15), 3.02 (tdapp, 1H, 3J5ax,5eq 4.5, 2J6ax 16.3, H-6eq), 2.75 (ddd(d), 1H, 4J18cis,trans < 0.5, 3J15 5.0, 3J19 7.8, 3J21 8.8, H-20), 2.32 (ddd, 1H, 3J3/15 3.0, 3J3/15 12.1, 2J14proS 15.0, H-14proR), 2.23 (ddd, 1H, 3J3/15 3.7, 3J3/15 11.8, 2J14proR 15.2, H-14proS); 13C NMR (methanol-d4, 125 MHz) δ171.40, 157.06, 136.70, 135.54, 132.19, 128.65, 126.51, 123.80, 119.97, 118.84, 113.67, 109.03, 107.19, 100.54, 97.41, 78.95, 78.12, 74.79, 71.87, 63.15, 53.05, 52.74, 45.50, 42.63, 34.82, 32.68, 19.52; ESI-MS (C27H34N2O935Cl+): calculated m/z [M+H]+ 565.1953, found m/z 565.1959; (C27H34N2O937Cl+): calculated m/z [M+H]+ 567.1923, found m/z 567.1945.
NMR characterization of isositsirikine alkaloid analog 14m
1H NMR (600 MHz, methanol-d4) δ7.51 (d, 1H, 3J10 7.7, H-9), 7.38 (d, 1H, 3J11 7.6, H-12), 7.26 (t, 1H, 3J9,11 7.6, H-10), 7.09 (t, 1H, 3J10,12 7.4, H-11), 5.96 (q, 1H, 3J18 6.8, H-19), 4.94-4.87 (m, 1H, H-3), 4.20 (d, 1H, 2J21b 14, H-21a), 4.13 (d, 1H, 2J21a 14, H-21b), 4.04 (dd, 1H, 3J16 4.0, 2J17b 12, H-17a), 3.82 (dd, 1H, 3J16 4.7, 2J17a 12, H-17b), 3.99-3.85 (m, 2H, H-23a, H-23b), 3.76 (s, 3H, H-22), 3.67-3.58 (m, 1H, H-5), 3.20 (dd, 1H, 3J5 7.0, 2J6b 14, H-6a), 3.17-3.09 (m, 1H, H-16), 2.98 (dd, 1H, 3J5 7.0, 2J6a 15, H-6b), 2.87-2.82 (m, 1H, H-15), 2.59-2.42 (m, 2H, H-14), 1.87 (d, 3H, 3J19 6.8, H-18); ESI-MS (C22H29N2O4+): calculated m/z [M+H]+ 385.2127, found m/z 385.2134.
Supplementary Material
Acknowledgments
We thank Dr. Shi Chen for help with molecular biology techniques, the Department of Chemistry Instrumentation Facility (DCIF) and the MIT Biopolymer Facility for instrument resources. The Beckman Foundation, MIT and GM074820 are acknowledged for financial support. Respective contribution of authors: SOC and PB: design of experiments and preparation of manuscript, PB: implementation of assay, expression system and mutagenesis, identification and characterization of enzymes and enzyme products, feeding study of 8m, EM: feeding experiment of 8o.
Footnotes
Supplemental Data
Detailed protocol for STS gene construction and expression, representative data for kinetic analysis, protocol for generation of saturation mutation libraries, control experiments for assay and MS and NMR data for chromophores, protocol for enzyme screening in 96 well plates, protocols for enzymatic synthesis of strictosidine analogs, detailed procedure for feeding studies and LS-MS data of feeding experiments.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.van der Heijden R, Jacobs DI, Snoeijer W, Didier H, Verpoorte R. The Catharanthus alkaloids: Pharmacognosy and biotechnology. Curr Med Chem. 2004;11:607–628. doi: 10.2174/0929867043455846. [DOI] [PubMed] [Google Scholar]
- 2.O’Connor SE, Maresh J. Chemistry and biology of monoterpene indole alkaloid biosynthesis. Nat Prod Rep. 2006;23:532–547. doi: 10.1039/b512615k. [DOI] [PubMed] [Google Scholar]
- 3.Johnson IS, Wright HF, Svoboda GH. Experimental basis for clinical evaluation of antitumor principles from Vinca rosea Linn. J Lab Clin Med. 1959;54:830–838. [Google Scholar]
- 4.Svoboda GH. Alkaloids of Vinca rosea Linn. (Catharanthus roseus) 1X: Extraction and characterization of leurosidine and leurocristine. Llyodia. 1961;24:173–178. [Google Scholar]
- 5.Fahy J. Modifications in the “upper” or velbenamine part of the vinca alkaloids have major implications for tubulin interacting activities. Curr Pharm Des. 2001;7:1181–1197. doi: 10.2174/1381612013397483. [DOI] [PubMed] [Google Scholar]
- 6.Fahy J, Duflos A, Ribet JP, Jacquesy JC, Berrier C, Jouannetaud MP, Zunino F. Vinca alkaloids in superacidic media: a method for creating a new family of antitumor derivatives. J Am Chem Soc. 1997;119:8576–8577. [Google Scholar]
- 7.Kruczynski A, Hill BT. Vinflunine, the latest Vinca alkaloid in clinical development: A review of its preclinical anticancer properties. Oncology and Hematology. 2001;40:159–173. doi: 10.1016/s1040-8428(01)00183-4. [DOI] [PubMed] [Google Scholar]
- 8.McCoy E, O’Connor SE. Directed biosynthesis of alkaloid analogs in the medicinal plant periwinkle. J Am Chem Soc. 2006;128:14276–14277. doi: 10.1021/ja066787w. [DOI] [PubMed] [Google Scholar]
- 9.Kutchan TM. Strictosidine: From alkaloid to enzyme to gene. Phytochemistry. 1993;32:493–506. doi: 10.1016/s0031-9422(00)95128-8. [DOI] [PubMed] [Google Scholar]
- 10.Chen S, Galan MC, Coltharp C, O’Connor SE. Redesign of a central enzyme in alkaloid biosynthesis. Chem Biol. 2006;13:1137–1141. doi: 10.1016/j.chembiol.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 11.Ma X, Panjikar S, Koepke J, Loris E, Stockigt J. The structure of Rauvolfia serpentina strictosidine synthase is a novel six-bladed beta-propeller fold in plant proteins. The Plant Cell. 2006;18:907–920. doi: 10.1105/tpc.105.038018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Geerlings A, Redondo FJ, Memelink J, Contin A, van der Heijden R, Verpoorte R. Screening method for cDNAs encoding putative enzymes converting loganin into secologanin by a transgenic yeast culture. Biotech Tech. 1999;13:605–608. [Google Scholar]
- 13.Luijendijk TJC, Stevens LH, Verpoorte R. Reaction for the localization of strictosidine glucosidase activity on polyacrylamide gels. Phytochem Anal. 1996;7:16–19. [Google Scholar]
- 14.Brown RT, Leonard J. Biomimetic synthesis of cathenamine and 19-epicathenamine, key intermediates to heteroyohimbine alkaloids. J Chem Soc Chem Comm. 1979:877–879. [Google Scholar]
- 15.Geerlings A. PhD thesis. Leiden University; 1999. Strictosidine-β-D-glucosidase; and enzyme in the biosynthesis of pharmaceutically important indole alkaloids. [Google Scholar]
- 16.Heinstein P, Höfle G, Stöckigt J. Involvement of cathenamine in the formation of N-analogues of indole akaloids. Planta Med. 1979;37:349–357. [Google Scholar]
- 17.Brake AJ, Merryweather JP, Coit DG, Heberlein UA, Masiarz FR, Mullenbach GT, Urdea MS, Valenzuela P, Barr PJ. Alpha -factor-directed synthesis and secretion of mature foreign proteins in Saccharomyces cerevisiae. Proc Natl Acad Sci USA. 1984;81:4642–4646. doi: 10.1073/pnas.81.15.4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Knappik A, Pluckthun A. An improved affinity tag based on the FLAG peptide for the detection and purification of recombinant antibody fragments. Biotechniques. 1994;17:754–761. [PubMed] [Google Scholar]
- 19.de Waal A, Meijer AH, Verpoorte R. Strictosidine synthase from Catharanthus roseus: Purification and characterization of multiple forms. Biochem J. 1995;306:571–580. doi: 10.1042/bj3060571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Patrick WM, Firth AE, Blackburn JM. User friendly algorithms for estimating completeness and diversity in randomized protein encoding libraries. Prot Eng. 2003;16:451–457. doi: 10.1093/protein/gzg057. [DOI] [PubMed] [Google Scholar]
- 21.McCoy E, Galan MC, O’Connor SE. Substrate specificity of strictosidine synthase. Bioorg Med Chem Lett. 2006;16:2475–2478. doi: 10.1016/j.bmcl.2006.01.098. [DOI] [PubMed] [Google Scholar]
- 22.Miyaura N, Suzuki A. Palladium-catalyzed cross-coupling reactions of organoboron-compounds. Chem Rev. 1995;95:2457–2483. [Google Scholar]
- 23.Trouet ABL, Hannart JAA, Rao KSB. Vinblastin-23-oyl amino acid derivatives. US Pat 4,639,456 Chem Abs. 1987;107:40181. [Google Scholar]
- 24.Hutchinson CR, Heckendorf AH, Straughn JL, Daddona PE, Cane DE. Biosynthesis of camptothecin. 3 Definition of strictosamide as the penultimate biosynthetic precursor assisted by carbon-13 and deuterium NMR spectroscopy. J Am Chem Soc. 1979;101:3358–3369. [Google Scholar]
- 25.Kohl W, Witte B, Höfle G. Alkaloide aus Catharanthus roseus-Zellkulturen, II. Z Naturforsch B Anorg Chem Org Chem. 1981;36b:1153–1162. [Google Scholar]
- 26.Hallard D, van der Heijden R, Verpoorte R, Lopes Cardoso MI, Pasquali G, Memelink J, Hoge JHC. Suspension cultured transgenic cells of Nicotiana tabacum expressing tryptophan decarboxylase and strictosidine synthase cDNAs from Catharanthus roseus produce strictosidine upon secologanin feeding. Plant Cell Rep. 1997;17:50–54. doi: 10.1007/s002990050350. [DOI] [PubMed] [Google Scholar]
- 27.Kutney JP, Brown RT. The structural elucidation of sitsirikine, dihydrositsirikine and isositsirikine. Tetrahedron. 1966;22:321–336. doi: 10.1016/0040-4020(66)80133-3. [DOI] [PubMed] [Google Scholar]
- 28.Kohl W, Witte B, Höfle G. Alkaloide aus Catharanthus roseus-Zellkulturen, III. Z Naturforsch B Anorg Chem Org Chem. 1982;37b:1346–1351. [Google Scholar]
- 29.Hughes EHH, Seung-Beom, Shanks Jacqueline V, San Ka-Yiu, Gibson Susan I. Characterization of an inducible promoter system in Catharanthus roseus hairy roots. Biotechnology Progress. 2002;18:1183–1186. doi: 10.1021/bp025603o. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.