

Abstract
CD4+ T cells regulate adaptive responses to pathogens by secreting unique subsets of cytokines that mediate inflammatory processes. The innate cytokines IL-12 and IFN-α/β regulate type I responses and promote acute IFN-γ secretion through the activation of the STAT4 transcription factor. Although IL-12-induced STAT4 activation is a conserved pathway across species, IFN-α/β-dependent STAT4 phosphorylation does not occur as efficiently in mice as it does in human T cells. In order to understand this species-specific pathway for IFN-α/β-dependent STAT4 activation, we have examined the molecular basis of STAT4 recruitment by the human IFNAR. In this report, we demonstrate that the N-domain of STAT4 interacts with the cytoplasmic domain of the human, but not the murine IFNAR2 subunit. This interaction mapped to a membrane-proximal segment of the hIFNAR2 spanning amino acids 299–333. Deletion of this region within the hIFNAR2 completely abolishes IFN-α/β-dependent STAT4 tyrosine phosphorylation when expressed in human IFNAR2-deficient fibroblasts. Thus, the human IFNAR2 cytoplasmic domain serves to link STAT4 to the IFNAR as a pre-assembled complex that facilitates cytokine-driven STAT4 activation.
Keywords: T cells, cytokine receptors, signal transduction
1. Introduction
Instruction of adaptive T cell responses to pathogens is regulated by innate cytokines that are secreted by professional antigen presenting cells (APCs) (Trinchieri, 2003). The innate cytokine IL-12 drives adaptive type I immune responses by promoting IFN-γ secretion from NK, CD8+ and CD4+ T cells. For CD4+ T cells, IL-12 directs a series of developmental cues leading to their capacity to secrete high levels of IFN-γ (Szabo et al., 2003). This IL-12-dependent pathway of T helper type I (Th1) development is initiated through the conserved activation of a key transcription factor, STAT4 (Kaplan and Grusby, 1998; Murphy et al., 1999). STAT4 activation represents a key commitment step in the generation of Th1 cells, and the requirement for STAT4 in Th1 development and acute IFN-γ secretion in CD4+ T cells has been demonstrated conclusively in STAT4-deficient mice (Kaplan et al., 1996; Thierfelder et al., 1996).
IL-12-dependent Th1 development is a conserved pathway that exists in both humans and mice. This pathway is conserved, in part, due to the ability of both the murine and human IL-12 receptors to recruit and activate STAT4 (Bacon et al., 1995; Jacobson et al., 1995). In addition to IL-12, type I interferons (IFN-α/β) promote STAT4 activation and induce IFN-γ secretion from CD4+ T cells (Cho et al., 1996; Rogge et al., 1998). However, unlike IL-12, the effects of IFN-α/β on STAT4 activation, Th1 development, and acute IFN-γ secretion are not well conserved when comparing mouse and human CD4+ T cells. In human CD4+ T cells, IFN-α/β promotes STAT4 activation (Cho et al., 1996; Farrar et al., 2000b; Rogge et al., 1998) and can drive acute IFN-γ secretion in response to IFN-α/β + IL-18 stimulation (Brinkmann et al., 1993; Matikainen et al., 2001; Parronchi et al., 1992; Sareneva et al., 2000; Sareneva et al., 1998). In murine CD4+ T cells, IFN-α/β is inefficient at promoting STAT4 activation (Berenson et al., 2004; Farrar and Murphy, 2000; Farrar et al., 2000a; Farrar et al., 2000b; Persky et al., 2005; Rogge et al., 1998; Szabo et al., 1997), and as a consequence, murine CD4+ T cells fail to differentiate to Th1 cells and do not secrete IFN-γ in response to IFN-α/β + IL-18 in vitro (Berenson et al., 2004; Persky et al., 2005; Rogge et al., 1998; Wenner et al., 1996).
Previous studies of the human IFN-α/β receptor (hIFNAR) revealed an important role for STAT2 in IFN-α/β-dependent STAT4 activation. Here, IFN-α/β-induced STAT4 tyrosine phosphorylation was found to be dependent upon the presence of human STAT2 (Farrar et al., 2000b). Unlike other STAT family members, STAT2 is highly divergent between mouse and human, and this dissimilarity is particularly striking within the COOH-terminal transactivation domain (Farrar et al., 2000a; Park et al., 1999; Paulson et al., 1999). The sequence divergence in STAT2 is functionally relevant because expression of the murine STAT2 molecule failed to restore IFN-α/β-dependent STAT4 phosphorylation in human STAT2-deficient cells (Farrar et al., 2000a). However, expression of a chimeric murine/human STAT2 molecule, in which the COOH-domain was replaced with the human sequence, restored IFN-α/β-dependent STAT4 activation in STAT2-deficient human fibroblasts. Based on these studies, it was proposed that the human STAT2 COOH-terminus was a critical species-specific component of the human IFNAR complex that was necessary for IFN-α/β-dependent STAT4 tyrosine phosphorylation (Farrar and Murphy, 2000; Farrar et al., 2000a). This finding was followed by the construction of a knockin mouse that expressed a chimeric murine/human (m/h) Stat2 gene in place of the wild-type murine Stat2 (Persky et al., 2005). Unexpectedly, T cells from these m/hSTAT2 knock-in mice were unable to activate STAT4 or commit to Th1 development in response to IFN-α/β even though the m/hSTAT2 molecule was functionally activated by the murine IFNAR and capable of binding DNA. Thus, although the human STAT2 COOH-terminus was found to be necessary for STAT4 activation by IFN-α/β in human fibroblasts, this region of STAT2 was insufficient to restore this pathway when expressed in murine CD4+ T cells.
In this study, we have re-evaluated the molecular mechanism by which STAT4 is recruited to the human IFNAR. Based on the significant divergence in primary amino acid sequence between the murine and human IFNAR, we have studied the role of the cytoplasmic domains of the human IFNAR in cytokine-dependent STAT4 activation and induction of IFN-γ gene expression. Here, we demonstrate that the N-domain of STAT4 is involved in pre-associating STAT4 with the cytoplasmic domain of the human IFNAR2 subunit in a species-specific manner. This interaction mapped to a membrane-proximal segment within the IFNAR2 cytoplasmic domain, and this sequence was necessary for IFN-α/β-dependent STAT4 tyrosine phosphorylation.
2. Materials and Methods
2.1. cDNA Expression Constructs
Full length human IFNAR2 was amplified from a full length cDNA clone (Invitrogen, Carlsbad, CA), digested with SalI and cloned into the XhoI-digested retrovirus vector, GFPRV. A c-myc tag sequence was inserted by PCR into the extracellular domain within 6 amino acids downstream of the leader peptide. The c-myc tag was inserted with primers: 5-TCTGAAGAAGATCTGTACACAGATGAATCTT GCACTTTC, and 5-AATAAGTTTTTGTTCATCAGGCGAATCATATGAAATACC.
For GST pulldown assays, partial cDNA fragments encoding the COOH-domains of mIFNAR1, mIFNAR2, hIFNAR1, hIFNAR2, and the N-domain of mSTAT4 were amplified by PCR from full length cDNA-containing vectors. The human IFNAR1 cytoplasmic domain and murine STAT4 N-domain PCR products were digested with BamHI and ligated into the BamHI site of the prokaryotic expression vector pGEX-2TK (Amersham Biosciences, Piscataway, NJ). The human IFNAR2, murine IFNAR1 and IFNAR2 cytoplasmic domain PCR products were digested with EcoR1 and ligated into the EcoR1 site within pGEX-2TK.
For yeast-2 hybrid analysis, cDNA fragments encoding the COOH-domain of human IFNAR1 were amplified by PCR, digested with EcoR1 and BamHI and cloned as GAL4 fusion proteins into the EcoR1/BamHI-digested Bait vector pGADT7 (BD-Clontech, Mountain View, CA). The human IFNAR2 PCR product was digested with EcoRI and BamHI and ligated into the EcoRI/BamHI site within pGADT7. The N-domain of mSTAT4 PCR product was digested with BamHI and XhoI and ligated into the BamHI/SalI site within pGADT7. Additionally, cDNA fragments encoding the N-domain, coiled-coil domain, DNA binding domain, linker domain, SH2 domain, and the COOH-terminal domain of murine STAT4 were amplified by PCR, digested with EcoRI/XhoI or BamHI/XhoI and cloned into the EcoRI/SalI or BamHI/SalI sites within the yeast expression vector pFBL23 (Beranger et al., 1997). This vector allows for the expression of N-terminal fusion proteins with the DNA binding domain of the LexA transcription factor, and these constructs are referred to as Target vectors throughout this study.
2.2. GST Pulldown Assays
GST fusion proteins were expressed in E. coli strain BL21 CodonPlus-RIL (Stratagene, La Jolla, CA). Expression of recombinant proteins was induced by the addition of 0.6 mM IPTG to 300 ml cultures that achieved log-phase growth at 37°C. Upon induction, the cultures were incubated for 3 h at 25°C to maintain stability of recombinant proteins. Cells were disrupted by sonication in 15 ml of lysis buffer (50 mM Tris-Cl (pH 8.0), 1 mM EDTA, 150 mM NaCl, 0.1% Triton-X 100, 5 mM benzamadine, 5 μg/ml leupeptin, 1 mM phenyl methyl sulfonyl fluoride (PMSF), and 0.2 mg/ml lysozyme), and the supernatant was subsequently incubated with glutathione-Sepharose (Amersham Biosciences) at 4°C for 1 h. Following extensive washes, recombinant protein-bound microspheres were incubated in 0.5 ml of a glutathione-sepharose pre-cleared cell lysate derived from 2fTGH cells expressing murine STAT4 as described below. Binding reactions were incubated for 1 h at 4°C followed by extensive washes in wash buffer (10 mM Tris-Cl (pH 7.6), 1 mM EDTA, 150 mM NaCl, 0.5% Triton-X 100). Bound proteins were resolved by 7% SDS-PAGE and transferred to PVDF membranes. STAT4 was detected by immunoblotting with anti-STAT4 antibody (SC-486, Santa Cruz Biotechnology, Santa Cruz, CA) or with a STAT4-N-domain antibody (SC-7959, Santa Cruz) and Gt-anti-Rb-HRP secondary antibody (Jackson Immunoresearch, West Grove, PA) followed by chemilluminescence detection.
2.3. Yeast 2-Hybrid Analysis
S. cerevisiae strains L40 (MATa, trp1, leu2, his3, LYS::lexA-HIS3, URA3::lexA-lacZ) and AMR70 (MATα, trp1, lys2, leu2, his3, URA3::(lexAop)8-lacZ) were propagated to log phase growth in YPD medium (2% peptone, 1% yeast extract, and 2% dextrose). Expression vectors pFBL23 and pGADT7 were introduced into yeast strains AMR70 and L40, respectively, by Li acetate-mediated transformation, and plated onto selective medium. Single transformants of AMR70-pFBL23 were plated on dropout medium (BD-Clontech) lacking tryptophan and uracil. L40-pGADT7 transformants were plated on dropout medium lacking leucine, lysine and uracil at 30°C for 24–48 h. AMR70 and L40 single transformants were mated in 0.5 ml YPD cultures at 30°C for 20 h followed by replica plating on dropout medium lacking tryptophan, uracil, leucine, lysine and in the absence or presence of histidine. Replica mating plates were allowed to grow for 3–5 days at 30°C for detection of specific interactions in the absence of histidine.
2.4. Immunoprecipitation and Western Blotting
For direct immunoprecipitation, cells were lysed in RIPA buffer (150 mM NaCl, 1.0% NP-40, 0.5% (w/v) deoxycholate, 0.1% (w/v) SDS, and 50 mM Tris-Cl (pH 8.0)) and sequential immunoprecipitation was performed with polyclonal anti-STAT1 (SC-346) and anti-STAT4 (SC-486) antibodies. Phosphorylated STAT4 were detected with polyclonal anti-phospho-STAT4 (Tyr 693, Zymed Laboratories, South San Francisco, CA) and phosphorylated STAT1 was detected with anti-phospho-STAT1 (Tyr 701, Upstate, Lake Placid, NY) in combination with a polyclonal Gt-anti-Rb-HRP secondary antibody followed by chemilluminescence detection. Blots were subsequently probed with anti-STAT4 (SC-486) and anti-STAT1 (SC-346) to detect relative amounts of STAT immunoprecipitates.
Cell lysates for co-immunoprecipitation assays (Fig. 4) were prepared by solubilizing cells in digitonin buffer (50 mM Tris-Cl (pH 8.0), 150 mM NaCl, 0.8% (w/v) digitonin (Sigma-Aldrich, St. Louis, MO), supplemented with 10 mM phenylmethyl sulfonyl fluoride, 5 μg/ml leupeptin, and 5 mM benzamadine). Duplicate cell lysates were immunoprecipitated with anti-STAT4 (NB34) and anti-c-myc (clone 9E10, Santa Cruz Biotechnology) antibodies. Immunoprecipitates were washed extensively in lysis buffer prior to SDS-PAGE. Immunoblotting was performed as described above.
Fig. 4.
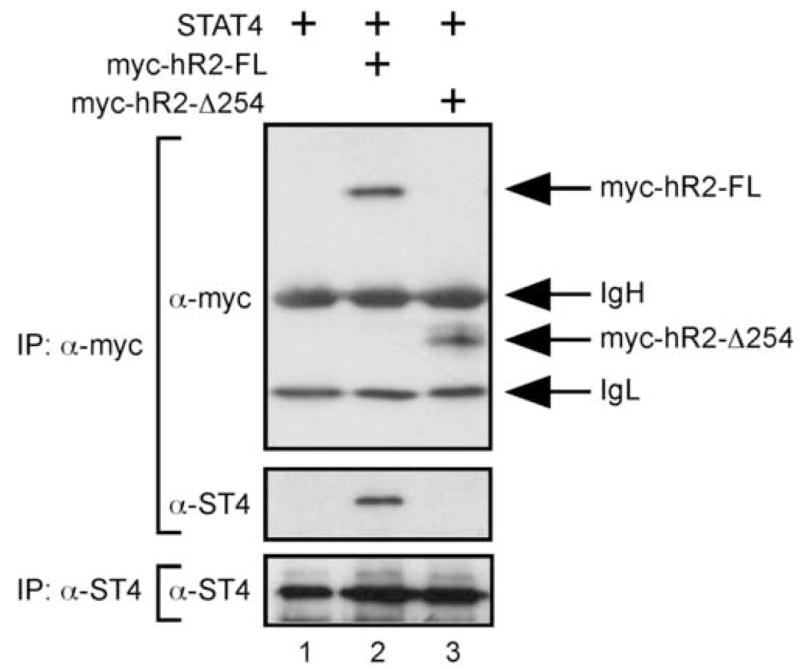
STAT4 interacts with the hIFNAR2 cytoplasmic domain in live cells. Cell lysates were prepared from U5A cells (IFNAR2-deficient) expressing STAT4 (lanes 1–3) and either a full-length c-myc-tagged hIFNAR2 (myc-hR2-FL, lane2) or a receptor molecule lacking the intracellular domain (myc-hR2-D254, lane 3). Samples were immunoprecipitated with α-c-myc followed by sequential immunoblotting for c-myc and murine STAT4 (upper panels). Duplicate samples were immunoprecipitated and immunoblotted with the α-STAT4 antibody as a control for STAT4 expression (lower panel). This experiment was performed twice with similar results.
3. Results
3.1. Species-specific interaction of the STAT4 N-domain with the human IFNAR2 subunit
Our previous studies demonstrated that although the human STAT2 C-terminus was required to promote IFN-α-dependent STAT4 activation in human fibroblasts (Farrar et al., 2000a), it was not sufficient to restore this pathway when expressed in murine T cells (Persky et al., 2005). This finding predicts that additional species-specific components of the IFNAR complex regulate STAT4 tyrosine phosphorylation. In addition to STAT2, the cytoplasmic domains of the IFNAR1 and IFNAR2 are poorly conserved when comparing the alignments of the murine and human cDNA sequences (Fig. 1). The murine and human IFNAR1 cytoplasmic domains share only 38.4% sequence identity (50.6% similarity, Fig. 1A), whereas the murine and human IFNAR2 cytoplasmic domains share 48.4% sequence identity (61.8% similarity, Fig. 1B). We previously demonstrated that none of the phosphorylated tyrosine residues within either the human IFNAR1 or IFNAR2 subunits could interact with the SH2 domain of STAT4 (Farrar et al., 2000b), thus eliminating the hypothesis that STAT4 was recruited to the human IFNAR based solely on a linear SH2/phosphotyrosine interaction. However, we have now reexamined how the IFNAR cytoplasmic domains contribute to IFN-α/β-dependent STAT4 activation based on recent findings implicating the STAT N-terminal domain (amino acids 1–130) in receptor-proximal tyrosine phosphorylation. These studies demonstrated that the STAT4 N-domain interacts as a homodimer (Chen et al., 2003; Vinkemeier et al., 1998), and mutations within the interface of that interaction disrupt dimer formation (Ota et al., 2004). Further, mutations that disrupt N-domain dimer formation also abrogate cytokine mediated STAT4 tyrosine phosphorylation (Ota et al., 2004). In addition, expression of a mutant STAT4 molecule lacking the N-domain, in cells or in transgenic mice, failed to support IL-12-induced STAT4 tyrosine phosphorylation (Chang et al., 2003).
Fig. 1.
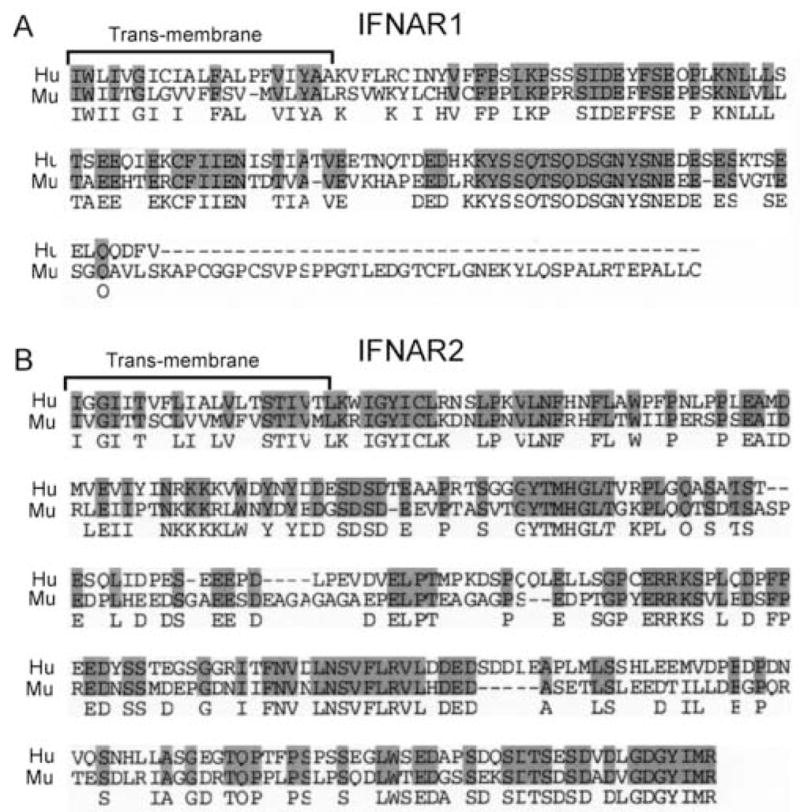
Alignment of the murine and human IFNAR1 and IFNAR2 transmembrane and cytoplasmic domains. Amino acid residues 437–557 (human, Hu) and 430–590 (murine, Mu) of the IFNAR1 (A), and residues 244–515 (Hu) and 243–513 (Mu) of the IFNAR2 (B) were aligned with the AlignX FAST algorithm within the VectorNTI software (Invitrogen, Carlsbad, CA).
Based on these recent findings, we wished to determine if the STAT4 N-domain could interact with the cytoplasmic domains of either the IFNAR1 or IFNAR2 subunits and whether any of these potential interactions were specific to the human IFNAR. To address this, we performed a series biochemical and genetic binding assays. First, the cytoplasmic domains of the human IFNAR1 and IFNAR2 were cloned as C-terminal fusions with GST. As controls for species specificity, we cloned the cytoplasmic domains of the murine IFNAR1 and IFNAR2 (by RT-PCR from murine cDNA) as fusion proteins with GST. These constructs were expressed in protease-deficient BL21 CodonPlus E. coli, and recombinant fusion proteins were isolated by glutathione-Sepharose chromatography. Fusion protein enrichments were routinely monitored by SDS-PAGE followed by silver staining (data not shown). Cell lysates, prepared from murine STAT4-transfected human 2fTGH cells (Farrar et al., 2000b), were incubated with equimolar quantities of purified fusion proteins bound to glutathione-Sepharose. Enriched GST was incapable of interacting with STAT4 (Fig. 2A, lane 1 and 2B, lane 3). As a positive control, we found that the N-domain of murine STAT4 was able to interact with full length murine STAT4, as expected (Fig. 2A, lane 2 and 2B, lane 4) (Ota et al., 2004). Further, we found that both the human IFNAR1 and IFNAR2 cytoplasmic domains were able to interact with STAT4 (Fig. 2A, lanes 3–4 and 2B, lanes 7–8). Interestingly, the murine IFNAR1 was also able to interact with STAT4 (Fig. 2B, lane 5), and the significance of this interaction with regard to STAT4 activation is unclear at this point. However, we found a species-specific interaction of STAT4 with the human IFNAR2 subunit that was not detected with the murine counterpart (Fig. 2B, compare lanes 6 and 8). Additional specificity was tested by performing pull-down assays from cell lysates derived from STAT1 over expressing U3A cells. None of the chimeric GST fusion proteins were able to interact with STAT1 (Fig. 2C) indicating that the interactions detected with STAT4 were specific.
Fig. 2.
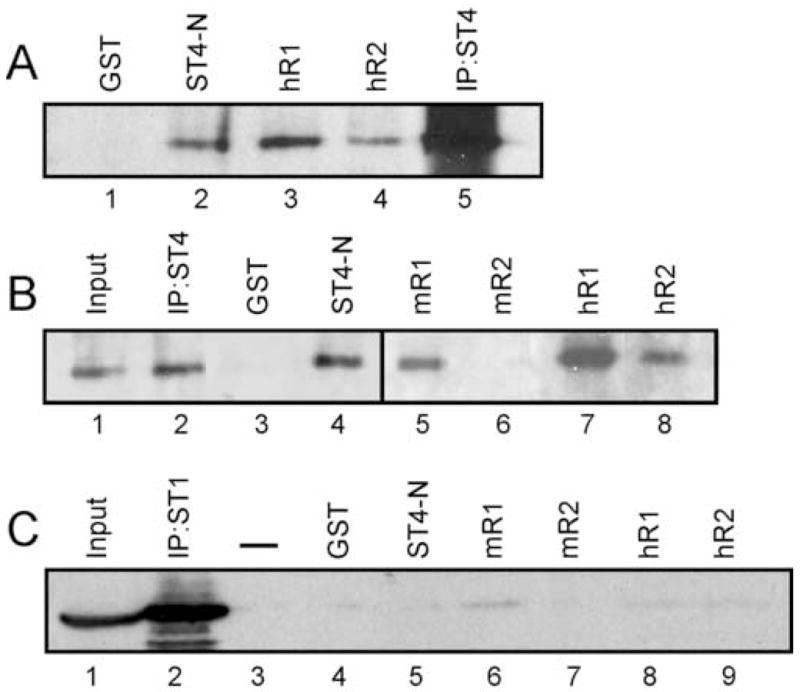
Species-specific interaction of STAT4 with the human IFNAR2 cytoplasmic domain. (A) Chimeric fusion proteins were bound to glutathione sepharose beads and incubated with lysates prepared from murine STAT4-expressing 2fTGH cells. STAT4 was also immunoprecipitated from these lysates as an additional control (lane 5). Bound proteins were immunoblotted for STAT4. (B) Chimeric fusion proteins were bound to glutathione-Sepharose beads, incubated with lysates prepared from STAT4-expressing 2fTGH cells, and immunoblotted for STAT4. (C) Chimeric fusion proteins were bound to glutathione-Sepharose beads, incubated with lysates prepared from STAT1-expressing U3A cells, and immunoblotted for STAT1. This experiment was performed 3 times with similar results.
In order to confirm these interactions, yeast-2 hybrid analyses were performed. For these experiments, the cytoplasmic domains of the human IFNAR1 and IFNAR2 were cloned as fusion proteins with the transactivation domain of Gal4 (Bait) within the pGADT7 vector. The N-domain of murine STAT4 (amino acids 1–130) was cloned as a N-terminal fusion protein with the DNA binding domain of LexA (Target) within the pFBL23 vector. The interaction of the STAT4 N-domain with the C-terminus of hIFNAR1 and hIFNAR2 was tested by the induction of the HIS3 gene driven by a LexA operator sequence. In these experiments, the yeast strain AMR70 (Matα) was transformed with the Target plasmids and mated to the L40 strain (Mata) transformed with the Bait plasmids (Fig. 3). The success of individual yeast matings was confirmed by growth of all colonies on selective medium in the presence of histidine (data not shown), whereas specific protein-protein interactions were detected by growth of yeast colonies on histidine-deficient medium (Fig. 3). As previously demonstrated (Ota et al., 2004), the STAT4 N-domains displayed homotypic interaction, and this was a relatively strong interaction as colonies began to appear within 24–48 h of incubation in the absence of histidine (Fig. 3, panel 3). There was no apparent interaction between the STAT4 N-domain and the hIFNAR1 cytoplasmic domain (Fig. 3, panel 1). However, an interaction was detected between the STAT4 N-domain and the cytoplasmic domain of the human IFNAR2 (Fig. 3, panel 2). This is likely to be a weaker interaction than the STAT4 N-domain/N-domain because colony growth was not apparent until 2 days following plating and were less numerous when compared to the positive control (Fig. 3, compare panels 2 and 3). In addition, we tested the ability of other domains of STAT4 (DNA binding domain, coiled-coil domain, linker domain, and the C-terminus) to interact with the human IFNAR1 and IFNAR2 cytoplasmic tails by this assay. The DNA-binding domain (Fig. 3, panels 4 and 5) and the linker domain of STAT4 (data not shown) displayed bait-dependent growth but did not interact with either the hIFNAR1 or hIFNAR2 subunits, thus supporting the observation that the weak interaction of the STAT4 N-domain with the hIFNAR2 was specific. The coiled-coil domain and the C-terminus of STAT4 displayed bait-independent growth in the absence of histidine (data not shown) thus precluding the ability to detect specific protein-protein interactions for these STAT4 domains.
Fig. 3.
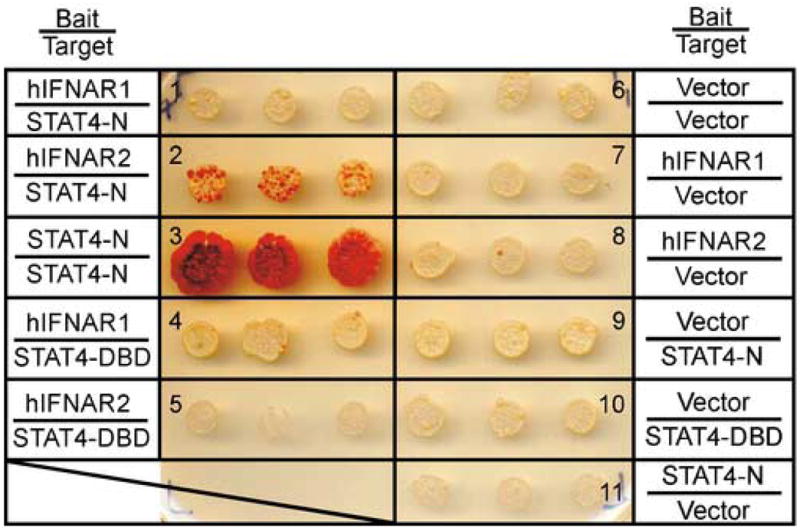
The murine STAT4 N-domain interacts with the human IFNAR2 subunit. Yeast strain L40 (Mata) were transformed with the indicated Bait vectors and mated to yeast strain AMR70 (Matα) that were transformed with the indicated Target vectors. Triplicate mating cultures were replica plated onto selective media without histidine and incubated for 3 days at 30°C. This experiment was performed 4 times with similar results. Abbreviations: C, cytoplasmic domain; N, amino terminus, DBD, DNA-binding domain.
Based on these findings that suggested a species-specific interactions between STAT4 and the hIFNAR2 subunit, we tested whether the STAT4- hIFNAR2 interaction could be observed in live cells by co-immunoprecipitation assays. For these experiments, a c-myc-tagged hIFNAR2 molecule was co-expressed along with full-length STAT4 in the human IFNAR2-deficient cell line U5A by retroviral transduction (Fig. 4). Immunoprecipitation of murine STAT4 with either a monoclonal (NB34 (Szabo et al., 1995)) or a polyclonal Rb-α-STAT4 antibody (SC-486, Santa Cruz) failed to co-precipitate the c-myc-tagged hIFNAR2 (data not shown), which may indicate that these antibodies disrupted the interaction of STAT4 with the hIFNAR2 cytoplasmic domain. However, both the c-myc-tagged hIFNAR2 and STAT4 were detected in immunoprecipitates utilizing an anti-c-myc antibody (Fig. 4, lane 2). In contrast, STAT4 was not detected in immunoprecipitates from cells that were either not transduced with the IFNAR2-expressing retrovirus (Fig. 4, lane 1) or transduced with retrovirus expressing a c-myc-tagged hIFNAR2 molecule that lacked the intracellular domain (Fig. 4, lane 3). Thus, association of STAT4 with the hIFNAR2 cytoplasmic domain is detectable within live cells. Taken together, these data suggest that the human IFNAR2 provides a species-specific molecular platform that mediates STAT4 association with the receptor complex prior to cytokine activation.
3.2. Functional mapping of hIFNAR2 sub-domains mediating STAT4 association and activation
Due to the significant sequence divergence between the murine and human IFNAR2 subunit, we wished to identify specific regions within the hIFNAR2 that contributed to both STAT4 N-domain binding and IFN-α-dependent STAT4 tyrosine phosphorylation. To probe this molecular interaction, a series of GST-pulldown experiments were performed with expression constructs encoding the full-length hIFNAR2 subunit and with serial C-terminal truncations (Fig. 5A). Enriched GST-fusion proteins (Fig. 5B) were incubated with lysates from 2fTGH cells expressing STAT4, and the binding of STAT4 with these proteins was detected by immunoblotting (Fig. 5C). In these experiments, STAT4 was capable of interacting with the full-length receptor, but not with GST alone (Fig. 5C, compare lanes 4 and 5). In addition, we found that sequences spanning amino acids 333 to 515 were dispensable for STAT4 preassociation (Fig. 5C, lanes 6–10). However, amino acids between residues 299 and 333 were required for STAT4 binding as the Δ299 deletion failed to interact with STAT4 (Fig. 5C, lane 11). Although the 2fTGH cells express endogenous STAT1, none of the GST fusion proteins precipitated STAT1 from these lysates (data not shown), demonstrating additional specificity for the STAT4-hIFNAR2 interaction.
Fig. 5.
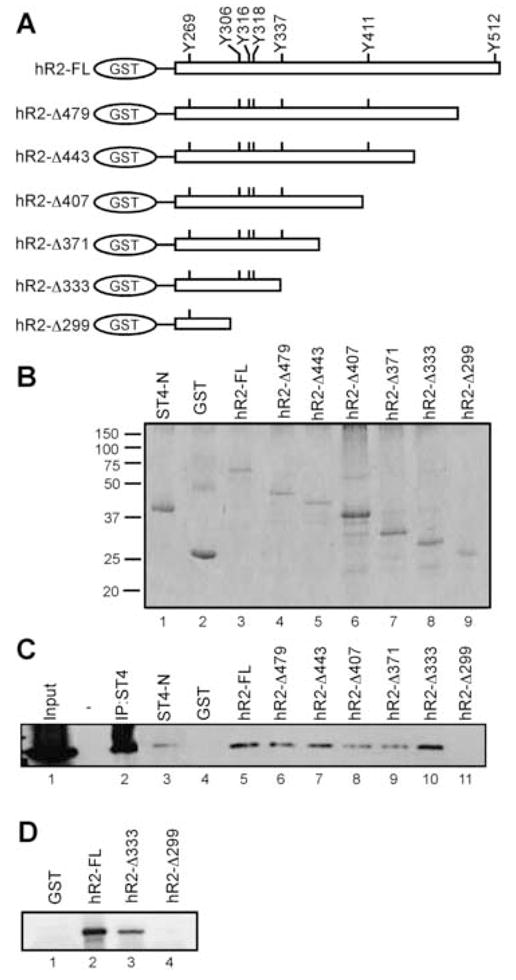
STAT4 interacts with a membrane-proximal region within the human IFNAR2 subunit. (A) GST-fusion constructs were designed to express the full-length intracellular domain of hIFNAR2 (hR2-FL) and C-terminal truncations that terminated at the indicated amino acid as denoted from the full-length receptor sequence (Accession #P48551). (B) GST-fusion proteins were isolated from E. coli lysates and analyzed by SDS-PAGE followed by coomassie blue staining. (C) Chimeric fusion proteins were incubated with lysates prepared from murine STAT4-expressing 2fTGH cells, and immunoblotted for STAT4. This experiment was performed 3 times with similar results. (D) Purified GST fusion proteins were incubated with 2fTGH cell lysates expressing amino acids 1–130 of the murine STAT4 N-domain. Precipitates were immunoblotted with α-STAT4 antibody raised against the STAT4 N-domain (Santa Cruz). This experiment was performed twice with similar results.
In order to determine if the N-domain was sufficient to mediate this interaction, 2fTGH cells were transduced with a retrovirus construct expressing the first 130 amino acids of STAT4 that constitutes the N-domain (Fig. 5D) (Chen et al., 2003; Ota et al., 2004; Vinkemeier et al., 1998). Similar to results obtained from lysates containing full-length STAT4, both the full-length hIFNAR2 cytoplasmic domain and the hIFNAR2-Δ333 deletion was found to interact with the STAT4 N-domain (Fig. 5D, lanes 2 and 3). However, hIFNAR2-Δ299 failed to interact with the STAT4 N-domain, suggesting that the STAT4 N-domain is sufficient to interact with the hIFNAR2 cytoplasmic domain upstream of Thr333.
STAT4 binding was correlated with IFN-α-dependent tyrosine phosphorylation within this region of the hIFNAR2 (Fig. 6). Here, a series of retrovirus vectors were constructed to express either the full-length IFNAR2 subunit or various C-terminal truncations corresponding to the regions that were identified to interact with STAT4 in Fig. 5. For these experiments, the human IFNAR2-deficient U5A cell line was utilized to probe the sufficiency of the hIFNAR2 to mediate IFN-α-dependent STAT4 activation. U5A cells were co-transduced with retrovirus vectors expressing STAT4 (within the CD4RV vector) and with either GFP alone (within the GFPRV vector, “Control”), or with GFPRV expressing the full-length hIFNAR2 and C-terminal truncations. Cells were activated with medium or with rhIFN-α (A) and both STAT4 and STAT1 were immunoprecipitated from cell lysates. Phosphorylated STAT4 and STAT1 as well as total STAT protein were detected by immunoblotting (Fig 6). As expected, IFN-α failed to promote STAT4 or STAT1 tyrosine phosphorylation in the absence of a functional IFNAR2 subunit, and co-expression of the full-length hIFNAR2 restored this activity (Fig. 6, lanes 1–4). A conservative deletion of 36 amino acids from the C-terminus of hIFNAR2 retained the ability to promote both STAT4 and STAT1 activation in response to IFN-α (Fig. 6, lanes 5–6). Truncation of hIFNAR2 at Pro-371 (Δ371) completely abrogated STAT1 phosphorylation, whereas STAT4 phosphorylation was retained yet diminished with the expression of this deletion. However, expression of hIFNAR2 Δ333 and Δ299 failed to restore any detectable STAT4 or STAT1 tyrosine phosphorylation in response to IFN-α. Collectively, these data suggest that STAT4 pre-associates within a region of the hIFNAR2 cytoplasmic domain spanning amino acids 299–333. However, this region was not sufficient to fully restore IFN-α-dependent STAT4 activation in vivo, indicating that additional interactions are required for STAT4 phosphorylation.
Fig. 6.

A membrane-proximal region within the hIFNAR2 subunit is required for IFN-α-dependent STAT4 tyrosine phosphorylation. hIFNAR2-deficient U5A cells were co-transduced with retrovirus constructs expressing STAT4 and with constructs expressing either the full-length hIFNAR2 subunit (hR2-FL) or with hIFNAR2 subunits that were truncated at specific amino acid residues as indicated in the figure. Cells were left resting or activated with rhIFN-α (A) (1000 U/ml) for 30 min at 37°C. STAT4 and STAT1 were sequentially immunoprecipitated from cell lysates and analyzed for phospho-STAT4 and phospho-STAT1 by immunoblotting. Blots were subsequently stripped and re-probed for STAT4 and STAT1 protein as indicated in the figure. This experiment was performed 3 times with similar results.
4. Discussion
The unexpected discovery of a species-specific pathway for STAT4 activation by type I interferons (Farrar and Murphy, 2000; Sinigaglia et al., 1999) has provided a unique opportunity to uncover general molecular mechanisms that regulate receptor-mediated STAT recruitment and subsequent tyrosine phosphorylation. In general, STAT recruitment and activation involves the direct binding of receptor-associated phosphorylated tyrosine residues by STAT SH2 domains (Greenlund et al., 1995; Heim et al., 1995; Wiederkehr-Adam et al., 2003; Yan et al., 1996), and this mechanism is involved in the activation of STAT4 by the IL-12 receptor (Naeger et al., 1999). Further, receptor specificity for STAT activation is constrained in part by the affinity of each STAT SH2 domain for specific amino acids adjacent to each tyrosine motif within receptor cytoplasmic domains (Greenlund et al., 1995; Krishnan et al., 1998; Yan et al., 1996). As neither the IFNAR1 nor R2 subunits are well conserved between mice and humans, the paradigm of direct STAT recruitment was the basis of previous studies that determined the ability of STAT4 to interact directly with phosphorylated tyrosine residues within the receptor COOH-termini (Farrar et al., 2000b). Here, STAT4 was unable to interact directly with any of the phosphorylated tyrosine residues within either the human IFNAR1 or R2 subunits. Thus, these studies initially ruled out the hypothesis that the IFNAR cytoplasmic domains were responsible for species-specific recruitment of STAT4 to the human IFNAR.
However, more recent studies have revealed a more complex mechanism by which STATs are recruited to receptor complexes. As discussed above, SH2-phosphotyrosine interactions are critical for cytokine-driven STAT phosphorylation. This is true for IFN-α-induced STAT4 activation because expression of a SH2-defective mutant STAT4 molecule fails to become phosphorylated in response to IFN-α in human fibroblasts (Farrar et al., 2000a). Yet, this single interaction fails to completely explain the rapid kinetics of STAT activation. For example, activation of STAT2 by the human IFNAR is dependent upon an interaction between the STAT2 SH2 domain and phosphorylated Y-466 within the human IFNAR1 subunit (Krishnan et al., 1998; Yan et al., 1996). Although this interaction is required, it is not sufficient for IFN-α/β-induced STAT2 phosphorylation. Stark and colleagues (Li et al., 1997) demonstrated that the N-terminal domain of STAT2 is also required for receptor-proximal recruitment of STAT2 to the human IFNAR. In these studies, the N-domain of STAT2 was shown to preassociate with the IFNAR2 subunit prior to cytokine activation. Recently, new studies have demonstrated a role for the N-terminal domain of STAT4 in receptor-mediated STAT4 tyrosine phosphorylation. Mutation analyses have revealed that the first 130 amino acids of STAT4 are required for both IL-12- and IFN-α/β-dependent tyrosine phosphorylation (Murphy et al., 2000; Ota et al., 2004). Further, transgenic expression of a STAT4 molecule lacking the N-terminal domain failed to mediate IL-12-dependent STAT4 tyrosine phosphorylation and IFN-γ secretion (Chang et al., 2003).
Based on the requirement for the STAT4 N-domain in IFN-α/β-dependent tyrosine phosphorylation, the present study has uncovered a unique mechanism for the species-specific activation of STAT4 by the human IFNAR. We have demonstrated that the STAT4 N-domain forms a preassociated complex with the cytoplasmic domain of the human IFNAR2 subunit. Further, this interaction is specific to the human IFNAR2, as the murine IFNAR2 does not bind STAT4. In addition, STAT4 was also observed to interact with the human and murine IFNAR1 subunit. However, the interaction with the hIFNAR1 was not confirmed in the yeast-2-hybrid assay. Thus, the nature of the STAT4-IFNAR1 interaction observed by GST-pulldown assays is unclear at this point and warrants further investigation. Based on these observations, we focused further analyses in this study on the STAT4-hIFNAR2 interaction because these two molecules interacted in a species-specific manner. We mapped the STAT4 N-domain interaction to a region of the hIFNAR2 spanning amino acids 299–333, which overall, is relatively well conserved between murine and human with regard to charge and hydrophobicity. This region contains 3 Tyr residues of which Y316 and Y318 are conserved at these positions in both the murine and human sequence. However, Y306 in the human IFNAR2 is mutated to Pro at that position within the murine receptor. Although it is unlikely that tyrosine modification would play a role in receptor pre-association, this particular mutation was intriguing as a Pro would introduce a significant structural alteration when compared to the human receptor. We tested the possibility that this mutation would alter the STAT4-hIFNAR2 interaction by point mutation. However, mutation of Y306 to Pro within the human IFNAR2 did not significantly affect STAT4 pre-association (data not shown). Thus it is likely that this region of the hIFNAR2 creates multiple point contacts with the STAT4 N-domain and that further structural mutations are responsible for the species-specific interaction.
This species difference in signaling has significant impact on our understanding of human responses to type I interferons. Exploiting the species differences among the molecular components of the IFNAR complex has shed significant light on the molecular mechanisms that regulate cytokine-driven STAT activation. The molecular insights gained from this study will allow for the reconstitution of the human pathway for IFN-α/β-dependent STAT4 activation in a murine model. In vivo reconstitution of this pathway in mice will allow for the analysis of host-pathogen interactions that more closely resemble the human condition.
Acknowledgments
The authors wish to thank K. Murphy, T. Murphy, and N. Ota for the kind gift of yeast plasmid vectors and for helpful discussions. We thank G. Stark for generously sharing the 2fTGH, U3A, and U5A cell lines. We thank N. Van Oers and J. Young for help with GST pull-down assays. We thank L. Hooper, A. Davis, and H. Ramos for critically reading the manuscript. This work was supported by a Special Fellows Award from the Leukemia and Lymphoma Society and an internal research grant from the American Cancer Society awarded to JDF.
ABBREVIATIONS
- GST
glutathione-sulfo-transferase
- IFNAR
IFN-α/β receptor
- STAT
signal transducer and activator of transcription
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bacon CM, Petricoin EF, 3rd, Ortaldo JR, Rees RC, Larner AC, Johnston JA, O’Shea JJ. Interleukin 12 induces tyrosine phosphorylation and activation of STAT4 in human lymphocytes. Proc Natl Acad Sci U S A. 1995;92:7307–11. doi: 10.1073/pnas.92.16.7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beranger F, Aresta S, de Gunzburg J, Camonis J. Getting more from the two-hybrid system: N-terminal fusions to LexA are efficient and sensitive baits for two-hybrid studies. Nucleic Acids Res. 1997;25:2035–6. doi: 10.1093/nar/25.10.2035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berenson LS, Farrar JD, Murphy TL, Murphy KM. Frontline: absence of functional STAT4 activation despite detectable tyrosine phosphorylation induced by murine IFN-alpha. Eur J Immunol. 2004;34:2365–74. doi: 10.1002/eji.200324829. [DOI] [PubMed] [Google Scholar]
- Brinkmann V, Geiger T, Alkan S, Heusser CH. Interferon alpha increases the frequency of interferon gamma-producing human CD4+ T cells. J Exp Med. 1993;178:1655–63. doi: 10.1084/jem.178.5.1655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang HC, Zhang S, Oldham I, Naeger L, Hoey T, Kaplan MH. STAT4 requires the N-terminal domain for efficient phosphorylation. J Biol Chem. 2003;278:32471–7. doi: 10.1074/jbc.M302776200. [DOI] [PubMed] [Google Scholar]
- Chen X, Bhandari R, Vinkemeier U, Van Den Akker F, Darnell JE, Jr, Kuriyan J. A reinterpretation of the dimerization interface of the N-terminal domains of STATs. Protein Sci. 2003;12:361–5. doi: 10.1110/ps.0218903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho SS, Bacon CM, Sudarshan C, Rees RC, Finbloom D, Pine R, O’Shea JJ. Activation of STAT4 by IL-12 and IFN-alpha: evidence for the involvement of ligand-induced tyrosine and serine phosphorylation. J Immunol. 1996;157:4781–9. [PubMed] [Google Scholar]
- Farrar JD, Murphy KM. Type I interferons and T helper development. Immunol Today. 2000;21:484–9. doi: 10.1016/s0167-5699(00)01710-2. [DOI] [PubMed] [Google Scholar]
- Farrar JD, Smith JD, Murphy TL, Leung S, Stark GR, Murphy KM. Selective loss of type I interferon-induced STAT4 activation caused by a minisatellite insertion in mouse Stat2. Nat Immunol. 2000a;1:65–9. doi: 10.1038/76932. [DOI] [PubMed] [Google Scholar]
- Farrar JD, Smith JD, Murphy TL, Murphy KM. Recruitment of Stat4 to the human interferon-alpha/beta receptor requires activated Stat2. J Biol Chem. 2000b;275:2693–7. doi: 10.1074/jbc.275.4.2693. [DOI] [PubMed] [Google Scholar]
- Greenlund AC, Morales MO, Viviano BL, Yan H, Krolewski J, Schreiber RD. Stat recruitment by tyrosine-phosphorylated cytokine receptors: an ordered reversible affinity-driven process. Immunity. 1995;2:677–87. doi: 10.1016/1074-7613(95)90012-8. [DOI] [PubMed] [Google Scholar]
- Heim MH, Kerr IM, Stark GR, Darnell JE., Jr Contribution of STAT SH2 groups to specific interferon signaling by the Jak-STAT pathway. Science. 1995;267:1347–9. doi: 10.1126/science.7871432. [DOI] [PubMed] [Google Scholar]
- Jacobson NG, Szabo SJ, Weber-Nordt RM, Zhong Z, Schreiber RD, Darnell JE, Jr, Murphy KM. Interleukin 12 signaling in T helper type 1 (Th1) cells involves tyrosine phosphorylation of signal transducer and activator of transcription (Stat)3 and Stat4. J Exp Med. 1995;181:1755–62. doi: 10.1084/jem.181.5.1755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan MH, Grusby MJ. Regulation of T helper cell differentiation by STAT molecules. J Leukoc Biol. 1998;64:2–5. doi: 10.1002/jlb.64.1.2. [DOI] [PubMed] [Google Scholar]
- Kaplan MH, Sun YL, Hoey T, Grusby MJ. Impaired IL-12 responses and enhanced development of Th2 cells in Stat4-deficient mice. Nature. 1996;382:174–7. doi: 10.1038/382174a0. [DOI] [PubMed] [Google Scholar]
- Krishnan K, Singh B, Krolewski JJ. Identification of amino acid residues critical for the Src-homology 2 domain-dependent docking of Stat2 to the interferon alpha receptor. J Biol Chem. 1998;273:19495–501. doi: 10.1074/jbc.273.31.19495. [DOI] [PubMed] [Google Scholar]
- Li X, Leung S, Kerr IM, Stark GR. Functional subdomains of STAT2 required for preassociation with the alpha interferon receptor and for signaling. Mol Cell Biol. 1997;17:2048–56. doi: 10.1128/mcb.17.4.2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matikainen S, Paananen A, Miettinen M, Kurimoto M, Timonen T, Julkunen I, Sareneva T. IFN-alpha and IL-18 synergistically enhance IFN-gamma production in human NK cells: differential regulation of Stat4 activation and IFN-gamma gene expression by IFN-alpha and IL-12. Eur J Immunol. 2001;31:2236–45. [PubMed] [Google Scholar]
- Murphy KM, Ouyang W, Szabo SJ, Jacobson NG, Guler ML, Gorham JD, Gubler U, Murphy TL. T helper differentiation proceeds through Stat1-dependent, Stat4-dependent and Stat4-independent phases. Curr Top Microbiol Immunol. 1999;238:13–26. doi: 10.1007/978-3-662-09709-0_2. [DOI] [PubMed] [Google Scholar]
- Murphy TL, Geissal ED, Farrar JD, Murphy KM. Role of the Stat4 N domain in receptor proximal tyrosine phosphorylation. Mol Cell Biol. 2000;20:7121–31. doi: 10.1128/mcb.20.19.7121-7131.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naeger LK, McKinney J, Salvekar A, Hoey T. Identification of a STAT4 binding site in the interleukin-12 receptor required for signaling. J Biol Chem. 1999;274:1875–8. doi: 10.1074/jbc.274.4.1875. [DOI] [PubMed] [Google Scholar]
- Ota N, Brett TJ, Murphy TL, Fremont DH, Murphy KM. N-domain-dependent nonphosphorylated STAT4 dimers required for cytokine-driven activation. Nat Immunol. 2004;5:208–15. doi: 10.1038/ni1032. [DOI] [PubMed] [Google Scholar]
- Park C, Lecomte MJ, Schindler C. Murine Stat2 is uncharacteristically divergent. Nucleic Acids Res. 1999;27:4191–9. doi: 10.1093/nar/27.21.4191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parronchi P, De Carli M, Manetti R, Simonelli C, Sampognaro S, Piccinni MP, Macchia D, Maggi E, Del Prete G, Romagnani S. IL-4 and IFN (alpha and gamma) exert opposite regulatory effects on the development of cytolytic potential by Th1 or Th2 human T cell clones. J Immunol. 1992;149:2977–83. [PubMed] [Google Scholar]
- Paulson M, Pisharody S, Pan L, Guadagno S, Mui AL, Levy DE. Stat protein transactivation domains recruit p300/CBP through widely divergent sequences. J Biol Chem. 1999;274:25343–9. doi: 10.1074/jbc.274.36.25343. [DOI] [PubMed] [Google Scholar]
- Persky ME, Murphy KM, Farrar JD. IL-12, but not IFN-alpha, promotes STAT4 activation and Th1 development in murine CD4+ T cells expressing a chimeric murine/human Stat2 gene. J Immunol. 2005;174:294–301. doi: 10.4049/jimmunol.174.1.294. [DOI] [PubMed] [Google Scholar]
- Rogge L, D’Ambrosio D, Biffi M, Penna G, Minetti LJ, Presky DH, Adorini L, Sinigaglia F. The role of Stat4 in species-specific regulation of Th cell development by type I IFNs. J Immunol. 1998;161:6567–74. [PubMed] [Google Scholar]
- Sareneva T, Julkunen I, Matikainen S. IFN-alpha and IL-12 induce IL-18 receptor gene expression in human NK and T cells. J Immunol. 2000;165:1933–8. doi: 10.4049/jimmunol.165.4.1933. [DOI] [PubMed] [Google Scholar]
- Sareneva T, Matikainen S, Kurimoto M, Julkunen I. Influenza A virus-induced IFN-alpha/beta and IL-18 synergistically enhance IFN-gamma gene expression in human T cells. J Immunol. 1998;160:6032–8. [PubMed] [Google Scholar]
- Sinigaglia F, D’Ambrosio D, Rogge L. Type I interferons and the Th1/Th2 paradigm. Dev Comp Immunol. 1999;23:657–63. doi: 10.1016/s0145-305x(99)00039-7. [DOI] [PubMed] [Google Scholar]
- Szabo SJ, Dighe AS, Gubler U, Murphy KM. Regulation of the interleukin (IL)-12R beta 2 subunit expression in developing T helper 1 (Th1) and Th2 cells. J Exp Med. 1997;185:817–24. doi: 10.1084/jem.185.5.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabo SJ, Jacobson NG, Dighe AS, Gubler U, Murphy KM. Developmental commitment to the Th2 lineage by extinction of IL-12 signaling. Immunity. 1995;2:665–75. doi: 10.1016/1074-7613(95)90011-x. [DOI] [PubMed] [Google Scholar]
- Szabo SJ, Sullivan BM, Peng SL, Glimcher LH. Molecular mechanisms regulating Th1 immune responses. Annu Rev Immunol. 2003;21:713–58. doi: 10.1146/annurev.immunol.21.120601.140942. [DOI] [PubMed] [Google Scholar]
- Thierfelder WE, van Deursen JM, Yamamoto K, Tripp RA, Sarawar SR, Carson RT, Sangster MY, Vignali DA, Doherty PC, Grosveld GC, Ihle JN. Requirement for Stat4 in interleukin-12-mediated responses of natural killer and T cells. Nature. 1996;382:171–4. doi: 10.1038/382171a0. [DOI] [PubMed] [Google Scholar]
- Trinchieri G. Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat Rev Immunol. 2003;3:133–46. doi: 10.1038/nri1001. [DOI] [PubMed] [Google Scholar]
- Vinkemeier U, Moarefi I, Darnell JE, Jr, Kuriyan J. Structure of the amino-terminal protein interaction domain of STAT-4. Science. 1998;279:1048–52. doi: 10.1126/science.279.5353.1048. [DOI] [PubMed] [Google Scholar]
- Wenner CA, Guler ML, Macatonia SE, O’Garra A, Murphy KM. Roles of IFN-gamma and IFN-alpha in IL-12-induced T helper cell-1 development. J Immunol. 1996;156:1442–7. [PubMed] [Google Scholar]
- Wiederkehr-Adam M, Ernst P, Muller K, Bieck E, Gombert FO, Ottl J, Graff P, Grossmuller F, Heim MH. Characterization of phosphopeptide motifs specific for the Src homology 2 domains of signal transducer and activator of transcription 1 (STAT1) and STAT3. J Biol Chem. 2003;278:16117–28. doi: 10.1074/jbc.M300261200. [DOI] [PubMed] [Google Scholar]
- Yan H, Krishnan K, Greenlund AC, Gupta S, Lim JT, Schreiber RD, Schindler CW, Krolewski JJ. Phosphorylated interferon-alpha receptor 1 subunit (IFNaR1) acts as a docking site for the latent form of the 113 kDa STAT2 protein. Embo J. 1996;15:1064–74. [PMC free article] [PubMed] [Google Scholar]