

Since 100 years ago scientists have become interested in the intracellular organelle mitochondrion, discovering its essentiality in life. Only half a century ago it was established that mitochondrial respiratory chains are responsible for energy metabolism/ATP production through the TCA cycle, coupling of oxidative phosphorylation (OXPHOS) and electron transfer 1. During the past two decades, it has become clear that other than energy generation, mitochondrion produces reactive oxygen species (ROS) as “side products” of respiration. Mitochondrial functions are also important in modulating cell cycle progression, cell survival and apoptosis 2.
1.1. Mitochondrial respiratory chain
The mitochondrial respiratory chain is consisted of 5 enzymatic complexes (I–V) of integral membrane proteins. They are NADH-CoQ reductase (complex I), Succinate-CoQ reductase (complex II), CoQ-cytochrome c reductase (complex III), cytochrome c oxidase (complex IV) and ATP synthase (complex V). Ubiquinone (CoQ) and cytochrome c are two freely diffusible molecules that mediate the transfer of electrons between complexes. Mitochondrial electron transport involves the passage of electrons from NADH or succinate to four inner-membrane protein complexes (I–IV) sequentially, coupling with the translocation of protons into the inter-membrane space. Electron flow through the electron-transport chain generates a proton gradient across the inner membrane that drives ATP synthesis 3.
ATP synthesis in the mitochondria could be inhibited by electron transport inhibitors, ATPase inhibitors and uncoupling agents. When electron transport is inhibited by pharmarcological agents (retenone, antimycin A, CN-, CO, myxothiazol and stigmatellin), ATP synthesis is reduced consequent to a decrease of proton gradient4. When mitochondrial ATPase is inhibited (i.e. by Oligomycin), no ATP is produced even when proton gradient is increased5. When electron transport is uncoupled (by 2,4-DNP), ATP synthesis is stoped but membrane permeability, proton gradient and oxygen consumption remain upregulated 4.
1.2. Mitochondrial DNA and nuclear DNA
Nuclear and mitochondrial DNA (mtDNA) has separate evolutionary origin. It is believed that mtDNA is derived from bacteria that were engulfed by early precursors of eukaryotic cells millions of years ago 6. Currently, there are 100–10,000 copies of human mtDNA per cell. Each copy of mtDNA (circular molecule) consists of 16,569 base pairs for 37 genes encoding 13 mitochondrial OXPHOS enzymes, 22 transfer RNA (tRNAs) and two ribosomal RNAs (rRNAs) 7. The latter two are invovled in expressional regulations of the OXPHOS enzymes in situ. These proteins/RNAs together constitute the core functional units of the mitochondria. However, nucleus DNAs predominantly encodes enzymes that regulate replication, repair, transcription and translation activities of mtDNA. Thus, the vast majority of proteins found in the mitochondria are encoded by nuclear DNA. It is believed that those nuclear DNA are of bacterial origin and were transferred to the nucleus during evolution 6.
When oxygen is present, glucose is metabolized into acetyl-CoA. It then enters the mitochondrial matrix for citric acid cycle (Krebs cycle), where it gets oxidized to CO2 by reducing NAD to NADH. NADH is utilized by the electron transport chain to creat further ATP as part of the OXPHOS. When one glucose molecule is fully oxidized, two acetyl-CoA is metabolized by the Krebs cycle to produce CO2 and H2O. Thirty molecules of ATP are produced by one molecule of glucose if all the reduced coenzymes are oxidized by the electron transport chain and used for OXPHOS 8.
mtDNA has two strands, a guanine-rich heavy (H) strand and a cytosine-rich light (L) strand. The replication of mtDNA is bidirectional but asynchronous9. There are no introns in mtDNA and all of the coding sequences are contiguous 10. The translation codes for mtDNA is slightly different from nuclear DNA because mtDNA and nuclear DNA are translated separately 11. In case of mitochondrion, AUA or AUU is sometimes used as an initiation codon instead of AUG. AGA and AGG of mtDNA, rather than being translated into argine, is used as a ‘stop’ signal. AUA of mtDNA encodes isoleucine whereas the same genetic code of nuclear DNA reads as methionine. UGA of mtDNA encodes tryptophan rather than a ‘stop’ signal for nuclear DNA/RNA translation12.
The unique location of mtDNA and the high copy number contribute to the unique features of mitochondria. First of all, mtDNA is maternally inherited. Secondly, mtDNA genes have a much higher mutation rate than nuclear DNA genes. Third, the copy numbers of mtDNA mutation are heteroplasmic and a certain threshold level of mutant mtDNA should be exceeding to manifest a disease. Therefore, gradual accumulation of somatic mtDNA mutations can finally cause mitochondrial dysfunction and loss of cellular energy production13. Reactive oxygen species (ROS) generated by mitochondria or elsewhere can cause damage to mtDNA. Indeed, mtDNA is highly susceptible to oxidative damage. This is because (1) mtDNA is in close proximity to the site of ROS/RNS production for being inside mitochondria, and that superoxide (O2•−) generated inside mitochondria is not permeable to cytosol (although its dismutation product hydrogen peroxide is); (2) mtDNA lacks histone proteins, which protects nuclear DNA from oxidative damage; and (3) mitochondrial polymerases lack specificity for base excision repair which is the major pathway eliminating oxidative DNA base lesions 14. Many of these DNA damages are mutagenic, contributing to cancer, aging, cardiovascular and neurodegenerative diseases 15.
1.3. Mitochondria and ROS production
Other than a major target for ROS-induced DNA damages, mitochondrion by itself can function as a primary source of ROS “leakage” in most tissues and cells. It has been estimated that about 0.2%–2% of oxygen consumed are converted into O2•− by eletronic transport chain (ETC) 16. The vast majority of basal cellular ROS (estimated at approximately 90%) are from mitochondria in healthy cells of high respiratory rates such as cardiomyocytes 17; although recent studies have shown that pre-assembled vascular NAD(P)H oxidases are the primary sources of ROS in vascular cells including endothelial cells and vascular smooth muscle cells 18–20. Mitochondrial electron transport generates O2•− as an inevitable by-product at two complexes, complex I and complex III 21, 22. Paradoxically, O2•− generation is enhanced at complex III during hypoxia via autoxidation of ubiquinone on both sides of mitochondrial inner membrane. Superoxide within intermebrane space, or released to cytosol via unidentified mechanisms, forms H2O2, which is responsible for the subsequent stabilization of the transcriptional factor, hypoxia induced factor-1α (HIF-1α) 23. HIF-1α could trigger diverse functional responses such as pulmonary vasoconstriction, induction of VEGF gene expression and enhanced angiogenesis, which would increase the cellular tolerability to hypoxic conditions 24. Therefore, mitochondria-derived ROS are involved in the oxygen sensing process in the pulmonary. However, the mechanism of hypoxia induced O2•− generation at complex III remain incompletely understood. It is proposed that the half life of ubisemiquinone is prolonged by the structural changes of complex III in response to hypoxia. This structural change increases accessibility of O2 to single electron. However, besides the net increase of O2•− production in complex III, more O2•− is directed to the intermembrane space over matrix compartments 25.
There is no consensus about the site of ROS production in complex I so far 26. Superoxide may be generated either by reverse electron transfer (RET) in the absence of NAD+-linked substrates upon succinate oxidation at complex I (succinate dehydrogenase) 27, or in much lower amounts in the forward electron transfer from the NAD+-linked substrates 28. However, O2•− generated at complex I can only be released to the matrix but not the cytosol 28. Moreover, seven other sites were also suggested to generate O2•− in the motochondria. They are cytochrome b5 reductase, monoamine oxidase, dihydroorotate dehydrogenase, dehydrogenase of α-glycerophosphate, succinate dehydrogenase, aconitase and α-ketoglutarate dehydrogenase complex 28.
1.4. Mitochondrial ROS elimination or detoxification
Mammalian mitochondria possess a multi-leveled ROS defense network of enzymes and non-enzymatic antioxidants. Even though a considerate amount of ROS is produced in mitochondrion constantly, the intrinsic antioxidant defense network within mitochondrion is so powerful that oxidative damage is almost completely abolished at baseline 26. One layer of ROS defenses is formed by enzymes scavenging O2•− and H2O2. Manganese containing superoxide dismutase (MnSOD) protect cells from O2•− attack by facilitating its dismutation into H2O2. H2O2 per se can be toxic and is detoxified into O2 and H2O spontaneously or catalyzed by enzymes including catalase and glutathione/glutathione peroxidases. Homozygous MnSOD knockout mice (30–80% decrease in MnSOD activity compared to wild type) do not survive longer than a few days after birth. The life span is shorter in these mice and the rate of aging is markedly increased. These mice also have more accumulated DNA damage, lower reduced glutathione (GSH) levels and more frequent cancer occurrence later in life 26.
Glutathione is considered the major antioxdiant molecule inside the cell. Approximately ~90% of glutathione is in its reduced form, GSH (γ-glutamylcysteinylglycine). GSH serves as a scavenger of hydroxyl radical thereby preventing free radical chain reaction 29. Only a small fraction of the total cellular pool of GSH is sequestered in mitochondria and the concentration of glutathione within mitochondria is in the range from 2 to 14 mmol/L 26. GSH reduces hydrogen- and organic-peroxides in the presence of glutathione peroxidase (GPx) and itself is oxidized to GSSG. Five different isoforms of GPx have been recently identified 30. They are GPx1, GPx2, GPx3, GPx4 and GPx6 (Table 1). GPx1 and GPx2 account for majority of the GPx activity in gastricintestin tract. Although GPx1 knockout mice appear normal in regular housing condition, the progression of atherosclerosis in ApoE knockout mice is promoted by GPx1 deficiency 31. Homozygotous double knockout mice deficient in both GPx1 and GPx2 showed symptoms of inflammtory bowel diseases, implicating oxidative stress-related damges 32. GPx-4 and α-tocopherol are effective in removing lipid peroxides. Among the five GPx, GPx2 and GPx4 have the higher ranking in the suppliance of selenium when it is limited. GPx4 null mice is embryonically lethal while the heterozygots are also more sensitive to oxidants compared to the WT 33. On the other hand, GSH could not be synthesized in mitochondria per se. It is replenished either by transported from cytosol or regenerated from GSSG in a reaction catalyzed by glutathione reductase (GR), utilizing NADPH as a cofactor. Although the GSH/GSSG ratio is a significant determinant of the redox status in mitochondria, it does not necessarily have an impact on H2O2 detoxification before GSH depletion exceeds ~50%. GSH also serves as a consumable in conjugation reactions to protect cells from lipid peroxidation products via an enzyme called glutathione-s transferase.
Table 1.
Mitochondrial antioxidant defense system
| Antioxidant system | Function | Significance | Ref |
|---|---|---|---|
| Enzymes | |||
| MnSOD | Facilitate dismutation of superoxide to H2O2 | Efficient solely dependent on its own activity not any cofactor | 84 |
| Catalase | Detoxify H2O2 | Less powerful than GSH | 85 |
| TrxR | Reduce Trx2 to Prx by utilizing NADPH | Similar to GR in GSH | 26 |
| Prx | Reduce H2O2 to oxygen using reducing equivalent from Trx | Similar to GPx in GSH system | 86 |
| γ-GST | Detoxify lipid peroxidation by consumption of GSH | Dependent on GSH availability | 87 |
| GR | Regenerate GSH utilizing NADPH | Catalyze the reaction to regenerate GSH | 88 |
| GPx | |||
| GPx1 | Cytosolic GPx, Reduce H2O2 to H2O by utilizing GSH | Protect cells from acute oxidative stress | 89, 90 |
| GPx2 | Gastrointestinal GPx, is upregulated in cancer cells and also a target of Nrf2 | Part of adaptive response to cancer | 91 |
| GPx3 | |||
| GPx4 | Reduce phospholipids hydroperoxides, H2O2, cholesterol peroxides | Interfere with NF-κB, reduces leukotriene and prostanoid biosynthesis, prevent COX-2 expression | 92 |
| GPx6 | Restrict to olfactory system | 30 | |
| Small molecules | |||
| Trx2 | Reduce H2O2 and lipid hydroperoxides | A supplement to GSH system | 93 |
| TP | Remove membrane lipid peroxide, and could be regenerated by CoQ or AA | It is the perimeter layer of ROS defense for mitochondrial | 94 |
| Glutathione | Detoxify H2O2, uptake from cytosol or regenerated by GR | Most important intracellular antioxidant | 95 |
| NADPH | Protect against oxygen centered radicals | Acting in accordance with other antioxidant network | 96 |
| Cytochrome c | Removal of superoxide | Contribute to generation of ATP | 97 |
MnSOD: Manganese superoxide dismutase; TrxR: Thioredoxin reductase; γ-Glutathione-s transferase; Prx: Peroxiredoxin; GSH: glutathione; GR: glutathione reductase; GPx: glutathione peroxidase; Trx: Thioredoxin; TP: a-tocopherol ; NADPH: Nicotinamide Adenine Dinucleotide Phosphate
Thioredoxin (Trx), NADPH and thioredoxin reductase (TrxR) comprise a thioredoxin system which share lots of similaries with GSH cycle. Trx2 is the major protein disulfide reductase in the cell. Similar to GR to GSH recycle, TrxR reduces the oxidized Trx-2 using NADPH as a cofactor. Similar to GPx, Peroxiredoxin (Prx) reduces H2O2 to molecular oxygen by using reducing equivalents from Trx. Trx supports GSH system when glutathione reductase (GR) is deficient. However they operate independently and have separate signaling roles. Mitochondria contain unique versions of Trx2, TrxR2, and Prx (the peroxidase that is recycled by thioredoxin-2 by consumption of NADPH). Trx2-knockout mice are embroyonically lethal consequent to massive apoptosis when respiration first begins. Thus it would seem that Trx2 is critical in preventing apoptosis from mitochondrial O2•− initated processes 34. However, it is not yet clear whether apoptosis is simply a result of excesssive ROS productions or alternatively, is partially attributed to the deficiencies in Trx2 function facilitating apoptosis.
Other antioxidants such as NAD(P)H and cytochrome c are also involved in the detoxification of mitochondrial ROS. Among all of them, the glutathione peroxidase/glutathione reductase cycle remains the main ROS removal system. Therefore, a loss of mitochondrial glutathione via the permeability transition pore would inevitably result in an oxidative stress. The mitochondrial antioxidant defense components are illustrated in Table 1.
1.5. Mitochondrial nitric oxide synthase (mtNOS), NO and Oxygen Consumption
Nitric oxide (NO) is a signaling molecule carrying out numerous roles but most notably regulation of local vascular tone and blood flow. By increasing vasodilation and blood flow, NO increases oxygen delivery and local tissue oxygen consumption. However, It is recently discovered that NO competes with oxygen for mitochondrial complex IV (cytochrome oxidase, COX), which has a high affinity for NO at 10–20 mmHg of tissue oxygen tension, to inhibit mitochondrial respiration. Oxygen may become available to the hydroxylases when NO is inhibiting cytochrome c oxidase, promoting the HIF-1α degradation. Conversely, when oxygen is not available, i.e., under hypoxic conditions or NO deficiencies, to block the complex IV access of oxygen,35 several pathways became activated to promote the phosphorylation of HIF-1α which would in turn result in HIF-1α dimerization and increased transcriptional activity 35.
On the other hand, NO has been well characterized to have anti-apoptotic effects on endothelial cells, and shown to protect against cell death induced by multiple stimuli, including TNF-α, serum starvation, hypoxia, and H2O2. The sensitivity to apoptosis is increased in eNOS knockout endothelium, suggesting an important role of eNOS-derived NO in cytoprotection 36. NO inhibits apoptosis is at least partially mediated via the downstream executioner caspase-3. In addition, NO can modulate apoptosis via mitochondrion. In isolated mitochondrion, NO effectively inhibits permeability transition and subsequent cytochrome c release 36. NO can also inhibit cytochrome c release from the mitochondrion indirectly through upregulation of Bcl-2 36. There is also emerging evidence that mitochondrial respiration is the target of NO thus mediating its cytoprotective effects. It was recently reported that the protective effect of endogenously produced NO against ROS is lost in cells deprived of respiring mitochondria, implying that NO plays a critical role in defense against oxidative stress through regulation of respiration.
More interestingly, several labs have recently reported that NO can be produced locally in mitochondria by mitochondrion-specific nitric oxide synthase (mtNOS) 37, 38. It indicates that mitochondrion per se can be a source of NO. It also means that the respiratory chain could be modulated by NO generated locally. The consequences of NOS activity regarding mitochondrial functions depend on the amount of NO that is produced. Low level of NO partially inhibits respiratory chain. This partial inhibition increases mitochondrial ROS production in short term which further leads to mitochondrial depolarization. This was associated with a significant reduction in mitochondrial calcium accumulation which in turn, increases cellular resistance to injury 39. However, if inhibition of cytochrome c oxidase is sustained, then deleterious effects such as inhibition of ATP synthesis, release of cytochrome c 37, increased oxygen radical production 40, and nitration of critical biomolecules may incur 41, 42. In addition, excessive NO generation may cause tissue damage. High concentrations of NO in the presence of relatively low tissue oxygen tensions may cause total inhibition of respiration. In this case, even if tissues are adequately supplied with oxygen, they are unable to use it. Meanwhile, more O2•− is generated which in turn interacts with NO to generate peroxynitrite, a highly reactive radical species which would further impair mitochondrial respiration, likely resulting in opening of the mitochondrial permeability transition pore and cell death. A fascinating discovery about NO and mitochondria has emerged recently. Nisoli et al. 43 showed that NO generation can stimulate the mitochondrial biogenesis and mice deficient in eNOS have deficiencies of mitochondrial enzymes. These data suggest that NO regulates mitochondrial function via genomic DNA 44.
1.6. Mitochondria and apoptosis
Two pathways have been characterized to mediate apoptosis: death receptor pathway and mitochondrial pathway 45. Mitochondrial pathway is triggered by the release of cytochrome c from mitochondria 45 (Figure 2). Both of these apoptotic pathways are mediated by cytosolic aspartate specific proteases termed Caspases. Caspase-8 activates disassembly in response to activation of death receptor pathway and caspase-9 activates disassembly in response to the mitochondrial pathway46. Both caspase-8 and caspase-9 can activate caspase-3 through proteolytic cleavage and caspase-3 is the one ultimately responsible for the most of apoptotic effects. Death receptor and mitochondrial pathways are linked via Bcl-2 (B-cell lymphocyticleukaemia proto-oncogene 2) family protein Bid. Once the mitochondrial apoptotic pathway was activated, the mitochondrial permeability transition pore is open due to elevated mitochondrial calcium levels and excessive ROS (generated at respiratory complex I and III sites). This is followed by cytochrome c release from mitochondria to cytosol with subsequent caspase activation initiating cell self-digestion and nuclear DNA fragmentation. Evidence suggests a significant role of mitochondria in cardiomyocyte apoptosis. In cardiomyocytes, the release of cytochrome c from mitochondria into cytosol has been reported in association with normal structural appearance and without DNA degradation. It is possible that myocytes require a certain threshold for cytochrome c release in the cytosol to start the apoptotic machinery or an alternative protein released from the mitochondria, such as apoptosis inducing factor (AIF). The release of AIF from the mitochondria also activates caspase-3 for the initiation of the apoptotic pathway 47.
Figure 2. Mitochondrial ROS and apoptosis.

ETC generated ROS induces oxidative stress which leads to upregulation of p53 and FAS gene expression. FAS activation will initiate death receptor pathway of apoptosis. However, p53 gene activation will trigger mitochondrial pathway of apoptosis by induction of cytochrome c release. Both death receptor and mitochondrial pathways are integrated into caspase cascade for apoptosis. Depending on the availability of intracellular ATP, the cell death pathway may switch from apoptosis to necrosis. While apoptosis is the predominant cell death pathway in the presence of adequate ATP 76.
2.1 Mitochondria and cardiovascular diseases
It has been suggested that patients with cardiovascular disease (CVD) have increased mtDNA damage in both heart and aorta 48. In a mouse model of myocardial infarction (MI, created by ligating of left coronary artery), increased ROS production was observed, which was accompanied by decreases in mtDNA copy numbers, mitochondrial-encoded gene transcripts, and related enzymatic activities (complexes I, III, and IV). Of note, nuclear-encoded genes (complex II) was not affected 49. Decreased vascular SOD activities have been associated with increased exposure to CVD risk factors and increased susceptibility to ischemia/reperfusion-mediated cardiac damages. In contrast, overexpression of mitochondrial antioxidants prevents these effects and increases cardiac tolerance to ischemia. Therefore oxidative stress caused by mitochondrial dysfunction may serve as the convergent pathway 50, 51. An interesting example is that mitochondrial specific overexpression of Prx-3 (an mitochondrial antioxidant protein) improved post-MI LV functions by restoring mitochondrial function such as DNA copy number and mitochondrial enzyme activity 52.
Numerous studies have reported that atherosclerosis is correlated with DNA damage. Moreover, meta analysis reveals that DNA-adduct level is a significant predictor of stage of atherogenesis after adjustment for CVD risk factors. Augmented DNA repair has been observed in atherosclerotic plaques compared to control tissue 51. Mitochondrial oxidant generation is increased in macrophage cells treated with oxLDL which further potentiates oxLDL formation 53. Cholesterol administration in rabbits is associated with impaired mitochondrial energetic function and decreased activity of mitochondrial dehydrogenases (e.g., SDH, etc.) 54. Moreover, mitochondrial membrane potential was decreased by free cholesterol that ativates mitochondrial apoptotic pathway 55. 8-oxoG immunoreactivity, DNA strand breaks and repair are increased in plaques of cholesterol-rich diet fed animals than in arteries from controls 56. Lipid lowering of fat diet retrived 8-oxoG levels and normalized DNA strand breaks. High-fat diets stimulate stress response (heat shock protein 70) and signal transduction genes (Ras, MAPK1) by inhibition of SOD and GPx gene expression which are involved in free radical scavenging 57. These effects could be prevented by scavengers of peroxides 53 and antioxidant supplementation of the high-fat diet and caloric restriction 57.
2.2 Heart failure
ROS is the integral signaling molecules in myocardial remodeling and failure 58. ROS induce hypertrophy and apoptosis in isolated cardiac myocytes. It has also been shown that ROS activates matrix metalloproteinase (MMP) in cardiac fibroblasts 59. Myocardial MMP activity is increased in the failing hearts. Left ventricle (LV) remodeling is dependent on activation of MMPs which is often secondary to increased ROS productions. It has been demonstrated that ROS scavengers inhibit development of LV remodeling and failure via inhibition of MMP activation, again implicating an important role of ROS in mediating development of heart failure via MMPs.
Recently, Rosenberger et al. reported that a decline in mitochondrial enzymatic activities and mtDNA copy numbers is associated with heart failure 59. Mitochondrial specific ROS production likely underlies these changes, as intermediate steps and possibly, one of the causal factors. Skeletal muscle-derived mitochondria in heart failure also produce more ROS production although paradoxically the complex I and III activities are decreased. Nonetheless, such defects in electron transfer function may lead to additional ROS production. The muscle atrophy and contractile dysfunction in heart failure patients may be due to the induction of apoptosis (Figure 2) 36. On the other hand, unlike nuclear DNA, the copy number of mtDNA has profound effects on the expression of mitochondrial-encoded genes, which is directly associated with RNA transcription, protein synthesis and consequently regulations of mitochondrial functions 58. Overexpression of mitochondrial transcription factor A (TFAM) in mice restores the mtDNA copy number and mitochondrial encoded transcripts to normal level, and at the same time attenuates post-MI LV remodeling and failure of the heart 60. Attenuation of oxidative stress have also been shown to more or less improve the exercise capacity of patient with heart failure 61.
2.3 Mitochondria, ischemia reperfusion and ischemia preconitioning
Ischemia reperfusion (I/R) is a serious heart problem due to death of cardiomyocytes. Exposure of cardiomyocytes to I/R lead to excessive apoptosis 62. Supplementation with mito Q and SOD inhibit apoptosis as well as associated ROS generation. Therefore, preserve of mitochondrial function is important to avoid serious damage from I/R. However, it has been well established that brief periods of ischemia provide protection for heart from prolonged ischemic insult, resulting in reduction in MI size, serverity of stuning and incidence of cardiac arrhythmias 63. This phenomenon is called ischemia preconditioning (IPC) and has been demonstrated in many animal models and cultured cells. During IPC, the cells were exposed to hypoxia and metabolic inhibition which affect the mitochondrial structure and function. Early ischemic damage increased mitochondrial swelling and the uncoupling of respiration and OXPHOS. Sustained ischemia leads to ATP depletion and subsequent de-energization of the cell resulting in necrotic cell death. IPC prevents ischemia-driven myocardial necrosis as well as myocardial apoptosis by inhibition of inflammatory cell activation and pro-apoptogenic protein expression such as BAX and Bad 64. Meanwhile, expression of antiapoptotic proteins that regulate both the mitochondria-mediated (Bcl-2 and Mcl-1) and the death-receptor-mediated (c-FLIP(L) and c-FLIP(S)) pathway of apoptosis are increased. As a result of the prosurvive signaling, the PT pore opening, Ca2+ influx to mitochondria and release of cytochrome c are all inhibited 65. Mitochondrial KATP channel is opened to due to increased ROS production, and mitogen-activated protein kinase (MAPK) or PKC activation which are underlying the mechanism of early phage of IPC (lasting 2–3 h) to protect heart from myocardial infarction but not stunning 66. In late phage of IPC (last 3–4 days), stress activated genes are induced to program heart genetically which protects heart from infarction as well as stunning. Antioxidant gene expression (etc, mitochondrial Mn-superoxide dismutase (MnSOD), eNOS) was upregulated by ROS via protein kinase mediated activation of antioxidant response element (ARE) which confers cytoprotection 67. It is also established that NO production is increased during late phage of IPC to protect heart from post-ischemic damage by interaction with electron transport chain and/or the mitochondrial permeability transition pore 64.
2.4 Mitochondrial ROS and intermediate hypoxia
It is difficult to determine the oxygen levels available to cells in the body. It has been shown that blood circulation has a partial pressure of oxygen (pO2) of 80–100 mmHg (1 mmHg = 133 Pa) which is equal to 10–12.5% O2 68. pO2 levels in the inner vascular retina are ~ 20mmHg which is equal to 3–6% O2. Thus, we estimate that in standard culture conditions (ambient 20% oxygen), cells are exposed to 2–5 fold higher concentrations of oxygen than they would likely encounter in vivo. Therefore, intermediate hypoxia is the oxygen tension defined as greater than 0.1% but less than 2% 69.
Oxygen supply is crucial to cell metabolism and viability. It was recently reported that mitochondrion is involved in oxygen sensing besides consuming oxygen as a major function. It was proposed that low oxygen tension decreases the rate of mitochondrial ROS generation, resulting in a decrease of oxidative stress. Of note, hypoxia was found to trigger a paradoxical increase of O2•− production at complex III 70. More interestingly, recent reports indicate that ROS regulate hypoxia-inducible transcription factor (HIF-1α) stability and transcriptional activity in hypoxic conditions 71. At the cellular level, oxygen partial pressure (pO2) is sensed by a family of protein hydroxylases (PH). It requires oxygen as a substrate which indicates it serves as an oxygen sensor in regulating HIF-1α activity. These enzymes covalently attached hydroxy groups to HIF-1α which regulates abundance and activity of the HIFs by degradation under normoxia. During anoxia, PH activity is fully abolished in the absence of oxygen. In addition to this highly specific and direct mechanism of oxygen sensing, mitochondria were proposed to sense oxygen and to transmit the signal by ROS generation via the electron transport chain at complex III as discussed in section 1.3. PH activity is partially inhibited by mitochondrial ROS that is released into cytosol because cytosolic not matrix catalase abrogates the HIF-1 stablization in hypoxia. Complex III but not complex I inhibitors abolished the effect suggesting complex III is the oringial source of ROS. However, the exact correlation between pO2 and ROS production, the precise downstream targets of ROS, and how ROS regulate these targets at the molecular level, remained unanswered 72.
HIF has also been shown to respond to non-hypoxic stimuli 73. The HIF is a master regulator of oxygen-sensitive gene expression. It not only plays a key role in adaptive response to hypoxia via the transactivation of genes encoding glucose transporters, glycolytic enzymes, VEGF and other key proteins, but also controls the establishment of essential physiologic systems during embryogenesis and their subsequent utilization during fetal and postnatal life 74. It is suggested that HIF regulation by oxygen tension may be involved in beneficial effect of IPC or contribute to cell death during I/R.
2.5 Mitochondria and diabetes
Several mtDNA mutations and depletions were reported to be associated with certain type of familiar diabetes such as mature onset diabetes of the young (MODY) which provides evidence for a direct link of mitochnondrial dysfunction with diabetes. The entry of glucose into the islet leads to flux through glycolysis which is used to generate ATP via mitochondrial ETC and eventually stimulate the release of insulin following membrane depolarization and Ca2+ influx. In diabetes, the glucose induced insulin secretion is impaired due to the disturbance of mitochondrial function and OXPHOS integrity. On the other hand, there are numerous recent reports that mitochondria are the main source of free radicals in the periphery tissues in diabetes 80. Brownlee et al 81 reported that hyperglycaemia is associated with a high mitochondrial membrane potential and an increase of ATP/ADP ratio due to excess accumulation of electron donors from the TCA cycle (e.g. NADH and FADH2). This could inhibit electron transport at complex III, increasing the half-life of free-radical intermediates of coenzyme Q (ubiquinone) which leads to reduction of O2 to superoxide anion. Excessive ROS production from mitochondria would induce oxidant stress in heart via the depletion of mtGSH in short term diabetes 82. This is associated with increased apoptosis via the increase in caspase-9 and -3 activity. More importantly, high glucose augments cellular oxidant stress by increasing flux through the hexosamine and polyol pathways, advanced glycation end-product (AGE) formation, and activation of protein kinase C (PKC) which damage the periphery tissues and organs 83.
2.6 Mitochondria and aging
An increasing amount of mutations in mitochondrial DNA have been gradually observed during aging 75. The levels of 8-OHDG mtDNA adducts and deletions increase exponentially with age75. In human and primate muscle, liver, and brain tissue, complex IV and mitochondrial OXPHOS enzyme activities decline with age. This is correlated with accumulation of mtDNA mutations, including deletions and base substitutions. As mutations accumulate, they exacerbate inherited OXPHOS defects until combined defects result in energetic failures 75.
It is also proposed that ROS governs the aging process. Oxidative damage to DNA causes modification of the purine and pyrimidine bases, single and double-strand breaks, and cross-links to other molecules. Many of these modifications in nuclear DNA and mtDNA of tissue cells are increased with age in mammals 76. It is further established that animals with lower metabolic rate which is accompanied with reduced oxidative damage have longer life span. Telomeres are located in the end of chromosomes, that protect chromosomes from degradation, fusion and recombination 76. A number of studies have shown that exposure to oxidant stress leads to faster telomere shortening and low ROS improves the telomere length 77. Selective targeting of antioxidants directly to the mitochondria counteracts telomere shortening and increases lifespan in fibroblasts under mild oxidative stress of cells 77. De la Asuncion et al 78 found mitochondrial glutathione markedly oxidized with aging in rats and mice. The oxidized to reduced glutathione ratio increases with age in the liver, kidney, and brain. Oral antioxidants protected against glutathione oxidation and mtDNA damage in rats and mice.
2.7 Mitochondria and neuronal diseases
Neurons are highly susceptible to oxidant stress and oxidation of proteins, lipids, and DNA have been found to be associated with neurodegeneration. Established mitochondrial disorders with identified mutations have found to be causes of neurological dysfunction. Substantial evidence implicates mitochondria in the pathogenesis of some neurodegenerative disorders, including Alzheimer disease, Parkinson disease, and Huntington disease 79. Secondary mitochondrial ETC dysfunction is also involved in the pathogenesis of other, more rare neurological disorders, including Freidreich ataxia and amyotropic lateral sclerosis. The common unifying theme is the production of ROS. The hypothesis is that damage to mtDNA and/or nDNA by ROS, which, in turn, cause mitochondrial ETC dysfunction and further DNA damage that could contribute to the onset of neoplasia.
Although regulations of mitochondrial respiratory chain and their contributions to human diseases are still under intensive investigations, data accumulated in the past decades, as discussed in the present review, have clearly established a critical role of mitochondrial dysfunction in regulating cell metabolic disorders, cell death, initiation and development of human pathologies including cardiovascular diseases, diabetes, aging and neurodegenerative disorders. Mitochondrion likely serves as a primary source for reactive oxygen species (ROS) productions in cells or organ systems with high respiratory rates such as cardiomyocytes. In other cell systems such as vascular endothelium and smooth muscle, it may serve as secondary system to help amplify ROS productions to result in sustained oxidant stress. By these related yet distinctive mechanisms, mitochondrion can thus contribute to the oxidant stress-related pathogenesis of heart diseases and atherosclerosis 18, 19. Antioxidants targeting different systems at different stages of the diseases may therefore prove critical in attenuating oxidant stress-related tissue injuries. Mitochondrial or cytosolic ROS may both be involved in causing mtDNA damages, which uniquely would result in OXPHOS dysfunction and additional ROS productions from mitochondria. Strategies specifically preserving mtDNA from oxidative damage may thus prove of significant therapeutic potential in treating cardiovascular diseases.
Figure 1. Mitochondrial ROS and antioxidant network.
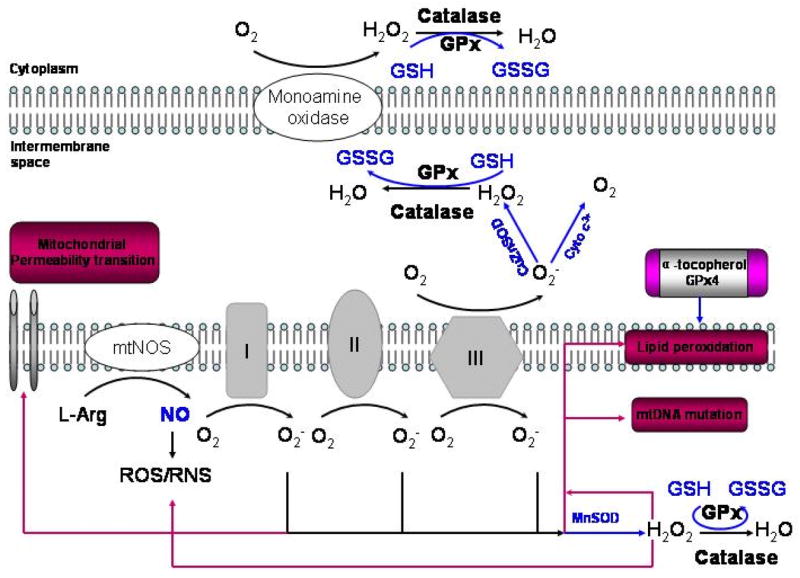
Electron leak to oxygen through complexes I and III can generate superoxide anion. The rate of O2− production is affected by mitochondrial metabolic state. Recent studies suggest that complex I releases O2− into the matrix while complex III can release O2− into the matrix as well as the intermembrane space. Superoxide anion can be converted to H2O2 by mitochondrial matrix enzyme MnSOD or by CuZnSOD in the intermembrane space. H2O2 is more stable than O2− and can diffuse out of the mitochondrion and into the cytosol. O2− can also react with another free radical, nitric oxide (NO−), formed by mitochondrial nitric oxide synthase, to generate the highly reactive peroxynitrite (ONOO−). However, mitochondria are normally protected from oxidative damage by a multilayer network of mitochondrial antioxidant system. H2O2 can be readily converted to water by mitochondrial glutathione peroxidase (GP), which oxidizes reduced glutathione (GSH) to oxidized glutathione (GSSG). In addition to GSH, mitochondria have other small thiols such as α-tocopherol and glutaredoxin which play important roles in thiol redox control and scavenging lipid peroxyl radicals. Data from 22.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES CITED
- 1.Lehninger AL. Phosphorylation coupled to oxidation of dihydrodiphosphopyridine nucleotide. J Biol Chem. 1951051951;190(1):345–359. [PubMed] [Google Scholar]
- 2.Kroemer G, Dallaporta B, Resche-Rigon M. The mitochondrial death/life regulator in apoptosis and necrosis. Annu Rev Physiol. 19981998;60:619–642. doi: 10.1146/annurev.physiol.60.1.619. [DOI] [PubMed] [Google Scholar]
- 3.Campbell NA, Reece JB. Biology. 6. San Francisco: Benjamin Cummings; 2002. [Google Scholar]
- 4.Lyamzaev KG, Izyumov DS, Avetisyan AV, Yang F, Pletjushkina OY, Chernyak BV. Inhibition of mitochondrial bioenergetics: the effects on structure of mitochondria in the cell and on apoptosis. Acta Biochim Pol. 20042004;51(2):553–562. [PubMed] [Google Scholar]
- 5.Lardy H, Reed P, Lin CH. Antibiotic inhibitors of mitochondrial ATP synthesis. Fed Proc. 1975071975;34(8):1707–1710. [PubMed] [Google Scholar]
- 6.Gray MW, Burger G, Lang BF. Genome Biol. 6. Vol. 2. 20012001. The origin and early evolution of mitochondria; p. REVIEWS1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Drew B, Leeuwenburgh C. Ageing and subcellular distribution of mitochondria: role of mitochondrial DNA deletions and energy production. Acta Physiol Scand. 2004122004;182(4):333–341. doi: 10.1111/j.1365-201X.2004.01371.x. [DOI] [PubMed] [Google Scholar]
- 8.Balaban RS. Regulation of oxidative phosphorylation in the mammalian cell. Am J Physiol. 1990031990;258(3 Pt 1):C377–C389. doi: 10.1152/ajpcell.1990.258.3.C377. [DOI] [PubMed] [Google Scholar]
- 9.Clayton DA. Replication of animal mitochondrial DNA. Cell. 1982041982;28(4):693–705. doi: 10.1016/0092-8674(82)90049-6. [DOI] [PubMed] [Google Scholar]
- 10.Zeviani M, Tiranti V, Piantadosi C. Mitochondrial disorders. Medicine (Baltimore) 1998011998;77(1):59–72. doi: 10.1097/00005792-199801000-00006. [DOI] [PubMed] [Google Scholar]
- 11.Wallace DC. Structure and evolution of organelle genomes. Microbiol Rev. 1982061982;46(2):208–240. doi: 10.1128/mr.46.2.208-240.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Montoya J, Ojala D, Attardi G. Distinctive features of the 5′-terminal sequences of the human mitochondrial mRNAs. Nature. 198104091981;290(5806):465–470. doi: 10.1038/290465a0. [DOI] [PubMed] [Google Scholar]
- 13.Larsen NB, Rasmussen M, Rasmussen LJ. Nuclear and mitochondrial DNA repair: similar pathways? Mitochondrion. 2005042005;5(2):89–108. doi: 10.1016/j.mito.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 14.Gutierrez G, Mendoza C, Montano LF, Lopez-Marure R. Ceramide induces early and late apoptosis in human papilloma virus+ cervical cancer cells by inhibiting reactive oxygen species decay, diminishing the intracellular concentration of glutathione and increasing nuclear factor-kappaB translocation. Anticancer Drugs. 200702007;18(2):149–159. doi: 10.1097/CAD.0b013e3280115111. [DOI] [PubMed] [Google Scholar]
- 15.Bohr VA, Stevnsner T, de Souza-Pinto NC. Mitochondrial DNA repair of oxidative damage in mammalian cells. Gene. 200203062002;286(1):127–134. doi: 10.1016/s0378-1119(01)00813-7. [DOI] [PubMed] [Google Scholar]
- 16.Balaban RS, Nemoto S, Finkel T. Mitochondria, oxidants, and aging. Cell. 200502252005;120(4):483–495. doi: 10.1016/j.cell.2005.02.001. [DOI] [PubMed] [Google Scholar]
- 17.Conn PM. Handbook of Models for Human Aging. Elsevier; 2006. [Google Scholar]
- 18.Cai H, Griendling KK, Harrison DG. The vascular NAD(P)H oxidases as therapeutic targets in cardiovascular diseases. Trends Pharmacol Sci. 2003 Sep;24(9):471–478. doi: 10.1016/S0165-6147(03)00233-5. [DOI] [PubMed] [Google Scholar]
- 19.Cai H. NAD(P)H oxidase-dependent self-propagation of hydrogen peroxide and vascular disease. Circ Res. 2005 Apr 29;96(8):818–822. doi: 10.1161/01.RES.0000163631.07205.fb. [DOI] [PubMed] [Google Scholar]
- 20.Li J-M, Shah AM. Endothelial cell superoxide generation: regulation and relevance for cardiovascular pathophysiology 10.1152/ajpregu.00124.2004. Am J Physiol Regul Integr Comp Physiol. 2004 November 1, 2004;287(5):R1014–1030. doi: 10.1152/ajpregu.00124.2004. [DOI] [PubMed] [Google Scholar]
- 21.Raha S, Robinson BH. Mitochondria, oxygen free radicals, disease and ageing. Trends Biochem Sci. 2000102000;25(10):502–508. doi: 10.1016/s0968-0004(00)01674-1. [DOI] [PubMed] [Google Scholar]
- 22.Turrens JF. Mitochondrial formation of reactive oxygen species. J Physiol. 200310152003;552(Pt 2):335–344. doi: 10.1113/jphysiol.2003.049478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guzy RD, Schumacker PT. Oxygen sensing by mitochondria at complex III: the paradox of increased reactive oxygen species during hypoxia 10.1113/expphysiol.2006.033506. Exp Physiol. 2006 September 1, 2006;91(5):807–819. doi: 10.1113/expphysiol.2006.033506. [DOI] [PubMed] [Google Scholar]
- 24.Waypa GB, Schumacker PT. Hypoxic pulmonary vasoconstriction: redox events in oxygen sensing 10.1152/japplphysiol.00722.2004. J Appl Physiol. 2005 January 1, 2005;98(1):404–414. doi: 10.1152/japplphysiol.00722.2004. [DOI] [PubMed] [Google Scholar]
- 25.Muller FL, Liu Y, Van RH. Complex III releases superoxide to both sides of the inner mitochondrial membrane. J Biol Chem. 200411192004;279(47):49064–49073. doi: 10.1074/jbc.M407715200. [DOI] [PubMed] [Google Scholar]
- 26.Andreyev AY, Kushnareva YE, Starkov AA. Mitochondrial metabolism of reactive oxygen species. Biochemistry (Mosc) 2005022005;70(2):200–214. doi: 10.1007/s10541-005-0102-7. [DOI] [PubMed] [Google Scholar]
- 27.Gyulkhandanyan AV, Pennefather PS. Shift in the localization of sites of hydrogen peroxide production in brain mitochondria by mitochondrial stress. J Neurochem. 2004072004;90(2):405–421. doi: 10.1111/j.1471-4159.2004.02489.x. [DOI] [PubMed] [Google Scholar]
- 28.Kushnareva Y, Murphy AN, Andreyev A. Complex I-mediated reactive oxygen species generation: modulation by cytochrome c and NAD(P)+ oxidation-reduction state. Biochem J. 200212012002;368(Pt 2):545–553. doi: 10.1042/BJ20021121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ji LL. Antioxidants and Oxidative Stress in Exercise. Proc Soc Exp Biol Med. 1999 December 1, 1999;222(3):283–292. doi: 10.1046/j.1525-1373.1999.d01-145.x. [DOI] [PubMed] [Google Scholar]
- 30.Brigelius-Flohe R. Glutathione peroxidases and redox-regulated transcription factors. Biol Chem. 2006 Oct–Nov;387(10–11):1329–1335. doi: 10.1515/BC.2006.166. [DOI] [PubMed] [Google Scholar]
- 31.Torzewski M, Ochsenhirt V, Kleschyov AL, et al. Deficiency of glutathione peroxidase-1 accelerates the progression of atherosclerosis in apolipoprotein E-deficient mice. Arterioscler Thromb Vasc Biol. 2007 Apr;27(4):850–857. doi: 10.1161/01.ATV.0000258809.47285.07. [DOI] [PubMed] [Google Scholar]
- 32.Esworthy RS, Aranda R, Martin MG, Doroshow JH, Binder SW, Chu FF. Mice with combined disruption of Gpx1 and Gpx2 genes have colitis. Am J Physiol Gastrointest Liver Physiol. 2001 Sep;281(3):G848–855. doi: 10.1152/ajpgi.2001.281.3.G848. [DOI] [PubMed] [Google Scholar]
- 33.Ran Q, Van Remmen H, Gu M, et al. Embryonic fibroblasts from Gpx4+/− mice: a novel model for studying the role of membrane peroxidation in biological processes. Free Radic Biol Med. 2003 Nov 1;35(9):1101–1109. doi: 10.1016/s0891-5849(03)00466-0. [DOI] [PubMed] [Google Scholar]
- 34.Patwari P, Lee RT. Thioredoxins, mitochondria, and hypertension. Am J Pathol. 2007032007;170(3):805–808. doi: 10.2353/ajpath.2007.061243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Giulivi C, Kato K, Cooper CE. Nitric oxide regulation of mitochondrial oxygen consumption I: cellular physiology. Am J Physiol Cell Physiol. 2006122006;291(6):C1225–C1231. doi: 10.1152/ajpcell.00307.2006. [DOI] [PubMed] [Google Scholar]
- 36.Davidson SM, Duchen MR. Endothelial mitochondria: contributing to vascular function and disease. Circ Res. 200704272007;100(8):1128–1141. doi: 10.1161/01.RES.0000261970.18328.1d. [DOI] [PubMed] [Google Scholar]
- 37.Ghafourifar P, Richter C. Nitric oxide synthase activity in mitochondria. FEBS Lett. 199712011997;418(3):291–296. doi: 10.1016/s0014-5793(97)01397-5. [DOI] [PubMed] [Google Scholar]
- 38.Dedkova EN, Ji X, Lipsius SL, Blatter LA. Mitochondrial calcium uptake stimulates nitric oxide production in mitochondria of bovine vascular endothelial cells. Am J Physiol Cell Physiol. 2004022004;286(2):C406–C415. doi: 10.1152/ajpcell.00155.2003. [DOI] [PubMed] [Google Scholar]
- 39.Rakhit RD, Mojet MH, Marber MS, Duchen MR. Mitochondria as targets for nitric oxide-induced protection during simulated ischemia and reoxygenation in isolated neonatal cardiomyocytes. Circulation. 200105292001;103(21):2617–2623. doi: 10.1161/01.cir.103.21.2617. [DOI] [PubMed] [Google Scholar]
- 40.Sarkela TM, Berthiaume J, Elfering S, Gybina AA, Giulivi C. The modulation of oxygen radical production by nitric oxide in mitochondria. J Biol Chem. 200103092001;276(10):6945–6949. doi: 10.1074/jbc.M007625200. [DOI] [PubMed] [Google Scholar]
- 41.Elfering SL, Haynes VL, Traaseth NJ, Ettl A, Giulivi C. Aspects, mechanism, and biological relevance of mitochondrial protein nitration sustained by mitochondrial nitric oxide synthase. Am J Physiol Heart Circ Physiol. 2004012004;286(1):H22–H29. doi: 10.1152/ajpheart.00766.2003. [DOI] [PubMed] [Google Scholar]
- 42.Traaseth N, Elfering S, Solien J, Haynes V, Giulivi C. Role of calcium signaling in the activation of mitochondrial nitric oxide synthase and citric acid cycle. Biochim Biophys Acta. 200407232004;1658(1–2):64–71. doi: 10.1016/j.bbabio.2004.04.015. [DOI] [PubMed] [Google Scholar]
- 43.Nisoli E, Clementi E, Paolucci C, et al. Mitochondrial biogenesis in mammals: the role of endogenous nitric oxide. Science. 200302072003;299(5608):896–899. doi: 10.1126/science.1079368. [DOI] [PubMed] [Google Scholar]
- 44.Momken I, Fortin D, Serrurier B, Bigard X, Ventura-Clapier R, Veksler V. Endothelial nitric oxide synthase (NOS) deficiency affects energy metabolism pattern in murine oxidative skeletal muscle. Biochem J. 200211152002;368(Pt 1):341–347. doi: 10.1042/BJ20020591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zimmermann KC, Green DR. How cells die: apoptosis pathways. J Allergy Clin Immunol. 2001102001;108(4 Suppl):S99–103. doi: 10.1067/mai.2001.117819. [DOI] [PubMed] [Google Scholar]
- 46.Ashkenazi A, Dixit VM. Death receptors: signaling and modulation. Science. 199808281998;281(5381):1305–1308. doi: 10.1126/science.281.5381.1305. [DOI] [PubMed] [Google Scholar]
- 47.Kumar D, Jugdutt BI. Apoptosis and oxidants in the heart. J Lab Clin Med. 2003112003;142(5):288–297. doi: 10.1016/S0022-2143(03)00148-3. [DOI] [PubMed] [Google Scholar]
- 48.Ballinger SW. Mitochondrial dysfunction in cardiovascular disease. Free Radic Biol Med. 200505152005;38(10):1278–1295. doi: 10.1016/j.freeradbiomed.2005.02.014. [DOI] [PubMed] [Google Scholar]
- 49.Tsutsui H. Oxidative stress in heart failure: the role of mitochondria. Intern Med. 2001 Dec;40(12):1177–1182. doi: 10.2169/internalmedicine.40.1177. [DOI] [PubMed] [Google Scholar]
- 50.Herdener M, Heigold S, Saran M, Bauer G. Target cell-derived superoxide anions cause efficiency and selectivity of intercellular induction of apoptosis. Free Radic Biol Med. 200012152000;29(12):1260–1271. doi: 10.1016/s0891-5849(00)00422-6. [DOI] [PubMed] [Google Scholar]
- 51.Madamanchi NR, Vendrov A, Runge MS. Oxidative stress and vascular disease. Arterioscler Thromb Vasc Biol. 2005012005;25(1):29–38. doi: 10.1161/01.ATV.0000150649.39934.13. [DOI] [PubMed] [Google Scholar]
- 52.Matsushima S, Ide T, Yamato M, et al. Overexpression of mitochondrial peroxiredoxin-3 prevents left ventricular remodeling and failure after myocardial infarction in mice. Circulation. 2006 Apr 11;113(14):1779–1786. doi: 10.1161/CIRCULATIONAHA.105.582239. [DOI] [PubMed] [Google Scholar]
- 53.Fuhrman B, Volkova N, Aviram M. Oxidative stress increases the expression of the CD36 scavenger receptor and the cellular uptake of oxidized low-density lipoprotein in macrophages from atherosclerotic mice: protective role of antioxidants and of paraoxonase. Atherosclerosis. 2002042002;161(2):307–316. doi: 10.1016/s0021-9150(01)00646-3. [DOI] [PubMed] [Google Scholar]
- 54.Mikaelian NP, Khalilov EM, Ivanov AS, Fortinskaia ES, Lopukhin I. [Mitochondrial enzymes in circulating lymphocytes during hemosorption for experimental hypercholesterolemia] Biull Eksp Biol Med. 1983091983;96(9):35–37. [PubMed] [Google Scholar]
- 55.Kinscherf R, Deigner HP, Usinger C, et al. Induction of mitochondrial manganese superoxide dismutase in macrophages by oxidized LDL: its relevance in atherosclerosis of humans and heritable hyperlipidemic rabbits. FASEB J. 1997121997;11(14):1317–1328. doi: 10.1096/fasebj.11.14.9409551. [DOI] [PubMed] [Google Scholar]
- 56.Martinet W, Knaapen MW, De Meyer GR, Herman AG, Kockx MM. Oxidative DNA damage and repair in experimental atherosclerosis are reversed by dietary lipid lowering. Circ Res. 200104132001;88(7):733–739. doi: 10.1161/hh0701.088684. [DOI] [PubMed] [Google Scholar]
- 57.Sreekumar R, Unnikrishnan J, Fu A, et al. Impact of high-fat diet and antioxidant supplement on mitochondrial functions and gene transcripts in rat muscle. Am J Physiol Endocrinol Metab. 2002052002;282(5):E1055–E1061. doi: 10.1152/ajpendo.00554.2001. [DOI] [PubMed] [Google Scholar]
- 58.Tsutsui H, Ide T, Kinugawa S. Mitochondrial oxidative stress, DNA damage, and heart failure. Antioxid Redox Signal. 2006 Sep–Oct;8(9–10):1737–1744. doi: 10.1089/ars.2006.8.1737. [DOI] [PubMed] [Google Scholar]
- 59.Rosenberger D, Moshal KS, Kartha GK, et al. Arrhythmia and neuronal/endothelial myocyte uncoupling in hyperhomocysteinemia*. Arch Physiol Biochem. 2006102006;112(4):219–227. doi: 10.1080/13813450601093443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ikeuchi M, Matsusaka H, Kang D, et al. Overexpression of mitochondrial transcription factor a ameliorates mitochondrial deficiencies and cardiac failure after myocardial infarction. Circulation. 2005 Aug 2;112(5):683–690. doi: 10.1161/CIRCULATIONAHA.104.524835. [DOI] [PubMed] [Google Scholar]
- 61.Kato K, Fukuma N, Kimura-Kato Y, et al. Improvement of sympathetic response to exercise by oral administration of ascorbic acid in patients after myocardial infarction. Int J Cardiol. 2006 Aug 10;111(2):240–246. doi: 10.1016/j.ijcard.2005.07.020. [DOI] [PubMed] [Google Scholar]
- 62.Neuzil J, Widen C, Gellert N, et al. Mitochondria transmit apoptosis signalling in cardiomyocyte-like cells and isolated hearts exposed to experimental ischemia-reperfusion injury. Redox Rep. 20072007;12(3):148–162. doi: 10.1179/135100007X200227. [DOI] [PubMed] [Google Scholar]
- 63.Chen Q, Camara AK, Stowe DF, Hoppel CL, Lesnefsky EJ. Modulation of electron transport protects cardiac mitochondria and decreases myocardial injury during ischemia and reperfusion. Am J Physiol Cell Physiol. 2007012007;292(1):C137–C147. doi: 10.1152/ajpcell.00270.2006. [DOI] [PubMed] [Google Scholar]
- 64.Jones SP, Bolli R. The ubiquitous role of nitric oxide in cardioprotection. J Mol Cell Cardiol. 2006 Jan;40(1):16–23. doi: 10.1016/j.yjmcc.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 65.Stein AB, Bolli R, Guo Y, et al. The late phase of ischemic preconditioning induces a prosurvival genetic program that results in marked attenuation of apoptosis. J Mol Cell Cardiol. 2007 Jun;42(6):1075–1085. doi: 10.1016/j.yjmcc.2007.03.908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stein AB, Tang XL, Guo Y, Xuan YT, Dawn B, Bolli R. Delayed adaptation of the heart to stress: late preconditioning. Stroke. 2004 Nov;35(11 Suppl 1):2676–2679. doi: 10.1161/01.STR.0000143220.21382.fd. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Campagne MV, Thibodeaux H, van Bruggen N, Cairns B, Lowe DG. Increased binding activity at an antioxidant-responsive element in the metallothionein-1 promoter and rapid induction of metallothionein-1 and -2 in response to cerebral ischemia and reperfusion. J Neurosci. 2000 Jul 15;20(14):5200–5207. doi: 10.1523/JNEUROSCI.20-14-05200.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Steurer J, Hoffmann U, Dur P, Russi E, Vetter W. Changes in arterial and transcutaneous oxygen and carbon dioxide tensions during and after voluntary hyperventilation. Respiration. 19971997;64(3):200–205. doi: 10.1159/000196671. [DOI] [PubMed] [Google Scholar]
- 69.Olive PL, Banath JP, Durand RE. The range of oxygenation in SiHa tumor xenografts. Radiat Res. 2002082002;158(2):159–166. doi: 10.1667/0033-7587(2002)158[0159:troois]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 70.Guzy RD, Hoyos B, Robin E, et al. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab. 2005 Jun;1(6):401–408. doi: 10.1016/j.cmet.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 71.Pouyssegur J, Mechta-Grigoriou F. Redox regulation of the hypoxia-inducible factor. Biol Chem. 2006102006;387(10–11):1337–1346. doi: 10.1515/BC.2006.167. [DOI] [PubMed] [Google Scholar]
- 72.Wenger RH. Mitochondria: oxygen sinks rather than sensors? Med Hypotheses. 20062006;66(2):380–383. doi: 10.1016/j.mehy.2005.08.047. [DOI] [PubMed] [Google Scholar]
- 73.Chandel NS, Budinger GR. The cellular basis for diverse responses to oxygen. Free Radic Biol Med. 200701152007;42(2):165–174. doi: 10.1016/j.freeradbiomed.2006.10.048. [DOI] [PubMed] [Google Scholar]
- 74.Semenza GL. Perspectives on oxygen sensing. Cell. 199908061999;98(3):281–284. doi: 10.1016/s0092-8674(00)81957-1. [DOI] [PubMed] [Google Scholar]
- 75.Huang H, Manton KG. The role of oxidative damage in mitochondria during aging: a review. Front Biosci. 200405012004;9:1100–1117. doi: 10.2741/1298. [DOI] [PubMed] [Google Scholar]
- 76.Lee HC, Wei YH. Oxidative stress, mitochondrial DNA mutation, and apoptosis in aging. Exp Biol Med(Maywood) 2007052007;232(5):592–606. [PubMed] [Google Scholar]
- 77.Liu L, Trimarchi JR, Smith PJ, Keefe DL. Mitochondrial dysfunction leads to telomere attrition and genomic instability. Aging Cell. 2002102002;1(1):40–46. doi: 10.1046/j.1474-9728.2002.00004.x. [DOI] [PubMed] [Google Scholar]
- 78.de la Asuncion JG, Del Olmo ML, Sastre J, et al. AZT treatment induces molecular and ultrastructural oxidative damage to muscle mitochondria. Prevention by antioxidant vitamins. J Clin Invest. 199807011998;102(1):4–9. doi: 10.1172/JCI1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.McKenzie M, Liolitsa D, Hanna MG. Mitochondrial disease: mutations and mechanisms. Neurochem Res. 2004032004;29(3):589–600. doi: 10.1023/b:nere.0000014829.42364.dd. [DOI] [PubMed] [Google Scholar]
- 80.Kowluru RA, Abbas SN. Diabetes-induced mitochondrial dysfunction in the retina. Invest Ophthalmol Vis Sci. 2003122003;44(12):5327–5334. doi: 10.1167/iovs.03-0353. [DOI] [PubMed] [Google Scholar]
- 81.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 200112132001;414(6865):813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 82.Ghosh S, Pulinilkunnil T, Yuen G, et al. Cardiomyocyte apoptosis induced by short-term diabetes requires mitochondrial GSH depletion. Am J Physiol Heart Circ Physiol. 2005082005;289(2):H768–H776. doi: 10.1152/ajpheart.00038.2005. [DOI] [PubMed] [Google Scholar]
- 83.Enns GM. The contribution of mitochondria to common disorders. Mol Genet Metab. 2003 Sep–Oct;80(1–2):11–26. doi: 10.1016/j.ymgme.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 84.Gardner PR, Raineri I, Epstein LB, White CW. Superoxide radical and iron modulate aconitase activity in mammalian cells. J Biol Chem. 1995 Jun 2;270(22):13399–13405. doi: 10.1074/jbc.270.22.13399. [DOI] [PubMed] [Google Scholar]
- 85.Radi R, Turrens JF, Chang LY, Bush KM, Crapo JD, Freeman BA. Detection of catalase in rat heart mitochondria. J Biol Chem. 1991 Nov 15;266(32):22028–22034. [PubMed] [Google Scholar]
- 86.Das KC, Pahl PMB, Guo X-L, White CW. Induction of Peroxiredoxin Gene Expression by Oxygen in Lungs of Newborn Primates. Am J Respir Cell Mol Biol. 2001 August 1, 2001;25(2):226–232. doi: 10.1165/ajrcmb.25.2.4314. [DOI] [PubMed] [Google Scholar]
- 87.Dringen R. Metabolism and functions of glutathione in brain. Prog Neurobiol. 2000 Dec;62(6):649–671. doi: 10.1016/s0301-0082(99)00060-x. [DOI] [PubMed] [Google Scholar]
- 88.Zoccarato F, Cavallini L, Alexandre A. Respiration-dependent removal of exogenous H2O2 in brain mitochondria: inhibition by Ca2+ J Biol Chem. 2004 Feb 6;279(6):4166–4174. doi: 10.1074/jbc.M308143200. [DOI] [PubMed] [Google Scholar]
- 89.Maiorino M, Chu FF, Ursini F, Davies KJ, Doroshow JH, Esworthy RS. Phospholipid hydroperoxide glutathione peroxidase is the 18-kDa selenoprotein expressed in human tumor cell lines. J Biol Chem. 1991 Apr 25;266(12):7728–7732. [PubMed] [Google Scholar]
- 90.Bao Y, Williamson G. Phospholipid hydroperoxide peroxidase activities in erythrocytes. Biochem Soc Trans. 1997 Nov;25(4):S557. doi: 10.1042/bst025s557. [DOI] [PubMed] [Google Scholar]
- 91.Arner ES, Holmgren A. The thioredoxin system in cancer. Semin Cancer Biol. 2006 Dec;16(6):420–426. doi: 10.1016/j.semcancer.2006.10.009. [DOI] [PubMed] [Google Scholar]
- 92.Yant LJ, Ran Q, Rao L, et al. The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic Biol Med. 2003 Feb 15;34(4):496–502. doi: 10.1016/s0891-5849(02)01360-6. [DOI] [PubMed] [Google Scholar]
- 93.Wood ZA, Schroder E, Robin Harris J, Poole LB. Structure, mechanism and regulation of peroxiredoxins. Trends Biochem Sci. 2003 Jan;28(1):32–40. doi: 10.1016/s0968-0004(02)00003-8. [DOI] [PubMed] [Google Scholar]
- 94.Parker C, 3rd, Vita JA, Freedman JE. Soluble adhesion molecules and unstable coronary artery disease. Atherosclerosis. 2001 Jun;156(2):417–424. doi: 10.1016/s0021-9150(00)00672-9. [DOI] [PubMed] [Google Scholar]
- 95.Pastore A, Federici G, Bertini E, Piemonte F. Analysis of glutathione: implication in redox and detoxification. Clin Chim Acta. 2003 Jul 1;333(1):19–39. doi: 10.1016/s0009-8981(03)00200-6. [DOI] [PubMed] [Google Scholar]
- 96.Kirsch M, De Groot H. NAD(P)H, a directly operating antioxidant? Faseb J. 2001 Jul;15(9):1569–1574. doi: 10.1096/fj.00-0823hyp. [DOI] [PubMed] [Google Scholar]
- 97.Pereverzev MO, Vygodina TV, Konstantinov AA, Skulachev VP. Cytochrome c, an ideal antioxidant. Biochem Soc Trans. 2003 Dec;31(Pt 6):1312–1315. doi: 10.1042/bst0311312. [DOI] [PubMed] [Google Scholar]