

Abstract
α-Internexin and the neurofilament triplet proteins (NF-L, NF-M, and NF-H) coassemble into intermediate filament networks in neurons. We have found that the RE1 silencing transcription factor (REST) plays a contributory role in the neuron-specific expression of the α-internexin, NF-H and NF-M genes. Chromatin immunoprecipitation and transient transfection experiments performed with neuronal CAD cells and non-neuronal NIH3T3 cells demonstrated that REST repressed transcription of these genes in NIH3T3 cells by binding and recruiting mSin3A, CoREST, HDAC1 and MeCP2 to the RE1 sites in the intron-1 of α-internexin and the 5′ flanking regions of NF-H and NF-M. No repression effect of the RE1 sites was observed in CAD cells, which express these neuronal genes but not REST. Treatment of NIH3T3 cells with trichostatin A (TSA), a HDAC inhibitor, relieved the REST-mediated repression and induced ectopic activation of α-internexin, NF-H and NF-M. The TSA treatment did not affect the levels of REST occupancy but caused coordinated changes in acetylation and methylation of histones around the RE1 sites of these genes in NIH3T3 cells consistent with a transition from transcriptional repression to transcriptional activation. Thus, REST regulates expression of these neuronal genes, partly by a HDAC-dependent epigenetic mechanism.
Keywords: α-internexin, neurofilament, REST, histone modification, transcription, neuronal intermediate filament
INTRODUCTION
α-Internexin and the neurofilament triplet proteins (NFTPs) NF-L, NF-M, and NF-H are neuronal intermediate filament (IF) proteins found in most neurons of the central and peripheral nervous systems (Ching & Liem 2006, Fliegner & Liem 1991). α-Internexin is found primarily in the central nervous system and minimally in the peripheral nervous system, while the NFTPs are expressed abundantly in both the central and peripheral nervous systems (Fliegner et al. 1994, Kaplan et al. 1990). α–Internexin can self-assemble and co-assemble with the NFTPs into filamentous networks, co-localizes with the NFTPs in most axons (Ching & Liem 1993, Ching & Liem 1998, Yuan et al. 2006), and may play a role in neuronal regeneration (McGraw et al. 2002). In contrast, NFTPs are obligate heteropolymers (Lee et al. 1993) and are determinants of axonal caliber (Jacomy et al. 1999, Rao et al. 2003, Xu et al. 1996). Abnormal accumulations and mutations of these neuronal IF proteins are associated with human neurodegenerative diseases (Ching & Liem 2006).
The RE1 silencing transcription factor REST (also known as neuron restrictive silencing factor NRSF) is a Kruppel-type zinc finger protein that binds a 21 bp DNA motif RE1 (also called NRSE) present in the regulatory regions of its target genes and silences or represses many neuronal genes, such as ion channels, neurotransmitter receptors and neurotrophic factors in nonneuronal cells (Bruce et al. 2004, Chong et al. 1995, Schoenherr & Anderson 1995, Schoenherr et al. 1996). In addition to its high levels of expression in non-neuronal cells, REST is expressed in embryonic stem cells and is posttranslationally degraded to minimal or undetectable levels during differentiation to neural progenitors and post-mitotic neurons, resulting in expression of neuronal genes (Ballas et al. 2005, Westbrook et al. 2008). However, REST is detected in certain mature neurons, where it regulates rather than silences gene expression (Calderone et al. 2003, Sun et al. 2005, Palm et al. 1998). Thus, REST appears to play diverse and complex roles in gene regulation in different cell types.
REST consists of three functional domains: a zinc-finger domain that binds the RE1 motif and two independent repression domains, one at the amino-terminus and one at the carboxyl-terminus (Tapia-Ramirez et al. 1997). The amino-terminal and carboxyl-terminal repression domains interact with corepressors mSin3A/B (Grimes et al. 2000, Huang et al. 1999, Naruse et al. 1999, Roopra et al. 2000) and CoREST (Andres et al. 1999, Ballas et al. 2001), respectively, which in turn, recruit histone deacetylases HDAC1 and HDAC2. CoREST also interacts with methyl-CpG-binding protein MeCP2, histone H3-lysine 4 demethylase LSD1, histone H3-lysine 9 methyltransferases G9a and chromatin-remodeling enzyme BRG1 (Battaglioli et al. 2002, Lunyak et al. 2002, Ooi et al. 2006, Roopra et al. 2004, Shi et al. 2004, Shi et al. 2003). Thus, REST regulates transcription of its target genes via chromatin modifications by recruiting multiple complexes of its cofactors to the RE1 site.
In eukaryotes, the basic unit of chromatin is the nucleosome, which consists of 147 bp DNA wrapped 1.65 turns around a histone octamer containing two copies of each histone: H2A, H2B, H3 and H4. The histone proteins are subjected to a variety of posttranslational modifications including acetylation, methylation, phosphorylation, ubiquitylation and sumoylation (reviewed in (Li et al. 2007)). Most, if not all, histone modifications are reversible. Acetylation and methylation of the lysine residues on histones H3 and H4 are the best-studied histone modifications. In general, acetylation of lysine residues on histones H3 and H4 by histone acetyl transferases correlates with active transcription while their deacetylation by HDACs reduces transcription and facilitates transcriptional repression (Dion et al. 2005, Kurdistani et al. 2004, Shahbazian & Grunstein 2007). Lysine residues of histones H3 and H4 can be mono-, di-, and trimethylated and unlike acetylation, lysine methylation can be associated with transcriptional activation or repression depending on the precise residues and contexts. For example, methylation of lysine 4 on histone H3 (H3K4) is associated with active transcription whereas di- and trimethylation of lysine 9 on histone H3 (H3K9) is associated with gene repression or silencing (Bernstein et al. 2005, Litt et al. 2001, Schubeler et al. 2004, Vakoc et al. 2006). Methylation of these lysine residues are regulated by histone methyl transferases and demethylases, and three of these enzymes, the H3K9 methyltransferase G9a and the H3K4 demethylases LSD1 and SMCX, are recruited as cofactor complexes by REST (Lee et al. 2005, Roopra et al. 2004, Shi et al. 2004, Tahiliani et al. 2007).
Little is known about the neuron-specific regulation of α–internexin and NFTPs. As the first step toward elucidating the mechanism that regulates their neuron-specific expression, we have undertaken the present study to show that REST represseses expression of these genes in nonneuronal cells via a HDAC-dependent pathway, involving crosstalk between acetylation and methylation of histones around the RE1 sites.
MATERIALS AND METHODS
Identification of RE1 motifs
α-Internexin and the neurofilament gene sequences were obtained from GenBank. To identify RE1 motifs, computational analysis of these genes, including 2–3 kb of the 5′ flanking regions, was performed using the MatInspector program that searches for transcription factor binding sites (http://www.genomatix.de/matinspector.html). The solution parameters of the MatInspector program were set for “matrix family” and ‘optimized” matrix similarity threshold.
Generation of Plasmid Constructs
The 5′ flanking sequences of rat α–internexin, mouse NF-H and NF-M genes were amplified by PCR using the following primers containing XhoI and HindIII cloning sites: 5′-CTCGAGGAATTCTGAACTACAAAGCAAAGCGCT-3′ and 5′-AAGCTTGGTGCCGGGGTCGGGGCCGGGTGCGCG-3′ for α internexin nucleotides −1219 to +73; 5′-CTCGAGCGTTAGGCATAGATTTAACCCTTCCCA-3′ and 5′-AAGCTTGGCCGGAGCAGGTGCGGCCGCCAG-3′ for NF-H nucleotides − 1392 to +130; 5′-CTCGAGCCACTAACACCGCTTTTGACCGGA-3′ and 5′-AAGCTTGGCCGGAGCAGGTGCGGCCGCCAG-3′ for NF-H nucleotides 1308 to +130; 5′-CTCGAGCTGGTAGGCTGCGAACCCAGCAGAATC-3′ and 5′-AAGCTTGGGTGTGTGGCTGTCACAGCGTTCTGC-3′ for NF-M nucleotides −943 to +17; 5′-CTCGAGGATCCTTGGGCGTCACAGCGCCTTAC-3′ and 5′-AAGCTTGGGTGTGTGGCTGTCACAGCGTTCTGC-3′ for NF-M nucleotides − 879 to +17. Genomic DNA extracted from mouse brain was used as DNA templates for NF-H and NF-M amplification and pSPN-αES16K (Ching et al. 1999) was used as a DNA template for α–internexin amplification. After restriction digestion and purification, the PCR fragments were inserted into the XhoI and HindIII cloning sites of the firefly luciferase-containing vector pGL3-basic (Promega, Madison, WI) to generate pα (−1219/+73)luc, pH(−1392/+130)luc, pH(−1308/+130)luc, pM(−943/+17)luc and pM(−879/+17)luc. To construct pα (−1219/+73)luc (1878/2012 RE) and pSV40-luc-α (1878/2012 RE), the α–internexin sequence of nucleotides 1878 to 2012 amplified by PCR using the forward primer 5′-GGATCCTTGTCCCTCTACTAAGCAAGCTTTGAG -3′ and the reverse primer 5′-GTCGACTAACTGGGACAATTTGGAAGCATAATG-3′ was inserted into the BamHI and SalI cloning sites downstream of the luciferase gene in pα (−1219/+73)luc and the SV40 promoter-containing vector pGL3-promoter (Promega), respectively. To generate pH(−1392/−1264 RE)-SV40-luc and pM(−943/−801 RE)-SV40-luc, the neurofilament gene sequences were PCR amplified with the following primers: 5′-GCTAGCCGTTAGGCATAGATTTAACCCTTC-3′ and 5′-CTCGAGGGCTCGTTCTTGGTTTGGTTTTCC-3′ for NF-H nucleotides –1392 to –1264; 5′-GCTAGCCTGGTAGGCTGCGAACCCAGCAGA-3′ and 5′-CTCGAGAAGTGGCACTCCAGTCTGCTTAAC-3′ for NF-M nucleotides −943 to −801. After restriction digestion and purification, these PCR fragments were inserted into the NheI and XhoI cloning sites upstream of the SV40 promoter in the pGL3-promoter vector. Mutations of the RE1 motifs in the luciferase-fusion plasmid constructs were performed using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA) with the following primers: 5′-GGTTAGGGCGCTGTAAGAGGTGCTGATCTC-3′ and 5′-GAGATCAGCACCTCTTACAGCGCCCTAACC-3′ for α–internexin; 5′-GCGGCTGCTGTTAGTGGTCATGAAG-3′ and 5′-CTTCATGACCACTAACAGCAGCCGC-3′ for NF-H; 5′-CCTGGGCGCTGTAGTTGGTCATGCC-3′ and 5′-GGCATGACCAACTACAGCGCCCAGG-3′ for NF-M (mutated nucleotides were underlined). The nucleotide sequences of all cloned fragments in the plasmid constructs were verified by DNA sequencing.
The rat REST cDNA sequences were PCR amplified from the adult rat brain Quick-Clone DNA (Clontech laboratories, Mountain View, CA) using the following primers: 5′-CACCATGGCCACCCAGGTGATGGGGCAGTC-3′ and 5′-CGTCCATCTGGGCAGGCTCTGTCTGAGCAA-3′ for the 2-kb 5′ end sequence; 5′-CCTGCTCTCACTCAGGCGGTGGTCACCCCA-3′ and 5′-CTCTACTCCTGCTCCTCAGCTGCTTCTTCA-3′ for the 1.3-kb 3′ end sequence. These PCR fragments were subcloned into the pCR 2.1 TOPO vector (Invitrogen, Carlsbad, CA). After digestion with EcoRI and SacI, the purified 1.9-kb REST 5′-end EcoRI-SacI fragment and 1.3-kb REST 3′-end EcoRI-SacI fragment were ligated to the EcoRI-digested, calf intestine alkaline phosphatase-treated pcDNA3 vector (Invitrogen) to generate pcDNA-REST. The nucleotide sequence of the REST clone was verified by DNA sequencing.
Cell cultures and transient transfection
CAD cells were grown in DMEM/F12 medium supplemented with 8% fetal bovine serum at 37°C and 5% CO2 as described previously (Qi et al. 1997). NIH3T3 cells were grown in DMEM medium supplemented with 10% calf serum at 37°C and 5% CO2 as described (ATCC, Manassas, VA). The cells were transiently transfected with equal molar amounts of the various luciferase-fusion constructs or pGL3-basic in serum-free growth media using LipofectAMINE-plus reagent according to the manufacturer’s protocol (Invitrogen). pRL-tK (Promega) containing the Renilla luciferase gene fused to the herpes simplex virus thymidine kinase promoter was cotransfected in order to normalize transfection efficiencies. For experiments that examine the effects of exogenously expressed REST, pCDNA3 or pCDNA-REST was cotransfected with the luciferase-fusion constructs and pRL-tK. After 3 h incubation with the DNA/LipofectAMINE plus complexes, the cells were grown in fresh media containing serum. The transfected cells were lysed 48 h after transfection and the luciferase activities of the cells lysates were determined using the dual-luciferase reporter assay system (Promega). For experiments that involve treatment of cells with trichostatin A (TSA; Sigma, St. Louis, MO), ethanol or TSA (150 ng/ml) was added 24 h after transfection and the cells were then lysed 24 h later. Transfections were performed in triplicates and each experiment was repeated at least three times. For TSA treatment experiments other than transfections, cells were grown for 24 h, treated with ethanol or TSA (150 ng/ml) for an additional 24 h and then harvested.
Electrophoretic mobility shift assays (EMSA)
Nuclear extracts were prepared as previously described (Slomiany et al. 2000). DNA fragments used as probes were labeled with α32P-dCTP (NEN Life Science Products, Boston, MA) and Klenow enzyme. DNA-protein binding reactions were carried out in a 20-μl final volume of binding buffer containing 20 mM Hepes (pH 7.9), 100 mM KCl, 4 mM MgCl2, 1 mM dithiothreitol, 1 mM EDTA, 5% glycerol and 2 μg of poly (dI-dc)•poly (dI-dc). Nuclear extract (5 μg) was preincubated with (in supershift experiments) or without anti-REST antibody P-18 (Santa Cruz Biotechnology, Santa Cruz, CA) in binding buffer on ice for 20 min. Radioactively labeled DNA probe was then added to each reaction and incubated at room temperature for an additional 20 min. For competition assays, a 100-fold molar excess of unlabeled double-stranded oligonucleotides was added to each reaction. The samples were subjected to electrophoresis in 4.5% nondenaturing polyacrylamide gel in 1X Tris-glycine-EDTA (TGE) buffer at 200 V and 4°C for 2h. To prepare the double-stranded oligonucleotides used in the competition assays, the following single-stranded oligonucleotides containing the wild-type or mutated RE1 motifs were purified by 8M urea-polyacrylamide gels and annealed with their purified complementary partners: 5′-GGGTTAGGGCGCTGTCCGAGGTGCTGATCTCGTG-3′ and 5′-GGGTTAGGGCGCTGTAAGAGGTGCTGATCTCGTG-3′ for α internexin; 5′-GGAGCGGCTGCTGTCCGTGGTGCTGAAGCGATAG-3′ and 5′-GGAGCGGCTGCTGTTAGTGGTCATGAAGCGATAG-3′ for NF-H; 5′-CACGCCTGGGCGCTGTCCTTGGTGCTGCCGGATCC-3′ and 5′-CACGCCTGGGCGCTGTAGTTGGTCATGCCGGATCC-3′ for NF-M (mutated nucleotides are underlined).
Chromatin immunoprecipitation (ChIP) assays
ChIP assays were performed according to the protocol of Upstate Biotechnology (Lake Placid, NY) except that crosslinking with 1% formaldehyde was done at room temperature for 10 min and was subsequently quenched with 125 mM glycine for 5 min. The chromatin samples were immunoprecipitated overnight with one of the following antibodies: anti-REST P18 (sc-15118x), anti-mSin3A (sc-767x), all from Santa Cruz; anti-H3-trimethylK4 (ab8580-100), anti-H3-dimethylK9 (ab1220), all from Abcam (Cambridge, MA); anti-CoREST (07-455), anti-MeCP2 (07-013), anti-HDAC1 (06-720), anti-acetylH3 (06-599), anti-acetylH4 (06-866), anti-H3-acetylK9 (06-942), rabbit normal Ig(12370) all from Upstate. Immune complexes were harvested with protein A/G Sepharose beads (Upstate). After decrosslinking, proteinase digestion, and purification, DNA samples were analyzed by PCR. For quantitative ChIPs, real-time PCR was performed using OmniMix HS PCR bead (Takara Bio Inc., Shiga, Japan) and SYBR-green (molecular Probe, Eugene, Oregon) in a Smart Cycler (Cepheid, Sunnyvale, CA). The abundance of the immunoprecipitated DNA in a sample was expressed as a percentage of the input DNA. The following PCR primers designed around each putative RE1 motif were used: 5′-GAGACACACGCGCAGTTTCCTTG-3′ and 5′-GAGGCCTAAACTTCCTAACTGGG-3′ for α–internexin; 5′-ACCTCCCTACTCCGTTAGGC-3′ and 5′-GCTTAGACTTGGAGCTCTTGC-3′ for NF-H; 5′-CCTCTGGACTCTGGTAGGC-3′ and 5′-GTGGCACTCCAGTCTGCTTAAC-3′ for NF-M; 5′-CGATCGATCACAGTCTGCGTC-3′ and 5′-CGTAGCCGAACGAACTCATGG-3′ for NF-L promoter; 5′-AGCGCCATGCAGGTATAGTACG-3′ and 5′-CCAAGCCCTCTGATAGCTGCTC-3′ for NF-L intron-1.
RNA extraction and reverse transcription-PCR (RT-PCR)
Total RNA was extracted as described (Chomczynski & Sacchi 1987). First-strand cDNA was synthesized with Superscript II reverse transcriptase and oligo (dT)12–18 primers (Invitrogen). PCR was performed with Herculase II fusion DNA polymerase (Stratagene). Real-time PCR was performed in a Smart Cycler using OmniMix HS PCR bead and SYBR-green. Primers were designed around exon/intron boundaries using Primer3 software (http://frodo.wi.mit.edu/cgi-bin/primer3/primer3_www.cgi). The following PCR primers were used: 5′-GCTACCAGGACAGCATTGG-3′ and 5′-ATTCAGCCCCGAGATGCTTA-3′ for α–internexin; 5′-CAGCTGGACAGTGAGCTGAG and 5′-CAAAGCCAATCCGACACTCT-3′ for NF-H; 5′-CGTCATTTGCGAGAATACCA-3′ and 5′-GTACAGAGGCCCGGTGATG-3′ for NF-M; 5′-CCATGCAGGACACAATCAAC-3′ and 5′-CGCCTTCCAAGAGTTTTCTG-3′ for NF-L; 5′-TGCACCACCAACTGCTTAG-3′ and 5′-GATGCAGGGATGATGTTC-3′ for mouse GAPDH. Relative changes in gene expression from real time RT-PCR experiments were analyzed using the 2−ΔΔCt method (Livak & Schmittgen 2001).
Western blot analysis
Proteins were electrophoresed in 8% polyacrylamide-SDS gels and then electrotransferred to Immobilon-P transfer membrane (Millipore Corp., Bedford, MA) as previously described (Ching et al. 1999). The blots were incubated with a rabbit polyclonal antibody to REST (07-579 from Upstate). After several washes, they were incubated with horse radish peroxidase-conjugated secondary antibody, treated with enhanced chemiluminescence (ECL) reagents (Amersham, Arlington Heights, IL) and exposed to x-ray films.
RESULTS
REST is detected at the RE1 motifs of the α-internexin, NF-H and NF-M genes
Computational analysis using the MatInspector program (http://www.genomatix.de/matinspector.html) shows the presence of the 21 bp RE1 motif in intron-1 of the mouse α–internexin gene (nucleotides 1995 to 2015) and in the 5′ flanking regions of the mouse NF-H (nucleotides −1354 to −1334) and mouse NF-M (nucleotides −901 to −881) genes. A RE1 motif that is less homologous to the RE1 consensus sequence is also found in the promoter (nucleotides −41 to −19) and intron-1 (nucleotides 1157 to 1177) of the mouse NF-L gene. These RE1 motifs are conserved between rodents and human in corresponding positions of these genes (Fig. 1A). A previous study has also shown the presence of a RE1 motif in the 5′ flanking region of the chicken NF-M gene (Schoenherr et al. 1996).
Figure 1.

Identification of the RE1 sites and differential gene expression patterns between NIH3T3 and CAD cells. A, DNA sequences of the RE1 motifs present in the α –internexin, NF-H and NF-M genes were identified by the MatInspector program. The RE1 motifs of the NF-L gene are less homologous to the RE1 consensus sequence as determined by the MatInspector program. B, RT-PCR assays performed on total RNAs from NIH3T3 cells (3T3) and CAD cells showed differential expression patterns of α –internexin, NF-H, NF-M and NF-L between the two cell types. C, Western blot analysis of nuclear extracts from NIH3T3 cells (3T3) and CAD cells with an antibody to REST. D, ChIP assays performed with REST antibody (R) showed that REST occupied the RE1 sites of α–internexin (α), NF-H (H) and NF-M (M) in NIH3T3 cells (3T3) but not in CAD cells. REST did not bind to the putative RE1 motifs in the promoter (pL) and intron-1 (iL) of the NF-L gene. I and Ig indicate input DNA sample and immunoprecipitation with sham antibody (IgG); respectively.
Functional analyses of the putative RE1 motifs identified above were performed using mouse neuronal CAD cells (Qi et al. 1997) and mouse NIH3T3 fibroblasts. CAD is a central nervous system-derived catecholaminergic cell line that expresses neuron-specific proteins and bears processes and varicosities similar to those of neurons (Qi et al. 1997). CAD cells express α–internexin, NF-H, NF-M and NF-L (Fig. 1B), but not REST (Fig 1C). The levels of the neuronal IF mRNAs detected in CAD cells were α–internexin>NF-H>NF-M>NF-L. In contrast, NIH3T3 cells express REST but not the neuronal IF genes.
To verify that REST occupies the identified RE1 sites in vivo, chromatin immunoprecipitation (ChIP) analyses were performed. Immunoprecipitation with REST antibody and PCR amplification using primers specific for the corresponding RE-1 containing regions resulted in REST antibody-specific DNA fragments from the α–internexin, NF-H and NF-M genes in NIH3T3 cells, but not in CAD cells (Fig. 1D). These results demonstrate that REST occupies the RE1 sites of these endogenous genes in the REST-expressing nonneuronal cells. ChIP assays did not yield any REST antibody-specific DNA fragment from the NF-L gene, indicating that REST does not bind to the putative RE1 motifs in the promoter and intron-1 of the NF-L gene in NIH3T3 cells (Fig. 1D). Similar results were obtained from ChIP assays performed with mouse liver extracts (data not shown). These putative RE1 motifs of the NF-L gene were therefore not analyzed any further.
REST represses expression of the α–internexin, NF-H and NF-M genes
To investigate how the RE1 motifs function in cells, the DNA sequences of the rat α–internexin gene and the mouse NF-H and NF-M genes carrying the RE1 motifs were fused to the firefly luciferase reporter gene, and the resulting fusion constructs were transiently transfected into mouse neuronal CAD cells and mouse NIH3T3 fibroblasts. The effects of the RE1 motifs on gene expression were then determined by comparing the relative luciferase activities of these fusion constructs in the two cell lines.
Since the full-length rat α–internexin gene was cloned and characterized in our laboratory (Ching et al. 1999), it was chosen over the mouse gene for convenient clonings in the present study. The rat α–internexin intron-1 sequence at nucleotides 1878 to 2012, which carries the RE1 motif at nucleotides 1929 to 1949, has a 93% homology to the corresponding mouse α–internexin sequence at nucleotides 1944 to 2078, which carries the RE1 motif at nucleotides 1995 to 2015. The RE1 sequences are identical between the rat and mouse α–internexin genes (Fig. 1A). To investigate the effects of the intronic RE1 on the rat α–internexin promoter activity, pα (−1219/+73) luc and pα (−1219/+73) luc (1878/2012 RE) were constructed and their relative luciferase activities were compared. Pα (−1219/+73) luc was constructed by cloning the 1219 bp 5′ flanking region and the 73 bp 5′ untranslated region of the rat α–internexin gene upstream of the firefly luciferase reporter gene. To generate pα (−1219/+73) luc (1878/2012 RE), the rat α–internexin intron-1 sequence at nucleotides 1878 to 2012 was added downstream of the luciferase gene in pα (−1219/+73) luc. In transiently transfected NIH3T3 cells, the relative luciferase activity of pα (−1219/+73) luc (1878/2012 RE) was 4.5-fold less than that of pα (−1219/+73) luc carrying no RE1 (Fig. 2A). When the RE1 was mutated, the resulting pα (−1219/+73) luc (1878/2012 mtRE) showed similar activity as pα(−1219/+73) luc. In transiently transfected CAD cells, which do not express REST, these three constructs yielded similar luciferase activities (Fig. 2A). Taken together, these results showed that the α–internexin intronic RE1 motif functioned as a repressor cis element in REST-expressing cells, but not in REST-nonexpressing cells.
Figure 2.

Repression effects of the RE1 motifs in nonneuronal cells. Relative luciferase activities of the luciferase fusion constructs in transiently transfected NIH3T3 and CAD cells were compared. A, Relative luciferase activities of the α–internexin/luciferase constructs bearing mutation or deletion of RE1 were compared to those of pα(−1219/+73)luc(1878/2012 RE). B, Relative luciferase activities of the NF-H/luciferase constructs bearing mutation or deletion of RE1 were compared to those of pH(−1392/+130 RE)luc. C, Relative luciferase activities of the NF-M/luciferase constructs bearing mutation or deletion of RE1 were compared to those of pM(−943/+17 RE)luc.
To demonstrate that REST mediated repression via the α–internexin intronic RE1, the three α–internexin luciferase fusion constructs were each transiently cotransfected into CAD cells with either a REST-expressing vector pcDNA-REST or an empty vector pcDNA3. As expected, pα (−1219/+73) luc showed the same levels of luciferase activity regardless of whether the CAD cells were cotransfected with either pcDNA-REST or pcDNA3. In contrast, pα (−1219/+73) luc (1878/2012 RE) showed a 4-fold decrease in luciferase activity in the cells cotransfected with pcDNA-REST, but not in the cells cotransfected with pcDNA3. The RE1 mutation in pα (−1219/+73) luc (1878/2012 mtRE) abolished the REST-mediated repression and increased the luciferase activity to 90% of that of pα (−1219/+73) luc in the cells cotransfected with pcDNA-REST (Fig. 3A).
Figure 3.

Repression by REST through the RE1 sites. A, α–internexin/luciferase, B, NF-H/luciferase and C, NF-M/luciferase constructs were transiently cotransfected into CAD cells with either an empty vector pcDNA3 or a REST-expressing construct pcDNA3-REST and relative luciferase activities were compared.
The 21bp RE1 motif was found in the 5′ flanking region of the mouse NF-H gene at nucleotides − 1354 to − 1334. The 1.3 kb 5′ flanking region and 130 bp of 5′ untranslated region of the mouse NF-H gene were cloned upstream of the luciferase reporter gene and the resulting constructs were transiently transfected into NIH3T3 and CAD cells. In transfected NIH3T3 cells, but not in CAD cells, the levels of luciferase activity of pH(−1392/+130 mtRE) luc with a mutated RE1 and pH(−1308/+130 ΔRE) luc with RE1 deleted were approximately 2-fold that of pH (−1392/+130 RE) luc with a wild-type RE1 (Fig. 2B). When these NF-H/luciferase fusion constructs were transiently cotransfected with either pcDNA-REST or pcDNA3 into CAD cells, the RE1 mutation or the RE1 deletion diminished the REST-mediated repression in the cells cotransfected with pcDNA-REST but showed no significant effects in the cells cotransfected with pcDNA3 (Fig. 3B).
The 21 bp RE1 motif was found in the 5′ flanking region of the mouse NF-M gene at nucleotides − 901 to −881. The NF-M/luciferase fusion constructs were generated by cloning the 0.9 kb 5′ flanking region and 13 bp 5′ untranslated region of the mouse NF-M gene upstream of the luciferase gene and transiently transfected into NIH3T3 and CAD cells. In transfected NIH3T3 cells, but not in the transfected CAD cells, pM(−943/+17 mtRE) luc carrying a mutated RE1 showed a 3-fold higher luciferase activity than pM(−943/+17 RE) luc with the wild-type RE1. Further deletion of the 5′ flanking region to remove the RE1 resulted in a 4-fold higher luciferase activity as seen in pM(−879/+17 ΔRE) luc (Fig. 2C). Similarly, the RE1 mutation or the RE1 deletion in the NF-M/luciferase fusion constructs completely abolished the REST-mediated repression in the CAD cells cotransfected with pcDNA-REST (Fig. 3C).
To examine the effects of exogenously expressed REST on the endogenous mRNA levels of α–internexin, NF-H and NF-M, CAD cells were transiently transfected with either pcDNA3 or pcDNA-REST, and real-time RT-PCR assays were performed. In CAD cells transfected with pcDNA-REST, the endogenous mRNA levels of α–internexin, NF-H, and NF-M showed a 7-fold, 2-fold, and 2-fold decrease, respectively, compared to CAD cells transfected with pcDNA3 (Fig. 4). These data are consistent with those of the transfection experiments performed with the luciferase fusion constructs described above. Taken together, they demonstrate that REST represses these neuronal IF genes via their RE1 sites.
Figure 4.
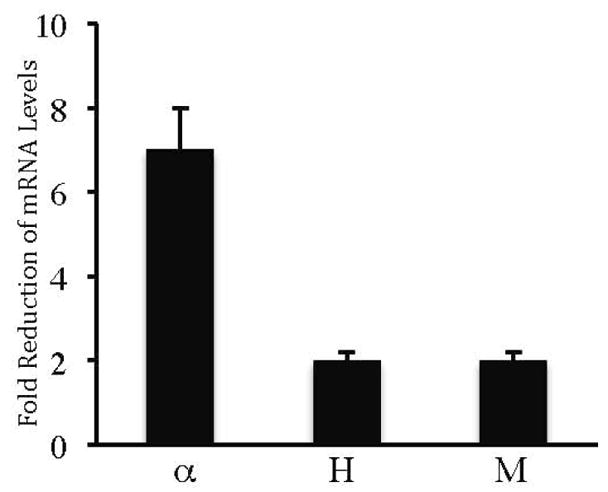
Effects of exogenously expressed REST on endogenous gene expression. Real time RT-PCR assays were performed on total RNAs isolated from CAD cells transiently transfected with either pcDNA3 or pcDNA-REST. Fold reduction of mRNA levels of α–internexin (α), NF-H (H) and NF-M (M) was determined by comparing the mRNA levels of pcDNA-REST transfected cells to those of the pcDNA3-transfected cells. Expression of each gene was normalized to endogenous levels of GAPDH, which is ubiquitously expressed.
The RE1 motifs of the neuronal IF genes repress a heterologous promoter
To determine whether the RE1 motifs of the neuronal IF genes are also able to repress a heterologous promoter in the REST-expressing NIH3T3 cell line, DNA fragments carrying a wild-type or a mutated RE1 motif were cloned into a plasmid vector pGL3-promoter, which contains a viral SV40 promoter linked to the firefly luciferase gene. pSV40-luc-α (1878/2012 RE) carrying the α–internexin wild-type RE1 downstream of the luciferase gene and pSV40-luc-α (1878/2012 mtRE) carrying the mutated RE1 showed 50% and 94% luciferase activities, respectively, relative to that of pGL3-promoter (Fig. 5A). pH(−1392/−1264 RE)-SV40-luc carrying the NF-H wild-type RE1 upstream of the SV40 promoter also showed a 50% reduction in luciferase activity whereas PH(−1392/−1264 mtRE)-SV40-luc carrying the mutated RE1 showed a 2-fold increase compared to pGL3-promoter. (Fig. 5B). Similarly, the NF-M wild-type RE1 upstream of the SV40 promoter decreased the luciferase activity of pM(−943/−801 RE)-SV40-luc to 30% of that of pGL3 promoter, whereas the mutated RE1 in pM(−943/−801 mtRE)-SV40-luc did not repress the SV40 promoter activity (Fig. 5C). Taken together, these results showed that the RE1 motifs of the α–internexin, NF-H and NF-M genes are able to act as a repressor cis element in a heterologous promoter.
Figure 5.

Repression effects of the RE1 motifs on a heterologous promoter. NIH3T3 cells were transiently transfected with SV40 promoter/luciferase fusion constructs carrying wild-type or mutated RE1 motifs of (A) α–internexin, (B) NF-H or (C) NF-M. Relative luciferase activitires were compared to those of pGL3-promoter, which carries the SV40 minimal promoter.
REST binds and recruits multiple cofactors to the RE1 sites of the neuronal IF genes
To examine the sequence-specific binding of REST to the RE1 sites of the α–internexin, NF-H and NF-M genes, electrophoretic mobility shift assays (EMSA) were performed using nuclear extracts from NIH3T3. The following DNA fragments carrying a wild-type RE1 were used as the probes: α-internexin intronic sequence at 1878 to 2012 (Fig. 6A), NF-H 5′ flanking sequence at − 1392 to − 1264 (Fig. 6B), and NF-M 5′ flanking sequence at − 943 to − 801 (Fig. 6C). Mutated RE1 oligonucleotide competitors were designed according to the mutated RE1 sequences in the luciferase fusion constructs used in the transfection experiments. Incubation of each probe with the NIH3T3 nuclear extracts yielded a specific REST/DNA complex, which was supershifted to higher mobility in the presence of the REST antibody and could be abolished by 100-fold molar excess of unlabeled wild-type RE1 oligonucleotide competitor, but not by unlabeled mutated RE1 oligonucleotide competitor. These results showed that REST was able to bind the wild-type, but not the mutated, RE1 of the α–internexin, NF-H and NF-M genes.
Figure 6.

Sequence-specific binding of REST to the RE1 sites. Electrophoretic mobility shift assays were performed with nuclear extracts (NE) from NIH3T3 cells. DNA fragments carrying a wild-type RE1 site from (A) α–internexin, (B) NF-H and (C) NF-M were used as the probes. Oligonucleotides carrying wild-type (Wt oligo) or mutated (Mt oligo) RE1 sites were used as competitors for REST binding. Arrow idicates the specific REST/DNA complex and arrowhead indicates the specific REST/DNA complex supershifted by the REST antibody (REST ab).
Although REST can recruit multiple cofactors to form the mSin3 and CoREST complexes, not all of these cofactors are recruited to the RE1 sites of all REST-target genes (Greenway et al. 2007, Lunyak et al. 2002). To determine whether REST recruits multiple cofactors to the identified RE1 sites of the α–internexin, NF-H and NF-M genes in vivo, chromatin immunoprecipitation (ChIP) analyses were performed. Immunoprecipitations with antibodies against mSin3A, CoREST, HDAC1 and MeCP2 and subsequent PCR amplification using primers specific for the corresponding RE1-containing regions resulted in DNA fragments of the appropriate sizes in NIH3T3 cells (Fig. 7), demonstrating that REST recruits these cofactors to the RE1 sites of these endogenous genes in REST-expressing cells.
Figure 7.
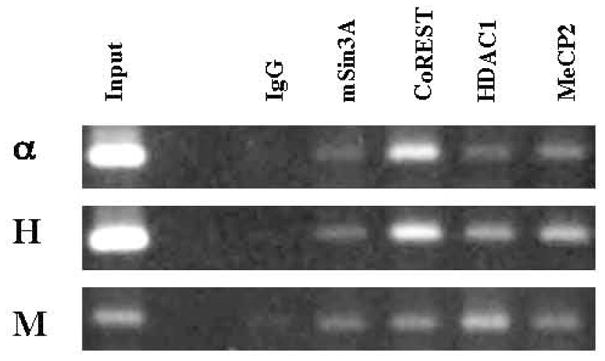
Recruitment of multiple cofactors to the RE1 sites in vivo. ChIP assays were performed with normal IgG and antibodies against mSin3A, CoREST, HDAC1 and MeCP2. Subsequent PCR assays were performed with primers specific for the corresponding RE1-containing regions of the α–internexin (α), NF-H (H) and NF-M (M) genes.
REST-mediated repression of α–internexin, NF-H and NF-M is relieved by TSA
Trichostatin A (TSA), a well-characterized, potent and specific inhibitor of HDAC (Yoshida & Horinouchi 1999), has frequently been used to study the effects of HDAC inhibition on REST-mediated repression. It was shown to derepress some but not all REST-target genes (Abderrahmani et al. 2001, Greenway et al. 2007, Huang et al. 1999, Lemonde et al. 2004, Lunyak et al. 2002, Otto et al. 2007). Since the α–internexin, NF-H and NF-M genes were shown to have HDAC recruited to their RE1 sites (Fig 7), we investigated whether their transcriptional repression could be relieved by TSA treatment. Exponentially proliferating NIH3T3 cells were treated with TSA (150 ng/ml) for 24 hours and subsequent RT-PCR analyses showed that TSA treatment induced ectopic activation of the endogenous α–internexin, NF-H and NF-M genes in NIH3T3 cells, albeit at low levels (Fig. 8A). In contrast, TSA treatment had no effect on expression of the control gene, GAPDH, which is ubiquitously expressed.
Figure 8.

Relief of REST-mediated repression of neuronal IF genes by TSA. A, RT- PCR assays were performed on total RNAs from CAD cells and NIH3T2 cells treated with (+) or without (−) TSA (150 ng/ml) for 24 h. α, H and M indicate α internexin, NF-H and NF-M, respectively. GAPDH was used as a control. B–D, NIH3T3 cells transiently transfected with the luciferase fusion constructs were treated with or without TSA (150 ng/ml) for 24 h. Luciferase activities of the TSA-treated cells were expressed relative to those of the untreated cells to show fold changes in transcriptional activities of the (B) α–internexin/luciferase, (C) NF-H/luciferase and (D) NF-M/luciferase constructs.
To investigate whether TSA mediated derepression of the α–internexin, NF-H and NF-M genes via the identified RE1 sites, NIH3T3 cells transiently transfected with the luciferase fusion constructs were treated for 24 hours with TSA (150 ng/ml). Luciferase activities of the TSA-treated cells transfected with pα (−1219/+73)luc(1878/2012 RE), pα (−1219/+73)luc(1878/2012 mtRE), or pα (−1219/+73)luc were 22.4-, 7.1-, and 7.8-fold, respectively, of those of the untreated transfected cells (Fig. 8B). These results showed that TSA enhanced transcriptional activity of pα (−1219/+73)Luc(1878/2012 RE) to a greater extent than the other two constructs by relieving the repression effects of the α–internexin wild-type RE1. Similarly, the luciferase fusion constructs of NF-H and NF-M containing the wild-type RE1 sites showed much greater extents of TSA-induced activation effects than those containing the RE1 mutation or deletion (Fig. 8C–D). Taken together, the data suggest that REST mediates repression of the α–internexin, NF-H and NF-M genes in part via a HDAC-dependent pathway.
TSA treatment leads to coordinated changes in histone modifications around the RE1 sites
In order to investigate the role of epigenetic mechanisms in REST-mediated repression of the α–internexin, NF-H and NF-M genes, the patterns of histone modifications around their RE1 sites were determined by quantitative ChIP assays and compared among the CAD cells, TSA-treated and untreated NIH3T3 cells. The RE1 sites of these three genes in the untreated NIH3T3 cells showed low levels of acetylated H4, acetylated H3, acetylated H3K9 and trimethylated H3K4, which are the histone marks correlated with transcriptional activation, and high levels of dimethylated H3K9, which is the histone mark associated with transcriptional repression (Fig. 9A). The histone modification patterns around these RE1 sites in the CAD cells are reciprocal to those in the untreated NIH3T3 cells: high levels of acetylated histones and trimethylated H3K4 and low levels of dimethylated H3K9. Thus, the histone modification patterns around the RE1 sites reflect the transcriptional states of these genes in the two cell types.
Figure 9.

TSA-induced changes in histone modifications around the RE1 sites. A, Quantitative ChIP assays were performed on chromatin samples from CAD cells and NIH3T3 cells treated with (+) or without (−) TSA (150 ng/ml) for 24 h. Chromatin samples were immunoprecipitated with normal IgG and antibodies against acetylated H4 (AcH4), acetylated H3 (AcH3), trimethylated H3K4 (TriMeH3K4), acetylated H3K9 (AcH3K9) and dimethylated H3K9 (DiMeH3K9), followed by real time PCR using primers corresponding to the RE1 regions of α–internexin (α), NF-H (H) and NF-M (M). Levels of immunoprecipitated DNA samples were normalized to those of normal IgG-immunoprecipitated DNA and were expressed as percentage of input DNA. B, Levels of REST occupancy were determined by quantitative ChIP assays performed with normal IgG and the anti-REST antibody as described above.
Comparison between the TSA-treated and untreated NIH3T3 cells showed that TSA treatment did not cause any significant changes in the levels of REST occupancy at the RE1 sites of the three genes (Fig. 9B). However, the histone modification patterns around these RE1 sites change significantly in response to TSA treatment (Fig. 9A). TSA treatment caused several folds of increase in the levels of acetylated H4, acetylated H3 and trimethylated H3K4. It caused the most dramatic increase (14-fold or more) in acetylated H3K9 at the levels comparable to those seen in the CAD cells. This increase was accompanied by a decrease in the levels of dimethylated H3K9. Overall, the data demonstrate that inhibition of HDAC activity by TSA causes coordinated changes in acetylation and methylation of histones around the RE1 sites of these neuronal intermediate filament genes that are consitent with a transition from the transcriptional repression state to the transcriptional activation state, resulting in ectopic expression of low levels of the neuronal mRNAs in NIH3T3 cells.
DISCUSSION
α–internexin and the neurofilament triplet proteins (NF-L, NF-M, and NF-H) are neuronal proteins that coassemble into intermediate filament networks in post-mitotic neurons. Although their assembly properties and functional roles have been studied extensively, little is known about regulation of their gene expression (reviewed in Ching and Liem, 2006). Previous studies using transgenic mice and transiently transfected cell lines did not identify and test any individual DNA elements or transcription factors for their neuron-specific transcriptional regulation, although the 5′ flanking region and introns of the NF-L gene were shown to confer neuron-specificity in some of the transgenic mouse lines (Beaudet et al. 1992, Leconte et al. 1994, Nakahira et al. 1990, Yazdanbakhsh et al. 1993). In the present study we performed experiments using the neuronal CAD cells and nonneuronal NIH3T3 cells and demonstrated that REST plays a contributory role in the neuron-specific expression of the α–internexin, NF-H and NF-M genes. Taken together, our results from transient transfections, EMSA and ChIP assays showed that REST represses the transcriptional activity of these genes in NIH3T3 cells by binding and recruiting mSin3A, CoREST, HDAC1 and MeCP2 to the RE1 site in the intron-1 of α–internexin and the RE1 sites in the 5′ flanking regions of NF-H and NF-M. No repression effects of these RE1 sites were observed in neuronal CAD cells, which lack REST. TSA treatment of NIH3T3 cells relieved REST-mediated repression and induced ectopic activation of these three genes, albeit at low levels. ChIP assays showed that inhibition of HDAC activity by TSA did not affect the levels of REST occupancy but caused coordinated changes of histone modifications around the RE1 sites that are consistent with a transition from transcriptional repression to transcriptional activation. Thus, our overall data indicate that REST mediates repression of the α–internexin, NF-H and NF-M genes, in part, via a HDAC-dependent epigenetic mechanism. Other factors may also be involved and contribute to the neuron-specific regulation of these genes. Recruitment of MeCP2 by REST to the RE1 sites of these neuronal IF genes suggest that DNA methylation may play a role in their regulation. Neuron-specific activators and enhancers that are not yet identified may be required for optimal/maximal levels of gene expression. These factors may partly explain why only low levels of neuronal IF mRNAs resulted from the TSA-induced ectopic activation of the three neuronal genes in NIH3T3 cells. Our computational analysis of the NF-L gene, including 3.65 kb of its 5′ flanking region and 3.0 kb of its 3′ flanking region, reveals two RE1 motifs that show moderate homology to the RE1 consensus sequence as determined by the MatInspector program (Fig. 1A). Although our ChIP assays did not show REST binding to these two putative RE1 sites of the NF-L gene, we cannot exclude the possibility of a role for REST in regulating NF-L expression, because it is possible that there are RE1 sites outside the regions we examined.
REST has been shown to mediate gene repression by recruiting multiple cofactors to form two distinct corepressor complexes, mSin3A/B and CoREST, at its amino-terminal and carboxyl-terminal domains, respectively. However, not all of these cofactors are recruited to the RE1 sites of all REST-target genes. Furthermore, different REST-target genes require one or both of the mSin3A/B and CoREST complexes for REST-mediated repression (Bingham et al. 2007, Greenway et al. 2007, Lunyak et al. 2002). The reason for these differences in cofactor requirements is poorly understood. Our ChIP assays showed that REST recruits mSin3A, CoREST, HDAC1 and MeCP2 to the RE1 sites of the α–internexin, NF-H and NF-M genes in NIH3T3 cells, indicating formation of two corepressor complexes, mSin3A/HDAC1 and CoREST/HDAC1/MeCP2.
Histone modifications play an important role in epigenetic regulation of gene expression. In CAD cells, which lack REST, high levels of acetylated H3, acetylated H4 and trimethlyated H3K4 and low levels of dimethlyated H3K9 are detected around the RE1 sites of the α–internexin, NF-H and NF-M genes. This pattern of histone modifications is consistent with active transcription of these genes in CAD cells. In contrast, in NIH3T3 cells, which express REST, low levels of acetylated H3, acetylated H4 and trimethlyated H3K4 and high levels of dimethlyated H3K9 are detected around the RE1 sites, consistent with silencing of these genes in the nonneuronal cell line. This pattern of histone modifications generates a repressive chromatin environment around the RE1 sites of these genes in NIH3T3 cells and presumably results from interaction of REST and its corepressor complexes, mSin3A/HDAC1 and CoREST/HDAC1, with G9a, LSD1 and SMCX. G9a, a methyltransferase responsible for mono- and dimethylation of H3K9 in euchromatin, is shown to generate a dimethylated H3K9-enriched domain around the RE1 sites of REST-target genes (Roopra et al. 2004, Shi et al. 2003). LSD1 demethylates mono- and dimethylated H3K4 and forms a multi-subunits complex that includes CoREST and HDAC 1/2 (Lee et al. 2005, Shi et al. 2003, Shi et al. 2005). SMCX demethylates di- and trimethylated H3K4 and is a component of a multi-subunits complex containing REST and HDAC 1/2 (Tahiliani et al. 2007). G9a is found in both the LSD1-complex and SMCX-complex.
Our ChIP assays showed that treatment of NIH3T3 cells with TSA, an inhibitor of HDAC activity, increased acetylation of histones H3 and H4 around the RE1 sites of the α–internexin, NF-H and NF-M genes. Concomitantly, it decreased levels of dimethylated H3K9 and increased levels of trimethylated H3K4. This pattern of histone modifications favors establishment of an open chromatin conformation for active transcription and may account for the ectopic activation of the three neuronal genes in TSA-treated NIH3T3 cells. Furthermore, these data indicate crosstalk between histone modifications around the RE1 sites of these genes and are consistent with recent studies that demonstrate interplay between histone modification enzymes. It has been shown that prior deacetylation of H3K9 is required for subsequent H3K9 methylation by G9a and that inhibition of HDAC 1/2 decreases dimethylation of H3K9 (Duan et al. 2005, Fang et al. 2007). In addition to providing deacetylated H3K9 substrate for G9a, HDAC 1/2 in the LSD1-complex may function to generate hypoacetylated histone substrate for LSD1 since the LSD1 complex is shown to preferentially demethylate hypoacetylated nucleosome. H3K9 acetylation as well as inhibition of HDAC1 by TSA diminish LSD1-mediated demethylation of H3K4. CoREST is required for the crosstalk between LSD1 and HDAC1 (Forneris et al. 2005, Lee et al. 2006, Lee et al. 2005, Shi et al. 2005). Physical as well as functional links between acetyltransferases and H3K4 methyltransferases are also observed. It has been shown that histone acetylation may be required for H3K4 methylation and that the degree of H3K4 methylation induced by HDAC inhibitors is correlated with acetylation levels of distinct H3 molecules (Nightingale et al. 2007, Zhang et al. 2004). MLL4, a H3K4 methyltransferase that is stimulated by acetylated substrates, is responsible for the crosstalk between H3 acetylation and H3K4 methylation (Nightingale et al. 2007). MLL1, another H3K4 methyltransferase that generates trimethylated H3K4, is found associated with mSin3A and HDAC 1/2 as well as with acetyltransferases and binds preferentially to acetylated histones (Milne et al. 2002, Nakamura et al. 2002). MLL1 is also found associated with MOF, which acetylates H4K16. MOF binds methylated H3K4 and acetylation of H4K16 by MOF depends on MLL1 (Dou et al. 2005).
REST and its target genes have been implicated in pathogenesis of Huntington’s disease. Wild-type huntingtin protein interacts with REST and sequesters it in the cytoplasm of neurons, thereby regulating its availability to RE1 sites in the nucleus. Expression of BDNF (brain-derived neurotrophic factor), which is important for neuronal survival, is downregulated in Huntington’s disease due to impaired ability of mutant huntingtin protein to interact with REST, resulting in higher levels of REST in the nucleus and repression of its target genes such as BDNF (Milne et al. 2002, Nakamura et al. 2002, Zuccato et al. 2007, Zuccato et al. 2003). Preliminary Western blot and DNA microarray data suggest that expression of NF-H is downregulated in rat striatal cultures that express mutant huntingtin protein (Zala et al. 2005). However, it is not known whether expression of neuronal IF proteins is downregulated in Huntington’s disease although neurofilamentous pathology and abnormal neurofilament phosphorylation patterns have been observed (DiProspero et al. 2004, Nihei & Kowall 1992). Changes in structure and levels of neuronal IFs affect axonal caliber and conduction velocities and may contribute to neuronal dysfunction and degeneration (Ching & Liem 2006). In light of our finding that expression of α–internexin, NF-H and NF-M is regulated by REST via a HDAC-dependent pathway, it would be interesting to determine whether expression of these neuronal proteins is downregulated in Huntington’s disease and whether HDAC inhibitors can be potential therapeutics for correcting this downregulation. Recent studies have shown that HDAC inhibitors provide neuroprotection and improve survival in transgenic mouse models of Huntington’s disease (Ferrante et al. 2003, Gardian et al. 2005).
The α–internexin, NF-H and NF-M genes are expressed in most neurons in the nervous systems. Our present study provides significant information on the regulatory DNA sequences and the epigenetic control for the neuron specificity of these genes. The RE1 sites of these genes were shown to exert repression effects in a REST-expressing neuronal cell line, but not in a REST-nonexpressing neuronal cell line. They were also shown to be capable of repressing a heterologous promoter. These repression effects could be relieved by a HDAC inhibitor. The RE1-containing regulatory DNA sequences of these genes are useful in generating neuron-target vectors for delivery of therapeutic agents to affected neurons in human diseases as well as for gene transfer to study neuronal gene function in animal models.
Acknowledgments
This work was supported by Grant NS15182 from the National Institutes of Health. We thank Ms. Laura Salvatierra for her assistance in the preparation of this manuscript.
Abbreviations
- NFTPs
neurofilament triplet proteins
- IF
intermediate filament
- EMSA
electrophoretic mobility shift assay
- ChIP
chromatin immunoprecipitation
- TSA
trichostatin A
- HDAC
histone deacetylase
References
- Abderrahmani A, Steinmann M, Plaisance V, Niederhauser G, Haefliger JA, Mooser V, Bonny C, Nicod P, Waeber G. The transcriptional repressor REST determines the cell-specific expression of the human MAPK8IP1 gene encoding IB1 (JIP-1) Mol Cell Biol. 2001;21:7256–7267. doi: 10.1128/MCB.21.21.7256-7267.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andres ME, Burger C, Peral-Rubio MJ, Battaglioli E, Anderson ME, Grimes J, Dallman J, Ballas N, Mandel G. CoREST: a functional corepressor required for regulation of neural-specific gene expression. Proc Natl Acad Sci U S A. 1999;96:9873–9878. doi: 10.1073/pnas.96.17.9873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballas N, Battaglioli E, Atouf F, et al. Regulation of neuronal traits by a novel transcriptional complex. Neuron. 2001;31:353–365. doi: 10.1016/s0896-6273(01)00371-3. [DOI] [PubMed] [Google Scholar]
- Ballas N, Grunseich C, Lu DD, Speh JC, Mandel G. REST and its corepressors mediate plasticity of neuronal gene chromatin throughout neurogenesis. Cell. 2005;121:645–657. doi: 10.1016/j.cell.2005.03.013. [DOI] [PubMed] [Google Scholar]
- Battaglioli E, Andres ME, Rose DW, Chenoweth JG, Rosenfeld MG, Anderson ME, Mandel G. REST repression of neuronal genes requires components of the hSWI.SNF complex. J Biol Chem. 2002;277:41038–41045. doi: 10.1074/jbc.M205691200. [DOI] [PubMed] [Google Scholar]
- Beaudet L, Charron G, Houle D, Tretjakoff I, Peterson A, Julien JP. Intragenic regulatory elements contribute to transcriptional control of the neurofilament light gene. Gene. 1992;116:205–214. doi: 10.1016/0378-1119(92)90517-s. [DOI] [PubMed] [Google Scholar]
- Bernstein BE, Kamal M, Lindblad-Toh K, et al. Genomic maps and comparative analysis of histone modifications in human and mouse. Cell. 2005;120:169–181. doi: 10.1016/j.cell.2005.01.001. [DOI] [PubMed] [Google Scholar]
- Bingham AJ, Ooi L, Kozera L, White E, Wood IC. The repressor element 1-silencing transcription factor regulates heart-specific gene expression using multiple chromatin-modifying complexes. Mol Cell Biol. 2007;27:4082–4092. doi: 10.1128/MCB.00269-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruce AW, Donaldson IJ, Wood IC, Yerbury SA, Sadowski MI, Chapman M, Gottgens B, Buckley NJ. Genome-wide analysis of repressor element 1 silencing transcription factor/neuron-restrictive silencing factor (REST/NRSF) target genes. Proc Natl Acad Sci U S A. 2004;101:10458–10463. doi: 10.1073/pnas.0401827101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderone A, Jover T, Noh KM, et al. Ischemic insults derepress the gene silencer REST in neurons destined to die. J Neurosci. 2003;23:2112–2121. doi: 10.1523/JNEUROSCI.23-06-02112.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ching GY, Chien CL, Flores R, Liem RK. Overexpression of alpha-internexin causes abnormal neurofilamentous accumulations and motor coordination deficits in transgenic mice. J Neurosci. 1999;19:2974–2986. doi: 10.1523/JNEUROSCI.19-08-02974.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ching GY, Liem RK. Assembly of type IV neuronal intermediate filaments in nonneuronal cells in the absence of preexisting cytoplasmic intermediate filaments. J Cell Biol. 1993;122:1323–1335. doi: 10.1083/jcb.122.6.1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ching GY, Liem RK. Roles of head and tail domains in alpha-internexin’s self-assembly and coassembly with the neurofilament triplet proteins. J Cell Sci. 1998;111 (Pt 3):321–333. doi: 10.1242/jcs.111.3.321. [DOI] [PubMed] [Google Scholar]
- Ching GY, Liem RKH. Neuronal Intermediate Filaments and Neurodegenerative Diseases. :Intermediate Filaments. Landes Biosciences. 2006:35–51. [Google Scholar]
- Chomczynski P, Sacchi N. Single-step method of RNA isolation by acid guanidinium thiocyanate-phenol-chloroform extraction. Anal Biochem. 1987;162:156–159. doi: 10.1006/abio.1987.9999. [DOI] [PubMed] [Google Scholar]
- Chong JA, Tapia-Ramirez J, Kim S, et al. REST: a mammalian silencer protein that restricts sodium channel gene expression to neurons. Cell. 1995;80:949–957. doi: 10.1016/0092-8674(95)90298-8. [DOI] [PubMed] [Google Scholar]
- Dion MF, Altschuler SJ, Wu LF, Rando OJ. Genomic characterization reveals a simple histone H4 acetylation code. Proc Natl Acad Sci U S A. 2005;102:5501–5506. doi: 10.1073/pnas.0500136102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiProspero NA, Chen EY, Charles V, Plomann M, Kordower JH, Tagle DA. Early changes in Huntington’s disease patient brains involve alterations in cytoskeletal and synaptic elements. J Neurocytol. 2004;33:517–533. doi: 10.1007/s11068-004-0514-8. [DOI] [PubMed] [Google Scholar]
- Dou Y, Milne TA, Tackett AJ, et al. Physical association and coordinate function of the H3 K4 methyltransferase MLL1 and the H4 K16 acetyltransferase MOF. Cell. 2005;121:873–885. doi: 10.1016/j.cell.2005.04.031. [DOI] [PubMed] [Google Scholar]
- Duan Z, Zarebski A, Montoya-Durango D, Grimes HL, Horwitz M. Gfi1 coordinates epigenetic repression of p21Cip/WAF1 by recruitment of histone lysine methyltransferase G9a and histone deacetylase 1. Mol Cell Biol. 2005;25:10338–10351. doi: 10.1128/MCB.25.23.10338-10351.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang S, Miao J, Xiang L, Ponugoti B, Treuter E, Kemper JK. Coordinated recruitment of histone methyltransferase G9a and other chromatin-modifying enzymes in SHP-mediated regulation of hepatic bile acid metabolism. Mol Cell Biol. 2007;27:1407–1424. doi: 10.1128/MCB.00944-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrante RJ, Kubilus JK, Lee J, et al. Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington’s disease mice. J Neurosci. 2003;23:9418–9427. doi: 10.1523/JNEUROSCI.23-28-09418.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fliegner KH, Kaplan MP, Wood TL, Pintar JE, Liem RK. Expression of the gene for the neuronal intermediate filament protein alpha-internexin coincides with the onset of neuronal differentiation in the developing rat nervous system. J Comp Neurol. 1994;342:161–173. doi: 10.1002/cne.903420202. [DOI] [PubMed] [Google Scholar]
- Fliegner KH, Liem RK. Cellular and molecular biology of neuronal intermediate filaments. Int Rev Cytol. 1991;131:109–167. doi: 10.1016/s0074-7696(08)62018-5. [DOI] [PubMed] [Google Scholar]
- Forneris F, Binda C, Vanoni MA, Battaglioli E, Mattevi A. Human histone demethylase LSD1 reads the histone code. J Biol Chem. 2005;280:41360–41365. doi: 10.1074/jbc.M509549200. [DOI] [PubMed] [Google Scholar]
- Gardian G, Browne SE, Choi DK, et al. Neuroprotective effects of phenylbutyrate in the N171-82Q transgenic mouse model of Huntington’s disease. J Biol Chem. 2005;280:556–563. doi: 10.1074/jbc.M410210200. [DOI] [PubMed] [Google Scholar]
- Greenway DJ, Street M, Jeffries A, Buckley NJ. RE1 Silencing transcription factor maintains a repressive chromatin environment in embryonic hippocampal neural stem cells. Stem Cells. 2007;25:354–363. doi: 10.1634/stemcells.2006-0207. [DOI] [PubMed] [Google Scholar]
- Grimes JA, Nielsen SJ, Battaglioli E, et al. The co-repressor mSin3A is a functional component of the REST-CoREST repressor complex. J Biol Chem. 2000;275:9461–9467. doi: 10.1074/jbc.275.13.9461. [DOI] [PubMed] [Google Scholar]
- Huang Y, Myers SJ, Dingledine R. Transcriptional repression by REST: recruitment of Sin3A and histone deacetylase to neuronal genes. Nat Neurosci. 1999;2:867–872. doi: 10.1038/13165. [DOI] [PubMed] [Google Scholar]
- Jacomy H, Zhu Q, Couillard-Despres S, Beaulieu JM, Julien JP. Disruption of type IV intermediate filament network in mice lacking the neurofilament medium and heavy subunits. J Neurochem. 1999;73:972–984. doi: 10.1046/j.1471-4159.1999.0730972.x. [DOI] [PubMed] [Google Scholar]
- Kaplan MP, Chin SS, Fliegner KH, Liem RK. Alpha-internexin, a novel neuronal intermediate filament protein, precedes the low molecular weight neurofilament protein (NF-L) in the developing rat brain. J Neurosci. 1990;10:2735–2748. doi: 10.1523/JNEUROSCI.10-08-02735.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurdistani SK, Tavazoie S, Grunstein M. Mapping global histone acetylation patterns to gene expression. Cell. 2004;117:721–733. doi: 10.1016/j.cell.2004.05.023. [DOI] [PubMed] [Google Scholar]
- Leconte L, Semonin O, Zvara A, Boisseau S, Poujeol C, Julien JP, Simonneau M. Both upstream and intragenic sequences of the human neurofilament light gene direct expression of lacZ in neurons of transgenic mouse embryos. J Mol Neurosci. 1994;5:273–295. doi: 10.1007/BF02736727. [DOI] [PubMed] [Google Scholar]
- Lee MG, Wynder C, Bochar DA, Hakimi MA, Cooch N, Shiekhattar R. Functional interplay between histone demethylase and deacetylase enzymes. Mol Cell Biol. 2006;26:6395–6402. doi: 10.1128/MCB.00723-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MG, Wynder C, Cooch N, Shiekhattar R. An essential role for CoREST in nucleosomal histone 3 lysine 4 demethylation. Nature. 2005;437:432–435. doi: 10.1038/nature04021. [DOI] [PubMed] [Google Scholar]
- Lee MK, Xu Z, Wong PC, Cleveland DW. Neurofilaments are obligate heteropolymers in vivo. J Cell Biol. 1993;122:1337–1350. doi: 10.1083/jcb.122.6.1337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemonde S, Rogaeva A, Albert PR. Cell type-dependent recruitment of trichostatin A-sensitive repression of the human 5-HT1A receptor gene. J Neurochem. 2004;88:857–868. doi: 10.1046/j.1471-4159.2003.02223.x. [DOI] [PubMed] [Google Scholar]
- Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128:707–719. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- Litt MD, Simpson M, Gaszner M, Allis CD, Felsenfeld G. Correlation between histone lysine methylation and developmental changes at the chicken beta-globin locus. Science. 2001;293:2453–2455. doi: 10.1126/science.1064413. [DOI] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(−Delta Delta C(T)) Method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- Lunyak VV, Burgess R, Prefontaine GG, et al. Corepressor-dependent silencing of chromosomal regions encoding neuronal genes. Science. 2002;298:1747–1752. doi: 10.1126/science.1076469. [DOI] [PubMed] [Google Scholar]
- McGraw TS, Mickle JP, Shaw G, Streit WJ. Axonally transported peripheral signals regulate alpha-internexin expression in regenerating motoneurons. J Neurosci. 2002;22:4955–4963. doi: 10.1523/JNEUROSCI.22-12-04955.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milne TA, Briggs SD, Brock HW, Martin ME, Gibbs D, Allis CD, Hess JL. MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol Cell. 2002;10:1107–1117. doi: 10.1016/s1097-2765(02)00741-4. [DOI] [PubMed] [Google Scholar]
- Nakahira K, Ikenaka K, Wada K, Tamura T, Furuichi T, Mikoshiba K. Structure of the 68-kDa neurofilament gene and regulation of its expression. J Biol Chem. 1990;265:19786–19791. [PubMed] [Google Scholar]
- Nakamura T, Mori T, Tada S, et al. ALL-1 is a histone methyltransferase that assembles a supercomplex of proteins involved in transcriptional regulation. Mol Cell. 2002;10:1119–1128. doi: 10.1016/s1097-2765(02)00740-2. [DOI] [PubMed] [Google Scholar]
- Naruse Y, Aoki T, Kojima T, Mori N. Neural restrictive silencer factor recruits mSin3 and histone deacetylase complex to repress neuron-specific target genes. Proc Natl Acad Sci U S A. 1999;96:13691–13696. doi: 10.1073/pnas.96.24.13691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nightingale KP, Gendreizig S, White DA, Bradbury C, Hollfelder F, Turner BM. Cross-talk between histone modifications in response to histone deacetylase inhibitors: MLL4 links histone H3 acetylation and histone H3K4 methylation. J Biol Chem. 2007;282:4408–4416. doi: 10.1074/jbc.M606773200. [DOI] [PubMed] [Google Scholar]
- Nihei K, Kowall NW. Neurofilament and neural cell adhesion molecule immunocytochemistry of Huntington’s disease striatum. Ann Neurol. 1992;31:59–63. doi: 10.1002/ana.410310111. [DOI] [PubMed] [Google Scholar]
- Ooi L, Belyaev ND, Miyake K, Wood IC, Buckley NJ. BRG1 chromatin remodeling activity is required for efficient chromatin binding by repressor element 1-silencing transcription factor (REST) and facilitates REST-mediated repression. J Biol Chem. 2006;281:38974–38980. doi: 10.1074/jbc.M605370200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto SJ, McCorkle SR, Hover J, et al. A new binding motif for the transcriptional repressor REST uncovers large gene networks devoted to neuronal functions. J Neurosci. 2007;27:6729–6739. doi: 10.1523/JNEUROSCI.0091-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm K, Belluardo N, Metsis M, Timmusk T. Neuronal expression of zinc finger transcription factor REST/NRSF/XBR gene. J Neurosci. 1998;18:1280–1296. doi: 10.1523/JNEUROSCI.18-04-01280.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi Y, Wang JK, McMillian M, Chikaraishi DM. Characterization of a CNS cell line, CAD, in which morphological differentiation is initiated by serum deprivation. J Neurosci. 1997;17:1217–1225. doi: 10.1523/JNEUROSCI.17-04-01217.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao MV, Campbell J, Yuan A, Kumar A, Gotow T, Uchiyama Y, Nixon RA. The neurofilament middle molecular mass subunit carboxyl-terminal tail domains is essential for the radial growth and cytoskeletal architecture of axons but not for regulating neurofilament transport rate. J Cell Biol. 2003;163:1021–1031. doi: 10.1083/jcb.200308076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roopra A, Qazi R, Schoenike B, Daley TJ, Morrison JF. Localized domains of G9a-mediated histone methylation are required for silencing of neuronal genes. Mol Cell. 2004;14:727–738. doi: 10.1016/j.molcel.2004.05.026. [DOI] [PubMed] [Google Scholar]
- Roopra A, Sharling L, Wood IC, Briggs T, Bachfischer U, Paquette AJ, Buckley NJ. Transcriptional repression by neuron-restrictive silencer factor is mediated via the Sin3-histone deacetylase complex. Mol Cell Biol. 2000;20:2147–2157. doi: 10.1128/mcb.20.6.2147-2157.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoenherr CJ, Anderson DJ. The neuron-restrictive silencer factor (NRSF): a coordinate repressor of multiple neuron-specific genes. Science. 1995;267:1360–1363. doi: 10.1126/science.7871435. [DOI] [PubMed] [Google Scholar]
- Schoenherr CJ, Paquette AJ, Anderson DJ. Identification of potential target genes for the neuron-restrictive silencer factor. Proc Natl Acad Sci U S A. 1996;93:9881–9886. doi: 10.1073/pnas.93.18.9881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubeler D, MacAlpine DM, Scalzo D, et al. The histone modification pattern of active genes revealed through genome-wide chromatin analysis of a higher eukaryote. Genes Dev. 2004;18:1263–1271. doi: 10.1101/gad.1198204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahbazian MD, Grunstein M. Functions of site-specific histone acetylation and deacetylation. Annu Rev Biochem. 2007;76:75–100. doi: 10.1146/annurev.biochem.76.052705.162114. [DOI] [PubMed] [Google Scholar]
- Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, Casero RA, Shi Y. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell. 2004;119:941–953. doi: 10.1016/j.cell.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Shi Y, Sawada J, Sui G, et al. Coordinated histone modifications mediated by a CtBP co-repressor complex. Nature. 2003;422:735–738. doi: 10.1038/nature01550. [DOI] [PubMed] [Google Scholar]
- Shi YJ, Matson C, Lan F, Iwase S, Baba T, Shi Y. Regulation of LSD1 histone demethylase activity by its associated factors. Mol Cell. 2005;19:857–864. doi: 10.1016/j.molcel.2005.08.027. [DOI] [PubMed] [Google Scholar]
- Slomiany BA, Kelly MM, Kurtz DT. Extraction of nuclear proteins with increased DNA binding activity. Biotechniques. 2000;28:938–942. doi: 10.2144/00285st08. [DOI] [PubMed] [Google Scholar]
- Sun YM, Greenway DJ, Johnson R, Street M, Belyaev ND, Deuchars J, Bee T, Wilde S, Buckley NJ. Distinct profiles of REST interactions with its target genes at different stages of neuronal development. Mol Biol Cell. 2005;16:5630–5638. doi: 10.1091/mbc.E05-07-0687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tahiliani M, Mei P, Fang R, Leonor T, Rutenberg M, Shimizu F, Li J, Rao A, Shi Y. The histone H3K4 demethylase SMCX links REST target genes to X-linked mental retardation. Nature. 2007;447:601–605. doi: 10.1038/nature05823. [DOI] [PubMed] [Google Scholar]
- Tapia-Ramirez J, Eggen BJ, Peral-Rubio MJ, Toledo-Aral JJ, Mandel G. A single zinc finger motif in the silencing factor REST represses the neural-specific type II sodium channel promoter. Proc Natl Acad Sci U S A. 1997;94:1177–1182. doi: 10.1073/pnas.94.4.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vakoc CR, Sachdeva MM, Wang H, Blobel GA. Profile of histone lysine methylation across transcribed mammalian chromatin. Mol Cell Biol. 2006;26:9185–9195. doi: 10.1128/MCB.01529-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westbrook TF, Hu G, Ang XL, et al. SCFbeta-TRCP controls oncogenic transformation and neural differentiation through REST degradation. Nature. 2008;452:370–374. doi: 10.1038/nature06780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Marszalek JR, Lee MK, Wong PC, Folmer J, Crawford TO, Hsieh ST, Griffin JW, Cleveland DW. Subunit composition of neurofilaments specifies axonal diameter. J Cell Biol. 1996;133:1061–1069. doi: 10.1083/jcb.133.5.1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yazdanbakhsh K, Fraser P, Kioussis D, Vidal M, Grosveld F, Lindenbaum M. Functional analysis of the human neurofilament light chain gene promoter. Nucleic Acids Res. 1993;21:455–461. doi: 10.1093/nar/21.3.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida M, Horinouchi S. Trichostatin and leptomycin. Inhibition of histone deacetylation and signal-dependent nuclear export. Ann N Y Acad Sci. 1999;886:23–36. doi: 10.1111/j.1749-6632.1999.tb09397.x. [DOI] [PubMed] [Google Scholar]
- Yuan A, Rao MV, Sasaki T, et al. Alpha-internexin is structurally and functionally associated with the neurofilament triplet proteins in the mature CNS. J Neurosci. 2006;26:10006–10019. doi: 10.1523/JNEUROSCI.2580-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zala D, Benchoua A, Brouillet E, Perrin V, Gaillard MC, Zurn AD, Aebischer P, Deglon N. Progressive and selective striatal degeneration in primary neuronal cultures using lentiviral vector coding for a mutant huntingtin fragment. Neurobiol Dis. 2005;20:785–798. doi: 10.1016/j.nbd.2005.05.017. [DOI] [PubMed] [Google Scholar]
- Zhang K, Siino JS, Jones PR, Yau PM, Bradbury EM. A mass spectrometric “Western blot“ to evaluate the correlations between histone methylation and histone acetylation. Proteomics. 2004;4:3765–3775. doi: 10.1002/pmic.200400819. [DOI] [PubMed] [Google Scholar]
- Zuccato C, Belyaev N, Conforti P, et al. Widespread disruption of repressor element-1 silencing transcription factor/neuron-restrictive silencer factor occupancy at its target genes in Huntington’s disease. J Neurosci. 2007;27:6972–6983. doi: 10.1523/JNEUROSCI.4278-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuccato C, Tartari M, Crotti A, et al. Huntingtin interacts with REST/NRSF to modulate the transcription of NRSE-controlled neuronal genes. Nat Genet. 2003;35:76–83. doi: 10.1038/ng1219. [DOI] [PubMed] [Google Scholar]