

Abstract
TGF-β is a pluripotent cytokine that mediates its effects through a receptor composed of TGF-β receptor type II (TGFBR2) and type I (TGFBR1). The TGF-β receptor can regulate Smad and nonSmad signaling pathways, which then ultimately dictate TGF-β's biological effects. We postulated that control of the level of TGFBR2 is a mechanism for regulating the specificity of TGF-β signaling pathway activation and TGF-β's biological effects. We used a precisely regulatable TGFBR2 expression system to assess the effects of TGFBR2 expression levels on signaling and TGF-β mediated apoptosis. We found Smad signaling and MAPK-ERK signaling activation levels correlate directly with TGFBR2 expression levels. Furthermore, p21 levels and TGF-β induced apoptosis appear to depend on relatively high TGFBR2 expression and on the activation of the MAPK-ERK and SMAD pathways. Thus, control of TGFBR2 expression and the differential activation of TGF-β signaling pathways appears to be a mechanism for regulating the specificity of the biological effects of TGF-β.
Keywords: TGF-β, Smad, nonSmad, p21, apoptosis, signaling
1. Introduction
Transforming growth factor β (TGF-β) is a secreted multifunctional cytokine that regulates a variety of cellular processes including growth inhibition secondary to G1/S phase arrest, differentiation, apoptosis, immunosuppression, stimulation of connective tissue deposition, secondary induction of angiogenesis, and probably maintenance of genomic stability. TGF-β induces these effects by activating the TGF-β receptor, a heterodimeric complex composed of type I (TGFBR1) and type II (TGFBR2) subunits, which are both serine-threonine kinases with single transmembrane domains [1]. Both TGFBRI and TGFBR2 appear to be essential for TGF-β induced effects on the cell [2, 3]. The TGF-β receptor is activated through a sequence of events that is initiated by the TGFBR2 subunit binding to the activated TGF-β ligand. The activated TGFBR2 associates with TGFBRI, and phosphorylates a glycine-serine rich sequence in TGFBRI. TGFBRI is then activated and can phosphorylate downstream targets [4]. The TGF-β receptor complex activates both the Smad signaling pathway, which includes Smad2, Smad3, and Smad4, and the nonSmad signaling cascades (e.g. PI3K/AKT, p38MAPK, MAPK-ERK, JNK, etc.) to produce the full spectrum of TGF-β responses [5-7].
With regard to the nonSmad signaling pathway, TGF-β can activate a variety of nonSmad signaling pathways, including the MAPK, JNK, p38MAPK, NFκB, RhoA, and PI3K pathways. TGF-β can regulate the nonSmad signaling pathways both directly and indirectly. For instance, direct physical interactions between the TGF-β receptor and TAB1 (TAK1-binding protein 1), which can regulate TAK1 (TGF-β Activated Kinase 1) and p38MAPK, and PI3K/AKT, have been shown [8]. Other nonSmad signaling pathways are also clearly TGF-β responsive but, in most cases, it has not been shown whether the effect is a direct or indirect effect of TGF-β. The nonSmad signaling pathways have been implicated in regulating TGF-β mediated apoptosis [8, 9], extracellular matrix production [10-12], and differentiation [11, 13].
The Smad signaling pathway is more completely understood than the nonSmad signaling pathways that are activated by the ligand bound TGF-β receptor. Ligand binding to the TGF-β receptor complex results in TGFBRI-mediated phosphorylation of Smad2 and Smad3 on two serine residues in a conserved Ser-Ser-X-Ser motif located at the C-terminus of the R-Smads [14, 15]. Phosphorylation of these serine residues activates the signaling pathway and is required for the function of Smads in transcription factor complexes [16, 17]. The phosphorylated Smad2 and Smad3 bind to Smad4, and the complex can then translocate into the nucleus to function as a transcription factor complex. The details of this pathway have been the subject of a recent review (see [18]).
Although initially perceived as a binary (i.e. “on” or “off”) and linear mechanism, it is now clear that the TGF-β signaling pathways' effects on the biological responses of a cell depend on the intensity and duration of the activation of the pathways [19]. Indeed, a substantial number of studies have demonstrated that the TGF-β signaling pathways are regulated by a variety of mechanisms that control the cellular localization, phosphorylation state, and expression levels of the post-receptor signaling elements [19, 20]. Moreover, these studies have provided a plausible explanation for why the cell state and cell type, referred to as the “cellular context” can have a profound effect on the way in which TGF-β affects a cell [21-23]. Recent data has shown that the degree of activation of the TGF-β signaling pathways is subject to regulation by a large number of intracellular and extracellular agonists and antagonists, including decorin, Smad7, Smurf, SARA, etc [24, 25].
The discovery of these regulatory mechanisms demonstrates that not only is the absolute activation state of the pathway important for determining TGF-β's effects on cells but that the degree of pathway activation and the duration of pathway activation is also likely important. In fact, it is plausible that the paradoxical effects of TGF-β on tumor cells could be at least partly explained through differences in the intensity of activation of the pathways or by differential activation of Smad vs. nonSmad signaling pathways. Regulation of the expression levels of TGFBR1 and TGFBR2 may be a mechanism through which the activation state of the TGF-β signaling pathways can be regulated [8]. Consistent with this model is the recent demonstration that levels of TGFBR1 have been shown to correlate with an increased risk of colorectal cancer [26].
In order to assess whether the regulation of TGFBR2 expression could be a mechanism for determining the response of cells to TGF-β, we generated a system using the RheoSwitch® inducible gene expression system to regulate the expression of TGFBR2. We have found that the level of TGFBR2 affects the ability of TGF-β to induce p21 and apoptosis in the V-400 colorectal cancer cell line. In addition, TGF-β's ability to activate the MAPK pathway is directly dependent on the expression level of TGFBR2. These results demonstrate that the regulation of the receptor level for TGF-β is a potential mechanism for determining the specificity of the response of a cell to TGF-β.
2. Materials and Methods
2.1. Cell Lines and inhibitors
V-400 is a microsatellite stable (MSS), TGF-β resistant colon cancer cell line, which was kindly provided by James K.V. Willson (UT Southwestern Medical School, Dallas, TX). It carries biallelic missense TGFBR2 mutations that inactivate the endogenous receptor, and it has an intact Smad signaling pathway [27]. The V-400 cell line was grown in Dulbecco's Modified Eagle Medium (Gibco, Grand Island, NY). The media was supplemented with 10% FBS (Cambrex, East Rutherford, NJ). The MEK1/2 inhibitor U0126 (cat# 662005), and the PIK3 inhibitor LY294002 (cat# 440204) were obtained from Calbiochem (San Diego, CA). The Smad7 plasmid was kindly provided by Neil Bhowmick (Vanderbilt University, Nashville, TN).
2.2. Inducible gene expression system
The inducible system used in this project was developed by RheoGene, Inc. (Norristown, PA), now Intrexon Corporation (Blacksburg, VA), and is a highly engineered system based on a modified version of the ecdysone receptor (EcR) [28, 29]. Important modifications of this system include mutations in the ecdysone receptor to increase potency to synthetic diacylhydrazine agonists; trans-positioning of the VP16 activation domain and the Gal4 DNA binding domain as fusions to the RXR and EcR components, respectively; and development of potent and bioavailable synthetic agonists. Two agonists, RG-102240 and RG-115819, were used in these studies. They are closely related diacylhydrazine analogs, with the latter being slightly more potent. These modifications significantly improve the precision of the transgene expression regulation by the synthetic agonist.
A single construct carrying both the RheoSwitch® receptor genes and an HA-tagged TGFBR2 transgene was made. The TGFBR2 transgene was inserted into the cloning site present in the lentiviral vector p5004, provided by RheoGene. (Figure 1) The TGFBR2 transgene was obtained from the vector pRC/CMV kindly provided by Dr. Sandford Markowitz (Case Western Reserve University, Cleveland, OH) [27]. The TGFBR2 gene was released from its original backbone using the enzymes NotI and EcoRI and then treated with Klenow. After cloning the TGFBR2 transgene into the p5004 vector, plasmids were screened for inserts with the correct orientation and were confirmed to carry a wild-type TGFBR2 transgene by sequencing
Figure 1. Schematic representation of the p5004 vector.
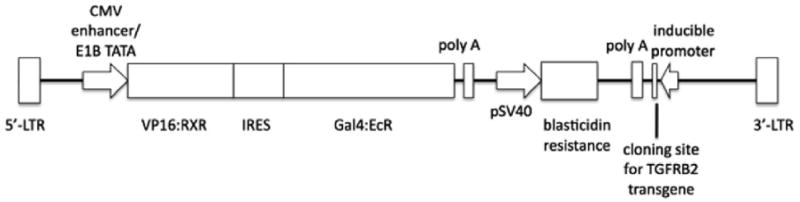
The functional elements in the vector are shown, including the location of the TGFBR2 transgene.
A lentivirus carrying the TGFBR2 transgene was produced in the Gene Therapy core lab at the Fred Hutchinson Cancer Research Center using a 293T-based packaging cell line. V-400 cells were infected with 0.5 ml of viral supernatant. The cells were incubated for 24 hours with the viral supernatant in presence of polybrene. The viral supernatant was then removed and Blasticidin (6 μg/ml) was added into the complete media (DMEM in presence of 10% FBS). The transduced cells were then subjected to cloning by limiting dilution. An evaluation to identify clones that displayed minimal expression in the baseline state but showed adequate induction of TGFBR2 after treatment with RG-115819 was performed in different clonal populations. The control cells were treated with DMSO vehicle alone.
2.3. Luciferase reporter assays
In order to evaluate TGF-β-mediated transcription, the cell lines were transiently transfected with the p3TP-lux reporter (kindly provided by Joan Massagué, Memorial Sloan-Kettering Cancer Center, New York, NY) or with the CAGA reporter assay (kindly provided by Bert Vogelstein, Johns Hopkins University, Baltimore, MA) concomitantly with the pRL-TK reporter construct (Promega, Madison, WI). The cells were treated with TGF-β1 (10ng/ml), and luciferase activity was evaluated 48h after transfection using the Dual Luciferase Reporter Assay System (Promega, Madison, WI) with a Veritas luminometer (Turner Biosystems, Sunnyvale, CA). The p21 expression was evaluated using a luciferase reporter assay (kindly provided by Dr. Xiao-Fan Wang's Laboratory) [30]. The luciferase activity was evaluated 24 and 48 hr post-transfection as noted above. All the transfections were performed using FuGENE 6 (Roche, Basel, Switzerland) following the protocol provided by the manufacturer. A 3:1 FuGENE-DNA ratio was used.
2.4. FACS analysis
V-400R2 and V-400 cells treated for 48 hours with RG-115819 and TGF-β1 were used for these studies. The cells were collected after being briefly treated with trypsin (0.125%) to detach them from the flasks, washed in media with 20% FBS (4°C) followed by 1X PBS (4°C), and then fixed in 3% formaldehyde for 15 minutes. The cells were then incubated with 10% normal horse serum for 30 minutes, washed with PBS 3 times, and incubated with an anti-HA antibody (1:100, Cat # 2367 Cell Signaling, Danvers, MA), followed by incubation with a goat-anti-mouse secondary antibody labeled with fluorescein (FITC) (1:100, Cat#:115-095-146, Jackson ImmunoResearch, West Grove, PA). The cells were then washed with PBS (3 times) and subjected to FACS analysis using FACScan (Becton Dickenson, Franklin Lakes, NJ). The results were analyzed using CellQuest software (Becton Dickenson, Franklin Lakes, NJ).
2.5. Western blotting
Cell lysates were prepared using RIPA buffer supplemented with a complete protease inhibitor cocktail (Roche) and phosphatase inhibitor cocktails 1 and 2 (Sigma-Aldrich, St. Louis, MO). The lysates were then used for SDS-PAGE (10% polyacrylamide gels and PVDF membranes, Pierce, Rockford, IL). Immunoblots were performed following the manufacturer's recommendations. The following antibodies were used: pERK1/2 (Cat # 9101 Cell Signaling, Danvers, MA 01923), ERK1/2 (Cat # 9102 Cell Signaling, Danvers, MA 01923), p21(Cat # OP68 EMD San Diego California), anti-TGFBR1 ((H-100 antibody; sc-904, 1;200), HRP-goat anti-mouse (Cat # SC 2031) and donkey anti-rabbit (Cat # SC 2317) (Santa Cruz Biotechnology). A commercial ECL kit (RPN 3004, GE Healthcare, Piscataway, NJ) was used to detect the HRP-labeled secondary antibodies and the signal was detected using autoradiography. Reagents used in conjunction with this kit included n-butyric acid sodium salt (Cat# B-5887), Luminol (Cat# A8511), and p-coumaric acid (Cat# C9008) (Sigma-Aldrich, St. Louis, MO).
2.6. PIK3/AKT specific ELISA
Phosphorylated AKT was detected using a commercially available ELISA assay (SuperArray, SABiosciences, Frederick, MD). Ten thousand cells/well were seeded in a 96 well plate, and were treated with different concentrations of RG-115819 in order to differentially induce the expression of the TGFBR2 transgene. TGF-β (10 ng/ml) was then added to the cells and the cells were harvested after 48 hours. The ELISA was performed following the manufacturer's recommendations (cat # FE-001 SuperArray), and read in a plate reader at 450 nm (VERSAmax, Molecular Devices, Sunnyvale, CA).
2.7. Quantitative RT-PCR
One μg of RNA extracted from V-400 and V-400R2, was reverse transcribed using oligo d(T) priming and Superscript-II reverse transcriptase (Invitrogen) and 2 nM dNTPs following the manufacturer's protocol. TaqMan On-Demand primers and probes were used to determine the relative expression levels of CDKN1A/p21 (Assay number Hs99999142_m1, Applied Biosystems, Foster City, CA) and 18S in all samples (Assay number Hs99999901_s1, Applied Biosystems,). The reactions were run in triplicate in the ABI Prism 7700 detection system (Applied Biosystems) and results were analyzed with SDS 2.1 software.
2.8. Apoptosis assays
Apoptosis was measured with two different assays: the Cell Death ELISA assay (Roche, Cat# 11 774 425 001), which detects histone-complexed DNA fragments, following the manufacturer's protocol, and the Caspase-Glo Luciferase-based reporter assay, which measures caspase 3 and 7 activity (Promega, Catalog number TB323) following the manufacturer's protocol.
3. Results
3.1. Generation of a colorectal cancer cell line with precisely regulatable TGFBR2 expression
In order to assess the effect of the expression level of TGFBR2 on the activation of TGF-β signaling pathways, we employed the RheoSwitch® gene regulation system that can precisely regulate the transgene of interest [28, 29, 31]. An HA-tagged TGFBR2 transgene was cloned into a lentiviral expression vector and used to generate lentivirus for transducing the target cell line, V-400. This cell line lacks a functional TGF-β receptor and becomes TGF-β responsive when it is reconstituted with TGFBR2 [27]. (See Materials and Methods section for information on the plasmids used.) The V-400 cells were transduced with TGFBR2 and clones expressing the RheoSwitch® receptor and TGFBR2 transgene were selected and then assessed for TGF-β responsiveness using the 3TP-lux or CAGA reporters. Two clones displayed a linear dose response of 3TP-Lux activity in relation to RG-102240 concentration. (Figure 2)
Figure 2. Assessment of TGF-β signaling in representative clones of V-400 after transduction with the inducible TGFBR2 transgene.
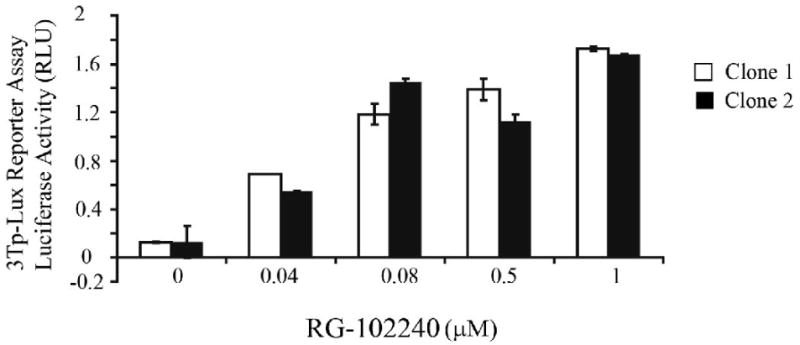
V-400R2 clones 1 and 2 display a linear dose response from 0-1.0 μM of RG-102240 with the greatest increase in 3TP-Lux activity occurring between 0 μM and 0.8 μM. 3TP-Lux activity was detected at low concentrations of RG-102240. Note that clone 1 has a wider dose-response range than clone 2.
The expression levels of the TGFBR2 transgene were then assessed by FACS analysis in order to determine the amount of TGFBR2 expressed on the surface of the transduced cells. (Immunoblotting of total protein lysates from the cells for the HA-tagged TGFBR2 transgene confirmed the results of the FACS analysis but does not demonstrate the same linear response to the RG-115819 because of the lower precision of immunoblotting to detect small differences in protein expression. (Figure 3A-B). Clones 1 and 2 showed a linear increase in 3TP-Lux induction and TGFBR2 transgene expression after treatment with increasing concentrations of RG-115819. (Figure 3A) A linear dose response for TGFBR2 expression between 0.04 μM to 1 μM RG-115819 was demonstrated, and thus this range of RG-115819 was used for the subsequent experiments. We did not observe any change in TGFBR1 expression with increasing concentrations of RG-115819, which led us to interpret that any RG-115819 dependent phenomenon were strictly the result of changes in the expression level of TGFBR2. (Figure 3C)
Figure 3. The expression of cell surface TGFBR2 in V-400 after treatment with RG-115819.
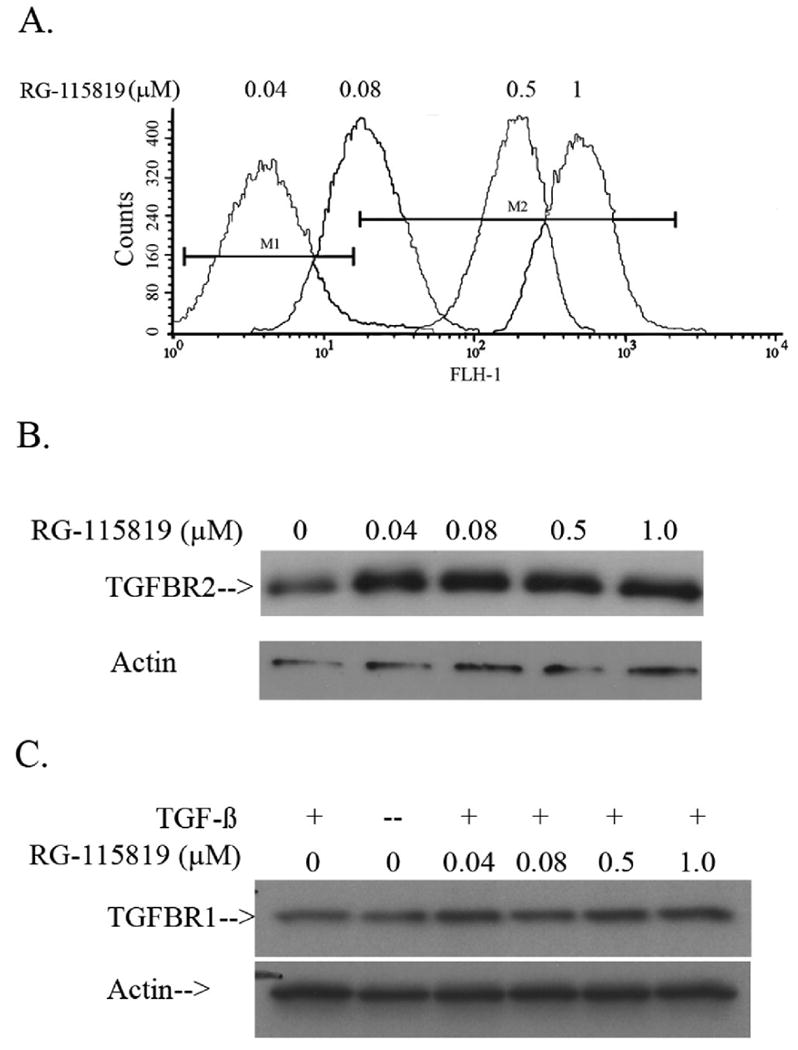
A. FACS analysis using an anti-HA antibody was performed after treatment with different doses of RG-115819 (0 μM-1.0 μM). An increased amount of cell surface TGFBR2 is detected with the increasing RG-115819 doses. M1 designates the fluorescence signal detected in the parental V-400 cell line. Of note, although no HA-tagged TGFBR2 can be detected with this technique after 0.04 μM RG-115819 treatment, as shown in figure 1, TGF-β induced gene expression is detected with this concentration of RG-115819 indicating that the transgenic receptor is present on the cell surface with this dose of RG-115819. The untreated V-400R2 clones showed the same level of fluorescence as that seen in the parental V-400 cell line. B. Immunoblotting using an anti-HA antibody was performed after treatment with different doses of RG-115819 (0 μM-1.0 μM). An increased amount of cell surface TGFBR2 is detected when comparing the untreated V-400R2 cell line to the cell line treated with the increasing RG-115819 doses although the resolution of immunoblotting does not reveal the changes in expression level as accurately as does flow cytometry. C. Immunoblotting using an anti-TGFBR1 antibody after treatment with different doses of RG-115819 (0μM-1.0μM) reveals no significant change in TGFBR1 expression with RG-115819 treatment. Expression of actin was assessed to control for protein loading.
3.2. The regulation of TGFBR2 expression levels affects the TGF-β mediated expression of CDKN1A/p21
TGF-β can regulate the expression of a variety of genes that control a myriad of cell responses including proliferation, apoptosis, differentiation, etc [30, 32, 33]. A significant question that arises is how the specificity of the response is determined. One of the TGF-β regulated genes that highlights the issues related to control of the specificity of the TGF-β response is the CDK inhibitor CDKN1A/p21, which plays an important role in several cellular processes including apoptosis, cell cycle arrest, induction of cell differentiation, and cellular senescence [32, 34]. Thus, we assessed the regulation of CDKN1A/p21 expression in the setting of different levels of TGFBR2 expression to determine if the regulation of the expression of the TGF-β receptor may be a mechanism through which TGF-β mediated responses are regulated. In order to determine how changes in the expression levels of TGFBR2 affect the expression of CDKN1A/p21, V-400R2 cells were treated for 24 and 48 hours with four different concentrations of RG-115819 (0.04, 0.08, 0.4 and 1 μM) and TGF-β (10ng/ml). The induction of CDKN1A/p21 expression was assessed using a CDKN1A/p21 luciferase reporter [35]. CDKN1A/p21 reporter activity was increased at 24 hours but no correlation between increasing TGFBR2 expression and reporter activity was observed at that time point. At 48 hours, a direct relation between TGFBR2 expression level and CDKN1A/p21 reporter activity was apparent (Figure 4A). Importantly, no basal CDKN1A/p21 reporter luciferase activity was detected in the V-400R2 without RG-115819 or in the parental cell line V-400. (Figure 4A) CDKN1A/p21 protein expression also correlated with the CDKN1A/p21 luciferase reporter activity. (Figure 4B) Localization of p21 was also assessed in light of the possibility that MAPK signaling could regulate p21 nuclear localization in these cells. We observed increased nuclear p21 in association with increased p21 expression induced by increased TGFBR2 expression. These results suggest that the increased p21 is being retained in the nucleus despite the presence of increased activated ERK and that p21 is acting to affect the behavior of the cell through actions on gene transcription (Supplemental Figure 1).
Figure 4. CDKN1A/p21 expression in V-400R2 cells after treatment with different concentrations of RG-115819.
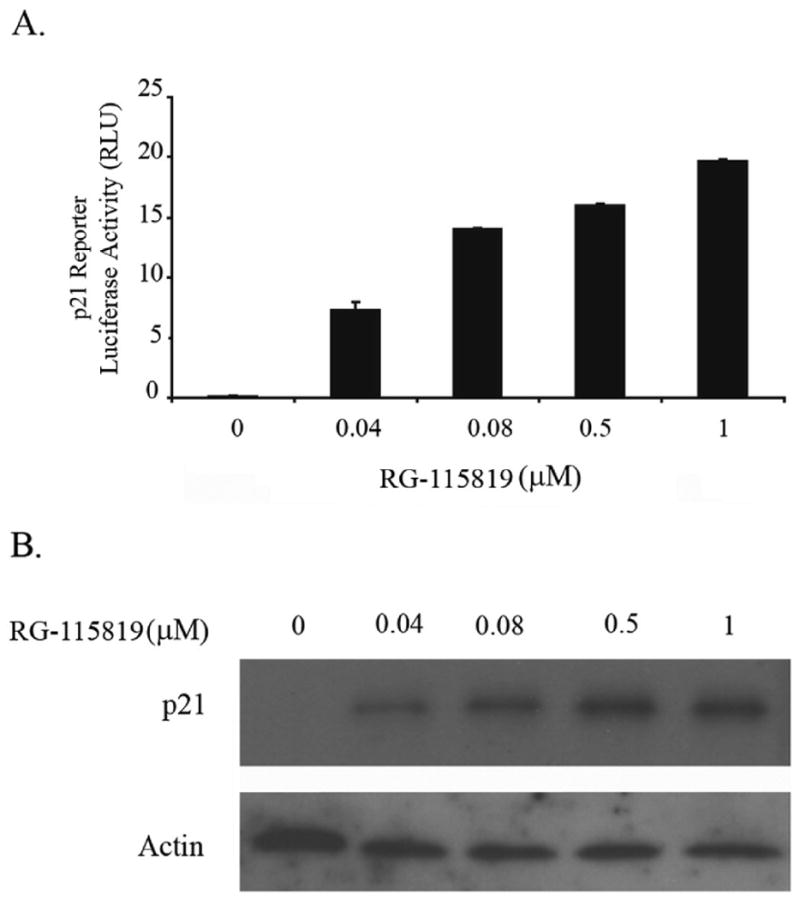
V-400R2 clone 1 was treated with four different concentrations of RG-115819 (0.04; 0.08; 0.4 and 1 μM) and then CDKN1A/p21 expression was assessed. A.CDKN1A/p21 luciferase reporter activity after 24 and 48 hours of RG-115819. Induction of CDKN1A/p21 is present at 24 and 48 hours with a linear dose-response being evident at 48 hours. B. After 48 hours of treatment with RG-115819, a dose related increase in CDKN1A/p21 protein expression is present, consistent with the CDKN1A/p21 reporter assay results. Densitometry was performed on the bands, and these results were normalized for loading using the densitometry values from the corresponding actin immunoblot. The densitometry revealed a linear increase in band density that correlated with increasing RG-115819 concentration [93 (0 μM), 90 (0.04 μM), 101 (0.08 μM), 121 (0.5 μM) and 159 (1 μM)]. Image J software (NIH) was used for the densitometry analysis.
3.3. TGFBR2 levels can differentially affect MAPK-ERK pathway activation and Smad pathway activation
The expression of CDKN1A/p21 is regulated by several signaling pathways and transcription factors, including the Smad signaling pathway, ERK signaling pathway, myc, etc. [23, 36-38]. In light of the differing effect of low and high levels of TGFBR2 expression on CDKN1A/p21 expression, we assessed the effect of different levels of TGFBR2 expression on the activation status of Smad and nonSmad signaling pathways. V-400R2 cells were treated with RG-115819 (0.4-1.0μM) and assessed for MAPK-ERK activation at 24 and 48 hours. ERK activation was not present at 24 hours but was present at 48 hours after treatment with RG-115819 and TGF-β. (Figure 5A) The level of phosphorylated ERK directly correlated with the expression level of TGFBR2 as well as with CDKN1A/p21 expression (Figure 5A). Of interest, we also assessed the effect of TGFBR2 expression on PI3K pathway activation and found that phosphorylated AKT is present in the parental cell line and that TGF-β does not induce an increase in phosphorylated AKT in the TGFBR2 reconstituted cell line at any level of TGFBR2 expression (data not shown). After assessing the effect of TGFBR2 on the MAPK-ERK pathway, we next assessed the activation state of the Smad signaling pathway. Unlike with the MAPK-ERK pathway, Smad2 phosphorylation was increased with moderate to high levels of TGFBR2 but not with low expression levels of the receptor. There was also a modest increase in phosphorylated Smad2 with increasing levels of TGFBR2 expression, but the range of increased phosphorylated Smad2 was more limited than that for phosphorylated ERK1/2. (Figure 5B) The functional effect of the increase in phosphorylated Smad2 can be observed by a linear increased in CAGA luciferase reporter activity, which is specific for Smad mediated transcription. (Figure 8)
Figure 5. ERK and Smad activation after treatment with RG-115819 demonstrates a direct dose relation of RG-115819 to activation of both pathways.
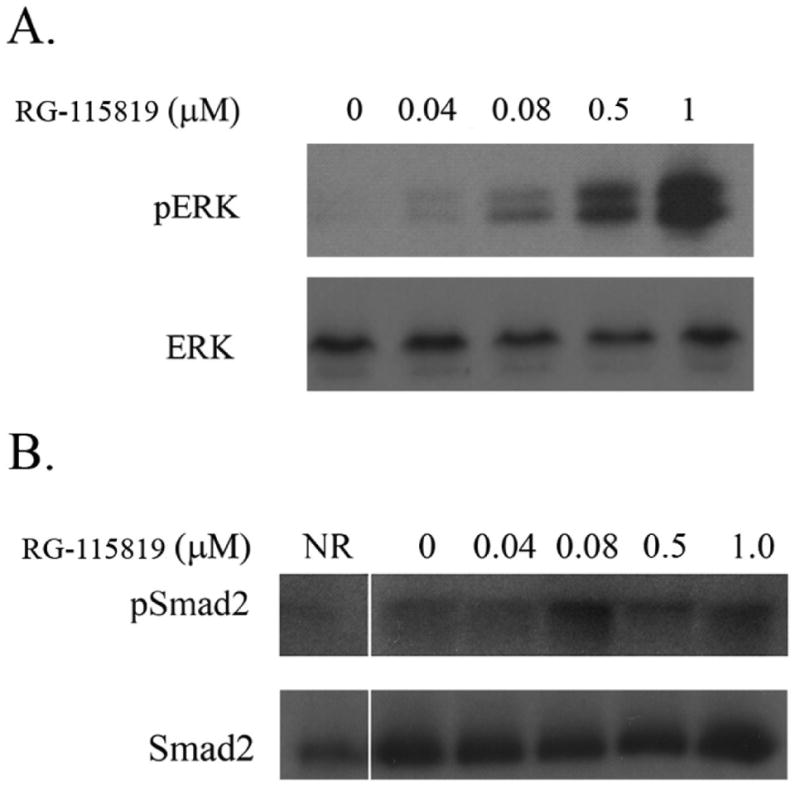
A. Immunoblotting for phosphorylated ERK1/2 (pERK) was performed on V-400R2 treated for 48 hours with RG-115819 (0 μM-1 μM). A dose-related increase in pERK in the cells is demonstrated. Total ERK levels reveal no differences with RG-115819 treatment. B. Immunoblot results of phosphorylated Smad2 (pSmad2) after 48 hours of treatment with RG-115819 also reveal a dose-related increase in the pSmad2 levels with doses greater than 0.08 μM. The amount of increase in pSmad2 is less than that observed for pERK. Densitometry was performed on the bands, and these results were normalized for loading using the densitometry values from the corresponding actin immunoblot. The densitometry revealed a linear increase in band density that correlated with increasing RG-115819 concentration [60 (0 μM), 90 (0.04 μM), 101 (0.08 μM), 121 (0.5 μM) and 159 (1 μM)]. Image J software (NIH) was used for the densitometry analysis.
Figure 8. TGFBR2 dependent apoptosis is suppressed by CDKN1A/p21 siRNA.
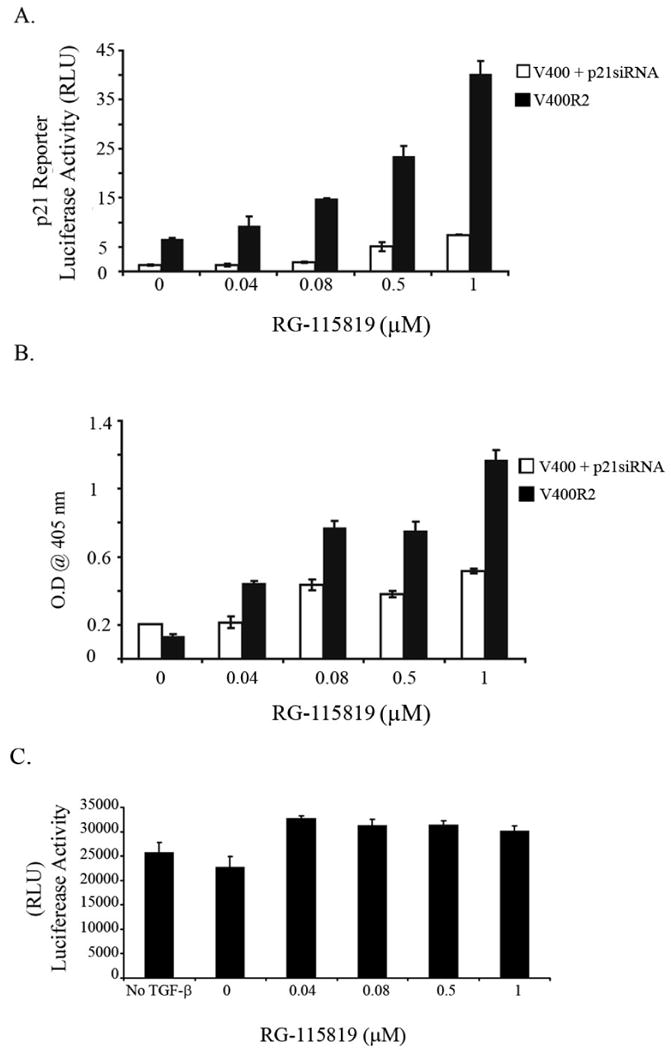
A. siRNA for CDKN1A/p21 suppresses CDKN1A/p21 reporter luciferase activity in the V-400R2 cell line. A dose related increase in luciferase reporter activity in the siRNA control treated cell line is demonstrated. B. Apoptosis in V-400R2 was measured using the Cell Death ELISA (Roche) after 48 hours of treatment with TGF-β (10ng/ml). Treatment with the CDKN1A/p21 siRNA inhibits the TGFBR2 dependent induction of apoptosis, which is seen in the siRNA control treated cells. C. TGF-β mediated apoptosis is inhibited by SMAD7. Apoptosis was induced by TGF-β in the V-400R2 cell line after transfection with SMAD7. SMAD7 abrogates the increased apoptosis observed in the cells V-400R2 cells treated with increasing concentrations of RG-115819.
3.4. TGF-β mediated regulation of CDKN1A/p21 expression is dependent on Smad and MAPK-ERK activation
It has been previously shown in other cell line systems that the MAPK-ERK pathway can control the expression of CDKN1A/p21 and that TGF-β can regulate CDKN1A/p21 through the MEK pathway [30, 39, 40]. Consequently we assessed the effect of inhibition of ERK activation on the ability of TGF-β to induce CDKN1A/p21 expression in the V-400R2 cell line in order to confirm that this was also true in this colorectal cancer cell line. TGF-β mediated activation of ERK was inhibited with the selective MAPK inhibitor U0126 (5μM) in V-400R2 cells treated with RG-115819 (0.04-1.0 μM) and TGF-β (10 ng/ml). The induction of TGFBR2 expression induced the expression of CDKN1A/p21, but blockade of the MAPK-ERK pathway prevented the differential increase in CDKN1A/p21 expression observed in the prior studies. These results suggest that CDKN1A/p21 expression is regulated by both MAPK-ERK and nonMAPK-ERK signaling pathways. (Figure 6A) We next assessed the effect of TGF-β mediated Smad pathway activation on the expression of CDKN1A/p21 using the inhibitory Smad, Smad7. Transfection of the V-400R2 cells with SMAD7 almost completely blocked the induction of the CAGA luciferase reporter and modestly suppressed the induction of CDKN1A/p21 reporter activity, but did not prevent the linear increase in CDKN1A/p21 expression observed with increasing TGFBR2 expression indicating the TGF-β mediated regulation of the precise expression levels of CDKN1A/p21 in V-400R2 is regulated predominantly by nonSmad signaling. (Figure 6B-C)
Figure 6. CDKN1A/p21 expression levels are regulated by MAPK/ERK in V-400R2 cells.
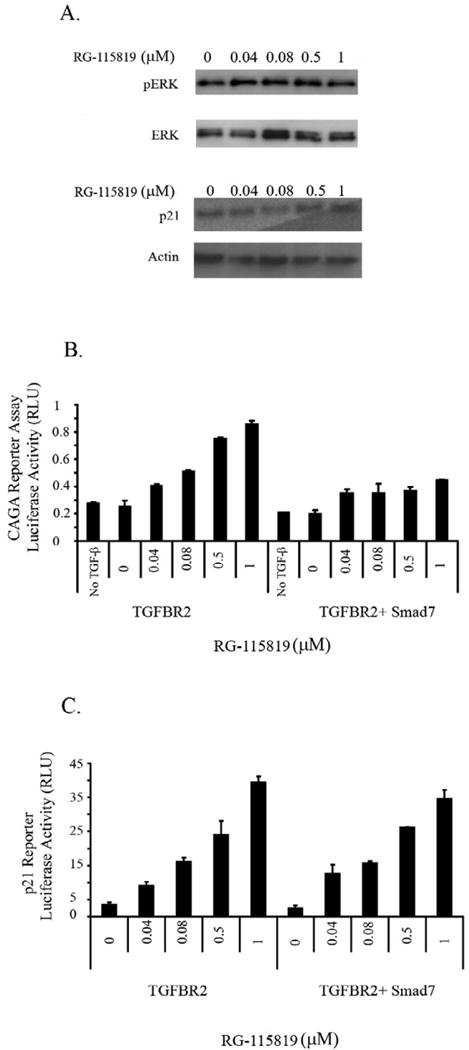
The V-400R2 cell line was cultured in the presence of RG-115819, TGF-β (10ng/ml) and the MAPK/ERK inhibitor U0126 for 48 hours. A. No increase in phosphorylated ERK (pERK) or in CDKN1A/p21 expression is detected in V-400R2 cells treated with the MAPK/ERK inhibitor demonstrating CDKN1A/p21's expression is regulated by ERK in a TGFBR2 dependent manner. The activation of ERK is considerably reduced due to the presence of the inhibitor. Of note, total levels of ERK were not altered. CDKN1A/p21 and pERK are detectable with vehicle only treatment, which is likely a consequence of a nonspecific effect of the U0126 vehicle. B. Inhibition of TGF-β mediated 3TPLux reporter activity by Smad7 occurs with all concentrations of ligand. V-400R2 cells were transfected with a plasmid expressing Smad7 in order to inhibit the Smad dependent pathway, and then transfected with the 3TP-Lux reporter. The dose dependent relation between luciferase activity and TGFBR2 is blocked by Smad7 demonstrating Smad7 is blocking Smad mediated transcription. C. Smad7 does not inhibit TGFBR2 dependent increases in CDKN1A/p21 luciferase reporter activity. V-400R2 cells were transfected with both Smad7 and the CDKN1A/p21 reporter assay. Interestingly, no significant changes in the activity of the CDKN1A/p21 luciferase reporter is detected after Smad7 transfection demonstrating that the expression level of TGFBR2 does not regulate CDKN1A/p21 expression through Smad signaling. The intensity of the bands was determined using densitometry as described in the prior figure legends. Densitometry for phosphorylated ERK revealed the following values: 120 (0 μM), 130 (0.04 μM), 126 (0.08μM), 145 (0.5 μM) and 131 (1 μM) and for p21: 130 (0 μM), 135 (0.04 μM), 142 (0.08 μM), 142 (0.5 μM) and 145 (1 μM).
3.5. TGFBR2 expression levels regulate the ability of TGF-β to induce CDKN1A/p21 mediated apoptosis
In light of our results showing that TGFBR2 expression directly correlates with increased ERK activation and CDKN1A/p21 expression, we carried out a series of studies to determine whether the expression level of TGFBR2 can directly affect the ability of TGF-β to induce apoptosis. The V-400R2 cell line was treated with RG-115819 in order to induce low and high levels of TGFBR2 and then assessed for TGF-β mediated apoptosis. A direct relationship between TGFBR2 expression and apoptosis was demonstrated showing that high levels of TGFBR2 are permissive for TGF-β mediated apoptosis. (Figure 7) These results suggest that TGF-β can mediate apoptosis in the V-400R2 cell line through the induction of the MAPK-ERK pathway and subsequent increase in CDKN1A/p21 expression. Thus, TGFBR2 levels may be a mechanism for determining whether cells are sensitive or resistant to TGF-β mediated apoptosis.
Figure 7. High expression levels of TGFBR2 induce apoptosis in V-400R2 cells.
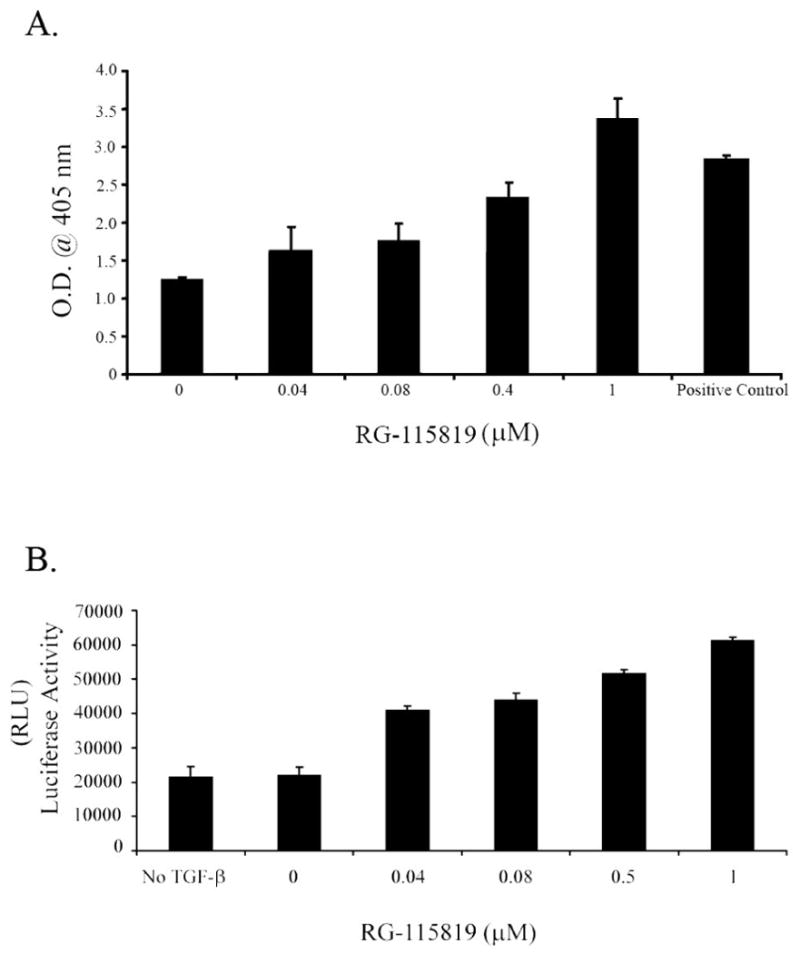
A. TGF-β mediated apoptosis was assessed using the Cell Death ELISA in V-400R2 treated with RG-115819 and TGF-β (10ng/ml). TGF-β mediated apoptosis is increased with doses of RG-115819 greater than 0.4 μM indicating that moderate-high levels of TGFBR2 and CDKN1A/p21 are needed for TGF-β to induce apoptosis in these cells. B. TGF-β mediated apoptosis assessed by caspase 3 and 7 activity confirms the results obtained with the Cell Death ELISA assay showing apoptosis directly correlates with TGFBR2 levels.
In order to establish if the induction of increased CDKN1A/p21 resulting from the increased TGFBR2 expression and ERK activation is a central mechanism for TGF-β mediated apoptosis in the V-400R2 cells, CDKN1A/p21 expression was inhibited with p21-siRNA in the presence of low and high levels or TGFBR2 and TGF-β (10ng/ml). (Figure 8A) Interestingly we found that inhibition of CDKN1A/p21 prevented the increase in apoptosis observed with increased TGFBR2 levels providing support for CDKN1A/p21 regulation being a mechanism for TGF-β mediated apoptosis in colon cancer. (Figure 8B) However, we also observed that inhibition of Smad signaling with SMAD7 impaired TGF-β mediated apoptosis presumably through a p21 independent mechanism, given that we did not observe an effect of SMAD7 on p21 expression. These results demonstrate TGFBR2 can regulate apoptosis through both p21 dependent and independent mechanisms. (Figure 8C).
4. Discussion
TGF-β can induce a variety of biological responses in cells, and these responses appear to be dependent on the cell type and context of the cell [41]. Some of the mechanisms that appear to play a role in determining the specificity of the response of a cell to TGF-β include signal pathway cross-talk, regulation of the expression levels of cytoplasmic and nuclear antagonists, and control of the composition of transcription factor complexes [20, 33]. In addition, over a decade ago, Derynck and Feng proposed the possibility of a receptor threshold model for determining the specificity of TGF-β's effects. This model proposes that there is a critical expression level of the TGF-β receptor that determines the specific TGF-β responses of a cell [42, 43]. Several lines of indirect evidence have supported this model, but to date there has been no direct assessment of this hypothesis [8, 43-47]. We now provide direct evidence that the regulation of the expression level of TGFBR2 can affect the specificity of the TGF-β response. We have shown that the activation of the Smad and nonSmad signaling pathways can be modulated by the expression level of TGFBR2 and that the activation state of the nonSmad signaling pathway principally determines whether TGF-β can induce CDKN1A/p21 mediated apoptosis in an epithelial cell line.
TGF-β and the TGF-β receptors are expressed in nearly all cell types and in developing and adult organisms. In embryonic development there is clear evidence that the concentration of active TGF-β and the extent of TGF-β signal pathway activation is tightly regulated and creates a gradient of responses that determines cell fate, among other things [41]. The majority of these studies have provided indirect and correlative evidence that the concentration of the ligands, receptors, and intracellular Smad proteins can determine the specific response of a cell to TGF-β. Further indirect evidence that the expression level can affect the specificity of the TGF-β response comes from studies of dominant negative TGFBR2 transgenes and chimeric receptors [22, 24, 48-50]. Our studies now provide direct evidence that the TGFBR2 expression level can regulate the intensity of activation of the Smad and nonSmad signaling pathways. Furthermore, the differences in activation of the Smad and MAPK-ERK pathways at different TGFBR2 expression levels suggests that not only can the level of TGFBR2 determine the intensity of pathway activation but also which pathways are activated [33]. The differential regulation of the post-TGF-β receptor pathways would be expected to lead to differences in transcription factor activation and transcription factor complex formation, which could then induce different patterns of gene expression.
Although the model above predicts that TGFBR2 expression levels will cause the differential expression of a variety of genes, we chose to specifically study CDKN1A/p21 because of its well-demonstrated regulation by TGF-β and because of its clear biological role in the inhibition of proliferation and induction of apoptosis. In addition, CDKN1A/p21 is regulated by TGF-β both by Smad and nonSmad signaling pathways and thus allows an assessment of the effect of Smad and nonSmad signaling on gene regulation [10, 51]. Our data suggest that CDKN1A/p21 is regulated by both the MAPK-ERK and Smad signaling pathways, which is true of other genes, such as FURIN [32]. These pathways may crosstalk at the level of Smad phosphorylation, nuclear localization of Smads, or at the level of transcription factor complex formation [33, 52, 53]. The differential activation of MAPK-ERK and Smad pathways suggests that at low levels of TGF-β receptor activation the nonSmad signaling pathways predominate over the Smad signaling pathways in determining the effect of TGF-β on the cell. The differences in activation of the MAPK and Smad pathways likely reflects the fact that the MAPK signaling pathway is a catalytic pathway whereas the Smad pathway is non-catalytic [8]. An interesting observation related to this explanation for the differential activation of the Smad and nonSmad pathways is the fact that in V-400R2 the MAPK-ERK pathway appears to be the predominant pathway regulating the level of CDKN1A/p21 expression whereas the Smad pathway has a more modest effect. These results are consistent with those of Hu et al who observed a similar phenomenon in the HaCaT keratinocyte cell line, but are in contrast to other studies that have shown that both Smad and SP1 mediated signaling are necessary for CDKN1A/p21 expression [20, 30, 54, 55]. These differences may reflect cell line specific differences in the expression of other regulators of CDKN1A/p21, such as c-Myc or MIZ-1 [56, 57].
There are several limitations related to our studies of the effect of TGFBR2 expression levels on TGF-β signaling. At this time, it is not clear whether the effects we have observed involve direct or indirect mechanisms for regulating the Smad and nonSmad signaling pathways. This limitation reflects our incomplete understanding of the mechanisms through which TGF-β affects the nonSmad signaling pathways [20]. In addition, we have not assessed the effect of TGFBR2 expression levels on the regulation of other signaling pathways and cannot exclude the possibility that these pathways are also playing a role in the regulation of CDKN1A/p21 and apoptosis that is affected by the expression level of the TGF-β receptor. Nonetheless, our results demonstrate that TGF-β receptor levels can regulate MAPK-ERK activation and that the control of this pathway has a central role in regulating CDKN1A/p21 expression and CDKN1A/p21 mediated apoptosis in the V-400R2 cell line.
Our results have several implications for the role of TGF-β in regulating normal cellular responses and for the role of TGF-β in cancer. The relevance of the effect of TGFBR2 expression levels on the regulation of TGF-β's effects on cells is appreciated in light of prior studies that have shown decreased TGFBR2 expression in a variety of cancers and in some inflammatory states [58]. In fact, many cancers have suppressed TGFBR2 expression without detectable mutations in any of the TGF-β signal pathway genes [59] [60-62]. In these cancers with an intact TGF-β receptor that is expressed at a low level, TGF-β appears to have the potential to act as an oncogene as well as a tumor suppressor gene [63]. This paradoxical role of TGF-β has been associated with its ability to activate Smad independent pathways (MAPK, PI3K and Rho) and with the modulation of TGF-β signaling pathway activation through interactions with other signaling pathways induced by mutated oncogenes and tumor suppressor genes [8, 18, 21, 22]. Our results suggest that an additional mechanism by which TGF-β may mediate paradoxical effects on cells is through the down-regulation of the expression of TGFBR2, which could differentially activate signaling pathways and alter the gene expression patterns of the cells. Thus, our results not only demonstrate that the expression level of TGFBR2 can influence the pathway activation status of TGF-β signaling pathways and the specific response of cells to TGF-β, but also that the regulation of the expression level is a plausible mechanism for the paradoxical effects of TGF-β on cancer cells. In summary, regulation of TGFBR2 expression levels is an additional mechanism for controlling the specificity of TGF-β's effects on cells and is a potential mechanism for the paradoxical effects of TGF-β observed in cancer.
Supplementary Material
Supplemental Figure 1: Immunostaining for p21 in the V-400R2 cell line. The V-400R2 cell line was grown with increasing concentrations of RG-115819 and then immunostained with an anti-p21 antibody (OP#68, EMD) and detected with a FITC-tagged secondary antibody (SC2012 in a 1:1000 dilution). Predominantly cytoplasmic p21 is observed in the cells treated with 0 or 0.5 μM RG-115819 whereas nuclear p21 can be seen in the cells treated with 1μM RG-115819.
These studies were conducted as follows. The V-400R2 cells were seeded in a chambered slide and grown for 48 hr with RG-115819 and TGF-β (10ng/ml). Then cells were fixed in −10aC methanol for 5 minutes and washed 3 times with 1X PBS. The slides were blocked with 10% normal blocking serum diluted in PBS (20 minute incubation at room temperature) and the washed with 1X PBS. The slides were then incubated with the primary antibody for 60 minutes at room temperature, washed in 1XPBS (three times), incubated in the secondary antibody (30 minutes at room temperature), and washed in 1X PBS (three times). The slides were then subjected to fluorescence microscopy.
Acknowledgments
This work was supported by RO1 CA115513, VA Dept. of R&D Presidential Early Career Award for Scientists and Engineers, and Mallinckrodt Scholar Award from the Edward Mallinckrodt Jr Foundation (to WMG).
We wish to acknowledge Patty Trobridge for her technical assistance with immunoblotting studies.
Footnotes
Dr's Dean Cress and Malla Padidam performed this work while employees of RheoGene, which was acquired by Intrexon Corporation. Dr. Cress is an employee of Intrexon. Dr. William Grady and Dr. Andres Rojas have no conflicts of interest to report.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Markowitz S, Wang J, Myeroff L, Parsons R, Sun L, Lutterbaugh J, Fan R, Zborowska E, Kinzler K, Vogelstein B, Brattain M, Willson J. Inactivation of the type II TGF-β receptor in colon cancer cells with microsatellite instability. Science. 1995;268:1336–1338. doi: 10.1126/science.7761852. [DOI] [PubMed] [Google Scholar]
- 2.Lin HY, Wang XF, Ng-Eaton E, Weinberg RA, Lodish HF. Expression cloning of the TGF-β type II receptor, a functional transmembrane serine/threonine kinase. Cell. 1992;68:775–785. doi: 10.1016/0092-8674(92)90152-3. [DOI] [PubMed] [Google Scholar]
- 3.Henis YI, Moustakas A, Lin HY, Lodish HF. The types II and III transforming growth factor-beta receptors form homo-oligomers. J Cell Biol. 1994;126:139–54. doi: 10.1083/jcb.126.1.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Markowitz S, Roberts A. Tumor supressor activity of the TGF-β pathway in human cancers. Cytokine and Growth Factor Reviews. 1996;7:93–102. doi: 10.1016/1359-6101(96)00001-9. [DOI] [PubMed] [Google Scholar]
- 5.Derynck R, Akhurst RJ, Balmain A. TGF-beta signaling in tumor suppression and cancer progression. Nat Genet. 2001;29:117–29. doi: 10.1038/ng1001-117. [DOI] [PubMed] [Google Scholar]
- 6.Shin I, Bakin AV, Rodeck U, Brunet A, Arteaga CL. Transforming growth factor beta enhances epithelial cell survival via Akt-dependent regulation of FKHRL1. Mol Biol Cell. 2001;12:3328–39. doi: 10.1091/mbc.12.11.3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Engel ME, Datta PK, Moses HL. Signal transduction by transforming growth factor-beta: a cooperative paradigm with extensive negative regulation. J Cell Biochem Suppl. 1998;31:111–22. doi: 10.1002/(SICI)1097-4644(1998)72:30/31+<111::AID-JCB15>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 8.Wakefield LM, Roberts AB. TGF-beta signaling: positive and negative effects on tumorigenesis. Curr Opin Genet Dev. 2002;12:22–9. doi: 10.1016/s0959-437x(01)00259-3. [DOI] [PubMed] [Google Scholar]
- 9.Bakin AV, Rinehart C, Tomlinson AK, Arteaga CL. p38 mitogen-activated protein kinase is required for TGFbeta-mediated fibroblastic transdifferentiation and cell migration. J Cell Sci. 2002;115:3193–206. doi: 10.1242/jcs.115.15.3193. [DOI] [PubMed] [Google Scholar]
- 10.Hocevar BA, Brown TL, Howe PH. TGF-beta induces fibronectin synthesis through a c-Jun N-terminal kinase-dependent, Smad4-independent pathway. Embo J. 1999;18:1345–56. doi: 10.1093/emboj/18.5.1345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhao Y. Transforming growth factor-beta (TGF-beta) type I and type II receptors are both required for TGF-beta-mediated extracellular matrix production in lung fibroblasts. Mol Cell Endocrinol. 1999;150:91–7. doi: 10.1016/s0303-7207(99)00021-0. [DOI] [PubMed] [Google Scholar]
- 12.Wang H, Radjendirane V, Wary KK, Chakrabarty S. Transforming growth factor beta regulates cell-cell adhesion through extracellular matrix remodeling and activation of focal adhesion kinase in human colon carcinoma Moser cells. Oncogene. 2004;23:5558–61. doi: 10.1038/sj.onc.1207701. [DOI] [PubMed] [Google Scholar]
- 13.Shin I, Bakin AV, Rodeck U, Brunet A, Arteaga CL. Transforming Growth Factor beta Enhances Epithelial Cell Survival via Akt-dependent Regulation of FKHRL1. Mol Biol Cell. 2001;12:3328–3339. doi: 10.1091/mbc.12.11.3328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Y, Feng XH, Wu RY, Derynck R. Receptor-associated Mad homologues synergize as effectors of the TGF-β response. Nature. 1996;383:168–172. doi: 10.1038/383168a0. [DOI] [PubMed] [Google Scholar]
- 15.Kretzschmar M, Liu F, Hata A, Doody J, Massague J. The TGF-beta family mediator Smad1 is phosphorylated directly and activated functionally by the BMP receptor kinase. Genes Dev. 1997;11:984–95. doi: 10.1101/gad.11.8.984. [DOI] [PubMed] [Google Scholar]
- 16.Abdollah S, Macias-Silva M, Tsukazaki T, Hayashi H, Attisano L, Wrana JL. TbetaRI phosphorylation of Smad2 on Ser465 and Ser467 is required for Smad2-Smad4 complex formation and signaling. J Biol Chem. 1997;272:27678–85. doi: 10.1074/jbc.272.44.27678. [DOI] [PubMed] [Google Scholar]
- 17.Souchelnytskyi S, Tamaki K, Engstrom U, Wernstedt C, ten Dijke P, Heldin CH. Phosphorylation of Ser465 and Ser467 in the C terminus of Smad2 mediates interaction with Smad4 and is required for transforming growth factor-beta signaling. J Biol Chem. 1997;272:28107–15. doi: 10.1074/jbc.272.44.28107. [DOI] [PubMed] [Google Scholar]
- 18.Seoane J, Le HV, Shen L, Anderson SA, Massague J. Integration of Smad and forkhead pathways in the control of neuroepithelial and glioblastoma cell proliferation. Cell. 2004;117:211–23. doi: 10.1016/s0092-8674(04)00298-3. [DOI] [PubMed] [Google Scholar]
- 19.Itoh S, ten Dijke P. Negative regulation of TGF-beta receptor/Smad signal transduction. Curr Opin Cell Biol. 2007;19:176–84. doi: 10.1016/j.ceb.2007.02.015. [DOI] [PubMed] [Google Scholar]
- 20.Pardali K, Kowanetz M, Heldin CH, Moustakas A. Smad pathway-specific transcriptional regulation of the cell cycle inhibitor p21(WAF1/Cip1) J Cell Physiol. 2005;204:260–72. doi: 10.1002/jcp.20304. [DOI] [PubMed] [Google Scholar]
- 21.Derynck R, Zhang YE. Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature. 2003;425:577–84. doi: 10.1038/nature02006. [DOI] [PubMed] [Google Scholar]
- 22.Bhowmick NA, Ghiassi M, Bakin A, Aakre M, Lundquist CA, Engel ME, Arteaga CL, Moses HL. Transforming Growth Factor-β1 Mediates Epithelial to Mesenchymal Transdifferentiation through a RhoA-dependent Mechanism. Mol Biol Cell. 2001;12:27–36. doi: 10.1091/mbc.12.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Siegel P, Massague J. Cytostatic and apoptotic actions of TGF-beta in homeostasis and cancer. Nat Rev Cancer. 2003;3:807–21. doi: 10.1038/nrc1208. [DOI] [PubMed] [Google Scholar]
- 24.Zhu HJ, Sizeland AM. A pivotal role for the transmembrane domain in transforming growth factor-beta receptor activation. J Biol Chem. 1999;274:11773–81. doi: 10.1074/jbc.274.17.11773. [DOI] [PubMed] [Google Scholar]
- 25.Pardali K, Moustakas A. Actions of TGF-beta as tumor suppressor and pro-metastatic factor in human cancer. Biochim Biophys Acta. 2007;1775:21–62. doi: 10.1016/j.bbcan.2006.06.004. [DOI] [PubMed] [Google Scholar]
- 26.Valle L, Serena-Acedo T, Liyanarachchi S, Hampel H, Comeras I, Li Z, Zeng Q, Zhang HT, Pennison MJ, Sadim M, Pasche B, Tanner SM, de la Chapelle A. Germline allele-specific expression of TGFBR1 confers an increased risk of colorectal cancer. Science. 2008;321:1361–5. doi: 10.1126/science.1159397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grady WM, Myeroff LL, Swinler SE, Rajput A, Thiagalingam S, Lutterbaugh JD, Neumann A, Brattain MG, Chang J, Kim SJ, Kinzler KW, Vogelstein B, Willson JKV, Markowitz S. Mutational inactivation of transforming growth factor-beta receptor type II in microsatellite stable colon cancers. Cancer Res. 1999;59:320–324. [PubMed] [Google Scholar]
- 28.Palli SR, Kapitskaya MZ, Kumar MB, Cress DE. Improved ecdysone receptor-based inducible gene regulation system. Eur J Biochem. 2003;270:1308–15. doi: 10.1046/j.1432-1033.2003.03501.x. [DOI] [PubMed] [Google Scholar]
- 29.Karzenowski D, Potter DW, Padidam M. Inducible control of transgene expression with ecdysone receptor: gene switches with high sensitivity, robust expression, and reduced size. Biotechniques. 2005;39:191–2. 194, 196. doi: 10.2144/05392ST01. passim. [DOI] [PubMed] [Google Scholar]
- 30.Hu PP, Shen X, Huang D, Liu Y, Counter C, Wang XF. The MEK pathway is required for stimulation of p21(WAF1/CIP1) by transforming growth factor-beta. J Biol Chem. 1999;274:35381–7. doi: 10.1074/jbc.274.50.35381. [DOI] [PubMed] [Google Scholar]
- 31.Kumar MB, Potter DW, Hormann RE, Edwards A, Tice CM, Smith HC, Dipietro MA, Polley M, Lawless M, Wolohan PR, Kethidi DR, Palli SR. Highly flexible ligand binding pocket of ecdysone receptor: a single amino acid change leads to discrimination between two groups of nonsteroidal ecdysone agonists. J Biol Chem. 2004;279:27211–8. doi: 10.1074/jbc.M403839200. [DOI] [PubMed] [Google Scholar]
- 32.Blanchette F, Rivard N, Rudd P, Grondin F, Attisano L, Dubois CM. Cross-talk between the p42/p44 MAP kinase and Smad pathways in transforming growth factor beta 1-induced furin gene transactivation. J Biol Chem. 2001;276:33986–94. doi: 10.1074/jbc.M100093200. [DOI] [PubMed] [Google Scholar]
- 33.Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113:685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 34.Child ES, Mann DJ. The intricacies of p21 phosphorylation: protein/protein interactions, subcellular localization and stability. Cell Cycle. 2006;5:1313–9. doi: 10.4161/cc.5.12.2863. [DOI] [PubMed] [Google Scholar]
- 35.Wang T, Danielson P, Li B, Shah P, Kim S, Donahoe P. p21ras farnesyltransferase alpha subunit in TGF-β and activin signaling. Science. 1996;271:1120–1122. doi: 10.1126/science.271.5252.1120. [DOI] [PubMed] [Google Scholar]
- 36.Olson MF, Paterson HF, Marshall CJ. Signals from Ras and Rho GTPases interact to regulate expression of p21Waf1/Cip1. Nature. 1998;394:295–9. doi: 10.1038/28425. [DOI] [PubMed] [Google Scholar]
- 37.Seoane J, Le HV, Massague J. Myc suppression of the p21(Cip1) Cdk inhibitor influences the outcome of the p53 response to DNA damage. Nature. 2002;419:729–34. doi: 10.1038/nature01119. [DOI] [PubMed] [Google Scholar]
- 38.Park GT, Morasso MI. Bone morphogenetic protein-2 (BMP-2) transactivates Dlx3 through Smad1 and Smad4: alternative mode for Dlx3 induction in mouse keratinocytes. Nucleic Acids Res. 2002;30:515–22. doi: 10.1093/nar/30.2.515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ciccarelli C, Marampon F, Scoglio A, Mauro A, Giacinti C, De Cesaris P, Zani BM. p21WAF1 expression induced by MEK/ERK pathway activation or inhibition correlates with growth arrest, myogenic differentiation and onco-phenotype reversal in rhabdomyosarcoma cells. Mol Cancer. 2005;4:41. doi: 10.1186/1476-4598-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Park KS, Ahn Y, Kim JA, Yun MS, Seong BL, Choi KY. Extracellular zinc stimulates ERK-dependent activation of p21(Cip/WAF1) and inhibits proliferation of colorectal cancer cells. Br J Pharmacol. 2002;137:597–607. doi: 10.1038/sj.bjp.0704909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Munoz NM, Baek JY, Grady WM. TGF-beta has paradoxical and context dependent effects on proliferation and anoikis in human colorectal cancer cell lines. Growth Factors. 2008:1. doi: 10.1080/08977190802291667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Derynck R, Feng XH. TGF-β receptor signaling. Biochimica et Biophysica Acta. 1997;1333:F105–F150. doi: 10.1016/s0304-419x(97)00017-6. [DOI] [PubMed] [Google Scholar]
- 43.Fafeur V, O'Hara B, Bohlen P. A glycosylation-deficient endothelial cell mutant with modified responses to transforming growth factor-beta and other growth inhibitory cytokines: evidence for multiple growth inhibitory signal transduction pathways. Mol Biol Cell. 1993;4:135–44. doi: 10.1091/mbc.4.2.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Feng XH, Filvaroff EH, Derynck R. Transforming growth factor-beta (TGF-beta)-induced down-regulation of cyclin A expression requires a functional TGF-beta receptor complex. Characterization of chimeric and truncated type I and type II receptors. J Biol Chem. 1995;270:24237–45. doi: 10.1074/jbc.270.41.24237. [DOI] [PubMed] [Google Scholar]
- 45.Wang J, Sergina N, Ko TC, Gong J, Brattain MG. Autocrine and exogenous transforming growth factor beta control cell cycle inhibition through pathways with different sensitivity. J Biol Chem. 2004;279:40237–44. doi: 10.1074/jbc.M401665200. [DOI] [PubMed] [Google Scholar]
- 46.Geiser AG, Burmester JK, Webbink R, Roberts AB, Sporn MB. Inhibition of growth by transforming growth factor-beta following fusion of two nonresponsive human carcinoma cell lines. Implication of the type II receptor in growth inhibitory responses. J Biol Chem. 1992;267:2588–93. [PubMed] [Google Scholar]
- 47.Brand T, MacLellan W, Schneider M. Dominant-negative receptor for type β transforming growth factors created by deletion of the kinase domain. The Journal of Biological Chemistry. 1993;268:11500–11503. [PubMed] [Google Scholar]
- 48.Yamaguchi K, Shirakabe K, Shibuya H, Irie K, Oishi I, Ueno N, Taniguchi T, Nishida E, Matsumoto K. Identification of a member of the MAPKKK family as a potential mediator of TGF-beta signal transduction. Science. 1995;270:2008–11. doi: 10.1126/science.270.5244.2008. [DOI] [PubMed] [Google Scholar]
- 49.Muraoka-Cook RS, Dumont N, Arteaga CL. Dual role of transforming growth factor beta in mammary tumorigenesis and metastatic progression. Clin Cancer Res. 2005;11:937–43. [PubMed] [Google Scholar]
- 50.Dumont N, Arteaga CL. Transforming growth factor-beta and breast cancer: Tumor promoting effects of transforming growth factor-beta. Breast Cancer Res. 2000;2:125–32. doi: 10.1186/bcr44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Engel ME, McDonnell MA, Law BK, Moses HL. Interdependent SMAD and JNK signaling in transforming growth factor- beta-mediated transcription. J Biol Chem. 1999;274:37413–20. doi: 10.1074/jbc.274.52.37413. [DOI] [PubMed] [Google Scholar]
- 52.Kretzschmar M, Doody J, Timokhina I, Massague J. A mechanism of repression of TGFβ/Smad signaling in oncogenic Ras. Genes and Development. 1999;13:804–816. doi: 10.1101/gad.13.7.804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Massague J. How cell read TGF-β signals. Nat Rev Mol Cell Biol. 2000;1:169–78. doi: 10.1038/35043051. [DOI] [PubMed] [Google Scholar]
- 54.Kardassis D, Papakosta P, Pardali K, Moustakas A. c-Jun transactivates the promoter of the human p21(WAF1/Cip1) gene by acting as a superactivator of the ubiquitous transcription factor Sp1. J Biol Chem. 1999;274:29572–81. doi: 10.1074/jbc.274.41.29572. [DOI] [PubMed] [Google Scholar]
- 55.Moustakas A, Pardali K, Gaal A, Heldin CH. Mechanisms of TGF-beta signaling in regulation of cell growth and differentiation. Immunol Lett. 2002;82:85–91. doi: 10.1016/s0165-2478(02)00023-8. [DOI] [PubMed] [Google Scholar]
- 56.Seoane J, Pouponnot C, Staller P, Schader M, Eilers M, Massague J. TGFbeta influences Myc, Miz-1 and Smad to control the CDK inhibitor p15INK4b. Nat Cell Biol. 2001;3:400–8. doi: 10.1038/35070086. [DOI] [PubMed] [Google Scholar]
- 57.Ten Dijke P, Goumans MJ, Itoh F, Itoh S. Regulation of proliferation by smad proteins. Journal of Cellular Physiology. 2002;191:1–16. doi: 10.1002/jcp.10066. [DOI] [PubMed] [Google Scholar]
- 58.Blobe GC, Schiemann WP, Lodish HF. Role of transforming growth factor beta in human disease. N Engl J Med. 2000;342:1350–8. doi: 10.1056/NEJM200005043421807. [DOI] [PubMed] [Google Scholar]
- 59.Grady WM, Markowitz SD. In: The TGF-β Family. Derynck R, Miyazono K, editors. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, NY: 2008. pp. 889–938. [Google Scholar]
- 60.Kim SJ, Im YH, Markowitz SD, Bang YJ. Molecular mechanisms of inactivation of TGF-beta receptors during carcinogenesis. Cytokine Growth Factor Rev. 2000;11:159–68. doi: 10.1016/s1359-6101(99)00039-8. [DOI] [PubMed] [Google Scholar]
- 61.Hougaard S, Norgaard P, Abrahamsen N, Moses HL, Spang-Thomsen M, Skovgaard Poulsen H. Inactivation of the transforming growth factor beta type II receptor in human small cell lung cancer cell lines. Br J Cancer. 1999;79:1005–11. doi: 10.1038/sj.bjc.6690161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Musch A, Rabe C, Paik MD, Berna MJ, Schmitz V, Hoffmann P, Nischalke HD, Sauerbruch T, Caselmann WH. Altered expression of TGF-beta receptors in hepatocellular carcinoma--effects of a constitutively active TGF-beta type I receptor mutant. Digestion. 2005;71:78–91. doi: 10.1159/000084523. [DOI] [PubMed] [Google Scholar]
- 63.Bierie B, Moses HL. TGF-beta and cancer. Cytokine Growth Factor Rev. 2006;17:29–40. doi: 10.1016/j.cytogfr.2005.09.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1: Immunostaining for p21 in the V-400R2 cell line. The V-400R2 cell line was grown with increasing concentrations of RG-115819 and then immunostained with an anti-p21 antibody (OP#68, EMD) and detected with a FITC-tagged secondary antibody (SC2012 in a 1:1000 dilution). Predominantly cytoplasmic p21 is observed in the cells treated with 0 or 0.5 μM RG-115819 whereas nuclear p21 can be seen in the cells treated with 1μM RG-115819.
These studies were conducted as follows. The V-400R2 cells were seeded in a chambered slide and grown for 48 hr with RG-115819 and TGF-β (10ng/ml). Then cells were fixed in −10aC methanol for 5 minutes and washed 3 times with 1X PBS. The slides were blocked with 10% normal blocking serum diluted in PBS (20 minute incubation at room temperature) and the washed with 1X PBS. The slides were then incubated with the primary antibody for 60 minutes at room temperature, washed in 1XPBS (three times), incubated in the secondary antibody (30 minutes at room temperature), and washed in 1X PBS (three times). The slides were then subjected to fluorescence microscopy.