

Abstract
Glucose metabolism through glycolysis and hexosamine pathway has been shown to be altered in type 2 diabetes. However, its fate through the pentose phosphate pathway (PPP) is currently unclear. In this study, we determined whether the activity of glucose-6-phosphate dehydrogenase (G6PD), the rate-limiting enzyme in the PPP, is modulated in the liver of Zucker obese fa/fa rats (9–11 weeks old). We found that G6PD expression and activity, NADPH levels and 6-phosphogluconate generation was significantly increased in liver of fa/fa rats. Inhibition of PI3 kinase and Src kinases decreased (P<0.05) G6PD activity in the fa/fa but not in the lean rat liver, suggesting that G6PD activity is regulated by PI3/Src kinase signaling pathways. G6PD-derived NADPH increased (P<0.05) superoxide anion levels by 70–90% in the fa/fa vs the lean rat liver, which was inhibited by NADPH oxidase inhibitor, gp91ds-tat (50 μM), and G6PD inhibitors, 6-aminonicotinamide (1 mM) and dihydroepiandrosterone (100 μM); therefore, indicating that elevated G6PD activity may be responsible for mediating superoxide generation. Interestingly, we also found a positive correlation between liver hypertrophy/increased G6PD activity (r2=0.77; P=0.0009) and liver hypertrophy/superoxide production (r2=0.51; P=0.0091) in fa/fa rats. Increased G6PD and NADPH oxidase expression and activity, in young hyper-glycemic and -insulinemic rats prior to the development of diabetes, appear to be contributing factors for the induction of oxidative stress. Since inhibition of G6PD activity decreases oxidative stress, we conclude that it behaves as a pro-oxidant in the fa/fa rat liver, in type 2 diabetes.
Keywords: Diabetes, Src kinase, NADPH, NADPH oxidase, Oxidative stress, Superoxide anion, Hydrogen peroxide, Liver, Zucker obese fa/fa
Type 2 diabetes affects approximately 170 million people around the world and these numbers are rapidly rising every year [1]. This metabolic syndrome is now widely recognized as a risk factor for liver disorders, obesity and cardiovascular diseases.
Several studies have suggested that oxidative stress is elevated by chronic hyperglycemia in diabetic animal models as well as in diabetic patients [2–7]. Overproduction of superoxide anion (O2−) by the mitochondria, polyol pathway, NAD(P)H oxidase, uncoupled nitric oxide synthase, and reduced endogenous antioxidant enzymes have been proposed to increase oxidative stress in hyperglycemia. Yet, the underlying mechanism that activates reactive oxygen species generation in hyper-glycemia or -insulinemia is far from clear. Brownlee and colleagues [8], have proposed that an increase in advanced glycation end products, activation of protein kinase C (β isoform), and activation of aldosereductase in diabetes, elevate O2− generation from mitochondrial respiratory chain. Furthermore, other studies have shown that in streptozotocin-treated (type 1 diabetes) rat model there is over-expression of NAD(P)H oxidase (Nox-2 and Nox-4) in the vascular system and kidneys with a concomitant elevation in O2− production [2, 9].
In most tissue types, constitutively active NAD(P)H oxidase produces low levels of O2− [10]. Additionally, in pathophysiological conditions, activation of NADPH oxidase following stimulation by factors such as, angiotensin II, thrombin, and TNFα [10, 11], leads to subsequent elevation in O2− generation. Other investigators and we have shown that NADPH derived from glucose-6-phosphate dehydrogenase (G6PD), a rate limiting enzyme in the pentose phosphate pathway (PPP) that produces a major portion of NADPH in the cell, fuels NADPH oxidase and O2− production in the failing heart [12, 13], angiotensin-II and PKC agonist treated vasculature [14, 15], and neutrophiles in basal state [16] and during respiratory burst [17]. Since NADPH levels have been shown to be elevated in the endothelial cells of diabetic (type 1) BB rats [18], we postulated that an increase in G6PD-derived NADPH in type 2 diabetes could elevate NADPH oxidase-derived oxidative stress.
This study was, therefore, designed to investigate the implications of imbalance in glucose metabolism observed in type 2 diabetes, on oxidoreductases enzymes such as G6PD and NADPH oxidase in the liver of young Zucker fa/fa rats, an animal model of type 2 diabetes, resembling human diabetes. In particular we investigated whether; [i] the G6PD expression and activity is up-regulated, [ii] protein expression and activity of NAD(P)H oxidase is altered, and [iii] PI3Kinase and Src kinases-dependent signaling pathways and G6PD-derived NADPH evoke Nox-derived O2− in liver from young (9–11 weeks old animals) Zucker fa/fa rats as compared to the lean rats.
Materials & Methods
NADPH, NADH, NADP+, NAD+, glucose-6-phosphate, glucose-6-phosphate dehydrogenase, 6-phosphogluconolactone, 6-hosphogluconate dehydrogenase, bis(N-methyacridinium) nitrate (lucigenin), antimycin, rotenone, dehydroepiandrosterone, 6-aminonicotinamide and other salts were purchased from Sigma (St. Louis, MO, USA). The stock solutions of 6-aminonicotinamide and dehydroepiandrosterone were made in dimethyl sulfoxide (DMSO; Sigma), and final 1:1000 dilutions in buffered physiological salt solution were used in the study. 2-(4-Morpholinyl)-8-phenyl-4H-1-benzopyran-4-one (LY294002), 4-Amino-5-(4-chlorophenyl)-7-(t-butyl) pyrazolo [3,4-d] pyrimidine (PP2) and their controls were purchased from Calbiochem, Inc (Carlsberg, CA, USA).
Animal model
The Institutional Animal Use Committee approved all protocols and surgical procedures, which were in accordance with National Institutes of Health and American Physiological Society guidelines. To examine our hypothesis, experiments were performed with adult male Zucker fa/fa rat, a prototype model of hyperglycemia and type 2 diabetes, purchased from Charles River laboratories, Wilmington, MA, USA. All rats were exposed to a 12:12-h light-dark cycle and allowed free access to standard rat food and water.
Isolation of liver
Animals were anesthetized with Nembutal (10 mg/kg) and abdominal cavity was opened. The liver was cannulated through the portal vein and perfused with normal tyrode solution at 37°C for 5 minutes. The liver was excised and cut in small pieces. The minced liver was homogenized as described in individual protocols and used for biochemical analyses. In some experiments, the isolated liver was perfused with inhibitors of G6PD, NADPH oxidase, mitochondrial respiratory complex I and III, and PI3 kinase and Src kinases in normal tyrode solution for 30 minutes at 37°C and then used for biochemical analyses.
Isolation of hepatocytes
Hepatic cells were isolated from liver by slight modification of previously described method [19]. Briefly, liver was washed with ice cold Ca2+-free buffer, pH 7.6, consisting of NaCl (150 mM), KCl (2.8 mM), glucose (5.5 mM), and HEPES (25 mM) for 10 minutes. Next, liver was treated with same buffer containing CaCl2 (3.8 mM), collagenase type I (0.5 mg/ml) and soyabean trysin inhibitor (10 μg/ml) at 37°C for 1 hr. The liver cells were dispersed by tituration and centrifuged at 1×g for 5 minutes. Hepatocytes were separated from non-parenchymal cells by centrifugation in isotonic percoll (specific gravity=1.06) at 50×g for 5 minutes. Percoll was removed by two washes in buffer.
Estimation of glucose and insulin levels
Blood glucose levels were determined by standard glucose strips using a glucometer (Abbot Laboratory, Chicago, IL, USA) and insulin levels were quantified by ELISA kit purchased from Cayman Chemical Co., Ann Arbor, MI, USA.
Estimation of superoxide
Superoxide levels were determined by previously published protocols [12]; briefly tissue homogenates (10–15 μl) were incubated at 37°C in 20 mM MOPS buffer, pH 7.4 containing 250 mM sucrose for 10 minutes in absence or presence of drugs and O2− was detected by lucigenin (5 μM) chemiluminescence. Some experiments examined O2− production in the absence of the NADPH regenerating system to evaluate the role of NADPH and NADH. O2− in hepatocytes was detected in luminometer (Synergy 2, Biotek Instruments, MA, USA) instead of liquid scintillation counter described previously [12].
Estimation of superoxide dismutase activity
SOD activity was assessed by measuring the dismutation of superoxide radicals generated by xanthine oxidase and hypoxanthine in a convenient 96 well format using a kit from Cayman Chemical Co.
Estimation of glutathione peroxidase and glutathione reductase activity
GSH peroxidase and reductase activity was measured using a kit from Cayman Chemical Co. The oxidation of NADPH to NADP+ was accompanied by a decrease in absorbance measured at 340 nm. The rate of decrease in the A340 was directly proportional to the peroxidase activity and the rate of decrease in the absorbance at 340 nm was directly proportional to the reductase activity in the sample.
Estimation of triglycerides, free fatty acid, and liver function markers
The triglyceride, free fatty acid, alkaline phosphatase (ALP), alanine aminotransferase (ALT) and γ-glutamyl transpeptidase (γ-GT) levels were determined in the serum collected from Zucker lean and fa/fa rats using kits purchased from Teco Diagnostics, Anaheim, CA, USA.
Western blot analysis
Homogenates were prepared from frozen tissues and total protein content was measured as described previously. Thirty-five micrograms of total protein was analyzed by SDS-PAGE. Western blot analyses was performed using rabbit polyclonal anti-G6PD (Sigma Chemical), mouse monoclonal anti-Nox-1, anti-Nox-2, and anti-p67phox (Transduction Laboratory, San Jose, CA, USA), goat polyclonal anti-Nox-4 and anti- p47phox, mouse monoclonal anti-α-tubulin (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Bands were visualized by autoradiography and quantified using densitometric analyses. Densitometric values for specific proteins were normalized with α-tubulin and expressed as a ratio of the two proteins.
Estimation of glucose-6-phosphate dehydrogenase activity
Glucose-6-phosphate dehydrogenase (G6PD) activity was measured in liver homogenates by following the reduction of NADP+ to NADPH [14]. NADPH fluorescence was detected at 340 nm (Ex) and 460 nm (Em) using a Flx800 microplate fluorescence detector (BioTek Instruments, Winooski, Vermont, USA).
Estimation of NAD(P)H levels
Frozen tissues were homogenized in an extraction medium containing NaOH (0.02 N) and cysteine (0.5 mmol/L) at 0°C. The extracts were then heated at 60°C for 10 min and neutralized with 2 ml of 0.25 M glycylglycine buffer, pH 7.6 as described previously and NADPH levels were estimated by determining NADPH fluorescence at 340 (Ex) and 460 (Em) using a previously described recycling method [20].
Statistical analysis
Values are means ±SEM. Comparisons between groups were made with Student’s t-test or analysis of variance (ANOVA) with Scheffé’s post-hoc test for multiple comparisons. Differences were considered significant at p<0.05.
Results
Glucose and insulin levels are increased in young Zucker fa/fa rats
Nine to Eleven weeks old Zucker fa/fa rats (345±6 g; n=6) were obese (P<0.05) as compared to lean rats (264±15 g; n=6), and had high (P<0.05) non-fasting plasma glucose [127±11 (lean) and 341±12 (Zucker fa/fa) mg/dl] and insulin [0.67±0.07 (lean) and 3.04±0.22 (Zucker fa/fa) ng/ml] levels.
Glucose-6-phosphate dehydrogenase activity is elevated in type 2 diabetes
Since glucose metabolism is altered in type 2 diabetes, it is possible that G6PD expression and/or activity may also be modulated. We performed Western blot analysis to determine whether G6PD expression was changed in liver of Zucker fa/fa rats. The results indicated that the expression of G6PD increased (P< 0.05) by 117 % in liver (Fig 1 A top panel and B) and hepatocytes (Fig 1 A bottom panel) of Zucker fa/fa as compared to the lean rats. In addition, we found that the activity of G6PD was significantly higher in the liver and hepatocytes of Zucker fa/fa animals by 400 % (Fig 1 C) and 160 % (P< 0.05), respectively, as compared to age-matched lean rat. Additionally, NADP+, substrate, -dependent enzyme activity curve was also significantly shifted to the left in hepatic tissue from Zucker fa/fa rats (Fig 1 D). Consistently, the NADPH (Fig 1 E) and 6-phospho-gluconate (Fig 1 F), products of G6PD, levels were significantly increased in the liver of Zucker fa/fa rats as compared to lean rats.
Figure 1. Glucose-6-phosphate dehydrogenase activity is elevated in type 2 diabetic model.
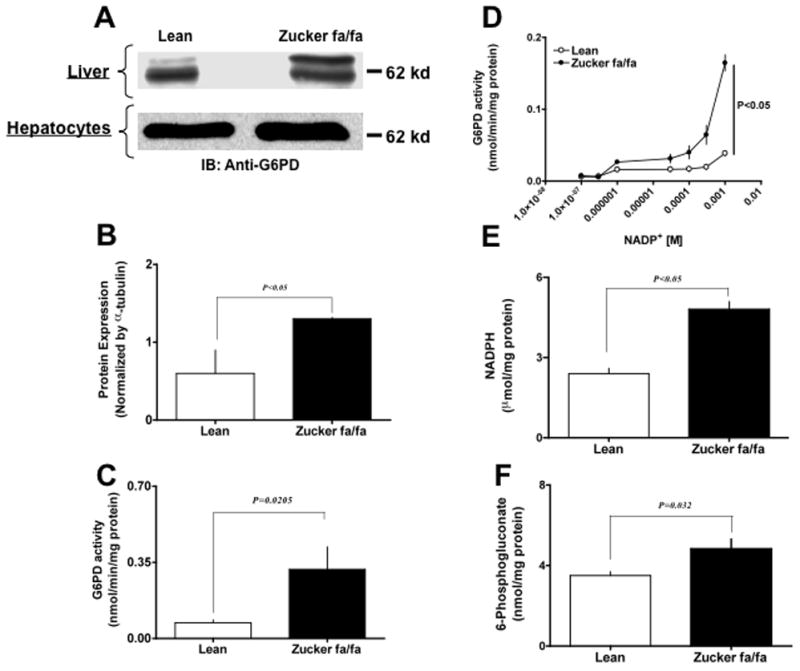
Zucker lean and fa/fa rat liver homogenates (Panel A top) and hepatocyte lysates (Panel A bottom) were analyzed on 9% SDS-PAGE. Western blot analysis was performed using rabbit polyclonal anti-G6PD antibody (A). All input lanes contain 35 μg of total protein content. Panel (A) represents one blot of six such independent experiments. (B) The graph represents G6PD protein expression in lean (n=6) and ZDF (n=6) rat liver as estimated by densitometric analysis (total: upper and lower band) and normalized with α-tubulin values. The graph represents G6PD activity (C), effect of NADP+-dependent on activity (D), NADPH levels (E) and 6-phospho-gluconate levels (F) from lean (n=6) and Zucker fa/fa (n=6) rat liver, respectively. Statistical analysis for (B–F) was performed using Student’s t-test and P<0.05 (lean versus Zucker fa/fa) was considered statistically significant.
Glucose-6-phosphate dehydrogenase activity is elevated by PI3 and Src kinases in type 2 diabetes
EGF associated tyrosine kinases-, PKC- and Src-dependent pathways have been shown to activate G6PD in renal cortical cells [21] and in the failing human hearts [22]. To elucidate how G6PD is activated in fa/fa, we studied the effects of kinases involved in the insulin signaling pathway such as, PI3 kinase [23] and Src family of protein tyrosine kinases [24], on G6PD activity. We found that there was an increase in the amount of total-Src (Fig 2 A & B) and phospho-Src416 (Fig. 2 A & C) in fa/fa vs. lean rats. Next, we treated liver with (i) PI3 kinase inhibitor-LY294002 (active form; 10 μM) and LY303511 (non-active form; 10 μM); and (ii) Src kinase inhibitor-PP2 (active form; 10 μM) and PP3 (non-active form; 10 μM), respectively, to determine the role of PI3 kinase and Src kinase in activating G6PD. The liver was perfused with the inhibitors for 30 minutes as described earlier (See Methods) and then the activity of G6PD was determined in liver homogenates in the presence of LY294002/LY303511 and PP2/PP3. Both active form of inhibitors attenuated (P<0.05) G6PD activity in fa/fa, but not in lean liver homogenate (Fig 2 D & E). Total-Src was increased in isolated hepatocytes (Fig 2 A), and treatment with PI3 kinase and Src kinase inhibitors decreased (P<0.05) G6PD activity (Control: 0.892±0.159; LY294002: 0.358±0.2893; PP2: 0.406±0.3802 nmol/min/mg protein) in hepatocytes from fa/fa rats.
Figure 2. Glucose-6-phosphate dehydrogenase is activated by Src kinase in the liver of Zucker fa/fa rats.
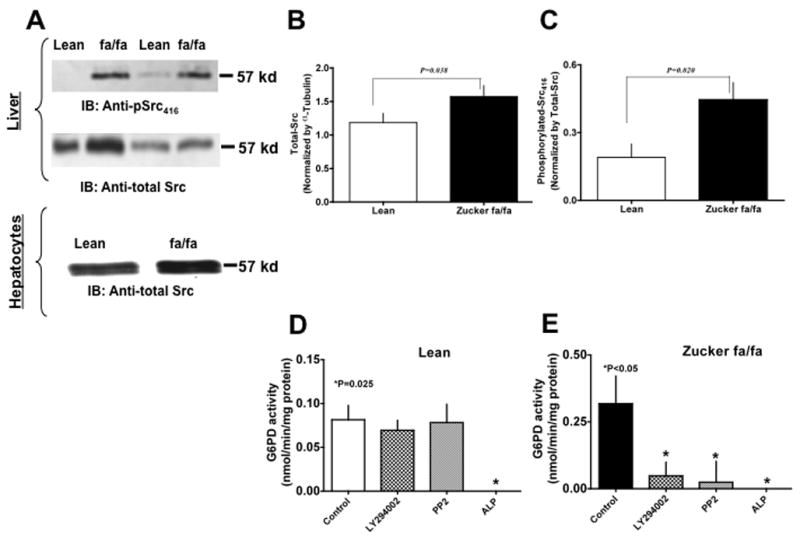
(A) Zucker lean (lanes 1 & 3) and fa/fa (lanes 2 & 4) rat liver homogenates (Panel A top) and hepatocyte lysates (Panel A bottom) were analyzed on 9% SDS-PAGE. Western blot analysis was performed using rabbit polyclonal anti-pSrc (first panel) and anti-total Src antibody (second panel). All input lanes contain 35 μg of total protein content. Panel (A) represents one blot of six such independent experiments. (B) & (C) Graphs represents total-Src (B) and phosphorylated-Src416 (C) in lean (n=6) and ZDF (n=6) rat liver as estimated by densitometric analysis and normalized with total-Src or α-tubulin values. (D) & (E) Lean (D) and Zucker fa/fa (E) liver tissues were incubated at 37°C for 10 minutes without and with PI3 kinase inhibitor, LY294002 (10 μM), Src kinase inhibitor, PP2 (10 μM), and alkaline phosphatase (200 U/ml), respectively. Graphs represents glucose-6-phosphate dehydrogenase activity in, Lean (D) and Zucker fa/fa (E) liver homogenates, respectively. Statistical analysis was performed using Student’s t-test and P<0.05 (lean versus Zucker fa/fa) was considered statistically significant.
Protein expression of NAD(P)H oxidase is altered in type 2 diabetic tissue
Considering the significant elevation of G6PD activity and subsequently NADPH levels, we performed experiments to determine whether expression of NADPH oxidase subunits were also altered in type 2 diabetes. We estimated expression of Nox-1, Nox-2, Nox-4, p47phox and p67phox in lean and fa/fa rat liver homogenates (Fig 3 A: top panel) and isolated hepatocyte lysate (Fig 3 A: bottom panel) by Western blot analysis. We were unable to detect Nox-2 (Fig 3 A; second panel), however, Nox-4 levels (Fig 3 A, third panel) were increased by approximately 2-fold (P<0.05; Fig 3 B) in the liver of fa/fa vs lean rats. Although, we observed a decreased expression of Nox-1 (P < 0.05, Fig 3 B), in fa/fa vs lean rats, increased levels of p47phox and p67phox (Fig 3 A, fourth and fifth panel, respectively) and significantly increased membrane-to-cytoplasmic ratio of p47phox (Fig 3 C), suggests an overall increase in the Nox-1/p47phox and p67phox complex, in the liver homogenates of fa/fa vs lean rat.
Figure 3. Protein expression of NAD(P)H oxidase is altered in Zucker fa/fa rat liver tissue.
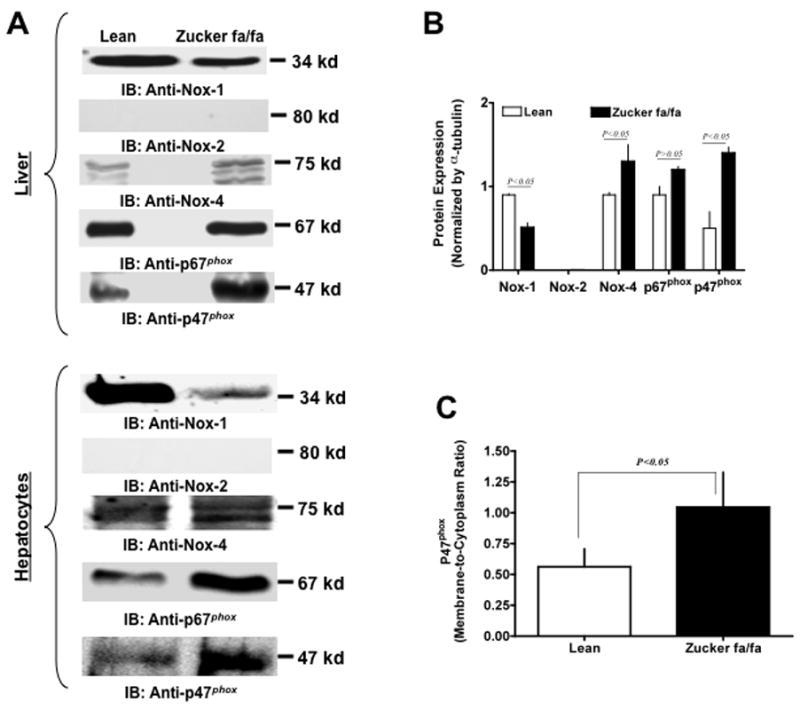
Lean and Zucker fa/fa rat liver homogenates (Panel A top) and hepatocyte lysates (Panel A bottom) were analyzed on 9% SDS-PAGE. Western blot analysis was performed using mouse monoclonal anti-Nox1 and -Nox2 (A; first & second panels), goat polyclonal anti-Nox4 (A; third panel), mouse monoclonal anti-p67phox (A; fourth panel) and goat polyclonal anti-p47phox (A; bottom panel) antibodies, respectively. All lanes contain 35 μg of total protein content. Panel (A) represents one blot of six such independent experiments. Panel B: Graph represents NADPH oxidase subunit expression in lean (n=6) and Zucker fa/fa (n=6) rat liver as estimated by densitometric analysis and normalized with α-tubulin values. Panel C: Lean and Zucker fa/fa rat liver tissue was fractionated by ultracentrifugation at 100,000×g to separate the membrane and cytoplasmic fractions. Graph represents levels of p47phox found in membrane as compared to cytoplasmic fraction in Zucker fa/fa rat liver. Statistical analysis was performed using Student’s t-test and P<0.05 (lean versus ZDF) was considered statistically significant.
Superoxide generated in type 2 diabetes is NADPH-dependent
Although studies have shown that O2− is elevated in diabetes, the source and mechanism of O2− generation are still unclear. Thus, to determine whether O2− derived from NAD(P)H oxidase or mitochondrial respiratory chain is elevated in type 2 diabetes, we estimated O2− levels in liver homogenates of lean and fa/fa rats. We found O2− generation was elevated (P<0.05) in the liver (Fig 4 A) and in isolated hepatocytes (lean: 7.85±2.69 and fa/fa: 13.34±3.93 units/mg protein) of fa/fa as compared to lean rats. Concurrently, SOD activity was also significantly increased (Fig 4 B), suggesting oxidative stress was increased in fa/fa liver. Furthermore, to rule out non-specific effects, homogenates were pretreated with superoxide dismutase (peg-SOD; 300 U/ml) to scavenge superoxide produced in the liver tissue. As expected, peg-SOD treatment decreased O2− levels by 60–80% (P<0.05) as compared to that of the untreated control. Consistently, by cytochrome reduction method, we found a 2-fold (P<0.05) elevation in O2− generation in fa/fa (76.9±5.4 nmol/mg protein) as compared to lean (32.5±8.1 nmol/mg protein) rats. Interestingly, there was a decrease in activity of GSH-Px (P<0.05; Fig 5 A) and GSH reductase (P<0.05; Fig 5 B) in fa/fa liver.
Figure 4. Superoxide generation is elevated in the liver of Zucker fa/fa rats.
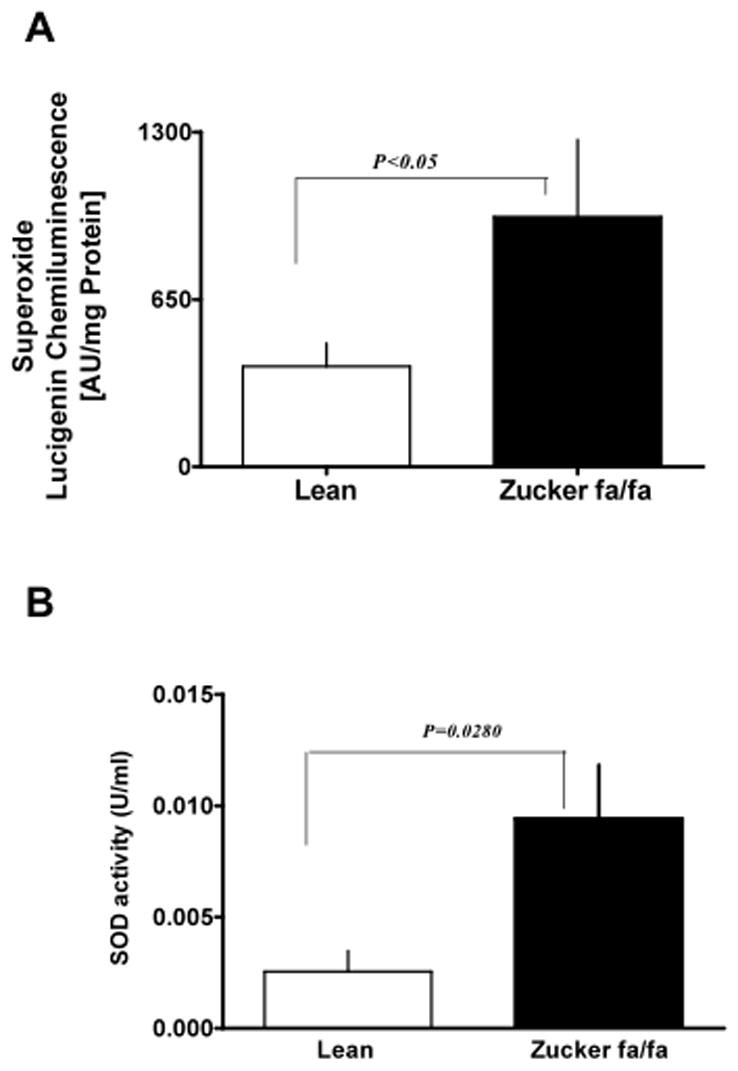
Liver homogenates obtained from lean (n=6) and Zucker fa/fa (n=6) rat were used to detected O2− production by lucigenin (5 μM) chemiluminescence (A); Superoxide dismutase activity (SOD; B). Statistical analysis was performed using Student’s t-test and P<0.05(lean versus Zucker fa/fa) was considered statistically significant.
Figure 5. Glutathione peroxidase and reductase activity is decreased in the liver of Zucker fa/fa rats.
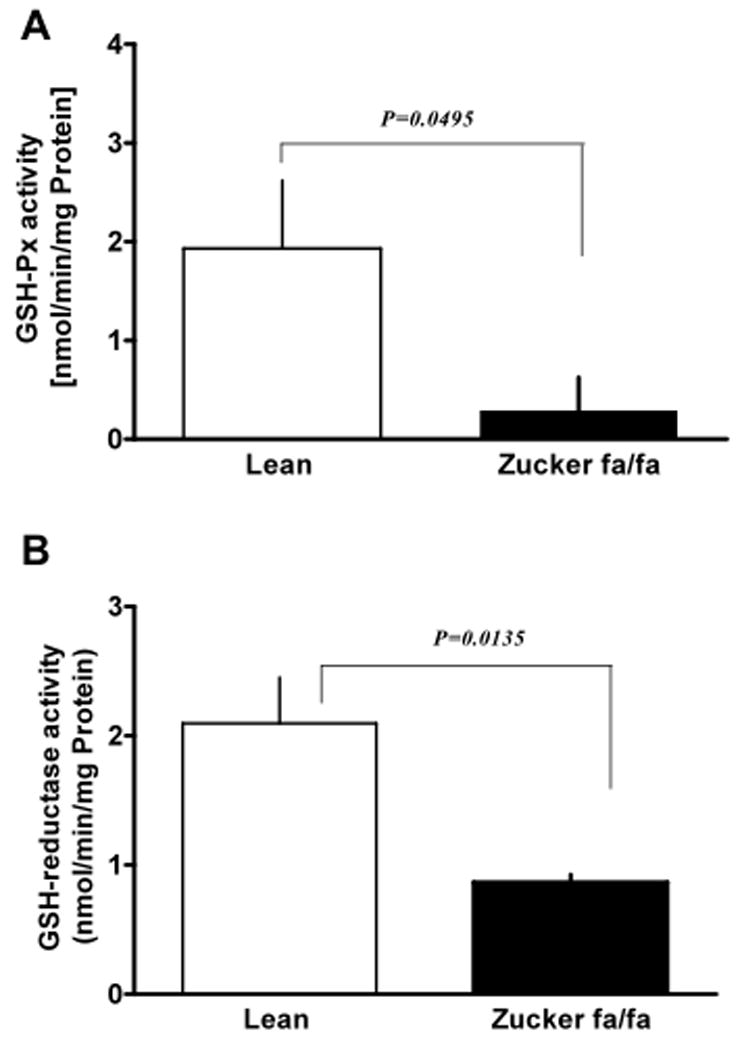
Liver homogenates obtained from lean (n=6) and Zucker fa/fa (n=6) rat were used to estimate glutathione peroxidase (GSH-Px; A) and glutathione reductase (GSH reductase; B) activities.
Additionally, we found that mitochondrial respiratory chain blockers, antimycin (10 μM) and rotenone (50 μM), did not decrease O2− production; while perfusion of the liver with an inhibitor of NADPH oxidase, gp91ds-tat, completely inhibited O2− generation in both lean and fa/fa rat liver (Fig 6 A). Scrambled gp91ds-tat peptide (50 μM), a negative control for gp91ds-tat, did not decrease O2− (981±120 Units/mg Protein). These experiments were performed in the presence of NADPH re-generating system.
Figure 6. Superoxide generated in liver is NADPH-dependent.
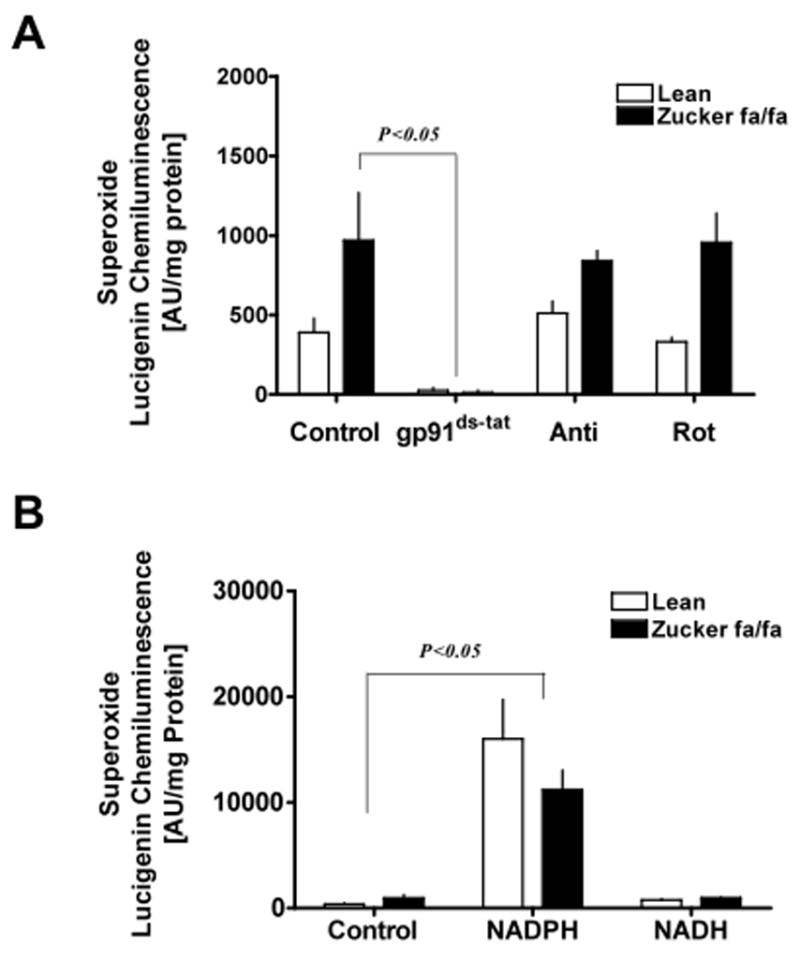
Liver homogenates obtained from lean (n=6) and Zucker fa/fa (n=6) animals were treated with: (A) either NADPH oxidase inhibitor, gp91ds-tat (50 μM), mitochondrial respiratory chain inhibitors, antimycin (10 μM) and rotenone (50 μM), and superoxide levels were determined by lucigenin (5 μM) chemiluminescence. (B) NADPH (100 μM) or NADH (100 μM) for 10 minutes at 37°C, and superoxide levels were determined as described above. These experiments were performed in the absence of NADPH regenerating system. Statistical analysis was performed using Student’s t-test and P<0.05 was considered statistically significant.
In general, O2− generation by NAD(P)H oxidase requires either NADPH or NADH; however, which of these reducing potential is required for O2− production in type 2 diabetes is unknown. Thus, to investigate whether NADPH or NADH was responsible to potentiate O2− generation in fa/fa liver tissues, we estimated O2− levels in presence of either NADPH (100 μM) or NADH (100 μM). The results suggested that O2− production was significantly augmented by NADPH, but not by NADH, therefore, demonstrating that NADPH is primarily utilized by NAD(P)H oxidase for O2− generation in the liver of fa/fa rats (Fig 6 B).
NADPH oxidase and glucose-6-phosphate dehydrogenase synergistically increases superoxide production in diabetic tissues
We have recently suggested that G6PD-derived NADPH fuels O2− production [12, 14, 22] and have demonstrated (Fig 6 B) that NADPH oxidase requires NADPH for activity. These observations taken together raised the question of whether NADPH oxidase and G6PD function in a coordinated manner to maintain elevated O2− in fa/fa rats. To test this hypothesis we first perfused the liver and incubated tissue homogenates in the absence and presence of PI3K and Src kinase inhibitors, since this signaling cascade is found to activate G6PD. We found that both types of inhibitors decreased (P<0.05) the O2− generation by ~50–60% in fa/fa rat liver (Fig 7 A) and abolished the O2− generation (under detectable limits) in hepatocytes. Next, we incubated the tissues with competitive and non-competitive inhibitors of G6PD, 6-aminonicotinamide (5 mM) and dihydroepiandrosterone (100 μM), respectively. The results indicated that O2− levels were decreased (P<0.05) by~67–78% by both 6-aminonicotinamide and dihydroepiandrosterone (Fig 7 B).
Figure 7. NADPH oxidase and glucose-6-phosphate dehydrogenase synergistically increases superoxide production in diabetic tissues.
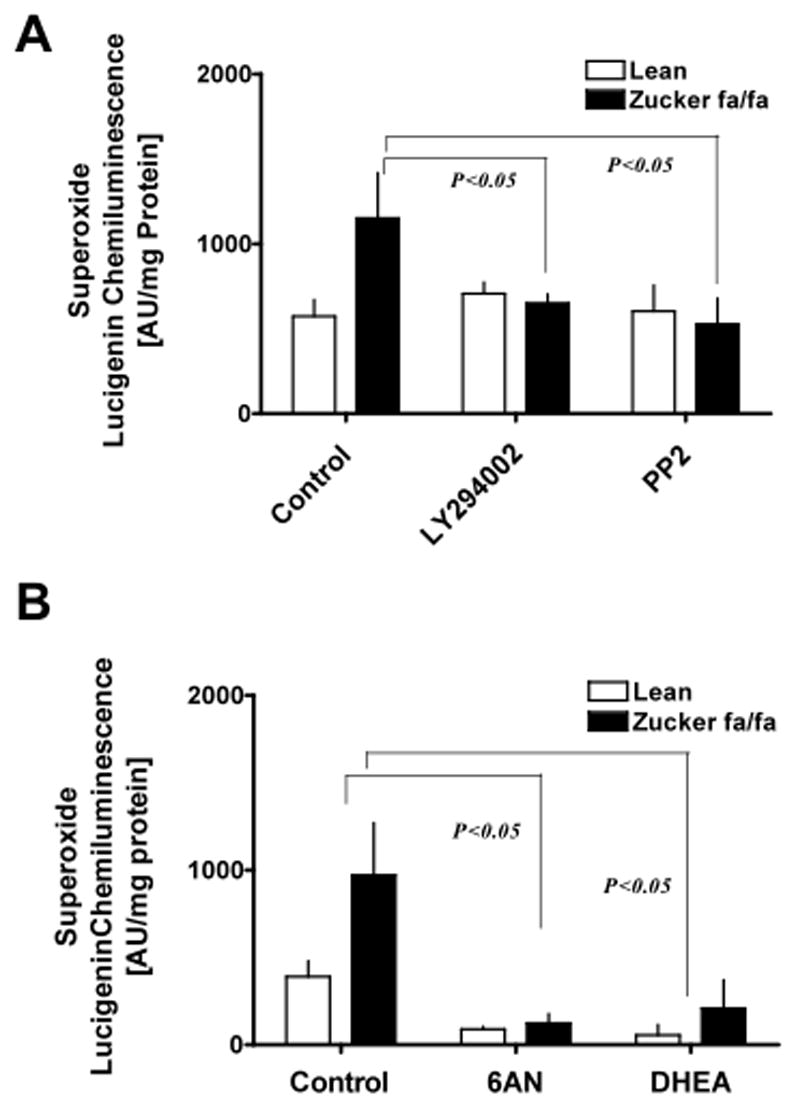
Liver tissues obtained from lean (n=6) and ZDF (n=6) animals were incubated (A) with PI3 kinase inhibitor, LY294002 (10 μM), and Src kinase inhibitor, PP2 (10 μM), for 10 minutes at 37°C and (B) either with 6-aminonicotinamide (5 mM) or dehydroepiandrosterone (DHEA; 100 μM), inhibitors of glucose-6-phosphate dehydrogenase, for 10 minutes at 37°C in the presence of NADPH regenerating system and superoxide levels were determined by lucigenin (5 μM) chemiluminescence. Statistical analysis was performed using Student’s t-test and P<0.05 (lean versus ZDF) was considered statistically significant.
A positive correlation exists between liver hypertrophy and increased G6PD activity/superoxide production in Zucker fa/fa rats
We found activity of γGT [1.17±0.15 (fa/fa) and 7.03±2.55 (lean) U/L], alkaline phosphatase [40.32±1.02 (fa/fa) and 142.40±28.40 (lean) U/L], and ALT [54.42±7.29 (fa/fa) and 263.00±100.70 (lean) U/L] in blood serum was significantly elevated in fa/fa vs lean rats. Furthermore, we also found that liver weight (Fig 8 A) and liver weight-to-body weight ratio (Fig 8 B) was significantly increased in fa/fa as compared to lean rats. We extracted DNA and estimated protein-to-DNA ratio (Fig 8 C) with a previously described method [25], and PCNA expression by Western blot analysis (Fig 8 D). Interestingly, we found that PCNA expression was down-regulated and protein-to-DNA ratio was elevated (P<0.05) in the liver of fa/fa as compared to lean rats suggesting decreased cell proliferation and a strong possibility of liver hypertrophy in fa/fa rats. Additionally, we found a positive correlation between G6PD activity (Fig 8 E) and O2− production with liver weight by regression analysis (Fig 8 F).
Figure 8. Liver hypertrophy occurs in young Zucker fa/fa rats.
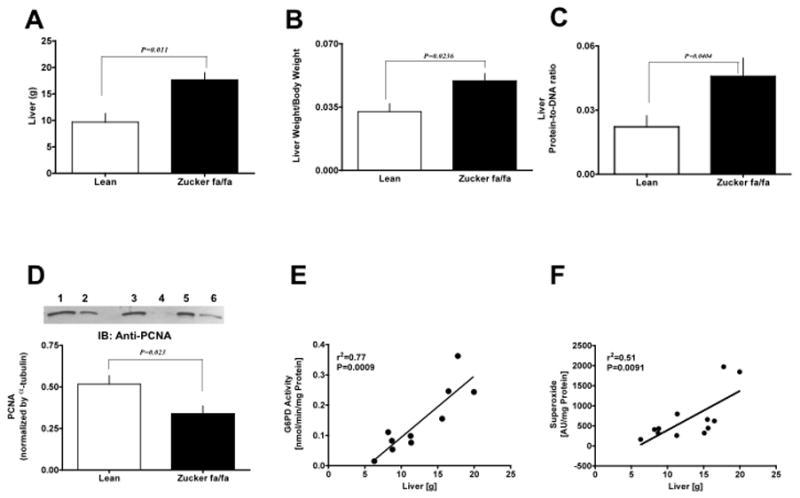
Liver weight (A) and liver-to-body weight (B) was also determined in these rats. Graph represents the liver protein-to-DNA ratio in lean and Zucker fa/fa rats (C). The expression of PCNA in lean (lanes 1, 3, and 5) and Zucker fa/fa (lanes 2, 4, and 6) liver was determined by western blot analysis using mouse monoclonal anti-PCNA antibody (D). Correlation between liver weight and G6PD activity (E) or superoxide levels (F) was determined by regression analysis. Statistical analysis was performed using Student’s t-test and P<0.05 was considered statistically significant.
Discussion
Type 2 diabetes is now widely recognized as a risk factor for liver dysfunction, obesity, cardiovascular diseases, and organ failure. This study was, therefore, undertaken to elucidate possible molecule(s)/mechanism(s) that may be responsible/involved for triggering liver dysfunction, in type 2 diabetes.
Glucose metabolism is considerably altered in various organs in type 2 diabetes. The fate of glucose through the PPP has not been well characterized and there is conflicting evidence about the activity of G6PD, the first/rate limiting enzyme in the PPP, in diabetes. For example, some studies suggest that G6PD activity is inhibited and NADPH is decreased in islets cells [26], Leydig cells [27], kidneys [28], lens [29], heart [30] and the liver [31] of type 1 diabetic rats (induced by streptozotocin- and alloxan-treatment); and in the liver [32], mononuclear leukocytes [33] and erythrocytes [34] of chronic diabetic patients. In contrast, recent studies have shown that G6PD is over-expressed in the adipocytes of obese (including db/db, ob/ob, and diet-induced obesity) mice, adipocytes and stromal-vascular cells of diabetic db/db mice, and adipocytes cultured, in vitro, under high glucose conditions [35, 36]. In lieu of these contrasting observations, we determined the expression and the activity of G6PD in liver and isolated hepatocytes of Zucker lean and fa/fa rats. Our data indicated that G6PD protein levels and activity was much higher in the liver of fa/fa vs lean rats, which was consistent with previous findings [37], and in addition we found that G6PD expression and activity was significantly higher in the isolated hepatocytes of fa/fa vs lean rats. Next, we proceeded to identify the pathways that could be responsible for increasing the expression and/or activity of G6PD in the fa/fa rat liver. Interestingly, we found significantly higher amounts of activated Src kinase (determined by phospho-Src416) in the fa/fa but not lean liver. Concomitantly, G6PD activity was significantly elevated in the fa/fa vs lean liver and hepatocytes by 4.4-fold and 1.6-fold, respectively. Inhibition of either PI3Kinase or Src kinase using LY294002 or PP2 inhibitors, respectively, significantly inhibited G6PD activity nearly to the levels observed in the lean liver and hepatocytes. However, no inhibitory effect of PI3kinase and Src kinase on G6PD activity was observed in the lean liver. Moreover, G6PD activity was completely abolished (Fig. 2 C & D) by alkaline phosphatase (50 U). This data provides an insight to the possibility that in fa/fa liver, G6PD activation may involve phosphorylation of G6PD via a signaling cascade that includes PI3Kinase and/or Src kinases. This notion is supported by the previous independent observations that induction of G6PD expression by insulin, in hepatocytes, requires PI3 kinase [23] and that EGF associated tyrosine kinases activate G6PD in renal cortical cells [21]. Hence it is possible that activation of G6PD by Src kinase probably occurs via phosphorylation of key tyrosine residues, located (as determined by scansite.mit.edu program) at its C-terminal, viz., Y279 (GRGGYFDEFG) and Y537 (YEGTYKWVN). Taken together, these findings indicate that PI3 kinase/Src kinase-dependent pathways could mediate an increase in G6PD expression as well as activity and this presumably shunts glucose through the PPP even though glucose uptake is impaired under hyperinsulinemic and/or hyperglycemic conditions prior to the development of diabetes in the fa/fa rats.
The major biological significance of G6PD is to generate NADPH, the cell’s most abundant reducing coenzyme. NADPH is an important cofactor for many enzymatic reactions in the cell, for example, fatty acid synthesis/oxidation, nitric oxide production and generation of GSH by glutathione reductase. We and others, have reported that G6PD-derived NADPH fuels NADPH oxidase-derived O2− production [14, 15]. Studies have suggested that increased oxidative stress is responsible for mutli-organ dysfunction and damage in streptozotocin- and alloxane-treated rats. However, our understanding about the mechanism(s) that increase(s) reactive oxygen species in type 2 diabetes is still unclear. This led us to examine G6PD-derived NADPH levels and its effect on oxidative stress. We found that inhibition of G6PD by 6-aminonicotinamide and dihydroepiandrosterone (G6PD inhibitors) significantly decreased O2− generation in fa/fa as well as lean rat liver. These findings taken together with the observations in figure 6B, suggest that NADPH, but not NADH, is consumed by constitutively active and by activated oxidases to produce O2− in lean and fa/fa liver and that G6PD-derived NADPH is an important co-factor in regulating NADPH oxidase activity and generating sub-maximal level of O2− in fa/fa rat liver. Interestingly, we also found that inhibition of PI3 kinase or Src kinase significantly decreased O2− production in liver of fa/fa, but not in lean, rats; thereby indicating that activation of G6PD and NADPH oxidase by Src kinase was responsible to increase O2− generation in fa/fa rat liver.
Over-expression of G6PD in the 3T3-L1 adipocytes, cultured in high glucose, in vitro, has been shown to increase oxidative stress by inducing the expression of Nox-2, p22phox, and p40phox subunits [36]. We found that Nox-4, p67phox and p47phox were significantly over-expressed in the fa/fa vs lean rat liver and hepatocytes, while we were unable to detect Nox-2 in both the rat livers. Increased levels of Nox-4 observed in this study and a significant increase in membrane-to-cytoplasmic ratio of p47phox, which may probably increase complex formation of Nox-1 with p67phox and p47phox, in turn maybe responsible for the elevation of O2− production in fa/fa rats. Furthermore, NADPH oxidase inhibitor, gp91ds-tat, that inactivates Nox-2 as well as Nox-1 by binding to p47phox binding sites [38], significantly attenuated O2− generation in the lean and fa/fa liver. However, no such effect was observed by the mitochondrial respiratory chain inhibitors; antimycin or rotenone. These results suggest that majority of O2− was generated by NADPH oxidase but not by the mitochondria. Interestingly, inactivation of anti-oxidant systems [39]; including GSH-Px activity (see figure 5), presumably led to the poor removal of reactive oxygen species, which could be one of the probable causes for elevating the oxidative stress observed in the hepatic tissue prior to the on-set of diabetes. Taken together, these observations provide novel evidence that elevation in G6PD activity in the liver of fa/fa rats, functions to sustain and compensate for increased NADPH consumption by activated NADPH oxidases and that G6PD-derived NADPH fuels NADPH oxidases to over-produce O2−, in hyper-glycemic and -insulinemic liver. Alternatively, increased reactive oxygen species could upregulate G6PD expression and increase intracellular NADPH, which may potentially be consumed by the activated pro-oxidant pathways. Additionally, G6PD-derived NADPH could promote GSH reductase activity and reduce oxidized GSH levels. However, in the event of a decrease in GSH peroxidase and reductase activity (figure 5), and a reduction in total GSH in liver [40], it is unlikely that NADPH may be involved in regulating the GSH redox cycling. In light of our observations, we propose that an increased G6PD-derived-NADPH, functions as a pro-oxidant and increases oxidative stress (O2−), instead of preventing oxidative stress, in the liver tissue in pathological conditions such as type 2 diabetes.
Studies have suggested that elevated NADPH oxidase-derived O2− found in the upper abdomen in the Zucker diabetic fatty rats [41], in the aorta and the kidney in streptozotocin-treated rats [2, 9], and in the diabetic human internal mammary artery and saphenous vein [3], could play a role in impairing organ function. Consistently, others have reported that over-expression of NADPH oxidase subunits (α-subunit of neutrophilic NADPH oxidase and p47phox [42, 43] in hepatic tissue plays a predominant role in the pathogenesis of early alcohol-induced hepatitis [44]; in the activation of mitogens in Kupfer cells by peroxisome proliferators [45]; in the NF-kB mediated production of mitogens that causes hepatocellular proliferation [46]; and in mediating the actions of Ang II on hepatic stellate cells and in liver fibrogenesis [47]. However, the precise signaling pathways involved in activating NADPH oxidases and NADPH oxidase-derived oxidative stress that cause liver dysfunction in diabetic animals are still elusive. The results presented in this study have elucidated a possible mechanism involving with two key players, G6PD and NADPH oxidase, which seem to be activated simultaneously in hepatic tissue and hepatocytes in a tyrosine kinase-dependent manner, prior to the on-set of diabetes (i.e. in 9–10 weeks old Zucker fa/fa rats), and operate in synergy to up-regulate O2−, perhaps contributing to the development of hyperglycemia/diabetes-associated hepatic complications.
We found an increase in liver weight in fa/fa as compared to lean rats, consistent with other studies [48, 49]. Our results show that expression of PCNA is down-regulated and the protein-to-DNA ratio is increased in fa/fa rats, thus suggesting that liver hypertrophy and not hyperplasia occurs in obese, hyper-insulinemic and -glycemic rats. Interestingly, we also found a positive correlation between liver weight and increased [1] G6PD activity and [2] O2− levels, indicating that concomitant activation of NADPH oxidase and G6PD, by hyperglycemia and/or hyperinsulinemia, could be a signal to either trigger or sustain hepatic cell growth in the congenital model of obesity and type 2 diabetes. In this regards, there is suggestive evidence that reactive oxygen species may play a role in the initiation of liver regeneration/growth, because of increased electron transport, resulting from the high metabolic load imposed on hepatocytes after partial hepatectomy of rat and mouse liver [50, 51]. Alternatively, elevated oxidants in conjunction with upregulation of G6PD activity could increase fatty acid synthesis and play a potential role in evoking either liver dysfunction or damage (determined by increase in the serum γGT, alkaline phosphatase and ALT levels) in fa/fa rats.
As summarized in the schematic model (figure 9), we have demonstrated that in liver of fa/fa rats G6PD expression and activity is increased by PI3 kinase- and Src kinase-dependent pathways; increase in NADPH oxidase-derived O2− generation is dependent on G6PD activity; and GSH-Px as well as GSH reductase activity is decreased. Interestingly, we have found that elevated G6PD activity and O2− levels have a positive correlation with hypertrophy of the liver; therefore, suggesting a plausible role for reactive oxygen species in triggering either liver dysfunction or growth. More importantly, suppression of G6PD activity by 6-aminonicotinamide and dihydroepiandrosterone decreased oxidative stress. Although it is unclear whether the increase in G6PD activity and O2− levels is a effect of hyper-insulinemia or -glycemia and further work is required to determine the consequences of their up-regulation on liver function, our results nevertheless imply that G6PD might be a novel target for therapeutic intervention to reduce oxidative stress, ameliorate organ dysfunction, and improve liver physiology in type 2 diabetes.
Figure 9. A schematic illustration of G6PD and NADPH oxidase activation in Zucker fa/fa rats.
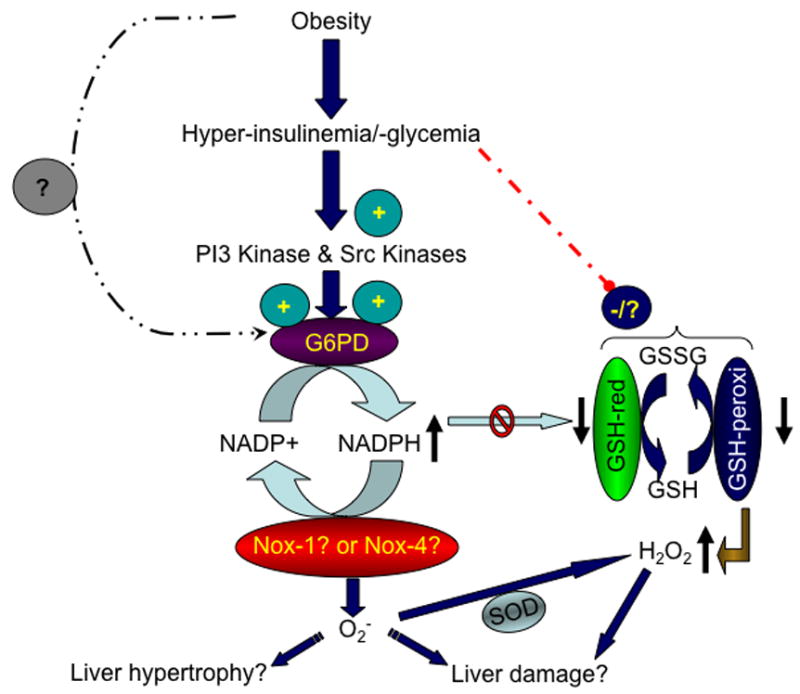
In the fa/fa rats, a congenital model of obesity and type 2 diabetes, changes in adipose tissue hormones and elevation in either insulin or glucose increases expression and activity of glucose-6-phosphate dehydrogenase (G6PD) and NADPH oxidases (Nox-1 and Nox-4). Conversely, glutathione reductase (GSH reductase) activity is decreased. This leads to an increase in superoxide (O2−) levels, which consequently either evokes liver growth or liver dysfunction.
Acknowledgments
This study was supported by AHA (grant: 0435070N), the National Heart, Lung, And Blood Institute (grant: R01HL085352) and NIH (grants: HL31069, HL43023, HL66331). A part of this work was presented in abstract form at Scientific Sessions of American Heart Association, Dallas, TX, 2005; and Experimental Biology Meeting, San Francisco, CA, 2006.
Abbreviation
- ALP
alkaline phosphatase
- ALT
alanine aminotransferase
- DMSO
dimethyl sulfoxide
- G6PD
glucose-6-phosphate dehydrogenase
- γ-GT
γ-glutamyl transpeptidase
- Nox-1
Nox-2 and Nox-4, NAD(P)H oxidase
- O2−
superoxide anion
- PPP
pentose phosphate pathway
- TNFα
Tumor necrotic factorα
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wild S, Roglic G, Green A, Sicree R, King H. Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care. 2004;27:1047–53. doi: 10.2337/diacare.27.5.1047. [DOI] [PubMed] [Google Scholar]
- 2.Hink U, Li H, Mollnau H, Oelze M, Matheis E, Hartmann M, Skatchkov M, Thaiss F, Stahl RA, Warnholtz A, Meinertz T, Griendling K, Harrison DG, Forstermann U, Munzel T. Mechanisms underlying endothelial dysfunction in diabetes mellitus. Circ Res. 2001;88:E14–22. doi: 10.1161/01.res.88.2.e14. [DOI] [PubMed] [Google Scholar]
- 3.Guzik TJ, Mussa S, Gastaldi D, Sadowski J, Ratnatunga C, Pillai R, Channon KM. Mechanisms of increased vascular superoxide production in human diabetes mellitus: role of NAD(P)H oxidase and endothelial nitric oxide synthase. Circulation. 2002;105:1656–62. doi: 10.1161/01.cir.0000012748.58444.08. [DOI] [PubMed] [Google Scholar]
- 4.Creager MA, Luscher TF, Cosentino F, Beckman JA. Diabetes and vascular disease: pathophysiology, clinical consequences, and medical therapy: Part I. Circulation. 2003;108:1527–32. doi: 10.1161/01.CIR.0000091257.27563.32. [DOI] [PubMed] [Google Scholar]
- 5.Giugliano D, Marfella R, Verrazzo G, Acampora R, Donzella C, Quatraro A, Coppola L, D’Onofrio F. Abnormal rheologic effects of glyceryl trinitrate in patients with non-insulin-dependent diabetes mellitus and reversal by antioxidants. Ann Intern Med. 1995;123:338–43. doi: 10.7326/0003-4819-123-5-199509010-00003. [DOI] [PubMed] [Google Scholar]
- 6.Kamata K, Ohuchi K, Kirisawa H. Altered endothelium-dependent and -independent hyperpolarization and endothelium-dependent relaxation in carotid arteries isolated from streptozotocin-induced diabetic rats. Naunyn Schmiedebergs Arch Pharmacol. 2000;362:52–9. doi: 10.1007/s002100000248. [DOI] [PubMed] [Google Scholar]
- 7.Hashim S, Li Y, Anand-Srivastava MB. G protein-linked cell signaling and cardiovascular functions in diabetes/hyperglycemia. Cell Biochem Biophys. 2006;44:51–64. doi: 10.1385/CBB:44:1:051. [DOI] [PubMed] [Google Scholar]
- 8.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–20. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 9.Etoh T, Inoguchi T, Kakimoto M, Sonoda N, Kobayashi K, Kuroda J, Sumimoto H, Nawata H. Increased expression of NAD(P)H oxidase subunits, NOX4 and p22phox, in the kidney of streptozotocin-induced diabetic rats and its reversibity by interventive insulin treatment. Diabetologia. 2003;46:1428–37. doi: 10.1007/s00125-003-1205-6. [DOI] [PubMed] [Google Scholar]
- 10.Lassegue B, Clempus RE. Vascular NAD(P)H oxidases: specific features, expression, and regulation. Am J Physiol Regul Integr Comp Physiol. 2003;285:R277–97. doi: 10.1152/ajpregu.00758.2002. [DOI] [PubMed] [Google Scholar]
- 11.Li JM, Mullen AM, Yun S, Wientjes F, Brouns GY, Thrasher AJ, Shah AM. Essential role of the NADPH oxidase subunit p47(phox) in endothelial cell superoxide production in response to phorbol ester and tumor necrosis factor-alpha. Circ Res. 2002;90:143–50. doi: 10.1161/hh0202.103615. [DOI] [PubMed] [Google Scholar]
- 12.Gupte SA, Levine RJ, Gupte RS, Young ME, Lionetti V, Labinskyy V, Floyd BC, Ojaimi C, Bellomo M, Wolin MS, Recchia FA. Glucose-6-phosphate dehydrogenase-derived NADPH fuels superoxide production in the failing heart. J Mol Cell Cardiol. 2006;41:340–9. doi: 10.1016/j.yjmcc.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 13.Zuurbier CJ, Eerbeek O, Goedhart PT, Struys EA, Verhoeven NM, Jakobs C, Ince C. Inhibition of the pentose phosphate pathway decreases ischemia-reperfusion-induced creatine kinase release in the heart. Cardiovasc Res. 2004;62:145–53. doi: 10.1016/j.cardiores.2004.01.010. [DOI] [PubMed] [Google Scholar]
- 14.Gupte SA, Kaminski PM, Floyd B, Agarwal R, Ali N, Ahmad M, Edwards J, Wolin MS. Cytosolic NADPH may regulate differences in basal Nox oxidase-derived superoxide generation in bovine coronary and pulmonary arteries. Am J Physiol Heart Circ Physiol. 2005;288:H13–21. doi: 10.1152/ajpheart.00629.2004. [DOI] [PubMed] [Google Scholar]
- 15.Matsui R, Xu S, Maitland KA, Hayes A, Leopold JA, Handy DE, Loscalzo J, Cohen RA. Glucose-6 phosphate dehydrogenase deficiency decreases the vascular response to angiotensin II. Circulation. 2005;112:257–63. doi: 10.1161/CIRCULATIONAHA.104.499095. [DOI] [PubMed] [Google Scholar]
- 16.Kindzelskii AL, Ueki T, Michibata H, Chaiworapongsa T, Romero R, Petty HR. 6-phosphogluconate dehydrogenase and glucose-6-phosphate dehydrogenase form a supramolecular complex in human neutrophils that undergoes retrograde trafficking during pregnancy. J Immunol. 2004;172:6373–81. doi: 10.4049/jimmunol.172.10.6373. [DOI] [PubMed] [Google Scholar]
- 17.Paul BB, Strauss RR, Jacobs AA, Sbarra AJ. Function of H(2)O(2), Myeloperoxidase, and Hexose Monophosphate Shunt Enzymes in Phagocytizing Cells from Different Species. Infect Immun. 1970;1:338–344. doi: 10.1128/iai.1.4.338-344.1970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Meininger CJ, Marinos RS, Hatakeyama K, Martinez-Zaguilan R, Rojas JD, Kelly KA, Wu G. Impaired nitric oxide production in coronary endothelial cells of the spontaneously diabetic BB rat is due to tetrahydrobiopterin deficiency. Biochem J. 2000;349:353–6. doi: 10.1042/0264-6021:3490353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carver RS, Sliwkowski MX, Sitaric S, Russell WE. Insulin regulates heregulin binding and ErbB3 expression in rat hepatocytes. J Biol Chem. 1996;271:13491–6. doi: 10.1074/jbc.271.23.13491. [DOI] [PubMed] [Google Scholar]
- 20.Lowry OH, Roberts NR, Kapphahn JI. The fluorometric measurement of pyridine nucleotides. J Biol Chem. 1957;224:1047–64. [PubMed] [Google Scholar]
- 21.Stanton RC, Seifter JL, Boxer DC, Zimmerman E, Cantley LC. Rapid release of bound glucose-6-phosphate dehydrogenase by growth factors. Correlation with increased enzymatic activity. J Biol Chem. 1991;266:12442–8. [PubMed] [Google Scholar]
- 22.Gupte RS, Vijay V, Marks B, Levine RJ, Sabbah HN, Wolin MS, Recchia FA, Gupte SA. Upregulation of glucose-6-phosphate dehydrogenase and NAD(P)H oxidase activity increases oxidative stress in failing human heart. J Card Fail. 2007;13:497–506. doi: 10.1016/j.cardfail.2007.04.003. [DOI] [PubMed] [Google Scholar]
- 23.Wagle A, Jivraj S, Garlock GL, Stapleton SR. Insulin regulation of glucose-6-phosphate dehydrogenase gene expression is rapamycin-sensitive and requires phosphatidylinositol 3-kinase. J Biol Chem. 1998;273:14968–74. doi: 10.1074/jbc.273.24.14968. [DOI] [PubMed] [Google Scholar]
- 24.Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ, Pollok BA, Connelly PA. Discovery of a novel, potent, and Src family-selective tyrosine kinase inhibitor. Study of Lck- and FynT-dependent T cell activation. J Biol Chem. 1996;271:695–701. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- 25.Franch HA, Shay JW, Alpern RJ, Preisig PA. Involvement of pRB family in TGF beta-dependent epithelial cell hypertrophy. J Cell Biol. 1995;129:245–54. doi: 10.1083/jcb.129.1.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Akpan JO, Wright PH, Dulin WE. Effect of diabetogenic nitrosourea on the activity of the pentose phosphate hunt in isolated islets. Acta Diabetol Lat. 1982;19:37–47. doi: 10.1007/BF02581184. [DOI] [PubMed] [Google Scholar]
- 27.Calvo JC, Biella de Souza Valle L, Baranao JL, Tesone M, Charreau EH. NADPH generating enzymes in Leydig cells from diabetic rats. Horm Metab Res. 1979;11:161–4. doi: 10.1055/s-0028-1092701. [DOI] [PubMed] [Google Scholar]
- 28.Xu Y, Osborne BW, Stanton RC. Diabetes causes inhibition of glucose-6-phosphate dehydrogenase via activation of PKA, which contributes to oxidative stress in rat kidney cortex. Am J Physiol Renal Physiol. 2005;289:F1040–7. doi: 10.1152/ajprenal.00076.2005. [DOI] [PubMed] [Google Scholar]
- 29.Hothersall JS, Muirhead RP, Taylaur CE, Jones RH. Anti-oxidant status in an in vitro model for hyperglycaemic lens cataract formation: competition for available nicotinamide adenine dinucleotide phosphate between glutathione reduction and the polyol pathway. Biochem Int. 1992;27:945–52. [PubMed] [Google Scholar]
- 30.Tarach JS. Some histochemical observations on the myocardial metabolism in experimental conditions. Part II. Acta Histochem. 1978;61:273–86. doi: 10.1016/S0065-1281(78)80073-7. [DOI] [PubMed] [Google Scholar]
- 31.Aragno M, Tamagno E, Gatto V, Brignardello E, Parola S, Danni O, Boccuzzi G. Dehydroepiandrosterone protects tissues of streptozotocin-treated rats against oxidative stress. Free Radic Biol Med. 1999;26:1467–74. doi: 10.1016/s0891-5849(99)00012-x. [DOI] [PubMed] [Google Scholar]
- 32.Cedola N, Cabarrou A, Auciello N, Doria I, Ponce de Leon H, Baylon N. The liver in human diabetes. Concentration of some induced enzymes. Acta Diabetol Lat. 1975;12:263–71. doi: 10.1007/BF02581099. [DOI] [PubMed] [Google Scholar]
- 33.Muggeo M, Moghetti P, Querena M, Cacciatori V, Zoppini G, Zenere M, Tosi F, Travia D, Bonora E. Mononuclear leukocytes from obese patients with type II diabetes have reduced activity of hexokinase, 6-phosphofructokinase and glucose-6-phosphate dehydrogenase. Horm Metab Res. 1993;25:160–4. doi: 10.1055/s-2007-1002068. [DOI] [PubMed] [Google Scholar]
- 34.Kher MM, Grover SD. Erythrocyte glucose-6-phosphate dehydrogenase deficiency in diabetes (maturity onset) J Assoc Physicians India. 1974;22:601–4. [PubMed] [Google Scholar]
- 35.Park J, Rho HK, Kim KH, Choe SS, Lee YS, Kim JB. Overexpression of glucose-6-phosphate dehydrogenase is associated with lipid dysregulation and insulin resistance in obesity. Mol Cell Biol. 2005;25:5146–57. doi: 10.1128/MCB.25.12.5146-5157.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Park J, Choe SS, Choi AH, Kim KH, Yoon MJ, Suganami T, Ogawa Y, Kim JB. Increase in glucose-6-phosphate dehydrogenase in adipocytes stimulates oxidative stress and inflammatory signals. Diabetes. 2006;55:2939–49. doi: 10.2337/db05-1570. [DOI] [PubMed] [Google Scholar]
- 37.Shepherd A, Cleary MP. Metabolic alterations after dehydroepiandrosterone treatment in Zucker rats. Am J Physiol. 1984;246:E123–8. doi: 10.1152/ajpendo.1984.246.2.E123. [DOI] [PubMed] [Google Scholar]
- 38.Lambeth JD. NOX enzymes and the biology of reactive oxygen. Nat Rev Immunol. 2004;4:181–9. doi: 10.1038/nri1312. [DOI] [PubMed] [Google Scholar]
- 39.Liu Y, Gutterman DD. The coronary circulation in diabetes: influence of reactive oxygen species on K+ channel-mediated vasodilation. Vascul Pharmacol. 2002;38:43–9. doi: 10.1016/s1537-1891(02)00125-8. [DOI] [PubMed] [Google Scholar]
- 40.Loven D, Schedl H, Wilson H, Daabees TT, Stegink LD, Diekus M, Oberley L. Effect of insulin and oral glutathione on glutathione levels and superoxide dismutase activities in organs of rats with streptozocin-induced diabetes. Diabetes. 1986;35:503–7. doi: 10.2337/diab.35.5.503. [DOI] [PubMed] [Google Scholar]
- 41.Sonta T, Inoguchi T, Tsubouchi H, Sekiguchi N, Kobayashi K, Matsumoto S, Utsumi H, Nawata H. Evidence for contribution of vascular NAD(P)H oxidase to increased oxidative stress in animal models of diabetes and obesity. Free Radic Biol Med. 2004;37:115–23. doi: 10.1016/j.freeradbiomed.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 42.Bachmann S, Ramasubbu K. Immunohistochemical colocalization of the alpha-subunit of neutrophil NADPH oxidase and ecto-5′-nucleotidase in kidney and liver. Kidney Int. 1997;51:479–82. doi: 10.1038/ki.1997.66. [DOI] [PubMed] [Google Scholar]
- 43.Gorlach A, Holtermann G, Jelkmann W, Hancock JT, Jones SA, Jones OT, Acker H. Photometric characteristics of haem proteins in erythropoietin-producing hepatoma cells (HepG2) Biochem J. 1993;290(Pt 3):771–6. doi: 10.1042/bj2900771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kono H, Rusyn I, Yin M, Gabele E, Yamashina S, Dikalova A, Kadiiska MB, Connor HD, Mason RP, Segal BH, Bradford BU, Holland SM, Thurman RG. NADPH oxidase-derived free radicals are key oxidants in alcohol-induced liver disease. J Clin Invest. 2000;106:867–72. doi: 10.1172/JCI9020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rusyn I, Kadiiska MB, Dikalova A, Kono H, Yin M, Tsuchiya K, Mason RP, Peters JM, Gonzalez FJ, Segal BH, Holland SM, Thurman RG. Phthalates rapidly increase production of reactive oxygen species in vivo: role of Kupffer cells. Mol Pharmacol. 2001;59:744–50. doi: 10.1124/mol.59.4.744. [DOI] [PubMed] [Google Scholar]
- 46.Rusyn I, Yamashina S, Segal BH, Schoonhoven R, Holland SM, Cattley RC, Swenberg JA, Thurman RG. Oxidants from nicotinamide adenine dinucleotide phosphate oxidase are involved in triggering cell proliferation in the liver due to peroxisome proliferators. Cancer Res. 2000;60:4798–803. [PubMed] [Google Scholar]
- 47.Bataller R, Schwabe RF, Choi YH, Yang L, Paik YH, Lindquist J, Qian T, Schoonhoven R, Hagedorn CH, Lemasters JJ, Brenner DA. NADPH oxidase signal transduces angiotensin II in hepatic stellate cells and is critical in hepatic fibrosis. J Clin Invest. 2003;112:1383–94. doi: 10.1172/JCI18212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Singh N, Kamath V, Rajini PS. Attenuation of hyperglycemia and associated biochemical parameters in STZ-induced diabetic rats by dietary supplementation of potato peel powder. Clin Chim Acta. 2005;353:165–75. doi: 10.1016/j.cccn.2004.10.016. [DOI] [PubMed] [Google Scholar]
- 49.Aprikian O, Busserolles J, Manach C, Mazur A, Morand C, Davicco MJ, Besson C, Rayssiguier Y, Remesy C, Demigne C. Lyophilized apple counteracts the development of hypercholesterolemia, oxidative stress, and renal dysfunction in obese Zucker rats. J Nutr. 2002;132:1969–76. doi: 10.1093/jn/132.7.1969. [DOI] [PubMed] [Google Scholar]
- 50.Lee FY, Li Y, Zhu H, Yang S, Lin HZ, Trush M, Diehl AM. Tumor necrosis factor increases mitochondrial oxidant production and induces expression of uncoupling protein-2 in the regenerating mice [correction of rat] liver. Hepatology. 1999;29:677–87. doi: 10.1002/hep.510290320. [DOI] [PubMed] [Google Scholar]
- 51.Nakatani T, Inouye M, Mirochnitchenko O. Overexpression of antioxidant enzymes in transgenic mice decreases cellular ploidy during liver regeneration. Exp Cell Res. 1997;236:137–46. doi: 10.1006/excr.1997.3715. [DOI] [PubMed] [Google Scholar]