

Abstract
The nucleic acid binding protein TDP-43 was recently identified in normal myonuclei and in the sarcoplasm of inclusion body myositis (IBM) muscle. Here we found TDP-43 sarcoplasmic immunoreactivity in 23% of IBM myofibers, while other reported IBM biomarkers were less frequent, with rimmed vacuoles in 2.8%, fluorescent Congo red material in 0.57%, SMI-31 immunoreactivity in 0.83%, and focal R1282 beta-amyloid immunoreactivity in 0.00% of myofibers. The presence of as little as > 1% of myofibers with non-nuclear sarcoplasmic TDP-43 was highly sensitive (91%) and specific (100%) to IBM among 50 inflammatory myopathy patient samples, though some patients with hereditary inclusion body myopathies and myofibrillar myopathy also had sarcoplasmic TDP-43. TDP-43 mutations were sought, and none were identified. TDP-43 could be one of many nucleic acid binding proteins that are abnormally present in IBM sarcoplasm. They could potentially interfere with the normal function of extranuclear RNAs that maintain myofiber protein production.
Keywords: Inclusion body myositis, inflammatory myopathies, TDP-43
Inclusion body myositis (IBM) is a progressive inflammatory skeletal muscle disease with poorly understood pathogenesis. The first pathological studies of IBM muscle reported abnormalities of myonuclei that suggested nuclear degeneration was a specific aspect of this disease compared with other inflammatory myopathies.9 Subsequent studies led to the hypothesis that rimmed vacuoles in IBM muscle sections arose from the breakdown of myonuclei.8 Attempting (and failing) to confirm a report that beta-amyloid precursor protein (βAPP) transcript was present in some myofibers from patients with IBM,26 a subsequent study found instead non-specific binding of many nucleic acid probes to an unidentified DNA-binding protein in the sarcoplasm of myofibers.21 Recent reports have provided further evidence for myonuclear abnormalities in IBM, demonstrating the presence of nuclear membrane proteins lamin A/C,12 emerin,12,20 valosin-containing protein,13 and histone H120 in the lining of rimmed vacuoles. Electron microscopic studies of IBM muscle have emphasized visible accumulation of myonuclei adjacent to degenerating cytomembranous whorls, tubulofilaments in myonuclei,14 and the focal rupture of the nuclear membrane.12
The nucleic acid binding protein TDP-43 was recently identified in normal muscle nuclei and also in non-nuclear sarcoplasm and around some rimmed vacuoles in IBM and inclusion body myopathy with Paget's disease and frontotemporal dementia (IBMPFD).30 The potential diagnostic value of TDP-43 immunohistochemistry for IBM was also suggested in this study by a high sensitivity and specificity for its visualization in non-nuclear regions of myofibers. Here we provide quantitative data regarding TDP-43 immunoreactivity in comparison to other reported immunohistochemical biomarkers, discuss its diagnostic value, and further clarify its distribution in IBM muscle.
Methods
Patients and Samples
Muscle biopsy specimens from 50 patients with inflammatory myopathies (IBM N=23; polymyositis N=9; dermatomyositis N=18), 10 patients with genetically-determined myopathies (4 with hereditary inclusion body myopathies, 2 suspected and 1 confirmed VCP mutations, and 1 suspected GNE mutation; 2 with clinical and histopathological diagnoses of myofibrillar myopathy but with unconfirmed mutations; and 1 each with confirmed mutations in dystrophin, ZNF9, calpain, and ryanodine receptor), 3 patients with neurogenic atrophy, and 4 normals underwent immunohistochemical studies for TDP-43. Subsets of these and other samples were studied with additional methods. Patients with IBM fulfilled criteria for definite or possible IBM;14 patients with polymyositis (PM) or dermatomyositis (DM) fulfilled criteria for definite or probable PM or DM.15 No patient with IBM received corticosteroids for treatment of the myopathy at any time. Normal subjects had no symptoms, signs, laboratory findings, or pathological abnormalities of a neuromuscular disease. Muscle biopsies were performed for diagnostic purposes. Blood samples from 6 patients with IBM were analyzed for the presence of TDP-43 mutations. Patients provided informed consent for research studies, as approved by our institutional review boards.
Serial sections and counting methods used in studies
From muscle samples of 50 patients with inflammatory myopathies, we performed serial 10 μm sections and stained 1 section with hematoxylin and eosin (H&E) and an adjacent section for TDP-43 and DNA with fluorescent molecules in all samples. Varying numbers of further adjacent sections were stained for Congo red and the fluorescent combinations of TDP-43/SMI-31/DAPI, TDP-43/MHCf/DAPI, and TDP-43/R1282/DAPI. For quantitation of the number of myofibers with sarcoplasmic TDP-43, two investigators (MS and SG) independently examined microscopic sections at 400x, randomly choosing fields and counting all myofibers in each field until at least 150 myofibers per patient section were counted. Each investigator was blinded to the other's results.
Immunohistochemistry
Ten-micron cryostat sections were fixed in either cold (5°C) 4% paraformaldehyde (PFA) for 5 minutes and then soaked consecutively in cold (5°C) 0.05 M Tris buffer, pH 7.5, room temperature Tris buffer, or were fixed in cold acetone (−10° ± 5°C) for 5 minutes and soaked in Tris buffer at room temperature. Tissue sections were transferred to 0.05 M Tris-saline-Triton X-100 buffer (TBS-T), pH 7.5, supplemented with 4% porcine serum for immunohistochemistry or to TBS-T for immunofluorescence. The latter tissue sections were incubated for 30 minutes with Image-iT FX signal enhancer reagent (cat. no. I36933, Molecular Probes, Inc./Invitrogen, Eugene, OR), although omitting this step did not appear to diminish the fluorescence signal-to-noise ratio. These slides were rinsed and soaked in TBS-T, then soaked in 0.05 M Tris-Brij-35 buffer, pH 7.5, supplemented with 2% bovine serum albumin. Following all incubations, slides were rinsed and soaked in TBS-T, and soaked in the same Tris-porcine serum buffer or Tris-bovine serum albumin buffer, respectively, prior to a subsequent step.
The primary antibodies used were rabbit polyclonal anti-TDP-43 (antibody to TAR DNA-binding protein 43, cat. no. 10782−2-AP, ProteinTech Group, Inc., Chicago, IL), mouse monoclonal anti-myosin heavy chain-fast (MHC-fast, cat. no. NCL-MHCf, clone WB-MHCf, isotype IgG1, Novocastra/Vision BioSystems, Inc., Norwell, MA/Leica Microsystems), mouse monoclonal antibody (SMI-31, ascites fluid) to neurofilaments, phosphorylated epitope (cat. no. SMI-31R, clone SMI-31, isotype IgG1, Covance Research Products, Inc., Berkeley, CA), mouse monoclonal anti-emerin antibody (cat. no. VP-E602, clone 4G5, isotype IgG1, Novacastra Laboratories Ltd., Newcastle upon Tyne, UK; obtained from Vector Laboratories Inc., Burlingame, CA), and rabbit polyclonal antibody R1282 directed against beta-amyloid (provided by Dr. Dennis J. Selkoe).
IHC and IF studies with TDP-43 antibody (PFA fixation,1:2000, 0.27 μg/ml, overnight) were carried out in similar fashion. Secondary antibodies were horseradish peroxidase-conjugated polymer bound to goat anti-rabbit immunoglobulins (cat. no. DPVR-110HRP, 30 mins., anti-rabbit PowerVision, ImmunoVision Technologies, Co./Vision BioSystems, Inc./Leica Microsystems) and Alexa Fluor 555 (or 488)-labeled goat anti-rabbit immunoglobulins (1:400, 5 μg/ml, 65 mins., Molecular Probes, Inc./Invitrogen), respectively. Dual staining (IF) of PFA fixed tissue sections with TDP-43 (1:2000, 0.27 μg/ml, overnight) and MHC-fast (1:60 dilution of reconstituted lyophilized tissue culture supernatant, 1 hr.) was carried out in sequence, followed by incubation with an admixture of Alexa Fluor 555-labeled goat anti-rabbit immunoglobulins and Alexa Fluor 488-labeled goat anti-mouse immunoglobulins (each at 1:400 dilution and 5 μg/ml, 1 hr. incubation; Molecular Probes, Inc./Invitrogen). With the same protocol (IF), dual staining (PFA fixation) with TDP-43 and mouse monoclonal anti-Emerin (1:100, 0.84 μg/ml, 90 mins.) was followed by incubation with an admixture of Alexa Fluor 488-labeled goat anti-rabbit immunoglobulins and Alexa Fluor 555-labeled goat anti-mouse immunoglobulins (each at 1:400 dilution and 5 μg/ml, 1 hr. incubation; Molecular Probes, Inc./Invitrogen).
Similarly, IHC and IF staining with SMI-31 (PFA or no fixation, 1:10,000, overnight) utilized secondary antibody horseradish peroxidase-conjugated polymer bound to goat anti-mouse immunoglobulins (cat. no. DPVM-110HRP, 30 mins, anti-mouse PowerVision, ImmunoVision Technologies, Inc.) and Alexa Fluor 488-labeled goat anti-mouse immunoglobulins (1:400 dilution, 5 μg/ml, 65 mins., Molecular Probes, Inc./Invitrogen), respectively. Dual staining (IF) of PFA fixed tissue sections with TDP-43 antibody was carried out overnight. An admixture of SMI-31 and TDP-43 antibodies contained each antibody at a final dilution as previously used. Secondary antibodies were an admixture of Alexa Fluor 555-labeled goat anti-rabbit immunoglobulins and Alexa Fluor 488-labeled goat anti-mouse immunoglobulins (each at 1:400 dilution and 5 μg/ml, 65 mins. incubation; Molecular Probes, Inc./Invitrogen). IF staining with R1282 antibody (1:1000, 20 hrs.) utilized secondary antibody Alexa Fluor 555-labeled goat anti-rabbit immunoglobulins (1:400, 5 μg/ml, 2 hrs., Molecular Probes, Inc./Invitrogen).
Congo red histochemistry
Frozen muscle sections were stained with Congo red (cat. no. C-580, Certified Biological Stain, total dye content 98%, C. I. no. 22120, Fisher Scientific, Pittsburgh, PA) based on the procedure of Puchtler et al.25 as described by Mendell et al.18
Immunoblots
Whole muscle lysates (WML) were prepared using 5 mg of cryostat sectioned muscle dounce homogenized in 200 μl of lysis buffer (containing 20 mM Tris pH 7.6, 2% SDS, 5 mM DTT), centrifuged at 10,000 g for 10 min at 4°C and the supernatant removed. The micro BCA assay (Pierce, Rockford, IL) was used to determine protein concentration, and the fractions were stored at −80°C. For SDS-PAGE, 30 μg of WML from each sample was diluted with NuPAGE LDS Sample Buffer (4X) (Invitrogen, Carlsbad, CA), reduced with 10 mM DTT, heated at 95°C for 10 minutes, centrifuged at 2000 g for 10 minutes, loaded onto 4−12% Bis-Tris Gels (Invitrogen), and resolved using MOPS running buffer (Invitrogen) at a voltage of 100−150 mV. The gels were transferred to a nitrocellulose membrane using NuPAGE Transfer Buffer (Invitrogen) at 30 mV for 1.5 hours, washed in PBS including 0.1% Tween-20 (PBST0.1%), blocked for 1 hr in 5% fat free milk in PBST0.1% (5%milk/PBST0.1%) and stored at 4°C.
Immunoblotting was carried out by incubating the membranes with rabbit anti-TDP-43 (cat# 10782−2-AP, ProteinTech Group; 1:1000 dilution overnight at 4°C), and after washing, with goat anti-rabbit HRP (cat # ab6721, Abcam, Cambridge, MA; 1:5000 dilution for 1 hour at room temperature). After stripping the blots using Restore Western Blot Stripping Buffer (cat #21062, Pierce) they were incubated with rabbit anti-actin (cat # sc-1616, Santa Cruz, Santa Cruz, CA; 1:10,000 dilution for 1 hour at room temperature), and after washing, with goat anti rabbit HRP (cat # ab6721, Abcam; 1:10,000 dilution for 1 hour at room temperature). SuperSignal West Pico Chemiluminescent Substrate (Pierce, Rockford IL) and Kodak films were used for visualization of the bands.
TDP-43 transcript measurement by microarrays
A subset of patients had muscle samples available for additional microarray experiments. Microarray experiments were performed on 25 inflammatory myopathy (IBM N=9, PM N=6, and DM N=10) and 10 normal muscle samples as previously described using Affymetrix HU-133A arrays representing approximately 18,000 genes.11 Gene expression levels were calculated using GC-Content Robust Multichip Analysis (GCRMA).31 Affymetrix probeset 200020_at representing TARDBP was used for TDP-43 transcript abundance.
TDP-43 gene sequencing
DNA was purified from 50 μl of human peripheral blood mononuclear cells (PBMCs) from 6 patients with IBM using the Qiagen DNeasy Blood and Tissue kit (cat #69505). Purified DNA quality and concentration were assessed using a Beckman Coulter DU-800 spectrophotometer and requiring a 260/280 ratio of greater than 1.7. All the exons and the intron-exon boundaries of TARDBP gene were PCR amplified with intronic primers, and sequencing of the amplified fragments was performed using the Big Dye Terminator Cycle Sequencing Ready Reaction Kit (Applied Biosystems, Wellesley, MA) using standard protocols. Reactions were run on an ABI3130, and mutation analysis was performed using Sequencher software v4.6 (Gene Codes Corporation, Ann Arbor, MI).
Results
Normal myonuclear localization of TDP-43 immunoreactivity
In all normal (N=4) muscle specimens, visible light microscopy showed the presence of TDP-43 immunoreactivity in myonuclei indicated by their colocalization with the DNA stain methyl green (Figure 1). The localization of TDP-43 to myonuclei was further confirmed in immunofluorescent studies through colocalization with DNA-binding fluorescent DAPI and SMI-31, reported in a separate manuscript as having nuclear immunoreactivity (Figure 1). TDP-43 localized internally to the nuclear membrane as shown in triple stained TDP-43, emerin, and DAPI sections (Figure 1). TDP-43 immunoreactivity was present in 98% of 1000 DAPI fluorescent myonuclei counted (250 in each of 4 normal sections; myonuclei were clearly distinguished from inflammatory cell nuclei by their presence internal to the sarcolemma). Autofluorescence was excluded by visualization of sections in both fluorescent channels. No staining of myonuclei was present with normal rabbit serum (for TDP-43) or tris with secondary fluorescent labeled antibodies (Supplementary Figure 1).
Figure 1. Normal myonuclear localization of TDP-43.

(A,B) TDP-43 light microscopic images. The pattern of staining suggests nuclear localization and colocalization with DNA staining methyl green confirms this. (C1−3) Colocalization of TDP-43, SMI-31, and DAPI in a single normal muscle section with triple immunofluorescent studies further confirms nuclear localization of both of these proteins. (D1−4) TDP-43 localizes internally to the nuclear membrane as shown in triple immunofluorescent studies with the nuclear membrane protein emerin and DAPI.
Non-nuclear sarcoplasmic accumulation of TDP-43 in IBM muscle
Across 23 IBM samples, a mean of 23% of IBM myofibers showed non-DAPI associated multiple curvilinear filamentous foci of bright TDP-43 immunoreactivity (Table 1). One investigator found 25% of myofibers affected, and the other found 22% in a blinded review of the same sections. These foci were not associated with SMI-31 or R1282 beta-amyloid immunoreactivity or Congo red fluorescence (Figure 2). Artifact was excluded through TDP-43 detection in separate sections stained with each of 2 immunofluorescent labels. These were then visualized with distinct filter sets to ensure that fluorescent signal was present only in the expected filter set (Supplementary Figure 2). Tissue autofluorescent signal was similarly excluded (Supplementary Figure 3).
Table 1. Non-nuclear sarcoplasmic TDP-43 and other histochemical myofiber biomarkers.
The mean percentage of affected myofibers and number of patients studied with TDP-43 is listed. Only a subset of patients was studied with each additional technique: rimmed vacuoles (N=15); Congo red (N=8); R1282 (N=4); SMI-31 (N=5). NS = not studied.
| Disease | # Patients for TDP-43 studies | TDP-43 sarcoplasmic | Rimmed vacuoles (H&E) | Congo red fluorescent | R1282 "beta-amyloid" | SMI-31 |
|---|---|---|---|---|---|---|
| Inflammatory Myopathy | ||||||
| IBM | 23 | 25% | 2.8% | 0.57% | 0.0% | 0.83% |
| PM | 9 | 0.16% | 0.0% | NS | NS | 0.0% |
| DM | 18 | 0.0% | 0.0% | NS | NS | 0.0% |
| Dystrophy | ||||||
| hIBM | 4 | 11.0% | NS | NS | NS | NS |
| Myofibrillar | 2 | 0.0%, 25.0%* | NS | NS | NS | NS |
| Other | 4 | 0.0% | NS | NS | NS | NS |
| Neurogenic | 3 | 0.0% | 0.0% | NS | 2.0% | 0.0% |
| Normal | 4 | 0.0% | 0.0% | NS | NS | 0.0% |
*-Mean value not representative of a population; only 2 samples from patients with myofibrillar myopathies were studied, neither genetically confirmed), one with no TDP-43 detected and one with 25% of affected myofibers.
Figure 2. Relationship of sarcoplasmic TDP-43 immunoreactivity to other features.

(A1-A3) Adjacent sections from an IBM sample show sarcoplasmic areas of intense TDP-43 staining that are not immunoreactive for SMI31 and do not show fluorescence after Congo red staining. (B1-B3) Adjacent sections from an IBM sample show TDP-43 sarcoplasmic staining distinct from SMI31 and DAPI nuclear staining. (C,D) High magnification of TDP-43 immunoreactivity in a myofiber from IBM, with adjacent section showing H&E appearance of this myofiber.
As necrotic myofibers typically show binding to many antibodies in histochemical studies, the comparison of TDP-43 immunoreactive myofibers with adjacent H&E-stained sections is especially important. Non-nuclear sarcoplasmic accumulation of TDP-43 occurred in non-necrotic myofibers in H&E-stained adjacent sections (Figure 3).
Figure 3. Paired H&E and TDP-43 images from IBM sections.

(A-B) TDP-43 sarcoplasmic distribution occurs in non-necrotic myofibers that may have mild abnormalities (enlarged rounded fiber #1; small or angulated fibers #2 and #3) or (C) substantial abnormalities (many slightly basophilic small fibers, some with enlarged nuclei; the vacuolated fiber #4) present on H&E staining.
In all IBM muscle samples, myofiber vacuoles were present and were sometimes lined with TDP-43 immunoreactive material and DAPI (Figure 4). Most punctate and curvilinear TDP-43 accumulations lacked visible SMI-31 immunoreactivity (Figure 4). Most myofibers with rimmed vacuoles did not contain TDP-43 sarcoplasmic accumulations in nearby sections. Overall correlation between the number of myofibers with rimmed vacuoles and the number with TDP-43 sarcoplasmic accumulations was marginal (correlation coefficient = 0.56).
Figure 4. Rimmed vacuole lining reactive with anti-TDP-43, SMI-31, and DAPI in IBM muscle.

(A1-A3) Triple stained immunofluorescent section shows vacuoles lined with reactivities for TDP-43, SMI-31, and DAPI. The arrow indicates a myofiber with sarcoplasmic TDP-43, whose nucleus (identified by SMI31 and DAPI staining) does not contain visible TDP-43. (B) Higher magnification of panel A-1, outlining TDP-43 lined vacuoles (arrowheads) and a myofiber with sarcoplasmic non-nuclear TDP-43 (arrow).
TDP-43 sarcoplasmic accumulations occurred without preference to myosin heavy chain-based fiber typing. Of 100 such affected myofibers across 4 IBM patient samples, 56% were type 1 and 44% either type 2 or hybrid (P=0.36) based on the absence or presence of fast myosin heavy chains on doubly-stained TDP-43 and MHCf immunofluorescent sections.
Relationship of nuclear and non-nuclear TDP-43 immunoreactivity in affected myofibers
Comparison of TDP-43 immunoreactivity with DAPI in dual fluorescent-stained sections of all 23 IBM samples showed that in myofibers containing sarcoplasmic TDP-43, nuclei typically were devoid of TDP-43 (Figure 5). Across 5 IBM samples, in 50 myofibers that lacked sarcoplasmic TDP-43 accumulation, 98.9% of nuclei (N=368 nuclei; identified by DAPI fluorescence) contained TDP-43 immunoreactivity. In these same IBM samples, in 50 myofibers containing TDP-43 sarcoplasmic accumulations only 12% of nuclei (N=251 nuclei) contained TDP-43 immunoreactivity. The loss of TDP-43 from nuclei in affected fibers was statistically significant (P<0.0001, Chi-square test). Nuclear TDP-43 staining was normal in PM and DM disease samples, with 97−98% (depending on the specific disease) of DAPI identified nuclei containing TDP-43 immunoreactivity.
Figure 5. Myofibers with sarcoplasmic TDP-43 typically show absent nuclear TDP-43 staining.

(A1−3) A triangular fiber #1 shows abundant sarcoplasmic linear TDP-43 accumulation. Nuclei (marked with arrowheads) are devoid of TDP-43. In contrast, the adjacent rounded myofiber #2 lacks sarcoplasmic accumulation and has normal TDP-43 nuclear immunoreactivity (arrows). Inflammatory cells are present between the two fibers. (B1−3) TDP-43 sometimes clusters around myonuclei (arrowheads) in addition to multifocally within the sarcoplasm. A-1 and B-1 are merged images of TDP-43 (A-2 and B-2) and DAPI (A-3 and B-3) fluorescent signals.
TDP-43 immunoblots in inflammatory myopathies
Immunoblots of 6 IBM, 9 other inflammatory myopathy, and 4 normal samples showed prominent 43 kDa bands in all samples (Figure 6). This band was larger than controls in all IBM samples, but whether this was due to increased numbers of TDP-43-carrying invading inflammatory cells or increased TDP-43 protein within myofibers could not be determined from these experiments. Three IBM samples had lower molecular weight TDP-43 immunoreactive bands, suggesting TDP-43 fragments, that were not present in any other samples. These 2 samples had among the highest percentage of affected myofibers in immunohistochemical studies (patient 372 with 75% and patient 354 with 49%). No other disease-specific bands were seen. The previously reported30 approximately 50 kDa band in IBM was present in all PM and DM samples as well. Its interpretation was further confounded by the presence of immunoglobulin heavy chain at this weight. It was detected by the secondary anti-immunoglobulin antibody as shown in blots performed with omission of primary antibody (Figure 6, lane 1). No 25 kDa fragment as previously reported in frontotemporal dementia brain was identified.22
Figure 6. TDP-43 immunoblots of inflammatory myopathy and normal muscle.

Immunoblots probed with either secondary anti-IgG antibody only (no primary antibody) or antibodies against TDP-43 or actin followed by secondary anti-IgG antibodies. Sample ID for each lane labeled in the bottom panel. An approximately 43 kDa band at the appropriate molecular weight for TDP-43 (black arrow) is seen in all muscle samples and was larger in all IBM samples, despite slightly less actin, than in other inflammatory myopathy samples (DM = dermatomyositis; NM = necrotizing myopathy, PM = polymyositis). The interpretation of the additional band previously reported in IBM muscle30 at approximately 50 kDa (white arrow) is confounded by the presence of abundant immunoglobulin heavy chains present in IBM muscle also at this molecular weight, as shown in the 1st lane stained only with secondary anti-IgG, without primary anti-TDP-43 antibody. TDP-43 immunoreactivity beyond IgG immunoreactivity may be present at this weight (compare sample 372 without and with primary antibody) but is not disease-specific, as it is present in DM, NM, and PM samples. Immunoreactivity at all other weights also appeared to be not disease-specific, except for 2 IBM samples (354 and 372) that showed greater intensity of multiple lower molecular weight TDP-43 fragments (*).
TDP-43 transcript abundance
Microarray experiments measured TDP-43 transcript in 25 inflammatory myopathy muscle samples (IBM N=9, PM N=6, and DM N=10) compared to 10 normal muscle samples. These showed mean increases of TDP-43 transcript compared to normal of 2.4-fold in IBM, compared with 1.7 for PM and 1.4 for DM. The increase in IBM was significant compared with PM (P=0.05). What cannot be determined from these experiments is whether the increased TDP-43 transcript in IBM was due to increased numbers of invading inflammatory cells or other cell types (i.e., fibroblasts) that might produce this transcript.
Non-nuclear sarcoplasmic accumulation of TDP-43 is a highly sensitive and specific abnormality in IBM muscle among inflammatory myopathies but also is present in some patients with hereditary inclusion body myopathies and myofibrillar myopathy
Except for 2 IBM samples with 0% and 1% affected myofibers, the range of the number of affected myofibers for the remaining 21 samples was 10−75%. The presence of even a single myofiber with non-nuclear sarcoplasmic accumulation of TDP-43 was highly sensitive (96%) and specific (85%) for IBM among 50 inflammatory myopathy patient samples (Table 2). The presence of greater than 1% of such myofibers was 91% sensitive and 100% specific for IBM. It was present in 21 of 23 IBM samples and none of 27 PM or DM samples.
Table 2.
Sensitivity, specificity, positive predictive value (PPV), and negative predictive value (NPV) of non-nuclear sarcoplasmic TDP-43 accumulation in IBM among inflammatory myopathies (50 samples)
| Cutoff | Sensitivity | Specificity | PPV | NPV |
|---|---|---|---|---|
| > 0% of myofibers | 96% | 85% | 85% | 96% |
| > 1% of myofibers | 91% | 100% | 100% | 93% |
A small number of muscle samples with a range of muscular dystrophies were also examined for TDP-43 immunoreactivity. Abundant TDP-43 accumulation was seen in 3 of 4 samples from patients with hereditary inclusion body myopathies and 1 of 2 patients with myofibrillar myopathy (Supplementary Figure 4). These diseases are accompanied by the formation of rimmed vacuoles. No accumulations were seen in 4 other muscular dystrophy samples with known mutations, 1 each with dystrophin (Becker muscular dystrophy), zinc finger 9 (myotonic dystrophy type 2), calpain-3 (limb-girdle muscular dystrophy 2A), and ryanodine receptor 1 (central core disease). No sarcoplasmic TDP-43 was present in 3 samples with neurogenic atrophy.
Fluorescent congophilic material does not localize with TDP-43 immunoreactivity
For 8 samples examined in paired sections for Congo red fluorescent and TDP-43 immunoreactive material, we found no myofibers that showed colocalization (Figure 2 and Supplementary Figure 5). Congo red fluorescent abnormalities were present in only 2 of 8 patient samples, in 0.7% and 4.4% of myofibers counted. Overall, Congo red fluorescent material was found in a mean of 0.57% of myofibers (11 myofibers out of 1917 counted). TDP-43 myofiber abnormalities were present in all 8 of these samples (mean 33% of myofibers, range 10−63% of myofibers, 547 myofibers of 1917 counted).
Lack of exonic mutations in TDP-43 in patients with IBM
Six patients underwent sequencing of TDP-43; no sequence variants were found in its exons. Two patients had heterozygous IVS5+69insG variants, a previously described common variant.10
Discussion
In this study, we found that extranuclear sarcoplasmic immunoreactivity of the normally nuclear protein TDP-43 is a prominent and highly sensitive and specific feature of IBM among the inflammatory myopathies. It is present in non-necrotic myofibers typically with minimal morphological abnormalities. Across 23 IBM samples, a mean of 23% of IBM myofibers showed non-nuclear multiple curvilinear filamentous TDP-43 immunoreactivity. The presence of even >1% of such affected myofibers in a muscle biopsy specimen was 91% sensitive and 100% specific for IBM in 50 patients with inflammatory myopathies. Sarcoplasmic TDP-43 was further seen in several patients with hereditary inclusion body myopathies and 1 patient with a clinical and histopathological (but not genetically confirmed) diagnosis of myofibrillar myopathy, potentially further linking rimmed vacuole disorders to myonuclear abnormalities.8,9,12,13,20,30 The findings described here are largely consistent with the recent report in which TDP-43 sarcoplasmic accumulations were reported in 21 (78%) of 27 patients with IBM and 1 (8%) of 12 patients with steroid-responsive PM, as well as 100% of 5 patients with hereditary inclusion body myopathies (h-IBM) associated with VCP mutations.30 The areas of TDP-43 immunoreactivity in that report did not show ubiquitin immunoreactivity, which previous quantitative studies have found to be only sparsely present in IBM (0.7% of myofibers).6
Abnormal inclusions of TDP-43 were recently identified as a consistent pathological feature of sporadic and familial frontotemporal lobar degeneration with ubiquitin-positive inclusions (FTLD-U) and also in sporadic and familial amyotrophic lateral sclerosis (ALS).22 In the brain, TDP-43 is normally immunohistochemically visible predominantly in the nuclei of neurons and some glial cells, whereas in FTLD-U and ALS, TDP-43 is typically redistributed from the nucleus to the cytoplasm.17 TDP-43 is a 414 amino acid nuclear protein encoded by the TARDBP gene that is highly conserved and ubiquitously expressed.7 TDP-43 has the classic domain architecture of a heterogeneous ribonuclear protein (hnRNP). It contains two RNA recognition motifs and a glycine-rich C-terminal region that allow it to bind single-stranded nucleic acid and proteins, respectively.7 Initially cloned as a human protein capable of binding to the TAR DNA of HIV-1, where it acts as a transcription repressor,23 TDP-43 was subsequently identified as part of a complex involved in splicing the cystic fibrosis transmembrane conductance regulator and the apolipoprotein A-II genes. TDP-43 has also been shown to act as a scaffold for nuclear bodies through an interaction with survival motor neuron protein.29 Although it is predominantly localized to the nucleus, dynamic studies performed in vitro have shown that TDP-43 shuttles between the nucleus and cytoplasm similar to many other hnRNPs.5 The abnormal accumulation of TDP-43 in the cytoplasm in some diseases may reflect a defect in nucleocytoplasmic shuttling. Indeed in IBM, we found that sarcoplasmic accumulation of TDP-43 was accompanied by its nuclear depletion (present in 12% of myonuclei of such fibers compared to 99% of myonuclei in fibers lacking sarcoplasmic accumulation), suggesting redistribution of this molecule from the myonucleus to the sarcoplasm. In IBM, multiple nuclear morphological abnormalities are present;8,9,12,20,21 but their relationship to sarcoplasmic TDP-43 accumulation is uncertain. Regardless of mechanism, the abnormal accumulation of extra-nuclear TDP-43 may lead to deleterious interaction with mRNAs or other RNA-binding proteins and may lead to impaired gene expression in affected cells. TDP-43 could be one of many nucleic acid binding proteins abnormally present in IBM sarcoplasm, similar to the unidentified nucleic acid binding protein described 15 years ago.21
A previous study also observed an approximately 50 kDa band on TDP-43 immunoblots in IBM muscle and suggested that this band was phosphorylated TDP-43 similar to what had been seen in frontotemporal dementia brain.22 We found this 50 kDa band, which appears at a higher molecular weight than the phosphorylated band in frontotemporal dementia brain,22 not just in IBM but also in 1 normal and in all PM and DM samples. Because inflammatory myopathy muscle samples contain abundant immunoglobulin molecules,11 and immunoglobulin heavy chains have molecular weights of approximately 50 kDa, it is important to exclude the possibility that 50kDa bands may be a result only of the secondary anti-immunoglobulin antibody reagents reacting to such endogenous human immunoglobulin. Indeed, we found after omission of primary antibody that some portions of these bands are the result of such endogenous immunoglobulin, not modified species of TDP-43.
We found no exonic mutations in TDP-43 in blood samples from 6 IBM patients. While our findings suggest that mutations in TDP-43 are not frequently associated with IBM, only studies of larger numbers of patients can adequately address this issue.
Major roles of beta-amyloid and tau in the pathogenesis of IBM have been theorized based on reported accumulations of Congo red fluorescent material,4 beta-amyloid immunoreactivity,1-3 and SMI-31 immunoreactivity19 in IBM muscle. We therefore examined these reactivities in comparison to TDP-43 in the same or adjacent sections (Figure 7). We found fluorescent Congo red material in only 0.57% of myofibers, and it was only present at all in only 2 of 8 samples. A likely pathogenic role of such material, the identity of which has not been established, has been argued for in studies that remarkably lack any quantitative data regarding its abundance.4 The rarity of the Congo red fluorescent material in our study is in agreement with the experience of all other studies that have reported quantitative data; they have found the number of affected myofibers detectable by this technique ranges from 0.02−0.82%,24 0.22−2.10%,6 and 0.50−4.40%.16 In data from 17 patients it was present in 0 fibers in 5 patients, 1−5 fibers in 8 patients, and 6−8 fibers in 4 patients, presumably out of typical sections containing thousands of myofibers.21 We also point out that use of this technique should be accompanied by exclusion of rounded autofluorescent material (“lipofuscin”), visualized with both red and green filter sets.
Figure 7.
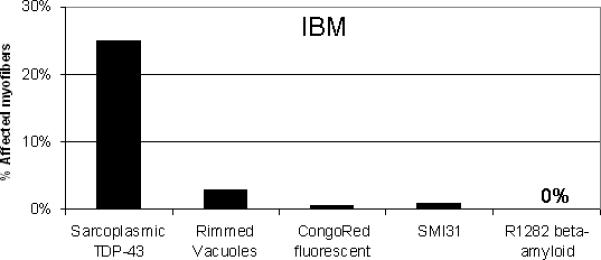
Comparison of sarcoplasmic TDP-43 abnormalities with other IBM histopathological biomarkers.
We also found that the R1282 antibody directed against beta-amyloid showed no immunoreactive accumulations in IBM. This is similar to what was previously reported with this antibody and with 8 other anti-beta-amyloid and beta-amyloid precursor protein (βAPP) antibodies in one study of 16 patients with IBM.27 The only accumulations of beta-amyloid observed in this study were seen in one control sample from a patient with peripheral neuropathy and neurogenic atrophy. This patient had increased R1282 immunoreactivity in target lesions of multiple fibers (Supplementary Figure 6). SMI-31 abnormalities were seen in only 0.83% of IBM myofibers in this study, in agreement with the only previously published quantitative data of 0.69%28 and 1.95%6 of myofibers. Alpha-B-crystallin (αBC) is another biomarker that has been reported in one immunohistochemical study present in a mean of 9.8% of IBM myofibers, compared with 0.78% of typical PM myofibers.6 αBC was also found in perifascicular myofibers in DM. It is possible that the percentage of abnormal αBC immunoreactive myofibers would be even higher if quantitated with immunofluorescence, the technique we used given its generally greater sensitivity than peroxidase-based immunohistochemistry. The degree of myofibers with visible abnormalities in the expression of αBC and TDP-43 and the specificity of these abnormalities for IBM compared to PM seem to set these findings apart from other reported biomarkers.
Supplementary Material
Supplementary Figure 1. Negative controls and exclusion of autofluorescence with TDP-43 studies. (A1−3) TDP-43 identified in an IBM myofiber with AF488 secondary antibody in the green channel (arrow) and appropriately not visible in the red channel or with tris+AF488 secondary. The arrowhead shows autofluorescent material present in both red and green channels. (B1−3) Muscle from a different patient with IBM showing TDP-43 sarcoplasmic material, lacking autofluorescence and not staining with control primary antibody normal rabbit serum.
Supplementary Figure 2. Exclusion of artifact for TDP-43 sarcoplasmic immunoreactivity. Studies were performed using both Alex fluorochrome 488 (green) and Alexa fluorochrome 555 (red) labels and examining sections with both filter sets individually. TDP-43 labeled with AF555 examined in the red channel (A-1) and TDP-43 labeled with AF488 examined in the green channel (B-1) show myofibers with sarcoplasmic TDP-43 (arrows); examination of these sections in the other channels (A-2 green and B-2 red) shows no fluorescent signal in these myofibers. In contrast, autofluorescent material (arrowheads), not interpreted as TDP-43 immunoreactivity, show fluorescence in both channels.
Supplementary Figure 4. TDP-43 non-nuclear accumulation in myofibrillar myopathy and hereditary inclusion body myopathies (h-IBM). (A) Myofibrillar myopathy. (B) h-IBM, autosomal recessive, suspected GNE mutation. (C and D) Autosomal dominant h-IBM, one confirmed and one suspected VCP mutation.
Supplementary Figure 5. Adjacent sections stained for TDP-43, Congo red, and beta-amyloid. (A1-A3; B1-B3) Myofibers with sarcoplasmic TDP-43 immunoreactivity (arrowheads) do not show Congo red fluorescence or beta-amyloid immunoreactivity on adjacent sections. Rare myofibers with Congo red fluorescence (panel B-2, arrow) do not show TDP-43 or beta-amyloid immunoreactivities on adjacent sections. (C and D) Positive control demonstrating correctness of Congo red staining method.
Supplementary Figure 6. Lack of R1282 beta-amyloid focal immunoreactivity in IBM and presence of R1282 focal immunoreactivity in neurogenic atrophy. (A-D) Sections from 4 patients with IBM; no focal accumulations of immunoreactive R1282 were seen. (E,F) Sections from a patient with neurogenic atrophy; R1282 staining is present in target lesions. The target of this immunoreactivity is uncertain. Together with the checkerboard pattern of staining seen in IBM myofibers in (A) and (D), this suggests the antibody is immunoreactive against some protein other than beta-amyloid.
Supplementary Figure 3. Tissue autofluorescence not counted as SMI-31 or TDP-43 immunoreactive accumulations. Unstained frozen muscle viewed with green and red fluorescent optics shows typical rounded autofluorescent material. Tissue autofluorescence was identified by rounded fluorescent material visible in both green (top panel) and red (bottom panel) channels.
Acknowledgments
Supported by grants to S.A.G. from the Muscular Dystrophy Association MDA3878 and the National Institutes of Health (R01 NS043471 and R21 NS057225). JPT was supported by a grant from the Packard Foundation.
Abbreviations
- IBM
inclusion body myositis
- PM
polymyositis
- DM
dermatomyositis
- IBMPFD
inclusion body myopathy with Paget disease and frontotemporal dementia
References
- 1.Askanas V, Engel WK, Alvarez RB. Light and electron microscopic localization of beta-amyloid protein in muscle biopsies of patients with inclusion-body myositis. Am J Pathol. 1992;141(1):31–36. [PMC free article] [PubMed] [Google Scholar]
- 2.Askanas V, Engel WK, Alvarez RB, Glenner GG. beta-Amyloid protein immunoreactivity in muscle of patients with inclusion-body myositis. Lancet. 1992;339(8792):560–561. doi: 10.1016/0140-6736(92)90388-j. [DOI] [PubMed] [Google Scholar]
- 3.Askanas V, Alvarez RB, Engel WK. beta-Amyloid precursor epitopes in muscle fibers of inclusion body myositis. Ann Neurol. 1993;34(4):551–560. doi: 10.1002/ana.410340408. [DOI] [PubMed] [Google Scholar]
- 4.Askanas V, Engel WK, Alvarez RB. Enhanced detection of congo-red-positive amyloid deposits in muscle fibers of inclusion body myositis and brain of Alzheimer's disease using fluorescence technique. Neurology. 1993;43(6):1265–1267. doi: 10.1212/wnl.43.6.1265-a. [DOI] [PubMed] [Google Scholar]
- 5.Ayala YM, Zago P, D'Ambrogio A, Xu YF, Petrucelli L, Buratti E, Baralle FE. Structural determinants of the cellular localization and shuttling of TDP-43. J Cell Sci. 2008;121(Pt 22):3778–3785. doi: 10.1242/jcs.038950. [DOI] [PubMed] [Google Scholar]
- 6.Banwell BL, Engel AG. AlphaB-crystallin immunolocalization yields new insights into inclusion body myositis. Neurology. 2000;54(5):1033–1041. doi: 10.1212/wnl.54.5.1033. [DOI] [PubMed] [Google Scholar]
- 7.Buratti E, Baralle FE. Multiple roles of TDP-43 in gene expression, splicing regulation, and human disease. Front Biosci. 2008;13:867–878. doi: 10.2741/2727. [DOI] [PubMed] [Google Scholar]
- 8.Carpenter S, Karpati G, Heller I, Eisen A. Inclusion body myositis: a distinct variety of idiopathic inflammatory myopathy. Neurology. 1978;28(1):8–17. doi: 10.1212/wnl.28.1.8. [DOI] [PubMed] [Google Scholar]
- 9.Chou SM. Myxovirus-like structures and accompanying nuclear changes in chronic polymyositis. Arch Pathol. 1968;86(6):649–658. [PubMed] [Google Scholar]
- 10.Gijselinck I, Sleegers K, Engelborghs S, Robberecht W, Martin JJ, Vandenberghe R, Sciot R, Dermaut B, Goossens D, van der Zee J, De Pooter T, Del-Favero J, Santens P, De Jonghe P, De Deyn PP, Van Broeckhoven C, Cruts M. Neuronal inclusion protein TDP-43 has no primary genetic role in FTD and ALS. Neurobiol Aging. 2007 doi: 10.1016/j.neurobiolaging.2007.11.002. ePub. [DOI] [PubMed] [Google Scholar]
- 11.Greenberg SA, Bradshaw EM, Pinkus JL, Pinkus GS, Burleson T, Due B, Bregoli L, O'Connor KC, Amato AA. Plasma cells in muscle in inclusion body myositis and polymyositis. Neurology. 2005;65(11):1782–1787. doi: 10.1212/01.wnl.0000187124.92826.20. [DOI] [PubMed] [Google Scholar]
- 12.Greenberg SA, Pinkus JL, Amato AA. Nuclear membrane proteins are present within rimmed vacuoles in inclusion-body myositis. Muscle Nerve. 2006;34(4):406–416. doi: 10.1002/mus.20584. [DOI] [PubMed] [Google Scholar]
- 13.Greenberg SA, Watts GD, Kimonis VE, Amato AA, Pinkus JL. Nuclear localization of valosin-containing protein in normal muscle and muscle affected by inclusion-body myositis. Muscle Nerve. 2007;36(4):447–454. doi: 10.1002/mus.20823. [DOI] [PubMed] [Google Scholar]
- 14.Griggs RC, Askanas V, DiMauro S, Engel A, Karpati G, Mendell JR, Rowland LP. Inclusion body myositis and myopathies. Ann Neurol. 1995;38(5):705–713. doi: 10.1002/ana.410380504. [DOI] [PubMed] [Google Scholar]
- 15.Hoogendijk JE, Amato AA, Lecky BR, Choy EH, Lundberg IE, Rose MR, Vencovsky J, de Visser M, Hughes RA. 119th ENMC international workshop: trial design in adult idiopathic inflammatory myopathies, with the exception of inclusion body myositis, 10−12 October 2003, Naarden, The Netherlands. Neuromuscul Disord. 2004;14(5):337–345. doi: 10.1016/j.nmd.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 16.Hutchinson DO. Inclusion body myositis: abnormal protein accumulation does not trigger apoptosis. Neurology. 1998;51(6):1742–1745. doi: 10.1212/wnl.51.6.1742. [DOI] [PubMed] [Google Scholar]
- 17.Kwong LK, Uryu K, Trojanowski JQ, Lee VM. TDP-43 proteinopathies: neurodegenerative protein misfolding diseases without amyloidosis. Neurosignals. 2008;16(1):41–51. doi: 10.1159/000109758. [DOI] [PubMed] [Google Scholar]
- 18.Mendell JR, Sahenk Z, Gales T, Paul L. Amyloid filaments in inclusion body myositis. Novel findings provide insight into nature of filaments. Arch Neurol. 1991;48(12):1229–1234. doi: 10.1001/archneur.1991.00530240033013. [DOI] [PubMed] [Google Scholar]
- 19.Mirabella M, Alvarez RB, Bilak M, Engel WK, Askanas V. Difference in expression of phosphorylated tau epitopes between sporadic inclusion-body myositis and hereditary inclusion-body myopathies. J Neuropathol Exp Neurol. 1996;55(7):774–786. doi: 10.1097/00005072-199607000-00003. [DOI] [PubMed] [Google Scholar]
- 20.Nakano S, Shinde A, Fujita K, Ito H, Kusaka H. Histone H1 is released from myonuclei and present in rimmed vacuoles with DNA in inclusion body myositis. Neuromuscul Disord. 2008;18(1):27–33. doi: 10.1016/j.nmd.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 21.Nalbantoglu J, Karpati G, Carpenter S. Conspicuous accumulation of a single-stranded DNA binding protein in skeletal muscle fibers in inclusion body myositis. Am J Pathol. 1994;144(5):874–882. [PMC free article] [PubMed] [Google Scholar]
- 22.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314(5796):130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 23.Ou SH, Wu F, Harrich D, Garcia-Martinez LF, Gaynor RB. Cloning and characterization of a novel cellular protein, TDP-43, that binds to human immunodeficiency virus type 1 TAR DNA sequence motifs. J Virol. 1995;69(6):3584–3596. doi: 10.1128/jvi.69.6.3584-3596.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pruitt JN, 2nd, Showalter CJ, Engel AG. Sporadic inclusion body myositis: counts of different types of abnormal fibers. Ann Neurol. 1996;39(1):139–143. doi: 10.1002/ana.410390122. [DOI] [PubMed] [Google Scholar]
- 25.Puchtler H, Sweat F, Levine M. On the binding of Congo red by amyloid. J Histochem Cytochem. 1962;10:355–364. [Google Scholar]
- 26.Sarkozi E, Askanas V, Johnson SA, Engel WK, Alvarez RB. beta-Amyloid precursor protein mRNA is increased in inclusion-body myositis muscle. Neuroreport. 1993;4(6):815–818. doi: 10.1097/00001756-199306000-00055. [DOI] [PubMed] [Google Scholar]
- 27.Sherriff FE, Joachim CL, Squier MV, Esiri MM. Ubiquitinated inclusions in inclusion-body myositis patients are immunoreactive for cathepsin D but not beta-amyloid. Neurosci Lett. 1995;194(1−2):37–40. doi: 10.1016/0304-3940(95)11718-c. [DOI] [PubMed] [Google Scholar]
- 28.van der Meulen MF, Hoogendijk JE, Moons KG, Veldman H, Badrising UA, Wokke JH. Rimmed vacuoles and the added value of SMI-31 staining in diagnosing sporadic inclusion body myositis. Neuromuscul Disord. 2001;11(5):447–451. doi: 10.1016/s0960-8966(00)00219-4. [DOI] [PubMed] [Google Scholar]
- 29.Wang HY, Wang IF, Bose J, Shen CK. Structural diversity and functional implications of the eukaryotic TDP gene family. Genomics. 2004;83(1):130–139. doi: 10.1016/s0888-7543(03)00214-3. [DOI] [PubMed] [Google Scholar]
- 30.Weihl CC, Temiz P, Miller SE, Watts G, Smith C, Forman M, Hanson PI, Kimonis V, Pestronk A. TDP-43 accumulation in IBM muscle suggests a common pathogenic mechanism with frontotemporal dementia. J Neurol Neurosurg Psychiatry. 2008;79(10):1186–1189. doi: 10.1136/jnnp.2007.131334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wu Z, Irizarry RA. Stochastic models inspired by hybridization theory for short oligonucleotide arrays. J Comput Biol. 2005;12(6):882–893. doi: 10.1089/cmb.2005.12.882. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. Negative controls and exclusion of autofluorescence with TDP-43 studies. (A1−3) TDP-43 identified in an IBM myofiber with AF488 secondary antibody in the green channel (arrow) and appropriately not visible in the red channel or with tris+AF488 secondary. The arrowhead shows autofluorescent material present in both red and green channels. (B1−3) Muscle from a different patient with IBM showing TDP-43 sarcoplasmic material, lacking autofluorescence and not staining with control primary antibody normal rabbit serum.
Supplementary Figure 2. Exclusion of artifact for TDP-43 sarcoplasmic immunoreactivity. Studies were performed using both Alex fluorochrome 488 (green) and Alexa fluorochrome 555 (red) labels and examining sections with both filter sets individually. TDP-43 labeled with AF555 examined in the red channel (A-1) and TDP-43 labeled with AF488 examined in the green channel (B-1) show myofibers with sarcoplasmic TDP-43 (arrows); examination of these sections in the other channels (A-2 green and B-2 red) shows no fluorescent signal in these myofibers. In contrast, autofluorescent material (arrowheads), not interpreted as TDP-43 immunoreactivity, show fluorescence in both channels.
Supplementary Figure 4. TDP-43 non-nuclear accumulation in myofibrillar myopathy and hereditary inclusion body myopathies (h-IBM). (A) Myofibrillar myopathy. (B) h-IBM, autosomal recessive, suspected GNE mutation. (C and D) Autosomal dominant h-IBM, one confirmed and one suspected VCP mutation.
Supplementary Figure 5. Adjacent sections stained for TDP-43, Congo red, and beta-amyloid. (A1-A3; B1-B3) Myofibers with sarcoplasmic TDP-43 immunoreactivity (arrowheads) do not show Congo red fluorescence or beta-amyloid immunoreactivity on adjacent sections. Rare myofibers with Congo red fluorescence (panel B-2, arrow) do not show TDP-43 or beta-amyloid immunoreactivities on adjacent sections. (C and D) Positive control demonstrating correctness of Congo red staining method.
Supplementary Figure 6. Lack of R1282 beta-amyloid focal immunoreactivity in IBM and presence of R1282 focal immunoreactivity in neurogenic atrophy. (A-D) Sections from 4 patients with IBM; no focal accumulations of immunoreactive R1282 were seen. (E,F) Sections from a patient with neurogenic atrophy; R1282 staining is present in target lesions. The target of this immunoreactivity is uncertain. Together with the checkerboard pattern of staining seen in IBM myofibers in (A) and (D), this suggests the antibody is immunoreactive against some protein other than beta-amyloid.
Supplementary Figure 3. Tissue autofluorescence not counted as SMI-31 or TDP-43 immunoreactive accumulations. Unstained frozen muscle viewed with green and red fluorescent optics shows typical rounded autofluorescent material. Tissue autofluorescence was identified by rounded fluorescent material visible in both green (top panel) and red (bottom panel) channels.