

Abstract
We report a concise and efficient method to make a circular single-stranded DNA containing a defined DNA lesion. In this protocol, phagemid DNA containing Uracil is used as a template to synthesize a complementary DNA strand using T7 DNA polymerase and an oligonucleotide primer including a site-specific DNA lesion. The ligated lesion-containing strand can be recovered after the phage-derived template DNA is degraded by treatment with E.coli Uracil DNA glycosylase and Exonucleases I and III. The resulting product is a circular single-stranded DNA containing a defined DNA lesion suitable for in vitro translesion replication assays.
Keywords: Translesion DNA synthesis, Mutagenesis, DNA Repair, Cyclobutane pyrimidine dimer, UV-light, Y-family DNA polymerase
1. Introduction
Replicative DNA polymerases are unable to bypass bulky lesions in DNA. As a result, the replication fork may collapse leading to cell cycle arrest and possibly cell death. To avoid such deleterious consequences, organisms possess specialized DNA polymerases that can circumvent DNA lesions in a process referred to as “Translesion DNA synthesis” (TLS). E.coli possesses three TLS polymerases and humans possibly as many as ten TLS polymerases [1]. The biological importance of TLS is typified by E.coli polV, which is responsible for the majority of mutagenic TLS in vivo [2] and DNA polymerase η (polη), which protects humans from UV-induced cancers by accurately replicating past cis-syn cyclobutane pyrimidine dimers (CPDs). Indeed, defects in human POLH, encoding polη, lead to the sunlight-sensitive and cancer-prone Xeroderma pigmentosum variant (XP-V) syndrome [3,4].
The TLS polymerases gain access to a nascent primer terminus via an interaction with the cell's replicative, ring-shaped, clamp (β-clamp in E.coli and PCNA in eukaryotes). The process is initiated by a clamp loader (γ-complex in E.coli and replication factor C in eukaryotes), which recognizes the DNA primer terminus and opens and assembles the clamp around the nascent DNA [5]. Each clamp has two (prokaryotes), or three (eukaryotes) potential DNA polymerase binding sites and may, therefore, engage multiple polymerases simultaneously. Indeed, such interactions are believed to be critical for switching between replicative and TLS polymerases [6].
In vitro studies investigating the effects of the replicative clamps on TLS have been hampered because the clamps readily slide off of linear DNA substrates. One option is to cap the DNA ends using large biomolecules such as Streptavidin beads linked to biotinylated oligonucleotides. However, this imposes large steric constraints and may affect the ability of the DNA polymerase to access the primer terminus. Circular, single-stranded templates are, therefore, more likely to provide more informative data on the effects of the replicative clamps on TLS and polymerase switching in vitro.
Protocols to generate such substrates have previously been reported by the Lawrence [7] and Fuchs laboratories [8]. Lawrence et al., used a long oligomer scaffold to anneal and subsequently ligate a short lesion-containing oligonucleotide into M13 DNA. Using a similar “scaffold” approach, Napolitano and Fuchs hybridized two related plasmids of slightly different sizes, so as to generate a gapped double-stranded substrate, to which the lesion-containing oligonucleotide was annealed and ligated. The non-adducted DNA strand was subsequently degraded, so as to generate a single-stranded lesion-containing substrate [8]. While both protocols achieve their desired goal, both are somewhat time consuming and include labor-intensive steps.
We were therefore interested in developing a protocol for the rapid and efficient purification of circular, single-stranded DNA containing a defined lesion. To achieve our goal, we took advantage of the methodology previously described by Kunkel and colleagues to generate site-directed mutations in DNA [9,10]. In the Kunkel protocol, a primer containing the desired mutation is annealed to a ssDNA template containing Uracil. After primer extension and ligation, the dsDNA is used to transfect ung+ E.coli wherein the Uracil-containing DNA is degraded. The Uracil-free ssDNA is converted back into duplex DNA by host polymerases and in the process, the nucleotide change in the original oligonucleotide primer is fixed as a mutation in vivo. We have used a conceptually similar approach in our protocol, but instead of using a primer with a mutation, we used a primer containing a site-specific DNA lesion and instead of degrading the Uracil-containing template in vivo, it is degraded in vitro using the combined actions of E.coli Uracil DNA glycosylase and Exonucleases I and III. The final product is a circular, single-stranded DNA molecule containing a defined lesion that can be used for in vitro replication and repair assays.
2. Materials and methods
2.1 Reagents
E.coli DNA polymerase I (Klenow Fragment) [pol I (Kf)], E.coli Exonuclease III, E.coli Exonuclease I, E.coli Uracil DNA glycosylase, E.coli RecA, T7 DNA polymerase, T4 Polynucleotide kinase, and M13KO7 helper phage were all purchased from New England Biolabs (Ipswich, MA). ATP was from Roche Applied Science (Indianapolis, IN). T4 DNA ligase and deoxyribose nucleoside triphosphates (dNTPs) were purchased from Invitrogen (Carlsbad, CA). Polyethylene glycol 6000 and 8000 was purchased from Sigma-Aldrich (St. Louis, MO).
2.2 DNAs
The two 60mer oligonucleotides U60T and U60B, were synthesized by Lofstrand Laboratories (Gaithersburg, MD). The cis-syn cyclobutane pyrimidine dimer (CPD)-containing oligonucleotide TTC48P, was synthesized by Phoenix BioTechnologies (Hunstville, AL). pBluescript II KS(+) was purchased from Stratagene (San Diego, CA). The QIAquick PCR purification kit was purchased from QIAGEN (Valencia, CA).
3. Results
3.1 Construction of a single-stranded circular DNA containing a CPD lesion
The first step of the protocol is to clone a short insert into a double-stranded plasmid vector capable of producing single-stranded phagemid DNA, so that one can subsequently anneal a lesion-containing oligonucleotide to the phagemid-derived ssDNA. In our case, we synthesized two 60mer oligonucleotides, U60T (5′-AAT TCG ATT CGA TAC TGG TAC TAA TGA TTA ACG AAT TAA GCA CGT CCG TAC CAT CGA TCA-3′) and U60B (5′-AGC TTG ATC GAT GGT ACG GAC GTG CTT AAT TCG TTA ATC ATT AGT ACC AGT ATC GAA TCG-3′), which were annealed to each other and cloned into EcoRI-HindIII digested pBluescript II KS(+) to generate a 3010 bp plasmid, pAVR88.
pAVR88 was introduced into E.coli strain CJ236 (dut, ung1), which allows for high levels of dUMP incorporation into DNA. A fresh single colony of CJ236 harboring pAVR88 was inoculated into 200 ml of LB broth with 0.75 μg/ml of Uridine and incubated at 37 °C with aeration for several hours. When the culture became slightly turbid, 1 × 1010 pfu of M13KO7 helper phage was added and cultured for an additional 1 hour prior to the addition of kanamycin (50 μg/ml) and subsequent overnight growth at 37 °C. The overnight culture was centrifuged at 13,000 × g for 15 min and the phage containing supernatant was collected and passed through a 0.22μm filter to eliminate any remaining CJ236 host cells. Polyethylene glycol 6000 (PEG-6000) and NaCl was added to a final concentration of 5% and 0.5M respectively. After incubation at 4 °C for 1 hour, phage particles were recovered by centrifugation at 13,000 × g for 15 min. The phage pellet was washed three times in 4 ml of TE, followed by sedimentation of insoluble debris and re-precipitation of phage particles. The purified phage were suspended in 2 ml TE and DNA was extracted with an equal volume of phenol:chloroform:isoamyl alcohol (25:24:1), until no debris was observed at the interface. The DNA was subsequently extracted with an equal volume of chloroform: isoamyl alcohol (24:1). After ethanol precipitation, the DNA pellet was suspended in 400 μl of TE. This approach usually gave a yield of ∼1-2 μg of ssDNA per milliliter of initial starting culture. The single-stranded DNA containing Uracil is hereafter referred to as sspAVR88.
The next step in the protocol is the generation of a complementary copy of sspAVR88 carrying a single, site-directed DNA lesion (Fig. 1). This is achieved by using an oligonucleotide containing a defined lesion as a primer for 2nd strand DNA synthesis in vitro. In our studies, we used a 48mer oligonucleotide, TTC48P (5′-TCG ATA CTG GTA CTA ATG ATT AAC GAC T-TA AGC ACG TCC GTA CCA TCG-3′), containing a CPD lesion (indicated by T-T in bold font). 30 pmol of TTC48P was 5′-phosphorylated by mixing the oligonucleotide with 1× T4 Polynucleotide Kinase (PNK) buffer, 10mM ATP and 40U of T4 PNK (160 μl reaction volume) and incubated at 37 °C for 30 min. This step is required for the ligation of the newly synthesized strand into a circular substrate. 20 pmol of sspAVR88 and annealing buffer (20 mM Tris-HCl pH 7.5, 2 mM MgCl2, 50 mM NaCl) were added directly to the reaction mixture (800 μl reaction volume) and incubated at 75 °C for 5 min before slowly cooling to room temperature. This inactivates T4 PNK and allows the primer to anneal to the sspAVR88 template. The primer was in slight excess over template (3:2 p/t), to ensure that the entire template was utilized for DNA synthesis. T4 DNA ligase buffer, 0.15 mM dNTPs, 5% PEG-8000, 160U native T7 DNA polymerase and 32U T4 DNA ligase were added (1080 μl final volume) and incubated at 33 °C for 20 min, followed by inactivation at 75 °C for 20 min. Under these conditions, most of the product was double-stranded, closed-circular DNA (Fig. 2B, lane 3, lower band), with a small portion of open-circular DNA (Fig. 2B, lane 3, upper band). The open circular molecules are likely derived from unligated products due to incomplete phosphorylation of the primer. Native T7 DNA polymerase was chosen to perform the ∼3 kb primer extension reaction because of its strong processivity and limited strand-displacement activity [11,12]. Addition of PEG-8000 to the reaction also improved the catalytic activity of T7 DNA polymerase and T4 DNA ligase [13]. After cooling to room temperature, 24U Uracil DNA glycosylase, 80U Exonuclease I and 400U Exonuclease III, were added to the synthesized double-strand DNA and incubated at 37°C for 60 min (1089 μl reaction volume). Uracil DNA glycosylase excises Uracil incorporated in the sspAVR88-derived strand leaving apurinic/apyrimidinic (AP) sites. Exonuclease III cleaves the AP site and degrades the nicked strand through its duplex-specific 3′→5′ exonuclease activity. Exonuclease I degrades any excess TTC48P primer. As a consequence, the double-stranded DNA was efficiently converted to a single-strand (Fig. 2B, lane 4) and the amount of product observed was approximately half of the input sspAVR88 (Fig. 2B, lanes 1, 4). The single-stranded circular DNA was subsequently purified using a QIAquick PCR purification kit according to the manufacturer's instructions (Fig. 2B, lanes 5, 6) and it is estimated that ∼ 10 μg of lesion-containing single-stranded circular substrate can be obtained per preparation following this protocol. The purified single-stranded circular (circular = O) DNA containing a CPD, was designated as pSOcpd.
Fig. 1.
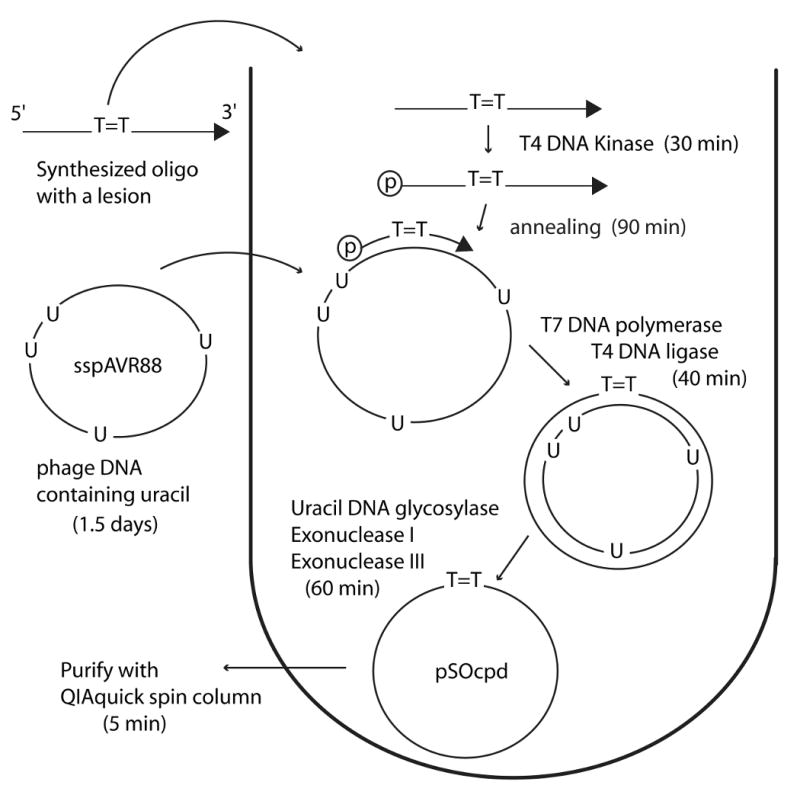
Flow chart for the construction of pSOcpd. All reactions were performed in one tube, except for the preparation of phage DNA and the purification of the final product. The approximate time to complete each step is shown in parentheses.
Fig. 2.

Verification of each step during second strand DNA synthesis and glycosylase/nuclease treatments. Samples were separated on 1% agarose gel containing 2 μg/ml ethidium bromide. Lane M, DNA size markers. Lane 1, 200ng of sspAVR88. Lane 2, product after annealing of the 2nd strand primer. Lane 3, product after second strand synthesis and ligation. Lane 4, product after glycosylase and nuclease treatment. Lane 5 and 6 are different amounts of purified pSOcpd. Equal portions corresponding to 200ng of sspAVR88 were loaded in lanes 1-5.
3.1 Replication assays with pSOcpd
We then used pSOcpd as a template for in vitro translesion replication assays. In particular, we assayed the ability of two A-family polymerases (E. coli pol I (Kf) and T7 DNA polymerase) and two Y-family TLS polymerases (polV(R391) and human polη), to bypass the CPD lesion in vitro. A 17mer primer, M13-TT (5′-32P -GCT CGA TGG TAC GGA CG-3′), was annealed to the pSOcpd template in a molar ratio of 4:5 (p/t) (Fig. 3A). The primer template mix (in 50 mM Tris-HCl pH 8.0, 5 mM MgCl2, 50 μg/ml BSA, 1.42 mM β-mercaptoethanol) was heated to 95 °C for 5 min and then slowly cooled to room temperature. 0.05 pmol of the primed pSOcpd was mixed with 200 μM dNTPs and 5 mM ATP in replication reaction buffer (50 mM Tris-HCl pH 7.5, 5 mM MgCl2, 2 mM DTT, 4% glycerol). Reactions were initiated by adding 0.025U pol I (Kf), 0.04U T7 DNA polymerase, 400 nM polV(R391) and 256 nM polη. Where indicated, 2 μM RecA, 50 nM β-clamp and 20 nM γ-complex were added to the reactions. After incubating at 30 °C for 30 min, the reaction was stopped by adding an equal volume of loading buffer (48.5% formamide, 5 mM EDTA, 0.05% xylene cyanol, 0.05% bromophenol blue). The products were denatured at 100 °C for 2 min and immediately resolved on a denaturing polyacrylamide gel (8 M urea, 12% acrylamide). The gel image was visualized with a Fuji image analyzer FLA-5100.
Fig. 3.
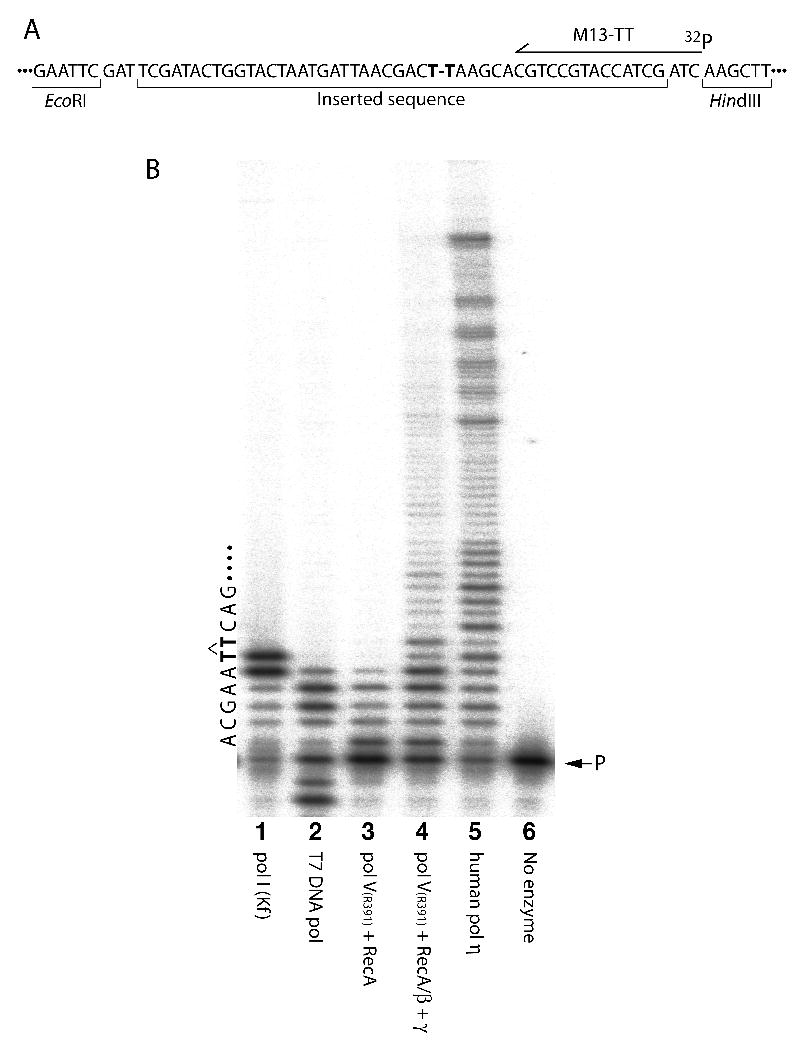
(A) Sequence of pSOcpd surrounding the cis-syn CPD (indicated as T-T in bold font). The binding site of the labeled primer M13-TT, is shown as an arrow. (B) In vitro DNA replication assay with pSOcpd. The TLS reactions were performed in the presence of pol I (Kf) (lane 1), T7 DNA polymerase (lane 2), pol V(R391) + RecA protein in the absence (lane 3), or presence of the β-clamp and γ-clamp loader complex (lane 4), or human polη (lane 5). The position of the labeled primer (lane 6), is shown on the right of the gel (P), while the local template sequence context and position of the T-T CPD (in bold font) is shown on the left side of the gel.
As noted in Fig. 3 (lanes 1, 3, 4, 5, 6), there was no detectable degradation of the radiolabeled primer, indicating that the final template product is not contaminated with Exo I, or Exo III proteins utilised in earlier steps of the protocol. However, significant degradation of the primer was observed in reactions containing T7 DNA polymerase (Fig. 3, track 2), but this is to be expected, given the robust 3′→5′ exonuclease activity of T7 DNA polymerase [12]. As expected, both A-family polymerases had difficulty in traversing the single CPD lesion (Fig. 3B, lanes 1 & 2). E.coli pol I (Kf) inserted a base opposite the 3′T of the CPD, but could not extend it further, while T7 DNA polymerase terminated synthesis immediately before the CPD. Both observations are consistent with previous in vitro studies with a CPD-containing oligonucleotide template [14]. However, longer exposures of the gels (data not shown), revealed very low levels of apparent TLS products with both pol I (Kf), and T7 DNA polymerases. We believe that such products probably do not reflect bona fide TLS events per se, but rather replication of very low-levels of contaminating undamaged oligonucleotide used to prepare pSOcpd. In contrast, the two Y-family TLS polymerases (polV(R391) and polη), were able to bypass the CPD (Fig. 3B, lanes 4 & 5). Limited polV(R391)-dependent TLS was observed in the absence of accessory factors (Fig. 3B, lane 3), but robust polV(R391)-dependent TLS was observed in the presence of the γ-clamp loader and β-clamp (Fig. 3B, c.f., lanes 3 and 4). Such observations are entirely consistent with previous studies indicating an important role for the β-clamp in TLS [15,16] and provide independent evidence that the β-clamp is correctly loaded on the circular, single-lesion template, by the γ-complex.
4. Summary
We describe a protocol to quickly and efficiently obtain a circular, single lesion-containing DNA substrate that can be used in vitro for replication assays. Indeed, once the initial phagemid template is obtained, all reactions can be performed in a single tube and the final product obtained in a matter of a few hours. In the present study, we obtained circular, single-stranded template containing a single cis-syn cyclobutane pyrimidine dimer in one particular sequence context. We expect that this protocol can easily be applied to a variety of modified DNA bases in different sequence contexts by simply making minor changes to the sequence inserted in the phagemid vector and the primer used for 2nd strand DNA synthesis.
Acknowledgments
This work was supported by funds from the NIH/NICHD Intramural Research Program. K.K. was also a recipient of a research fellowship from the Japan Society for the Promotion of Science. A.V. was supported by funds of the Programa Ramón y Cajal (Ministerio de Ciencia e Innovación, Spain).
Abbreviations
- pol
DNA polymerase
- TLS
Translesion DNA synthesis
- XP-V
Xeroderma Pigmentosum Variant
Footnotes
Conflict of interest statement: The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bebenek K, Kunkel TA. Family growth: the eukaryotic DNA polymerase revolution. Cell Mol Life Sci. 2002;59:54–57. doi: 10.1007/s00018-002-8405-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kato T, Shinoura Y. Isolation and characterization of mutants of Escherichia coli deficient in induction of mutations by ultraviolet light. Mol Gen Genet. 1977;156:121–131. doi: 10.1007/BF00283484. [DOI] [PubMed] [Google Scholar]
- 3.Masutani C, Kusumoto R, Yamada A, Dohmae N, Yokoi M, Yuasa M, Araki M, Iwai S, Takio K, Hanaoka F. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase |. Nature. 1999;399:700–704. doi: 10.1038/21447. [DOI] [PubMed] [Google Scholar]
- 4.Johnson RE, Kondratick CM, Prakash S, Prakash L. hRAD30 mutations in the variant form of Xeroderma Pigmentosum. Science. 1999;285:263–265. doi: 10.1126/science.285.5425.263. [DOI] [PubMed] [Google Scholar]
- 5.Kelman Z, Hurwitz J. Protein-PCNA interactions: a DNA-scanning mechanism? Trends Biochem Sci. 1998;23:236–238. doi: 10.1016/s0968-0004(98)01223-7. [DOI] [PubMed] [Google Scholar]
- 6.Plosky BS, Woodgate R. Switching from high-fidelity replicases to low-fidelity lesion-bypass polymerases. Curr Opin Genet Dev. 2004;14:113–119. doi: 10.1016/j.gde.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 7.Banerjee SK, Christensen RB, Lawrence CW, LeClerc JE. Frequency and spectrum of mutations produced by a single cis-syn thymine-thymine dimer in a single-stranded vector. Proc Natl Acad Sci USA. 1988;85:8141–8145. doi: 10.1073/pnas.85.21.8141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Napolitano RL, Fuchs RP. New strategy for the construction of single-stranded plasmids with single mutagenic lesions. Chem Res Toxicol. 1997;10:667–671. doi: 10.1021/tx970018w. [DOI] [PubMed] [Google Scholar]
- 9.Kunkel TA. Rapid and efficient site-specific mutagenesis without phenotypic selection. Proc Natl Acad Sci U S A. 1985;82:488–492. doi: 10.1073/pnas.82.2.488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kunkel TA, Bebenek K, McClary J. Efficient site-directed mutagenesis using uracil-containing DNA. Methods Enzymol. 1991;204:125–139. doi: 10.1016/0076-6879(91)04008-c. [DOI] [PubMed] [Google Scholar]
- 11.Bebenek K, Kunkel TA. The use of native T7 DNA polymerase for site-directed mutagenesis. Nucleic Acids Res. 1989;17:5408. doi: 10.1093/nar/17.13.5408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tabor S, Richardson CC. Selective inactivation of the exonuclease activity of bacteriophage T7 DNA polymerase by in vitro mutagenesis. J Biol Chem. 1989;264:6447–6458. [PubMed] [Google Scholar]
- 13.Pheiffer BH, Zimmerman SB. Polymer-stimulated ligation: enhanced blunt- or cohesive-end ligation of DNA or deoxyribooligonucleotides by T4 DNA ligase in polymer solutions. Nucleic Acids Res. 1983;11:7853–7871. doi: 10.1093/nar/11.22.7853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith CA, Baeten J, Taylor JS. The ability of a variety of polymerases to synthesize past site-specific cis-syn, trans-syn-II, (6-4), and Dewar photoproducts of thymidylyl-(3′->5′)-thymidine. J Biol Chem. 1998;273:21933–21940. doi: 10.1074/jbc.273.34.21933. [DOI] [PubMed] [Google Scholar]
- 15.Tang M, Pham P, Shen X, Taylor JS, O'Donnell M, Woodgate R, Goodman M. Roles of E. coli DNA polymerases IV and V in lesion-targeted and untargeted SOS mutagenesis. Nature. 2000;404:1014–1018. doi: 10.1038/35010020. [DOI] [PubMed] [Google Scholar]
- 16.Fujii S, Fuchs RP. Defining the position of the switches between replicative and bypass DNA polymerases. EMBO J. 2004;23:4342–4352. doi: 10.1038/sj.emboj.7600438. [DOI] [PMC free article] [PubMed] [Google Scholar]