

Abstract
KSHV infection is associated with the development of three proliferative diseases: Kaposi sarcoma (KS), primary effusion lymphoma (PEL), and multicentric Castleman disease (MCD). These conditions are also intimately associated with human immunodeficiency virus (HIV) infection, and important synergistic interactions between these two viruses have been described. Despite differences in viral gene expression patterns for each condition, KSHV encodes similar oncogenic proteins which promote the activation of sequential and parallel signaling pathways. Therapeutic strategies have been implemented to target these unique signaling pathways, and such molecular targeting is the focus of many current research efforts. The scope of this review is to present contemporary knowledge about the epidemiology, virology, and immunology of KSHV, as well as highlight several key oncogene products, which may be targets for chemotherapy.
Keywords: Kaposi sarcoma, Castleman disease, herpesvirus 8, Kaposi sarcoma associated herpesvirus, Primary Effusion Lymphoma
Introduction
In 1994 a previously unrecognized γ-herpesvirus was discovered by Chang and Moore using representational difference analysis to identify DNA fragments of this virus from Kaposi sarcoma (KS) tissue samples.1 Named Kaposi sarcoma–associated herpesvirus (KSHV), and also known as human herpesvirus-8 (HHV-8), KSHV was subsequently also identified in primary effusion lymphoma (PEL) and multicentric Castleman disease (MCD) samples.2,3 More than a coincidental finding, KSHV infection is a requisite for the development of KS and PEL, and is the pathogenic stimulant for many cases of MCD, including all HIV-associated MCD. In addition to these three aforementioned ‘hallmark’ diseases, other conditions, such as hemophagocytic lymphohistiocytosis4 have been associated with KSHV.
Although infection with KSHV is necessary for the development of KSHV-associated disease, it is not sufficient. Both HIV coinfection and immunosuppression significantly increase the risk of KSHV-associated disease.
This review covers the basics of KSHV virology, epidemiology, immunology as it relates to the immunosuppressed host, and diagnosis. The key pathogenic mechanisms of KSHV that lead to tumorigenesis will be highlighted with an emphasis on those which utilize signaling pathways with known inhibitors. Additionally, the epidemiology, pathology, clinical and diagnostic features of the three classic KSHV-associated diseases will be discussed. The rationale for anti-viral therapy against KSHV in each of these diseases is addressed.
KSHV Epidemiology
Information about the epidemiology of KS comes from cancer registry data on the incidence of KS in various geographic and demographic groups, while serologic and molecular epidemiologic studies have characterized the epidemiology of KSHV infection.5,6,7 KS was initially described as an uncommon tumor among elderly Mediterranean men, and subsequently reported among African children in the 1960s.8,9 The association with immunodeficiency was first reported in patients undergoing solid organ transplantation, but in 1981, an epidemic of KS among young men who have sex with men in the United States served as the harbinger of a new immunodeficiency syndrome, subsequently identified as being caused by HIV.10,11 As the HIV epidemic progressed, KS was found almost exclusively among men who have sex with men.12 Coupled with epidemiologic data which found KS to be more common among persons at greater risk for sexually transmitted infections, an infectious etiology of KS was sought.
After the identification of KSHV as the etiologic agent of KS, the development of serologic assays allowed for seroepidemiologic studies that confirmed that KSHV prevalence varies widely, from approximately 1–3% of blood donors in North America to more than 70% in regions of Africa where KSHV is endemic.13 The seroprevalence of KSHV has been found to roughly mirror the incidence of KS, though populations with gross disparities between the two highlight the importance of other potential cofactors in the progression from chronic KSHV infection to KS.
Definitive data on the mode of KSHV transmission are lacking. Evidence for sexual, horizontal, and parenteral transmission may be found in the medical literature. The virus is shed frequently from the oropharynx of both immunocompetent and immunocompromised men, as well as women, in endemic areas.14–16 Behaviors associated with exposure to saliva are correlated with a higher risk of KSHV infection, implicating both sexual and horizontal transmission.17–19 A relatively high KSHV seroprevalence has been described among injection drug users, and an increased incidence of KSHV infection has been noted in transfusion recipients in endemic areas, suggesting that parenteral transmission may be possible.20 Finally, transmission of KSHV from donors of solid organs has been described.21–23 Taken together, these disparate data make it difficult to counsel persons at risk for KSHV-associated disease on methods to reduce their risk of KSHV acquisition.24
Diagnosis and virology
An individual may be diagnosed with KSHV infection by being diagnosed with KS or PEL. Additionally, many indirect serologic tests for KSHV are available, though few commercial assays exist. Serologic assays for KSHV infection are limited in both sensitivity and specificity, and conflicting data have been produced with different methodologies. Combined with the limited availability of testing, there are few clinical indications for KSHV-serologic testing apart from epidemiologic or research settings. Direct detection of KSHV DNA from clinical specimens via polymerase chain reaction may be reasonable in a restricted set of clinical conditions, as discussed below. Finally, in situ hybridization or immunohistochemistry may reveal KSHV proteins expressed in human tissue, often used adjunctively in the diagnosis of KS, PEL or MCD.
Like all herpesviruses, KSHV alternates between two phases of its life cycle. The lytic phase is hallmarked by active viral replication, and a wide range of KSHV-gene products are expressed.25 During latency, however, gene expression is extremely limited. The virus is maintained as episomes attached to the host chromosome, replicated with the host chromosome, and subsequently passed to daughter cells. These diseases vary in their degree of lytic replication. KS lesions have only a small quantity of lytic viral replication, MCD is associated with a very high degree of lytic replication, and PEL is intermediate.
Pathogenesis of KSHV-associated tumorigenesis
KSHV encodes for numerous, specific proteins postulated to play a role in the pathogenesis of KS, PEL, and MCD, many of which have been pirated from the human host in the course of viral evolution. KSHV produces molecules critical in the transduction of signals that stimulate cell proliferation and inhibit apoptosis. The latency-associated nuclear antigen (LANA, LNA-1) is one such protein which primarily functions to tether the viral genome to the infected host cells genome; however it also promotes cell survival and contributes to transformation of KSHV-infected cells by interacting and altering the function of the tumor suppressor proteins p53 and retinoblastoma protein.26–28 Another example is the viral G protein-coupled receptor (vGPCR), a lytic phase gene product sharing significant homology with the high-affinity IL-8 receptor. Its dysregulated expression leads to oncogenesis through numerous cellular proliferation, transformation, pro-angiogenic, and anti-apoptotic signaling pathways.29,30 The vGPCR leads to proangiogenic signals by the upregulation of hypoxia inducible factor 1-alpha and subsequent expression of vascular endothelial growth factor-A (VEGF-A) and activation of VEGF-receptor-2, which in turn activates the phophatidyl-inositol-3-kinase(PI3K)/AKT/mammalian target of rapamycin (mTOR) pathway.29 Additionally, blockade of vGPCR and inhibition of PI3K leads to inactivation of the transcription factor and anti-apoptotic protein nuclear factor-kappa B (NF-kB), thereby blocking transformation.29 A third oncogenic protein is the viral FLICE inhibitory protein (vFLIP) which is associated with constitutively activated NF-kB, and through the manipulation of this pathway is purported to function as an oncogene.31,32 Lastly, KSHV encodes for a human IL-6 (hIL-6) homologue, viral IL-6 (vIL-6) which stimulates the known hIL-6-induced signaling pathways via the shared cytokine signaling receptor gp130 coupled to the endogenous JAK/STAT pathway.33 KSHV-infected cells induce and secrete vIL-6 and can retain some proportion of this intracellularly, which then binds to gp130 and activates STAT3 in an autocrine fashion.29
Inhibitors to many of the above mentioned pathways exist, are FDA-approved for other indications, and may offer substantial therapeutic benefit in the treatment of KSHV-associated diseases. Anti-VEGF agents include bevacizumab, sunitinib and sorafenib; inhibitors of mTOR include rapamycin, temsirolimus, and everlimus; lastly, the proteosome inhibitor bortezomib blocks the effects of NF-kB. Inhibitors of the JAK and STAT pathways are being investigated in various diseases, though none are FDA-approved for any indication at this time. Additionally, antibodies to IL-6 may be effective in treating some patients with MCD.34
KSHV in the immunocompromised host
Clinical observations identify T-cells as playing an important role in the control of KS, as evidenced by the regression of KS with the reduction of immunosuppressive treatment following transplant, or clinical improvement and possible exacerbation (flare) of KS in subjects with immune reconstitution following HAART.35,36 Studies have found absent T-cell proliferative responses to KSHV among HIV-infected KSHV seropositive men and low quantities of KSHV-specific T-cells among HIV-positive and HIV-negative persons with KS.37,38 In addition, increased cytotoxic lymphocyte (CTL) responses to KSHV in HIV-infected persons receiving HAART has been shown, though the recent reporting of persistent KS despite effective HAART raises important questions about the mechanisms that control the progression of KSHV infection and KS.39,40
In experimental models, a relationship exists between KSHV and HIV, where the replication of each virus may be enhanced in the presence of the other. The HIV Tat gene for example, upregulates HIV gene expression, and has been shown to play a crucial role in the development of KS via interaction with KSHV gene products. Tat promotes the migration and proliferation of cytokine activated endothelial cells and stimulates KS cell growth in mouse models.41 Accordingly, when injected subcutaneously into nude mice, Tat causes KS-like lesions.42 Moreover, about 15% of male transgenic mice overexpressing the Tat gene develop skin tumors resembling KS at age 12–18 months.42
Similarly, KSHV infection may enhance HIV replication.43 LANA has been shown to activate long terminal repeats of HIV-1 through its association with Tat.44 Interestingly, HIV infection via Tat leads to increased KSHV infectivity.45 It is believed that these actions of Tat may account for the rather aggressive course of AIDS-KS compared with the more indolent behaviour of KS in the HIV-seronegative population.
KSHV-Associated Diseases: Clinical Presentation and Management Kaposi Sarcoma (KS)
KS occurs in several clinical-epidemiologic settings.46 Classic KS is a non-aggressive disease that usually affects elderly, Mediterranean men, is not associated with HIV infection, and presents with a limited number of cutaneous lesions on the lower extremities; disseminated disease is uncommon. Endemic KS affects persons from sub-Saharan Africa and is also not associated with HIV. This is a more aggressive and morbid disease than classic KS. Immunodeficiency clearly enables KS formation, and patients taking immunomodulatory agents, most notably in the context of solid-organ transplantation, often develop transplant-associated KS. Lastly, AIDS-associated, or “epidemic” KS, is the most common tumor in HIV-infected patients. It is characterized often by widely disseminated cutaneous disease, with advanced cases involving the oral mucosa, and viscera (most frequently the lungs and gastrointestinal tract).
A wide range of treatments for KS is available. Independent of any other clinical factor, all patients with AIDS-associated KS should receive highly active antiretroviral therapy (HAART).46 Effective antiretroviral regimens are associated with both a reduction in the incidence of AIDS-related KS, regression in size and number of existing lesions, and histological regression of existing KS lesions. Few data exist on the comparative efficacy of various HAART regimens in the treatment of KS, though experimental models and anecdotal data may support the use of solely protease-inhibitor-containing regimens.47 Several antiviral agents, including ganciclovir, foscarnet, and cidofovir, have been shown to inhibit KSHV replication in vitro. Antiviral therapy (with cidofovir) aimed at KSHV has not been shown to be effective by itself for the treatment of KS, perhaps in part due to the small amount of lytic KSHV in KS tumors.48,49 Antivirals may be effective as an adjunct to more conventional chemotherapy or in the treatment of diseases with a higher degree of lytic replication (MCD and PEL, as discussed below).
The clinical context of KS (ie. HIV status, transplant status, extent and site of disease) is crucial to selecting appropriate treatment. Patients with limited local disease may benefit from a variety of therapies including intralesional chemotherapy (vinblastine is most commonly used), topical ointments (alitretinoin gel), cryotherapy, laser therapy, photodynamic therapy, and infrequently excisional surgery. Radiation therapy can effectively palliate symptomatic disease that is not extensive enough to warrant systemic therapy, but is too extensive to be treated with intralesional chemotherapy. Patients with rapidly progressing, extensive cutaneous, and/or visceral disease should receive systemic therapy. Systemic treatments have traditionally involved cytotoxic chemotherapy. Though numerous chemotherapeutic agents have been shown to be effective, three are FDA-approved in the USA for this indication based on clinical effectiveness and a reasonable side-effect profile. They include the two liposomal anthracyclines (pegylated liposomal doxorubicin and liposomal daunorubicin) and the taxane paclitaxel. Non-traditional therapies have been the focus of recent clinical investigation in the field. Some have shown promise in early clinical trials (thalidomide, imatinib, COL3), or are actively being investigated in such trials (rapamycin, bevacizumab, sunitinib, sorafenib).46
Lastly, patients who develop KS on immunomodulatory agents should, when feasible, be placed on an immunosuppressive regimen that includes rapamycin or one of its analogues. This recommendation is based on a series of solid-organ transplant recipients in whom KS regressed when switched to rapamycin.50
Primary Effusion Lymphoma (PEL)
PEL is an unusual lymphoproliferative disorder, accounting for 2% or less of HIV-associated lymphomas, and is even more rarely encountered in the HIV-seronegative patient. PEL is divided into classic and solid variants. Classic PEL is characterized by lymphomatous involvement of the serosal surfaces, whereas solid PEL manifests initially with tissue-based tumors and no malignant effusions.51 They are similar by morphology, immunophenotype and molecular analysis.52 KSHV along with high levels of interleukin (e.g. IL-6) may be found in PEL tumor cells, and this is frequently necessary to demonstrate in order to aid in the diagnosis. The ramifications of large and typically recurrent pleural, pericardial, and peritoneal effusions are grave and are responsible for the high morbidity and mortality associated with this condition.51
PEL cells have a characteristic phenotype highlighted by CD45, CD30, CD38, CD138 and MUM1 coexpression.51 Classic B-cell markers (CD19, CD20) and T-cell markers (CD2, CD3, CD5, CD7) are not typically seen. Gene expression profiling has shown that PEL expresses a gene profile distinct from other lymphomas, but more akin to multiple myeloma cell lines.
There is no clear standard of care established in the treatment of PEL, and due to its low incidence, randomized clinical trials at present are not feasible. As with the other KSHV-associated diseases, if HIV co-infection is identified, antiretroviral therapy is critical, as spontaneous regression with the commencement of HAART has been described.51,53 Traditionally, the use of standard cytotoxic regimens used for non-Hodgkin lymphomas are suboptimal, and median survival in treated cohorts is poor.54 Induction of apoptosis with the inhibition of NF-kB in PEL cell lines has led to the investigation of proteasome inhibitors which decrease the activation of NF-kB and its anti-apoptotic effects.55 Bortezomib, a proteosome inhibitor that is FDA approved in the USA for use in multiple myeloma, has been shown to enhance the in vitro cytotoxic effects of doxorubicin and paclitaxel, and has been used successfully in combination with anthracycline-based cytotoxic chemotherapy regimens.56 Inhibition of mTOR with rapamycin is effective at decreasing in vitro PEL growth and in vivo mouse xenograft model tumor growth, and its increasing use in the treatment of PEL can be foreseen.57 Cases of prolonged survival in persons treated adjunctively with antiviral therapy (ganciclovir or cidofovir) have also prompted the adjunctive use of these drugs in PEL. Valproate to treat PEL induces lytic KSHV replication and leads to apoptosis in combination with antiviral agents.47
Multicentric Castleman Disease (MCD)
MCD is an aggressive lymphoproliferative disorder, characterized by constitutional symptoms, anemia, and generalized lymphadenopathy. Small series have shown that most MCD cases are driven by KSHV, including 100% of HIV-seropositive patients and the majority of HIV-negative patients.58 Failure to identify KSHV in all MCD lesions may either reflect technical limitations in KSHV detection, the ability for KSHV to induce MCD distant to the biopsied tissue, or an alternate etiology for a limited number of cases. On occasion, MCD may be associated with non-Hodgkin lymphoma, particularly the plasmablastic variant.33 Key in making the diagnosis is to suspect MCD in high risk individuals who present in the appropriate clinical context (i.e. immunosuppressed individual with KSHV infection or other KSHV-associated disease). Definitive diagnosis can only be made by pathologic examination of an involved lymph node or extranodal mass. Detection of KSHV in biopsied tissue or in the peripheral blood can aid in the diagnosis. C-reactive protein, KSHV-viral load, and serum IL-6 levels, if available, may be useful as markers of disease activity and response to therapy.33
In patients with MCD and HIV infection, treatment with antiretroviral therapy is necessary, but caution should be taken as life-threatening flares of MCD have been reported as a manifestation of immune reconstitution.59 Systemic therapy is the mainstay of treatment for patients with MCD and ranges from aggressive remission-induction chemotherapy regimens [CHOP(cyclophosphamide-doxorubicin-vincristine-prednisone); ABV(doxorubicin-bleomycin-vincristine)], single agent maintenance chemotherapy (oral etoposide, cyclophosphamide, vinblastine), immunomodulatory agents (thalidomide, interferon-α), and monoclonal antibodies against the IL-6 receptor (altizumab) and CD20 (rituximab).33
Among all of these treatments, rituximab has shown the most promise in inducing durable remissions. In a prospective study of 24 individuals with chemotherapy-dependent HIV-associated MCD, rituximab was associated with sustained remission off treatment at day 60 (the primary end point) in 22 patients (92%).60 More recently, the efficacy and safety of four weekly infusions of rituximab in 21 consecutive patients with previously untreated plasmablastic HIV-associated MCD has been investigated.61 All but one patient achieved clinical remission of symptoms, hematological and serum chemistry normalization, and 70% achieved a radiological response. In three patients who relapsed, re-treatment with rituximab was successful.62 The main adverse event seen in these patients is reactivation of KS, which is intriguing and may be due to a rapid B cell depletion that is observed during rituximab therapy, or an immune reconstitution inflammatory syndrome to hitherto latent antigens.63 Rituximab therapy in this study was shown to be associated with a decline in KSHV levels initially and at the successful treatment of relapse.
Given the lytic nature of KSHV in MCD, antiviral therapy is also a consideration. A recent randomized controlled trial demonstrated the efficacy of valganciclovir in reducing KSHV replication in individuals with KSHV infection but without evidence of KS, PEL, or MCD.64 In patients with MCD, ganciclovir and valganciclovir have been independently shown to induce remissions alone or in combination with other agents.65,66
Conclusion
Differential KSHV gene expression has the ability to promote the development of three distinct neoplastic conditions (KS, PEL, and MCD). Emerging knowledge about the various signal transduction pathways used by KSHV to mediate oncogenesis has helped identify numerous drugable targets. Promising therapeutic targets include mTOR and VEGF for KS; mTOR, NF-kB and VEGF for PEL; and CD20-positive B cells and KSHV-lytic replication for MCD. Further, continued basic research focusing on KSHV-gene products and their functions may uncover more telling details about the upregulation and utilization of molecular pathways that should provide additional and more efficient therapeutic targets.
Figure 1.
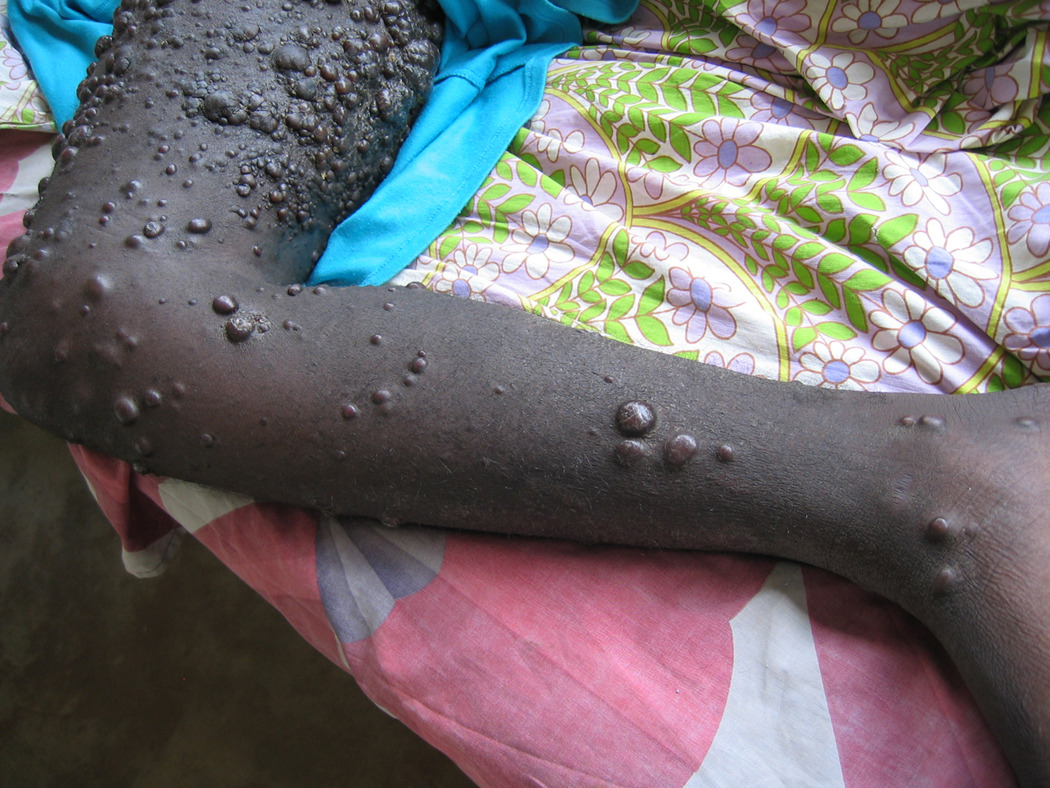
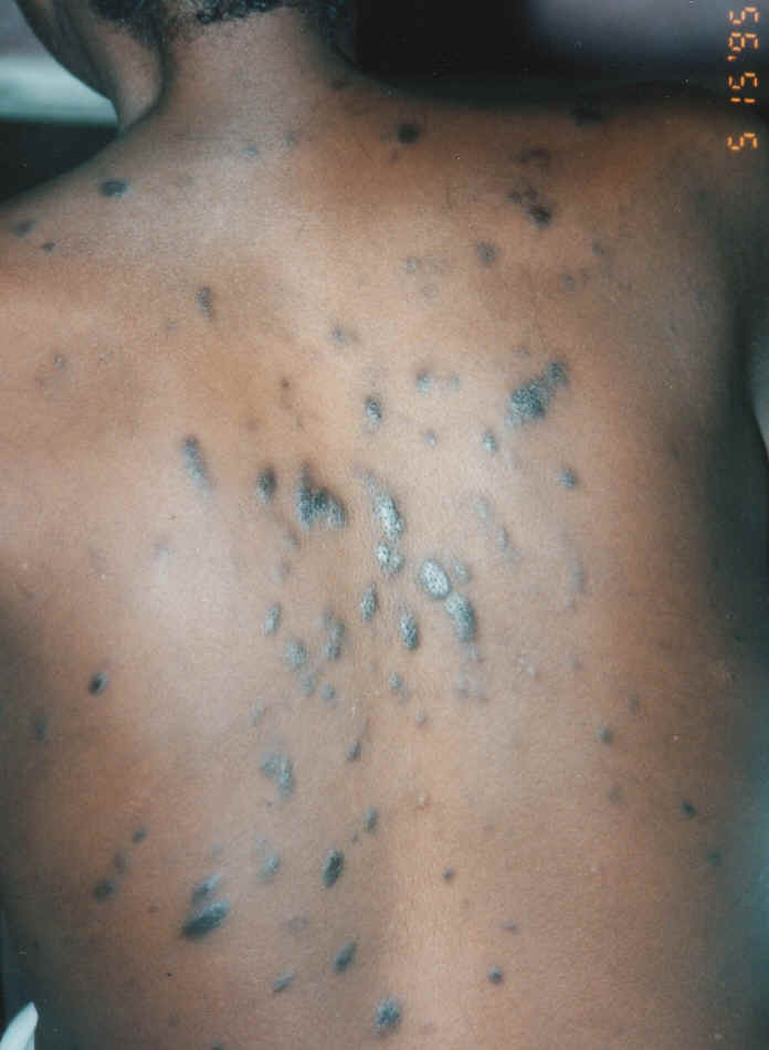
Extensive papular/nodular Kaposi sarcoma on leg (Fig 1a) and back (Fig 1b) in a Uganda patient. (Fig1c) Cutaneous KS of plaque stage (H&E stain, magnification x 200) (Fig 1d) Kaposi sarcoma cutaneous tumor in which the spindled tumor cells demonstrate HHV8 immunoreactivity (LNA-1 immunohistochemical stain).
Figure 2.
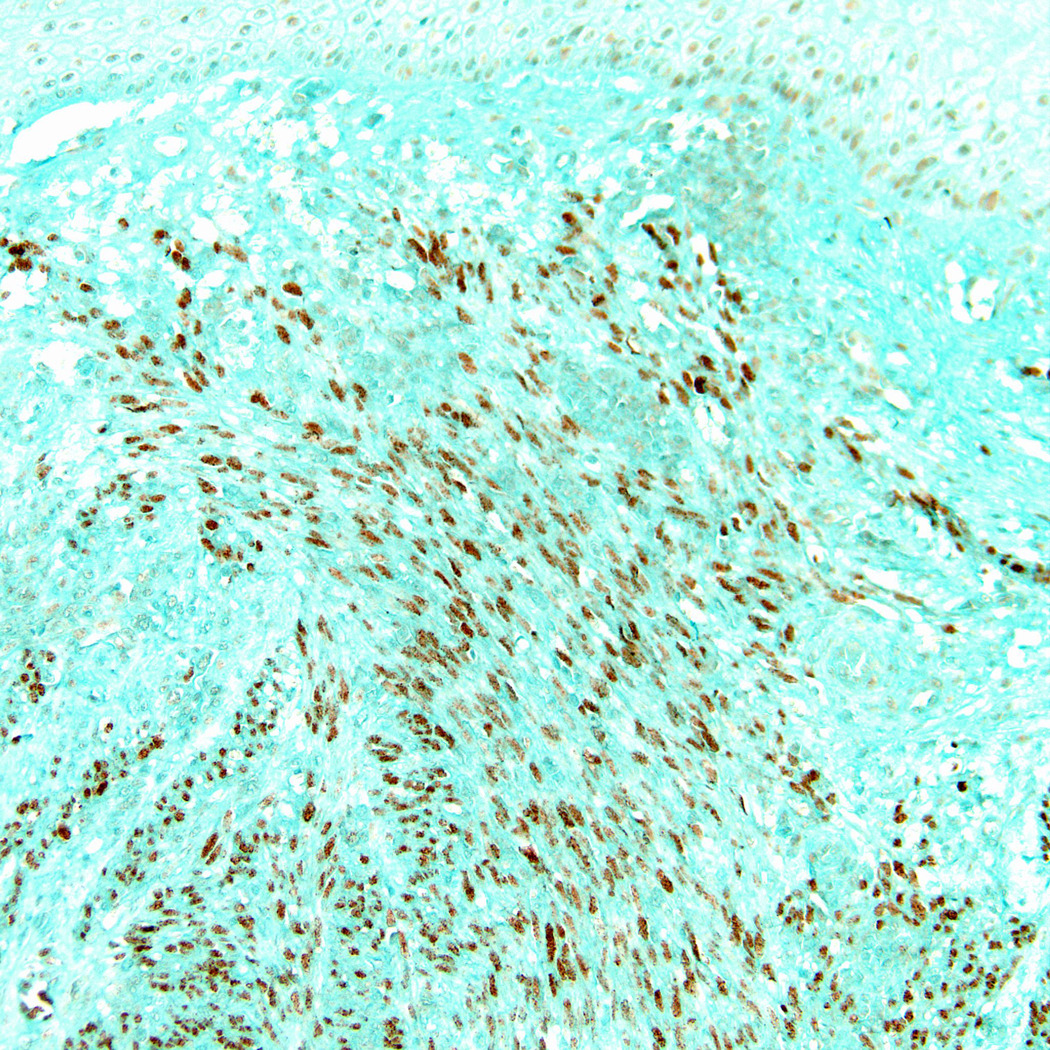
Lymph node with HIV-associated MCD showing multiple regressing follicles surrounded by an expanded and vascular interfollicular zone (H&E stain, magnification x 100).
Figure 3.
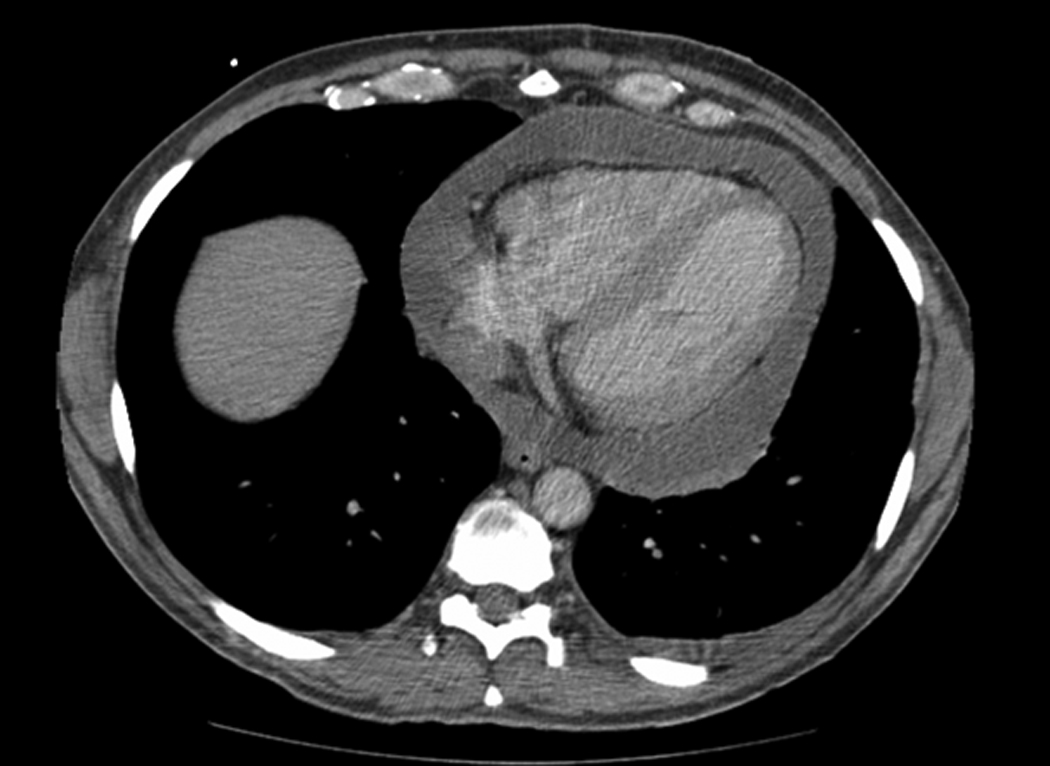
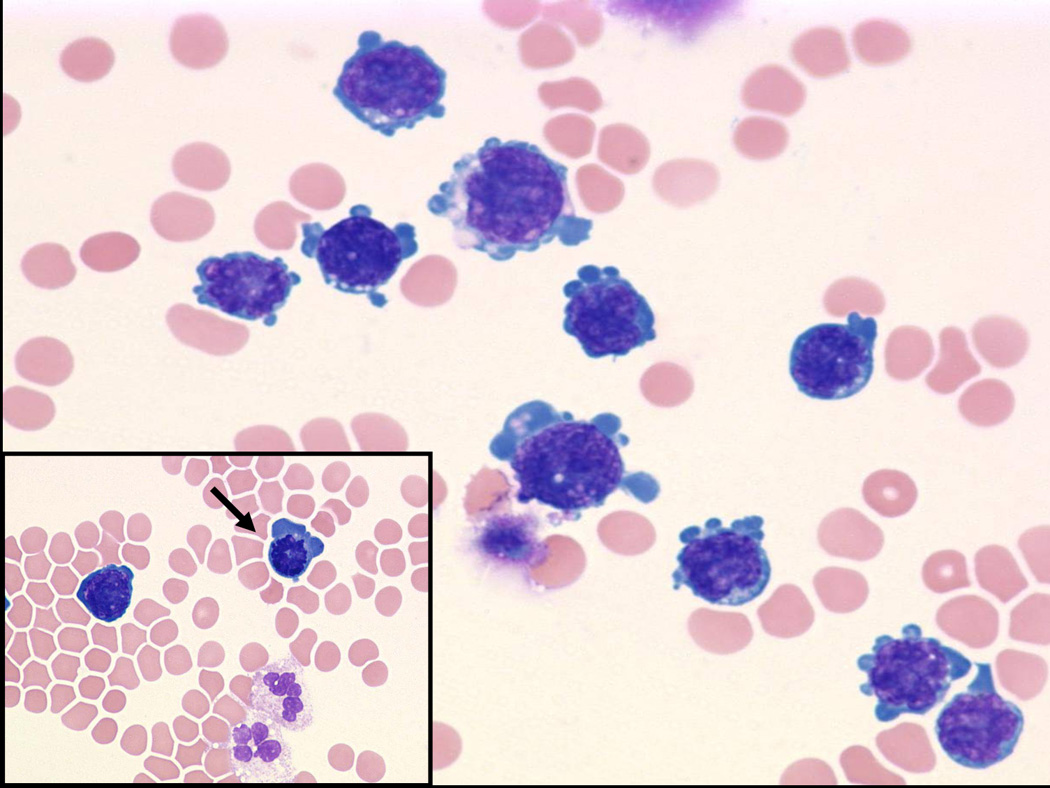
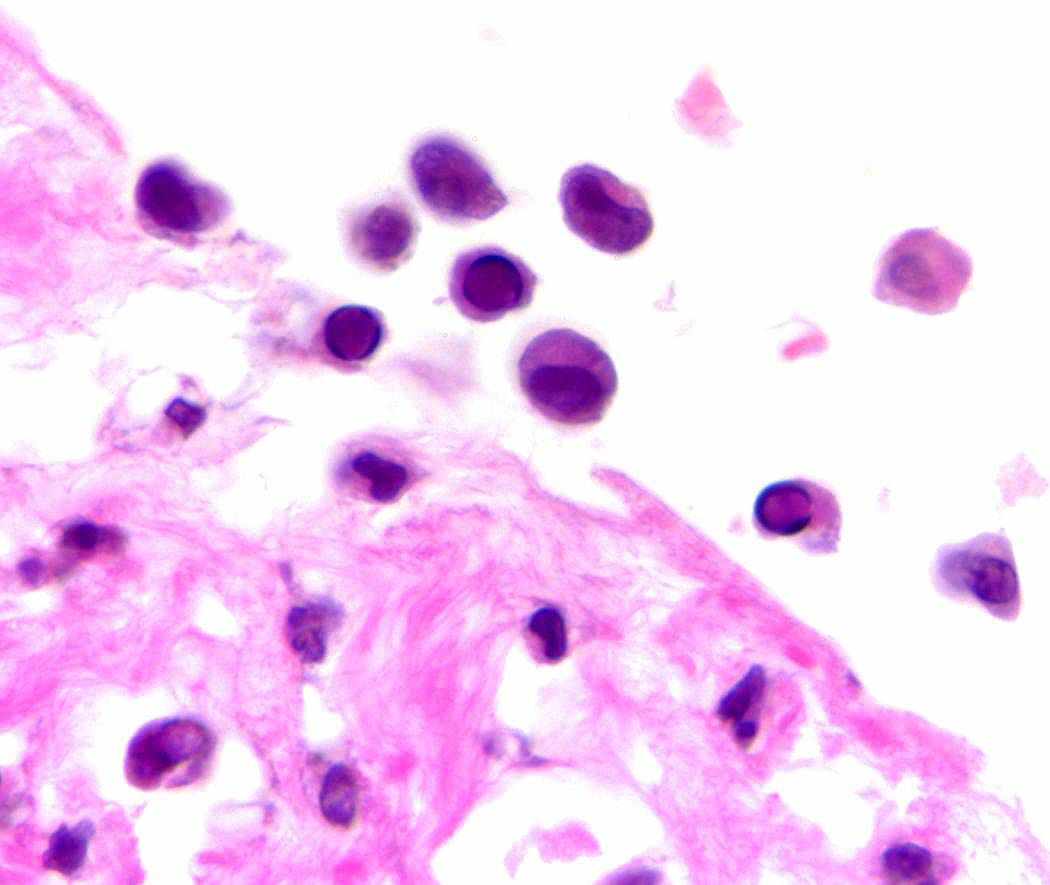
(Fig 3a) Pericardial primary effusion lymphoma (CT scan of the chest). (Fig 3b) Photomicrograph of pericardial fluid, cytospin preparation with atypical lymphoid cells with basophilic cytoplasm and vacuolization (Wright-Giemsa, original magnification x 600), Inset: prominent mitosis (arrow) in one of the lymphoma cells. (Fig 3c) HIV-associated PEL showing lymphoma cells with HHV8 viral nuclear inclusions (cell block preparation; H&E stain). [Figs 3a and 3b, copyright 2007, The AIDS Reader, CMPMedica., all rights reserved]
Acknowledgment
This work has been supported by a grant from the AIDS Malignancy Consortium/NCI to BJD.
References
- 1.Chang Y, Cesarman E, Pessin MS, et al. Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi's sarcoma. Science. 1994;266:1865–1869. doi: 10.1126/science.7997879. [DOI] [PubMed] [Google Scholar]
- 2.Cesarman E, Chang Y, Moore PS, et al. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas. N Engl J Med. 1995;332:1186–1191. doi: 10.1056/NEJM199505043321802. [DOI] [PubMed] [Google Scholar]
- 3.Soulier J, Grollet L, Oskenhendler E, et al. Kaposi's sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman's disease. Blood. 1995;86:1276–1280. [PubMed] [Google Scholar]
- 4.Grossman WJ, Radhi M, Schauer D, et al. Development of hemophagocytic lymphohistiocytosis in triplets infected with HHV-8. Blood. 2005;106(4):1203–1206. doi: 10.1182/blood-2005-03-0950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Eltom MA, Jemal A, Mbulaiteye SM, Devesa SS, Biggar RJ. Trends in Kaposi's sarcoma and non-Hodgkin's lymphoma incidence in the United States from 1973 through 1998. J Natl Cancer Inst. 2002;94:1204–1210. doi: 10.1093/jnci/94.16.1204. [DOI] [PubMed] [Google Scholar]
- 6.Chatlynne LG, Ablashi DV. Seroepidemiology of Kaposi's sarcoma-associated herpesvirus (KSHV) Semin Cancer Biol. 1999;9:175–185. doi: 10.1006/scbi.1998.0089. [DOI] [PubMed] [Google Scholar]
- 7.Dukers NH, Rezza G. Human herpesvirus 8 epidemiology: what we do and do not know. Aids. 2003;17:1717–1730. doi: 10.1097/01.aids.0000076337.42412.86. [DOI] [PubMed] [Google Scholar]
- 8.Kaposi M. Idiopathisches multiples Pigmentsarkom der Haut. Arch Dermatol Syphilol. 1872;4:265. [Google Scholar]
- 9.Taylor JF, Templeton AC, Vogel CL, Ziegler JL, Kyalwazi SK. Kaposi's sarcoma in Uganda: a clinico-pathological study. Int J Cancer. 1971;8:122–135. doi: 10.1002/ijc.2910080116. [DOI] [PubMed] [Google Scholar]
- 10.Stribling J, Weitzner S, Smith GV. Kaposi's sarcoma in renal allograft recipients. Cancer. 1978 Aug;42(2):442–446. doi: 10.1002/1097-0142(197808)42:2<442::aid-cncr2820420210>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 11.Hymes KB, Cheung T, Greene JB, et al. Kaposi's sarcoma in homosexual men-a report of eight cases. Lancet. 1981;2:598–600. doi: 10.1016/s0140-6736(81)92740-9. [DOI] [PubMed] [Google Scholar]
- 12.Beral V, Peterman TA, Berkelman RL, Jaffe HW. Kaposi's sarcoma among persons with AIDS: a sexually transmitted infection? Lancet. 2000;335(8682):123–128. doi: 10.1016/0140-6736(90)90001-l. [DOI] [PubMed] [Google Scholar]
- 13.Grossman Z, et al. Absence of Kaposi sarcoma among Ethiopian immigrants to Israel despite high seroprevalence of human herpesvirus 8. Mayo Clin Proc. 2002;77:905–909. doi: 10.4065/77.9.905. [DOI] [PubMed] [Google Scholar]
- 14.Casper C, Krantz E, Selke S, et al. Frequent and Asymptomatic Oropharyngeal Shedding of Human Herpesvirus 8 among Immunocompetent Men. J Infect Dis. 2007;195:30–36. doi: 10.1086/509621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Casper C, Redman M, Huang ML, et al. HIV Infection and Human Herpesvirus-8 Oral Shedding Among Men Who Have Sex with Men. J Acquir Immune Defic Syndr. 2004;35:233–238. doi: 10.1097/00126334-200403010-00003. [DOI] [PubMed] [Google Scholar]
- 16.Taylor MM, Chohan B, Lavreys L, et al. Shedding of human herpesvirus 8 in oral and genital secretions from HIV-1-seropositive and -seronegative Kenyan women. J Infect Dis. 2004;190:484–488. doi: 10.1086/421466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Plancoulaine S, Abel L, van Beveren M, et al. Human herpesvirus 8 transmission from mother to child and between siblings in an endemic population [In Process Citation] Lancet. 2000;356:1062–1065. doi: 10.1016/S0140-6736(00)02729-X. [DOI] [PubMed] [Google Scholar]
- 18.Casper C, Carrell D, Miller KG, et al. HIV serodiscordant sex partners and the prevalence of human herpesvirus 8 infection among HIV negative men who have sex with men: baseline data from the EXPLORE Study. Sex Transm Infect. 2006;82:229–235. doi: 10.1136/sti.2005.016568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Casper C, Wald A, Pauk J, et al. Correlates of prevalent and incident Kaposi's sarcoma-associated herpesvirus infection in men who have sex with men. J Infect Dis. 2002;185:990–993. doi: 10.1086/339605. [DOI] [PubMed] [Google Scholar]
- 20.Cannon MJ, Dollard SC, Smith DK, et al. Blood-borne and sexual transmission of human herpesvirus 8 in women with or at risk for human immunodeficiency virus infection. N Engl J Med. 2001;344:637–643. doi: 10.1056/NEJM200103013440904. [DOI] [PubMed] [Google Scholar]
- 21.Hladik W, Dollard SC, Mermin J, et al. Transmission of human herpesvirus 8 by blood transfusion. N Engl J Med. 2006;355:1331–1338. doi: 10.1056/NEJMoa055009. [DOI] [PubMed] [Google Scholar]
- 22.Luppi M, Barozzi P, Santagostino G, et al. Molecular evidence of organ-related transmission of kaposi sarcoma-associated herpesvirus or human herpesvirus-8 in transplant patients [In Process Citation] Blood. 2000;96:3279–3281. [PubMed] [Google Scholar]
- 23.Barozzi P, Luppi M, Facchetti F, et al. Post-transplant Kaposi sarcoma originates from the seeding of donor-derived progenitors. Nat Med. 2003;9:554–561. doi: 10.1038/nm862. [DOI] [PubMed] [Google Scholar]
- 24.2001 USPHS/IDSA guidelines for the prevention of opportunistic infections in persons infected with human immunodeficiency virus. HIV Clin Trials. 2001;2:493–554. doi: 10.1310/AQML-UABK-5LLB-E615. [DOI] [PubMed] [Google Scholar]
- 25.Ganem D. KSHV infection and the pathogenesis of Kaposi's sarcoma. Annu Rev Pathol. 2006;1:273–296. doi: 10.1146/annurev.pathol.1.110304.100133. [DOI] [PubMed] [Google Scholar]
- 26.Cotter MA, 2nd, Robertson ES. The latency-associated nuclear antigen tethers the Kaposi's sarcoma-associated herpesvirus genome to host chromosomes in body cavity-based lymphoma cells. Virology. 1999;264:254–264. doi: 10.1006/viro.1999.9999. [DOI] [PubMed] [Google Scholar]
- 27.Friborg J, Jr, Kong W, Hottinger MO, Nabel GJ. p53 inhibition by the LANA protein of KSHV protects against cell death. Nature. 1999;402:889–894. doi: 10.1038/47266. [DOI] [PubMed] [Google Scholar]
- 28.Radkov SA, Kellam P, Boshoff C. The latent nuclear antigen of Kaposi sarcoma-associated herpesvirus targets the retinoblastoma-E2F pathway and with the oncogene Hras transforms primary rat cells. Nat Med. 2000;6:1121–1127. doi: 10.1038/80459. [DOI] [PubMed] [Google Scholar]
- 29.Sullivan R, Dezube BJ, Koon HB. Signal transduction targets in Kaposi’s sarcoma. Curr Opin Oncol. 2006;18:546–462. doi: 10.1097/01.cco.0000239884.05914.13. [DOI] [PubMed] [Google Scholar]
- 30.Sodhi A, Montaner S, Gutkind JS. Does dysregulated expression of a deregulated viral GPCR trigger Kaposi's sarcomagenesis? FASEB J. 2004;18:422–427. doi: 10.1096/fj.03-1035hyp. [DOI] [PubMed] [Google Scholar]
- 31.Field N, Low W, Daniels M, et al. KSHV vFLIP binds to IKK-gamma to activate IKK. J Cell Sci. 2003;15(116):3721–3728. doi: 10.1242/jcs.00691. [DOI] [PubMed] [Google Scholar]
- 32.Sun Q, Zachariah S, Chaudhary PM. The human herpes virus 8-encoded viral FLICE-inhibitory protein induces cellular transformation via NF-kappaB activation. J Biol Chem. 2003;278(52):52437–52445. doi: 10.1074/jbc.M304199200. [DOI] [PubMed] [Google Scholar]
- 33.Stebbing J, Pantanowitz L, Dayyani F, et al. HIV-associated multicentric Castleman’s disease. Am J Hematol. 2008 Feb 7; doi: 10.1002/ajh.21137. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 34.Nishimoto N, Kanakura Y, Aozasa K, et al. Humanized anti-interleukin-6 receptor antibody treatment of multicentric Castleman disease. Blood. 2005;106(8):2627–2632. doi: 10.1182/blood-2004-12-4602. [DOI] [PubMed] [Google Scholar]
- 35.Nagy S, Gyulai R, Kemeny L, Szenohradszky P, Dobozy A. Iatrogenic Kaposi's sarcoma: HHV8 positivity persists but the tumors regress almost completely without immunosuppressive therapy. Transplantation. 2000;69:2230–2231. doi: 10.1097/00007890-200005270-00053. [DOI] [PubMed] [Google Scholar]
- 36.Paparizos VA, Kyriakis KP, Papastamopoulos V, Hadjivassiliou M, Stavrianeas NG. Response of AIDS-associated Kaposi sarcoma to highly active antiretroviral therapy alone. J Acquir Immune Defic Syndr. 2002;30:257–258. doi: 10.1097/00042560-200206010-00015. [DOI] [PubMed] [Google Scholar]
- 37.Strickler HD, Goedert JJ, Bethke FR, et al. Human herpesvirus 8 cellular immune responses in homosexual men. J Infect Dis. 1999;180:1682–1685. doi: 10.1086/315056. [DOI] [PubMed] [Google Scholar]
- 38.Guihot A, Dupin N, Marcelin AG, et al. Low T cell responses to human herpesvirus 8 in patients with AIDS-related and classic Kaposi sarcoma. J Infect Dis. 2006;194:1078–1088. doi: 10.1086/507648. [DOI] [PubMed] [Google Scholar]
- 39.Wilkinson J, Cope A, Gill J, et al. Identification of Kaposi's Sarcoma-Associated Herpesvirus (KSHV)-Specific Cytotoxic T-Lymphocyte Epitopes and Evaluation of Reconstitution of KSHV-Specific Responses in Human Immunodeficiency Virus Type 1-Infected Patients Receiving Highly Active Antiretroviral Therapy. J. Virol. 2002;76:2634–2640. doi: 10.1128/JVI.76.6.2634-2640.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Krown SE, Lee JY, Dittmer DP. AIDS Malignancy Consortium. More on HIV-associated Kaposi's sarcoma. N Engl J Med. 2008;358(5):535–536. doi: 10.1056/NEJMc072994. author reply 536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ensoli B, Gendelman R, Markham P, et al. Synergy between basic fibroblast growth factor and HIV-1 Tat protein in induction of Kaposi's sarcoma. Nature. 1994;371:674–680. doi: 10.1038/371674a0. [DOI] [PubMed] [Google Scholar]
- 42.Vogel J, Hinrichs SH, Reynolds RK, Luciw PA, Jay G. The HIV tat gene induces dermal lesions resembling Kaposi's sarcoma in transgenic mice. Nature. 1988;335:606–611. doi: 10.1038/335606a0. [DOI] [PubMed] [Google Scholar]
- 43.Aoki Y, Tosato G. HIV-1 Tat enhances Kaposi sarcoma-associated herpesvirus (KSHV) infectivity. Blood. 2004;104(3):810–814. doi: 10.1182/blood-2003-07-2533. [DOI] [PubMed] [Google Scholar]
- 44.Hyun TS, Subramanian C, Cotter MA, 2nd, et al. Latency-associated nuclear antigen encoded by Kaposi's sarcoma-associated herpesvirus interacts with Tat and activates the long terminal repeat of human immunodeficiency virus type 1 in human cells. J Virol. 2001;75(18):8761–8771. doi: 10.1128/JVI.75.18.8761-8771.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Caselli E, Galvan M, Cassai E, et al. Human herpesvirus 8 enhances human immunodeficiency virus replication in acutely infected cells and induces reactivation in latently infected cells. Blood. 2005;106(8):2790–2797. doi: 10.1182/blood-2005-04-1390. [DOI] [PubMed] [Google Scholar]
- 46.DiLorenzo G, Konstantinopoulos PA, Pantanowitz L, et al. Management of AIDS-related Kaposiߣs sarcoma. Lancet Oncol. 2007;8:167–176. doi: 10.1016/S1470-2045(07)70036-0. [DOI] [PubMed] [Google Scholar]
- 47.Casper C, Wald A. The use of antiviral drugs in the prevention and treatment of Kaposi sarcoma, multicentric Castleman disease and primary effusion lymphoma. Curr Top Microbiol Immunol. 2007;312:289–307. doi: 10.1007/978-3-540-34344-8_11. [DOI] [PubMed] [Google Scholar]
- 48.Little RF, Merced-Galindez F, Staskus K, et al. A pilot study of cidofovir in patients with kaposi sarcoma. J Infect Dis. 2003;187:149–153. doi: 10.1086/346159. [DOI] [PubMed] [Google Scholar]
- 49.Grundhoff A, Ganem D. Inefficient establishment of KSHV latency suggests an additional role for continued lytic replication in Kaposi sarcoma pathogenesis. J Clin Invest. 2004;113:124–136. doi: 10.1172/JCI200417803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Stallone G, et al. Sirolimus for Kaposi's sarcoma in renal-transplant recipients. N Engl J Med. 2005;352:1317–1323. doi: 10.1056/NEJMoa042831. [DOI] [PubMed] [Google Scholar]
- 51.Nador RG, Cesarman E, Chadburn A, et al. Primary effusion lymphoma: a distinct clinicopathologic entity associated with the Kaposi's sarcoma-associated herpes virus. Blood. 1996;88:645–656. [PubMed] [Google Scholar]
- 52.Chadburn A, Hyjek E, Mathew S, et al. KSHV-positive solid lymphomas represent an extra-cavitary variant of primary effusion lymphoma. Am J Surg Pathol. 2004;28:1401–1416. doi: 10.1097/01.pas.0000138177.10829.5c. [DOI] [PubMed] [Google Scholar]
- 53.Oksenhendler E, Clauvel JP, Jouveshomme S, et al. Complete remission of a primary effusion lymphoma with antiretroviral therapy. Am J Hematol. 1998;57:266. doi: 10.1002/(sici)1096-8652(199803)57:3<266::aid-ajh25>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 54.Simonelli C, Spina M, Cinelli R, et al. Clinical features and outcome of primary effusion lymphoma in HIV-infected patients: a single-institution study. J Clin Oncol. 2003;21:3948–3954. doi: 10.1200/JCO.2003.06.013. [DOI] [PubMed] [Google Scholar]
- 55.Keller SA, Schattner EJ, Cesarman E. Inhibition of NF-kappaB induces apoptosis of KSHV-infected primary effusion lymphoma cells. Blood. 2000;96(7):2537–2542. [PubMed] [Google Scholar]
- 56.An J, Sun Y, Fisher M. Rettig. Antitumor effects of bortezomib (PS-341) on primary effusion lymphomas. Leukemia. 2004;18:1699–1704. doi: 10.1038/sj.leu.2403460. [DOI] [PubMed] [Google Scholar]
- 57.Sin SH, Roy D, Wang L, et al. Rapamycin is efficacious against primary effusion lymphoma (PEL) cell lines in vivo by inhibiting autocrine signaling. Blood. 2007;109:2165–2173. doi: 10.1182/blood-2006-06-028092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Casper C. The aetiology and management of Castleman disease at 50 years: translating pathophysiology to patient care. Br J Haematol. 2005;129:3–17. doi: 10.1111/j.1365-2141.2004.05311.x. [DOI] [PubMed] [Google Scholar]
- 59.Aaron L, Lidove O, Yousry C, Roudiere L, Dupont B, Viard JP. Human herpesvirus 8-positive Castleman disease in human immunodeficiency virus-infected patients: the impact of highly active antiretroviral therapy. Clin Infect Dis. 2002;35:880–882. doi: 10.1086/342696. [DOI] [PubMed] [Google Scholar]
- 60.Gerard L, Berezne A, Galicier L, et al. Prospective study of rituximab in chemotherapy-dependent human immunodeficiency virus associated multicentric Castleman's disease: ANRS 117 CastlemaB Trial. J Clin Oncol. 2007;25:3350–3356. doi: 10.1200/JCO.2007.10.6732. [DOI] [PubMed] [Google Scholar]
- 61.Bower M, Powles T, Williams S, et al. Brief communication: rituximab in HIV-associated multicentric Castleman disease. Ann Intern Med. 2007;147:836–839. doi: 10.7326/0003-4819-147-12-200712180-00003. [DOI] [PubMed] [Google Scholar]
- 62.Powles T, Stebbing J, Montoto S, et al. Rituximab as retreatment for rituximab pretreated HIV-associated multicentric Castleman disease. Blood. 2007;110:4132–4133. doi: 10.1182/blood-2007-08-106187. [DOI] [PubMed] [Google Scholar]
- 63.Bower M, Nelson M, Young AM, et al. Immune reconstitution inflammatory syndrome associated with Kaposi's sarcoma. J Clin Oncol. 2005;23:5224–5228. doi: 10.1200/JCO.2005.14.597. [DOI] [PubMed] [Google Scholar]
- 64.Casper C, Krantz EM, Kuntz SR, et al. The Efficacy of Valganciclovir in the Suppression of Human Herpesvirus 8 (HHV-8) Replication: A Randomized, Double-Blind, Placebo-Controlled Trial. J Infect Dis. 2008 doi: 10.1086/588820. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Casper C, Nichols WG, Huang ML, Corey L, Wald A. Remission of HHV-8 and HIV-associated multicentric Castleman disease with ganciclovir treatment. Blood. 2004;103:1632–1634. doi: 10.1182/blood-2003-05-1721. [DOI] [PubMed] [Google Scholar]
- 66.Valencia ME, Moreno V, Martinez P, Casado I. Favorable outcome of Castleman's disease treated with oral valganciclovir. Med Clin (Barc) 2005;125(10):399. doi: 10.1157/13079178. [DOI] [PubMed] [Google Scholar]