

Abstract
Genetic instability and cellular proliferation have been associated with Aurora-kinase expression in several cancer entities, including multiple myeloma. Therefore, the expression of Aurora-A, -B and -C was determined by Affymetrix DNA-microarrays in 784 samples including two independent sets of 233 and 345 CD138-purified myeloma-cells from previously untreated myeloma-patients. Chromosomal aberrations were assessed by comprehensive iFISH and proliferation of primary myeloma-cells by propidium-iodine staining. The effect of the clinical Aurora-kinase inhibitor VX680 on proliferation of 20 human-myeloma-cell-lines and survival of 5 primary myeloma-cell-samples was tested. We found Aurora-A and -B to be expressed at varying frequencies in primary myeloma-cells of different patient-cohorts, Aurora-C in testis-samples only. Myeloma-cell samples with detectable vs. absent Aurora-A expression show a significantly higher proliferation rate, but neither a higher absolute number of chromosomal aberrations present (aneuploidy) nor of subclonal aberrations (chromosomal instability). VX680 induces apoptosis in all myeloma-cell-line- and primary myeloma-cell-samples tested. Presence of Aurora-A expression delineates significantly inferior event-free and overall-survival in two independent cohorts of patients undergoing high-dose chemotherapy, independent of conventional prognostic factors, i.e. serum-β2-microglobulin or ISS-stage. In conclusion, using gene expression profiling, Aurora-kinase inhibitors as promising therapeutic option for newly-diagnosed patients can be tailoredly given to patients with adverse prognosis, expressing Aurora-A.
Introduction
Multiple myeloma is an incurable malignant disease of clonal plasma cells which accumulate in the bone marrow causing clinical signs and symptoms related to the displacement of normal hematopoiesis, formation of osteolytic bone lesions, and production of monoclonal protein 1. Multiple myeloma cells (MMC) at the time of diagnosis are characterized by a low proliferation rate that increases in relapse 2. Presence of proliferation correlates with adverse prognosis 3;4. At the same time, myeloma cells harbor a high median number of chromosomal aberrations 5;6, often associated with genetic instability 6. Proliferation 7 and chromosomal instability 8 in turn are associated with the expression of Aurora-kinases. In several cancer entities, Aurora-kinases have been implicated in tumor formation and progression 9–14.
Aurora-kinases represent a family of conserved mitotic regulators comprising three closely related members, i.e. Aurora-A, -B and -C 9. Aurora-C expression is restricted to germ cells, where it regulates spermatogenesis 15. Aurora-A associates with the centrosome and spindle microtubules being required for centrosome separation and spindle assembly 7. Aurora-B is a member of the chromosomal passenger complex, as such sequentially recruited to centromeres, spindle midzone and midbody as mitosis progresses 16, and required for chromosome biorientation, the spindle assembly checkpoint, and cytokinesis 7. Inhibition of Aurora-A and -B exhibits distinct phenotypic features: loss of Aurora-A activity induces a centrosome separation defect and a monopolar spindle phenotype 17;18; inhibition of Aurora-B generates polyploidy through defects in cytokinesis, which ultimately leads to a loss in cell viability 19–22. Aurora-A and -B expression has been detected by quantitative real-time PCR (qRT-PCR) in myeloma cell lines 23;24 and small series of myeloma patients 23;24. Aurora-kinase inhibitors like VX680 have been shown to abrogate proliferation and induce apoptosis in human myeloma cells lines and primary myeloma cells 23–25.
We assess here the expression of Aurora-A, -B, and -C in 784 Affymetrix gene expression profiles of malignant plasma cells from previously untreated myeloma patients compared to normal bone marrow plasma cells (BMPC), their non-malignant proliferating precursors (polyclonal plasmablastic cells, PPC), and human myeloma cell lines (HMCL). We find that in our data set 24 % of previously untreated myeloma patients express Aurora-A. We show myeloma cells expressing Aurora-A kinase to have a higher proliferation-rate, whereas the number of chromosomal aberrations (aneuploidy) is not higher compared to myeloma cells with absent Aurora-A expression. The same holds true for subclonal aberrations (i.e. genetic instability), which are less frequent in myeloma cell-samples expressing Aurora-A. Aurora-A kinase expression in turn is significantly associated with an inferior event-free (EFS) and overall survival (OAS) in two independent cohorts of a total of 513 myeloma-patients treated with high-dose chemotherapy (HDT) and autologous stem cell transplantation (ASCT).
Aurora-kinase inhibitors (including VX680 tested here) are very active on human myeloma cell lines and primary myeloma cells and represent a promising weapon in the therapeutic arsenal against multiple myeloma. Gene expression profiling allows an assessment of Aurora-kinase expression and thus in turn a tailoring of treatment to patients expressing these kinases.
Materials and Methods
Patients and healthy donors
Patients presenting with previously untreated MM (n=233) or monoclonal gammopathy of unknown significance (MGUS, n=12) at the University Hospitals of Heidelberg and Montpellier and 14 healthy normal donors were included in the study approved by the institutional review board of the Medical Faculty of the Ruprecht-Karls-University Heidelberg, Germany, the institutional review board of the University of Arkansas for Medical sciences, AR, USA, and the institutional review board of the CHU Montpellier, France, for the respective patients. Written informed consent was obtained in accordance with the Declaration of Helsinki. The first 65 patients comprise the training group (TG), the 168 additional the independent validation group (VG). Patients were diagnosed, staged and response to treatment was assessed according to standard criteria 26–28. 168 patients underwent frontline HDT with 200 mg/m2 melphalan and ASCT according or in analogy to the GMMG-HD3-trial 29. Survival data were validated by an independent cohort of 345 patients treated within the total therapy 2 protocol 30. For clinical parameters, see Supplementary Table S1.
Samples
For an overview, see Supplementary Table S2. Plasma cells from the bone marrow were purified using CD138-microbeads (Miltenyi Biotech, Bergisch Gladbach, Germany) and purity was assessed by flow cytometry (FACSCalibur, Becton Dickinson, Heidelberg, Germany). An aliquot of unpurified (whole) bone marrow (WBM) of patients (n=57) and healthy donors (n=7) was obtained after NH4-lysis as published 31. An alternate aliquot was subjected to FACS-sorting (FACSAria, Becton Dickinson) in CD3+-, CD14+-, CD15+-, and CD34+-cells. Peripheral CD27+-memory B-cells (MBC) were generated as published 32. Testis-samples (n=5) were obtained from healthy donors. The HMCL XG-1, XG-2, XG-3, XG-4, XG-5, XG-6, XG-7, XG-10, XG-11, XG-12, XG-13, XG-14, XG-16, XG-19, XG-20 were generated at INSERM U847 as published 33–35. HG-1 was generated in the Multiple Myeloma Research Laboratory Heidelberg. U266, RPMI-8226, LP-1, OPM-2, SKMM-2, AMO, JJN-3, KMS-12-BM, L363, NCI, MOLP-8 (Deutsche Sammlung von Mikroorganismen und Zellkulturen GmbH, Braunschweig, Germany) were cultured as recommended. PPC (n=12) 36, osteoclasts 32 (OC; n=5) and mesenchymal stromal cells (MSC, n=19) 37 were generated and analyzed as previously published.
Chemicals
The 4,6 diaminopyrimidine VX680 (Cyclopropanecarboxylicarid{4[4(4-methyl-piperazin-1-yl)-6- (5-methyl-2H-pyrazol-3-ylamino)-pyrimidin-2-ylsulfanyl]-phenyl}-amide; ACC corporation, San Diego, CA, USA) was dissolved in dimethyl sulfoxide (DMSO; Merck, Darmstadt, Germany) and stored at a final concentration of 10 mM at −80 °C.
Interphase-FISH (iFISH)
Interphase-FISH-analysis was performed on CD138-purified plasma cells as described 5 using a set of probes for the chromosomal regions 1q21, 6q21, 8p21, 9q34, 11q23, 11q13, 13q14.3, 14q32, 15q22, 17p13, 19q13, 22q11, as well as the translocations t(4;14)(p16.3;q32.3) and t(11;14)(q13;q32,3), (Poseidon-probes, Madison, WI, USA). Ploidy-status, clonal/subclonal aberrations for a single aberration (i.e. present in ≥ 60 % vs. 20–59 % of assessed MMC) were defined as published 5. A modified copy number score (CS) 5;38 (excluding gains of 1q21) and the score of Wuilleme et al. 39 using chromosomes 5, 15, 19 (CSW) was used to assess ploidy. For the assessment of the absolute number of chromosomal aberrations, 105 patients were assessed for the translocations t(4;14) and t(11;14) as well as numerical aberrations of the chromosomal regions 11q13, 11q23, 1q21, 17p13, 13q14.3, 14q32. For the assessment of the presence of subclonal aberrations, a MMC-sample was considered to contain a subclonal aberration, if at least one aberration was detected in ≥ 70 % of the myeloma cells present, and at least one other aberration was detected in 20–59 % of assessed MMC.
Gene expression profiling (GEP)
RNA extraction was performed using the RNeasy kit (Qiagen, Hilden, Germany), the SV-total RNA extraction kit (Promega, Mannheim, Germany) and Trizol (Invitrogen, Karlsruhe, Germany). Labeled cRNA was generated using the small sample labeling protocol vII (Affymetrix, Santa Clara, CA, USA), and hybridized to U133 A+B GeneChip microarray (Affymetrix) for training, and U133 2.0 plus arrays for validation group, according to the manufacturer’s instructions. When different probe sets were available for the same gene, we chose the probe set yielding the maximal variance and the highest signal. Aurora-A (Hs00233470_m1), Aurora-B (Hs00177782_m1) and Aurora-C (Hs00152930_m1, all Applied Biosystems, Darmstadt, Germany) gene expression was analyzed by qRT-PCR using the ABI Prism 7700 Sequence Detection System 40. The expression data are deposited in ArrayExpress under the accession numbers E-MTAB-81 and E-GEOD-2658.
Measurement of proliferation by 3H-thymidine
Proliferation of 20 HMCL was investigated according to published protocols 41–43. Per well, 10.000 cells were cultured in 96-well-plates in RPMI-1640 (Biochrom, Berlin, Germany) containing 10 % FCS (Invitrogen) with or without (medium control) graded concentrations (10, 2, 0.4, 0.08, 0.016, 0.0032 μM) of VX680. DMSO at the highest concentration present in the 10 μM well served as DMSO-control. For the HG- and XG-lines, 2 ng/ml IL-6 (R&D Systems, Wiesbaden, Germany) was added. Proliferation was evaluated after 5 days of culture: cells were pulsed with 37 kBq of 3H-thymidine for 18 h, harvested and 3H-thymidine uptake measured.
Measurement of proliferation of primary myeloma cells by propidium iodine
The Plasma Cell Labeling Index, i.e. the percentage of MMC in S-phase, was determined by flow cytometry using a FACSCalibur. WBM (106 cells per tube) was incubated with 20 μl of either control IgG-FITC, CD38-FITC (both Beckman Coulter, Krefeld, Germany, clone A07795 and A07778) and CD138-FITC (Diaclone, Stamford, CT, U; 954.501.010), respectively. After NH4-lysis, cells were resuspended with propidium iodine (PI-) solution (1mg/ml PI in 1x citrate buffer containing 0.1 % Tween 1mg/ml RNase A (Sigma-Aldrich, Schnelldorf, Germany)) for 45 min at 4 °C. The percentage of CD138+ S-phase cells was determined using ModFit software (Verity Software House, Topsham, ME, USA) using a rectangular mathematical model for calculating the S-phase fraction in % of the selected CD138+ plasma cells.
Survival of primary myeloma cells
Primary MMC cultured together with their bone marrow microenvironment (negative fraction of plasma cell purification) of 5 newly-diagnosed patients were exposed to concentrations of 100, 20, 4, 0.8, 0.16, 0.032 μM VX680. Cell viability was measured by CD138-FITC (IQ products, Groningen, Netherlands, clone B-A38) β/PI (Pharmingen, Heidelberg, Germany) staining after 6 days of culture and referred to the medium and DMSO-control, respectively 44. One μl of PI with a concentration of 50 μg/ml was used.
Apoptosis induction
XG-1 and XG-10 were cultured in 24-well-plates at 105 cells per well in RPMI-1640 containing 10 % FCS and 2 ng/ml IL-6 with or without 1 μM VX680. After 8, 24, 48 and 72 h of culture, cells were stained for annexin V-FITC and PI according to the manufacturer’s instructions (Pharmingen) and analyzed on a FACSAria.
Intracellular staining for Aurora-A and -B
Intracellular Aurora-A (clone 35C1; Abcam, Cambridge, United Kingdom) and -B (Cell signaling technology, Danvers, MA, USA) expression of 10 HMCL was measured by flow cytometry using a fixation and permeabilization kit (eBioscience, San Diego, CA, USA). Overlays were established using the Infinicyt 1.1 Software (Cytognos, Salamanca, Spain).
Western blotting
Cells were pelleted and resuspended in lysis buffer containing 10mM Tris-HCl (pH 7.05), 50 mM sodium chloride, 30 mM sodium pyrophosphate decahydrate, 50 mM sodium fluoride, 5 μM zinc chloride, 1 % Triton-X 100, and a protease/phosphatase inhibitor cocktail (Complete mini tablets; Roche, Basel, Switzerland). After pelleting, supernatants were mixed with loading buffer (Roti, Carl Roth, Karlsruhe, Germany), heated for 5 min at 95 °C and separated on 10 % NuPAGE Bis-tris gels (Invitrogen). Immunodetection was performed using the WesternBreeze Kit (Invitrogen). Membranes were incubated with antibodies against Aurora-A, -B (see above) and β-actin (Ab5, Becton Dickinson) as loading control. HELA cells served as positive control.
Statistical analysis
Gene expression data were gcrma-normalized 45. To assess presence or absence of gene expression independently of Affymetrix-mismatch-probesets, the “Presence-Absence calls with Negative Probesets (PANP)” algorithm 46 was used. Differential gene expression was assessed using empirical Bayes statistics in linear models for microarray data 47. P-values were adjusted for multiple testing controlling the false-discovery-rate as defined by Benjamini and Hochberg at a level of 5 % 48. Expression profiles of 439 samples (233 MM, 14 BMPC, 12 PPC, 12 MGUS, 40 HMCL, 13 MBC, 64 WBM, 19 MSC, 5 CD3, 5 CD14, 5 CD15, 5 CD34, 7 OC, and 5 testis) divided in TG (n=113, MM n=65) and VG (n=257, MM n=168) were analyzed. As a further validation, 345 samples of newly-diagnosed myeloma patients from the Arkansas-group were analyzed.
Event-free survival 29 and overall survival 29 were investigated for the 168 patients (48 TG, 120 VG) undergoing HDT and ASCT using Cox’s proportional hazard model. Two groups of patients with presence and absence of Aurora-A expression were delineated. Findings were validated using the same strategy on the independent group of 345 patients from the Arkansas-group. For myeloma cells, association of chromosomal aberrations and clinical parameters with gene expression was calculated using two-sample t-statistic. Differences in clinical parameters between defined groups were investigated by analysis-of-variance (ANOVA). Correlation was measured using the Spearman correlation coefficient. Correlation with categorical variables was measured using the Kendall’s tau coefficient. For assessing the relationship between categorical variables, Fisher’s Exact Test was used. The centrosome-index was calculated as published by Chng et al. 49. For the calculation on the Arkansas-group, our 7 BMPC samples were normalized together with the 345 MMC-samples.
The gene expression based proliferation index is calculated as described in Supplementary Text S1. In all statistical tests, an effect was considered as statistically significant if the P-value of its corresponding statistical test was not greater than 5 %. All statistical computations were performed using R 50 version 2.7.0 and Bioconductor 51, version 2.2.
Results
Expression of Aurora-A, -B and -C
First, we assessed expression (Table 1,2) and differential expression (Supplementary Table S3A,B) of Aurora-A, -B, and -C in primary myeloma cells, normal bone marrow plasma cells, their precursors, as well as normal and myelomatous bone marrow. In our data set, Aurora-A and -B are expressed in 24 % and 3 % of primary myeloma cells and all PPC as well as HMCL. In the Arkansas-data, Aurora-A, -B, and -C are expressed in 48/345 (14 %), 12/345, (3 %) and 0/345 myeloma cell-samples, respectively. The mean expression of Aurora-A and -B is significantly and by several orders of magnitude higher in proliferating plasmablastic cells and cell lines compared to non-proliferating MBC, or BMPC (each P<.001 in TG and VG; Supplementary Table S3A,B, Figure 1A). Aurora-A and -B are expressed in almost all bone marrow samples of healthy individuals and myeloma patients (Table 1). Here, the mean expression of Aurora-B is significantly different (lower) in myelomatous compared to normal bone marrow (4.85 vs. 3.81 arbitrary units, P=.009). A significant stage dependent differential gene expression could be found for Aurora-A between myeloma cells from early- (MGUS and MMI) and advanced stage (MMII and MMIII) patients (P=.01 and .01, in TG and VG, respectively). Aurora-A and -B expression correlates significantly in the VG (rs=0.59, P<.001) and Arkansas-group (rs=0.61, P<.001).
Table 1. Presence of expression of Aurora-kinase A (AURKA), -B (AURKB), -C (AURKC) as judged by PANP.
in normal bone marrow plasma cells (BMPC), proliferating polyclonal plasmablastic cells (PPC), memory B-cells (MBC), multiple myeloma cells (MMC), cells from patients suffering from monoclonal gammopathy of unknown significance (MGUS), human myeloma cell lines (HMCL) and the bone marrow of normal donors (ND-WBM) as well as myeloma patients (MM-WBM).
| Gene Symbol | Probeset | MBC present [%] (n=13) | PPC present [%] (n=12) | BMPC present [%] (n=14) | MGUS present [%] (n=12) | MM present [%] (n=233) | HMCL present [%] (n=40) | ND-WBM present [%] (n=7) | MM-WBM present [%] (n=57) |
|---|---|---|---|---|---|---|---|---|---|
| AURKA | 208079_s_at | 0,0 | 100,0 | 0,0 | 0,0 | 24,0 | 100,0 | 100,0 | 100,0 |
| AURKB | 209464_at | 0,0 | 100,0 | 0,0 | 0,0 | 3,0 | 100,0 | 100,0 | 84,2 |
| AURKC | 211107_s_at | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
Table 2. Presence of expression of Aurora-kinase A (AURKA), -B (AURKB), -C (AURKC) as judged by PANP.
in subfractions of the bone marrow (mesenchymal stromal cells (MSC) from normal donors (ND-MSC), patients with monoclonal gammopathy of unknown significance (MSC-MGUS) as well as myeloma patients (MSC-MM); osteoclast (OC)), and testis-samples, respectively.
| Gene Symbol | Probeset | CD3 present [%](n=5) | CD14 present [%](n=5) | CD15 present [%](n=5) | CD34 present [%](n=5) | MSC-ND present [%](n=7) | MSC-MGUS present [%](n=5) | MSC-MM present [%](n=7) | OC present [%](n=7) | Testis [%](n=5) |
|---|---|---|---|---|---|---|---|---|---|---|
| AURKA | 208079_s_at | 0,0 | 60,0 | 100,0 | 100,0 | 100,0 | 100,0 | 100,0 | 100,0 | 100,0 |
| AURKB | 209464_at | 0,0 | 0,0 | 0,0 | 20,0 | 28,6 | 20,0 | 0,0 | 0,0 | 0,0 |
| AURKC | 211107_s_at | 0,0 | 0,0 | 0,0 | 0,0 | 0,0 | 0,0 | 0,0 | 0,0 | 100,0 |
Figure 1. Expression of Aurora-A (AURKA), -B (AURKB), and -C (AURKC) as determined by gene expression profiling.
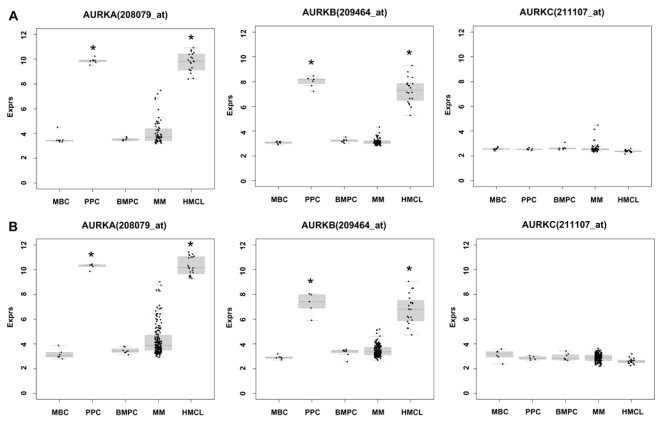
in memory B-cells (MBC), polyclonal plasmablastic cells (PPC), normal plasma cells (BMPC), myeloma cells (MMC), and myeloma cell lines (HMCL) within the (A) training and (B) validation group. An asterisk (*) indicates a significant difference of the indexed population compared to both, BMPC and MBC at a level of P<.001.
Validation of gene expression by qRT-PCR, western blotting and flow cytometry
To validate Aurora-kinase expression detected by gene expression profiling, we performed qRT-PCR, western blotting and flow-cytometric staining. Aurora-A expression in terms of “presence” or “absence” (Ct-value ≥ 35) by qRT-PCR is consistent with results by PANP in 10/11 primary myeloma cell-samples. One sample “absent” by qRT-PCR is judged “marginal” by PANP. Aurora-A expression by GEP strongly correlates with dCt value by qRT-PCR (rs= −0.87, P<.001). Aurora-B expression is consistent with results by PANP in 3/6 samples. All samples are “present” by qRT-PCR but three are judged “absent” by PANP. Aurora-B expression by GEP strongly correlates with dCt value by qRT-PCR (rs= −0.83, P=.06). Aurora-C expression by qRT-PCR is consistent with absence of expression detected by PANP in 5/6 samples. One sample “present” by qRT-PCR (Ct-value 34.8) is judged “absent” by PANP. Aurora-C expression by GEP strongly correlates with the dCt value obtained by qRT-PCR (rs= −0.94, P=.02). Aurora-A and -B expression in HMCL was further validated by western blotting (Figure 2B) and intracellular flow cytometry (exemplary data shown in Figure 2C).
Figure 2. Validation of gene expression by quantitative real-time PCR, western blotting and flow cytometry.
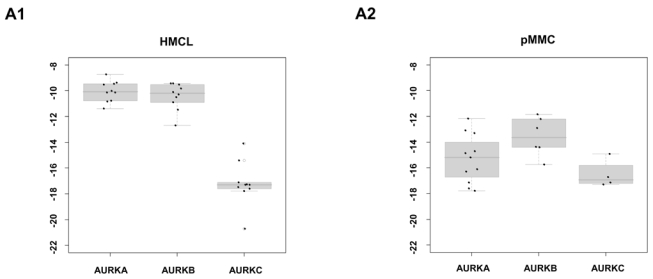
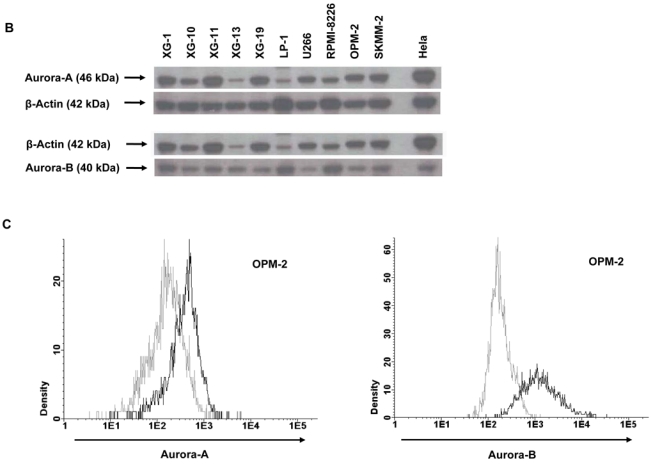
To validate gene expression data, quantitative real-time PCR for Aurora-A, -B, -C in (A1) 10 myeloma cell lines (HMCL) and (A2) 10 primary myeloma cells (pMMC) was performed. Shown are -dCt-values (reference gene 18S-RNA). (B) Gene expression data were further validated by western blotting. Shown are the blots of 10 cell lines for Aurora-A and -B with β-actin as loading control and HELA cells as positive control, respectively. (C) Intracytoplasmatic expression of Aurora-A and -B as determined by flow cytometry. Shown is the cell line OPM-2. Light grey line: control without primary antibody, black line: measurement with primary and secondary antibody.
Association of Aurora-kinase expression with proliferation and chromosomal aberrations
To investigate the biological impact of Aurora-kinase expression, we assessed the association with proliferation, chromosomal aberrations and presence of subclonal aberrations as detected by iFISH, and a published centrosome-index 49.
The expression of Aurora-A correlates with proliferation in terms of the plasma cell labeling index assessed by PI staining (n=66, rs=0.45, P<.001) and the gene expression based proliferation index in TG (rs=0.62, P<.001) and VG (rs=0.87, P<.001). The same holds true for the latter for Aurora-B in the VG (rs=0.64, P<.001).
The absolute number of chromosomal aberrations is not significantly different between myeloma cells expressing or not expressing Aurora-A (mean 5.1 and 5.1, n=105 patients, n=8 FISH-probes tested). Presence/absence of Aurora-A expression does not significantly interrelate to the presence/absence of hyperdiploidy as determined by either CS or CSW, neither does the presence/absence of Aurora-A expression interrelate to the presence of any of the single aberrations t(11;14), t(4;14), or numerical aberrations of 17p13, 9q34, 15q22, 19q13, 4p16, 14q32 or 22q11. Interestingly, in patients with presence of Aurora-A expression, gains of 11q13 (P=.03) and 11q23 (P=.004) are significantly less frequent compared to those with absent Aurora-A expression. The opposite holds true for patients with gain of 1q21 (P=.002) or deletion of 13q14 (P=.03) as well as deletion of 8p21 (P=.03): MMC of patients with present Aurora-A expression show a significantly higher number of these respective aberrations. For a gain 1q21, for which data were also available for the Arkansas-group, the same observation was made (n=244, P<.001). However, subclonal aberrations per se are significantly more frequent in MMC with absent Aurora-A expression (67 with vs. 37 without) compared to present Aurora-A expression (18 vs. 23, P =.03). For single aberrations, subclonal presence (vs. full-clonal or absence of the respective aberration) is significantly more frequent in MMC of patients with absence of Aurora-A expression for gains of 11q13 (P<.03), 11q23 (P<.001), and losses of 13q14 (P=.009). Losses of 17p13 marginally fail significance (P=.06). Losses of 8p21 are significantly (P=.04) more frequent in patients with presence of Aurora-A expression.
The centrosome-index correlates with Aurora A-expression in our series (TG r=.53, P<.001; VG r=.32, P<.001) and the Arkansas-data (r=.43, P<.001). The centrosome-index is significantly predictive for EFS and OAS in the Arkansas-group (EFS P=.005, OAS, P<.001) on which it has been derived, but not our series (EFS P=.09, OAS, P=.99; Supplementary Figure S1).
Prognostic value of Aurora-kinase-expression
Next, we investigated whether the presence of Aurora-kinase expression has a prognostic impact in newly-diagnosed myeloma patients treated with HDT and ASCT.
Presence of Aurora-A expression in MMC is an adverse prognostic factor in terms of EFS and OAS in our data (n=168, EFS P=.003, hazard ratio (HR) 2.02, confidence interval (CI) [1.26,3.25], OAS, P=.03, HR 2.31, CI [1.04,5.15]) and the Arkansas-group (n=345, P=.004, HR 1.74, CI [1.18,2.57], OAS, P=.008, HR 1.85, CI [1.16,2.95]), Figure 3. The expression-signal of Aurora-A as single continuous variable is significantly predictive for EFS in the VG (n=120, P<.001) and the Arkansas-group (P<.001). The same holds true for OAS in the Arkansas-group (P<.001) and it marginally failed significance for our VG (n=120, P=.06).
Figure 3. Prognostic relevance of Aurora-A expression for two independent groups of patients treated with high-dose therapy and autologous stem cell transplantation.
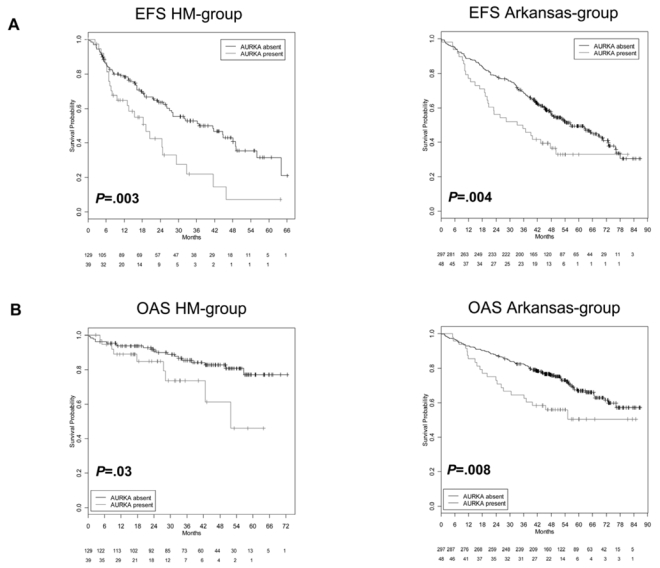
Shown are (A) event-free (EFS) and (B) overall survival (OAS) in our cohort of patients (left side) and the Arkansas-group (right side) for absence (black curve) vs. presence (grey curve) of Aurora-A expression in CD138-purified myeloma cells. Presence of Aurora-A expression in myeloma cells is an adverse prognostic factor in terms of EFS and OAS in both groups.
In a Cox-model tested with the international staging system (ISS), presence of Aurora-A expression appears as independent prognostic factor for EFS in our data (Aurora A-expression (P=.003), ISS (P=.009)), and the Arkansas-data (Aurora-A expression (P<.001), ISS (P<.001)). For OAS, Aurora-A expression marginally fails independence (P=.08), ISS (P=.009)) in our data-set, but is significantly independent in the Arkansas-data (Aurora-A expression (P<.001), ISS (P<.001)).
In a Cox-model tested with serum-β2-microglobulin (B2M) as continuous variable, presence of Aurora-A expression appears as independent prognostic factor for EFS in our data (Aurora-A expression (P=.002), B2M (P=.006)), and the Arkansas-data (Aurora-A expression (P<.001), B2M (P<.001)), and OAS (Aurora-A expression (P=.06), B2M (P<.001)), and the Arkansas-data (Aurora-A expression (P<.001), B2M (P<.001)).
Activity of VX680 on myeloma cell lines and primary myeloma cells
Given the expression and prognostic value of Aurora-kinases, we tested the activity of the pan- Aurora-kinase inhibitor VX680 that has previously shown activity on a small series of human myeloma cells, on a large series of 20 myeloma cell lines. VX680 significantly inhibits proliferation of all HMCL investigated (Figure 4A1). The maximum inhibition at 10 μM ranges from 64.4 % (HG-1) to 100 % (OPM-2). The concentration to reduce proliferation to half of the control value (IC50) is reached in all myeloma cell lines; it ranges from 0.003–2.715 μM, Figure 4A2. No significant correlation could be found between the expression of Aurora-A, -B or hyaluron mediated motility receptor (HMMR) and the IC50 of 12 myeloma cell lines tested (see discussion).
Figure 4. Inhibition of proliferation of myeloma cell lines as well as survival of primary myeloma cells and cells of the bone marrow microenvironment.
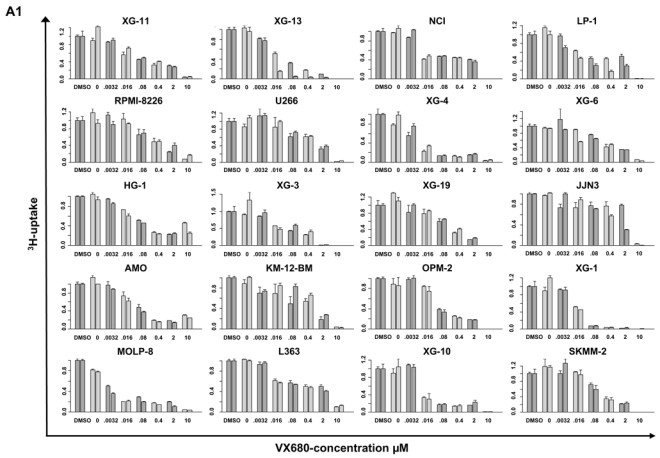
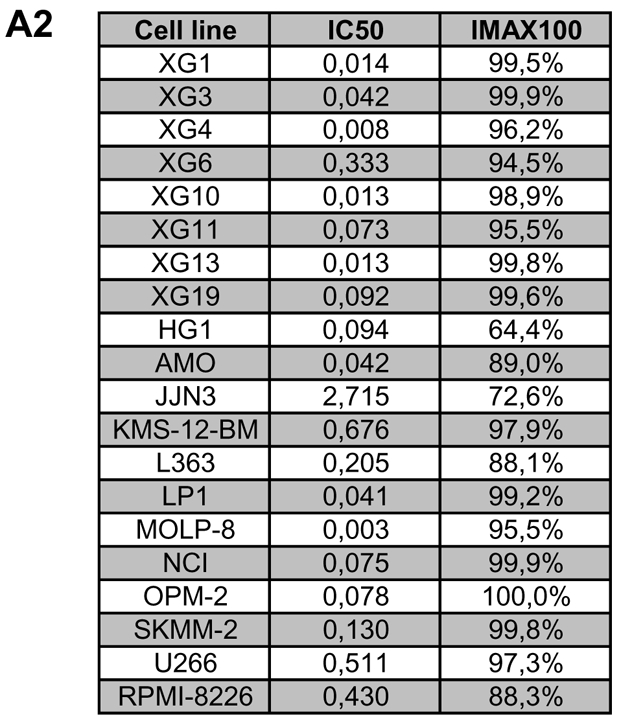
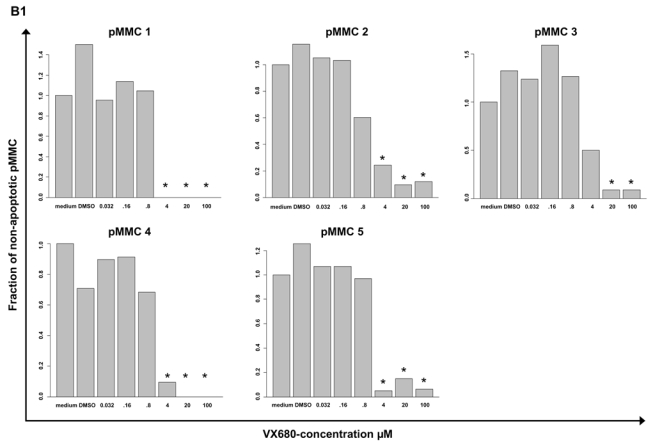
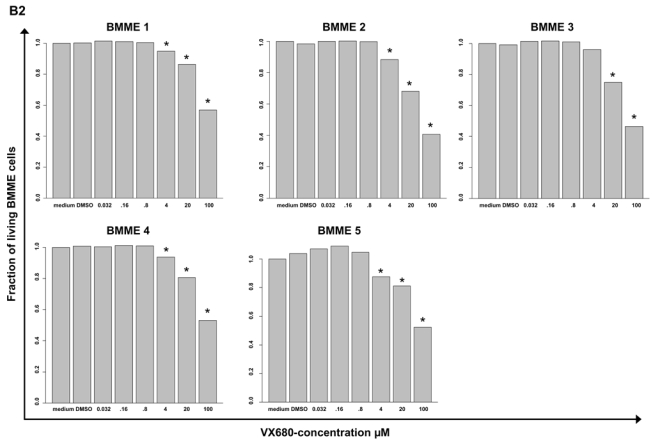
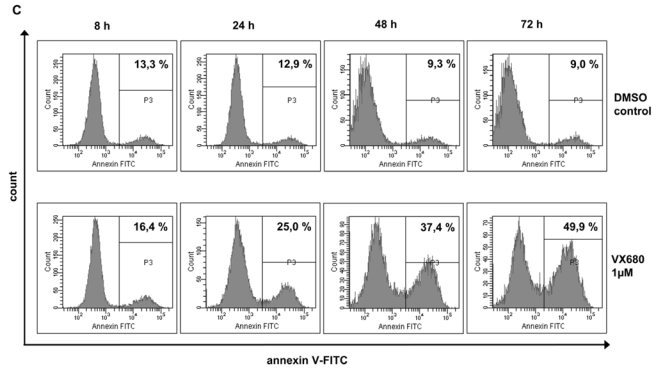
(A1) Inhibition of proliferation of 20 myeloma cell lines by the pan-Aurora-kinase inhibitor VX680 in graded concentrations vs. medium- and DMSO-control, respectively, measured by 3H-thymidine uptake. Two independent experiments were performed in triplicates. (A2) The IC50 (in μM) and maximal inhibition at 10 μM (IMAX10 in %) are shown. (B1) Survival of primary myeloma cells (pMMC) cultured within their bone marrow microenvironment (negative fraction of plasma cell purification) is significantly inhibited compared to the medium control as determined by staining with anti-CD138-FITC antibody and propidium iodine. An asterisk (*) indicates a significant decrease between the medium control and the respective VX680 concentration. (B2) Survival of cells within the bone marrow microenvironment (BMME cells; negative fraction of plasma cell purification) was determined as described above for pMMC. An asterisk (*) indicates a significant decrease between the medium control and the respective VX680 concentration. (C) Induction of apoptosis by VX680 at 1 μM as determined by annexin V-staining after 8, 24, 48 and 72 h.
VX680 significantly inhibits the survival of primary myeloma cells cultivated within their bone marrow microenvironment from 5/5 newly-diagnosed myeloma patients at a concentration of 4 μM (50 - 100 % inhibition, Figure 4B1). At the same dose level, VX680 induces significant but lower toxicity within the bone marrow microenvironment (Figure 4B2). Four of four samples, for which sufficient RNA was available, showed an expression of Aurora-A by qRT-PCR.
Next, XG-1 and XG-10 were cultured for 3 days with or without VX680. Cell viability and apoptosis were determined by flow-cytometric analysis of annexin V-binding and PI uptake after 8, 24, 48 and 72 h. Exposure of XG-1 and XG-10 against 1 μM VX680 induces apoptosis after 8 h (XG-1: 23.5 % vs. 24.1 %, XG-10: 16.4 % vs. 13.3 %) to 72 h (XG-1: 17.9 % vs. 85.5 %, XG-10: 9.0 % vs. 49.9 %), Figure 4C, exemplary data shown for XG-10.
Discussion
Expression of Aurora-kinases
In our set of previously untreated myeloma patients (n=233), expression of Aurora-A and -B could be detected in 24 % and 3 % of purified myeloma cell-samples. The same percentage of Aurora A and -B expression could be found for the subgroup of patients treated with HDT and ASCT (n=168, 23.2 % and 3.5 %, respectively). In the independent data set of Shaughnessy et al. (n=345), the same percentage of patients (3 %) expresses Aurora-B, but Aurora-A expression could only be detected in 14 % of patients. This observation could not be explained by the use of U133 A+B chips for part of our patients, as in these, the percentage of patients expressing Aurora-A is even lower (10.4 % of patients in the HDT-cohort). Likewise, the use of a double amplification (our data) instead of single amplification protocol (Arkansas-group) could not be taken as explanation, as one would rather expect a higher percentage of detection in the single-amplification group. In an additional set of patients treated within the GMMG-HD4 trial, an Aurora-A expression can be detected in 43/70 (61 %) of cases. Taken together, the percentage of patients expressing Aurora-A seems to be quite variable in different patient populations, indicating to assess Aurora-A expression when testing Aurora-inhibitors within clinical trials.
“Presence” of gene expression as determined by gene expression profiling based on PANP needs to be interpreted as presence above the background level (“threshold”) seen for unspecific hybridization for negative strand matching probesets. As such a background correction is not performed when analyzing qRT-PCR data, the latter might have a detection threshold within the background of gene expression. This is one possible explanation why in a previously published small patient series, Evans et al. found by qRT-PCR all CD138-purified myeloma samples to express Aurora-A (5/5) 23 and Aurora-B (7/7) 24. Alternate explanations are a more advanced (relapsed) patient population, or a contamination by other cell types, as in these series, purity of CD138-sorted plasma cells was only assessed by morphology, and expression of Aurora-A and -B could be detected in almost all our bone marrow samples (Table 1,2).
The lower frequency of Aurora-B compared to Aurora-A expression in the same sample as detected by GEP seems likewise to be related to the detection-threshold: (i) In normal plasma cells, the expression levels of Aurora-A and -B are of the same height (Supplementary Table S3A–B), but the differential expression of Aurora-A in proliferating plasmablastic cells and myeloma cell lines is higher compared to Aurora-B (Supplementary Table S3A,B). (ii) Despite of a tight correlation between qRT-PCR and gene expression profiling, qRT-PCR shows a comparable expression level for myeloma cell lines in terms of Aurora-A and -B expression (mean Ct 27.6 vs. 28.2). (iii) All samples expressing Aurora-B by PANP express likewise Aurora-A.
Biological implications
Aurora-kinases have been associated with proliferation 7 and genetic instability 8 in different cancer entities 9–14, including multiple myeloma 25.
Aurora-A and -B are expressed in all myeloma cell lines and proliferating 36 (non-malignant) plasmablastic cells, and are significantly higher expressed in both of these compared to memory B-cells or normal plasma cells. At the same time, expression of Aurora-A and -B correlates with the plasma cell labeling index determined by PI-staining as well as the gene expression based proliferation index. Furthermore, the percentage of primary myeloma cells of untreated patients expressing Aurora-A is in agreement with the low proliferative rate of these. The same holds true for the significant increase in Aurora-A expression from early- to late-stage plasma cell dyscrasias in both our training and validation group. Thus, Aurora-kinase expression is obviously associated with proliferation in multiple myeloma.
As chromosomal aberrations can be detected by iFISH in almost all primary myeloma cells 5;6, e.g. in all our patients tested, but only myeloma cells from a minor fraction of myeloma patients express Aurora-A or -B, Aurora-kinase expression in CD138-positive primary myeloma cells cannot be the cause of aneuploidy or ongoing genetic instability in myeloma. Two further strong arguments are given by the fact that Aurora-A or -B expression height (or presence of expression) neither correlates with the median number of chromosomal aberrations in an individual sample, nor the presence of subclonal (i.e. emerging) aberrations, but to the contrary, presence of subclonal aberrations at all is significantly associated with the absence of Aurora-expression. The same holds true if the presence of specific subclonal aberrations is considered (11q13, 11q23, 13q14) vs. clonal gain or normal copy number state with the exception of deletions of 8p21. It is interesting to denote that aberrations of 1q21 (gains), 13q14.3 (deletions) and 8p21 (deletions), the first two associated with advanced stages 52;53, are significantly more frequent in myeloma cells expressing Aurora-A. At the same time, gains of 11q13 and 11q23 are less frequent in myeloma cells expressing Aurora-A.
It cannot, however, be ruled out by our analysis that Aurora-expression is present in a small fraction of myeloma cell precursors, i.e. putative proliferating “myeloma stem cells” thereby creating genomic instability, with this presence of Aurora-expression not being maintained in the differentiated non-proliferating progeny. If this is the case, one would expect the presence of Aurora-expression within the myeloma stem cells leading to a higher number of clonal and subclonal chromosomal aberrations in advanced stage or relapsed compared to early-stage myeloma patients. At the same time, structural aberrations or point mutations in the Aurora-kinase genes of myeloma cells might be present as indicator for a respective role of Aurora-kinase expression in putative myeloma stem cells. Both investigations, being beyond the scope of this paper, are currently performed by our group.
Taken together, it is unlikely that Aurora-kinase expression in CD138-purified myeloma cells drives genetic instability in myeloma, but is, as a high labeling index and the presence of chromosomal aberrations associated with disease “progression”, rather a sign of proliferative, “advanced” myeloma cells.
Prognostic value of Aurora-kinase expression
Presence of Aurora-A expression in myeloma cells is an adverse prognostic factor in terms of EFS and OAS in our data (n=168) and the Arkansas-group (n=345, Figure 3), as is the expression-height of Aurora-A as single continuous variable. In a Cox-model tested with either ISS or B2M (as continuous variable), presence of Aurora-A expression appears as independent prognostic factor for EFS and OAS. Of note, Aurora-A (STK6) is also one of the genes within the gene expression based high-risk score for myeloma 54. Aurora-A kinase expression in our data set correlates with the gene expression based centrosome index that has likewise shown prognostic significance on the Arkansas-dataset 49 (Supplementary Figure S1). Thus, direct assessment of Aurora-A kinase expression allows identifying a poor-risk patient population independent of B2M or ISS.
Inhibition of proliferation of HMCL and primary MMC by Aurora-kinase inhibitors
Proliferation of all tested 20 myeloma cell lines is significantly inhibited by VX680 with an IC50 ranging from 0.003 to 2.715 μM (Figure 4A2). The observed minimal IC50 is in the range of data from Tyler et al. 19 who have shown an inhibition of in vitro kinase activity by VX680 in terms of phosphorylation of histone-H3 by Aurora-A, -B, and -C (with IC50-values of 36, 18, and 25 nM, respectively); Aurora-A and -B kinase activity was completely abrogated at 1 μM 19. VX680 induced a marked delay in mitosis, presumably due to Aurora-A dependent effects on spindle bipolarity, but did override a mitotic arrest imposed by several experimental agents, indicating an effect on the spindle assembly checkpoint, likely through Aurora-B inhibition 19. Our data are in agreement with VX680 having been shown to induce a dose-dependent inhibition of proliferation on myeloma cell lines 25. Inhibition of proliferation by VX680 was accompanied in our hands by apoptosis induction in myeloma cell lines and primary myeloma cell-samples in agreement with findings from Shi et al. 25. Driven by the published observation that forced over-expression of Aurora-A reduces 25, and downregulation by siRNA increases 23 the susceptibility of myeloma cell lines towards Aurora-kinase inhibitors, we investigated whether the IC50 in 12 myeloma cell lines tested might correlate with the expression level of Aurora-A. However, in our hands, it did not. The same observation was made in terms of expression of HMMR, for which a forced up-regulation was described to increase, and a down-regulation to decrease the sensitivity of the respective myeloma cell line 25, but does not in our data. This might be explained by a plethora of growth and survival-factors differentially expressed between HMCL causing a high inter-cell-line variance in terms of sensitivity against VX680 (Figure 4A2). Thus, only if Aurora-A or HMMR- expression is the single parameter varied (e.g. by siRNA) within one cell line, differences in the sensitivity of the respective HMCL might be seen.
Implications for myeloma treatment
The presence of multiple chromosomal aberrations in multiple myeloma suggests that, during the evolution of myeloma, a disruption of cell cycle checkpoints has occurred. This would normally arrest cells at the G1/S and G2/M transitions or at mitosis when DNA-damage or spindle abnormalities have occurred, thus allowing for potential damage-repair 25. Alternatively or in addition, these checkpoints might be “overruled” by intrinsic aberrant or increased expression of D-type cyclins and myeloma growth and survival factors (“strained” checkpoints). In both cases, myeloma cells might be particularly susceptible to the induction of apoptotic death in mitosis (the so-called mitotic catastrophe 55) when further assaults on the mitotic machinery are induced. Experimental evidence for the latter is given by the fact that VX680 inhibits proliferation of both, CD3/CD28- or phytohaemagglutinin-stimulated peripheral blood lymphocytes and myeloma cell lines, but induces apoptosis only in human myeloma cell lines 25. Thus, Aurora-kinase inhibitors are indicated in multiple myeloma not because myeloma cells show a genetic instability, but because Aurora-kinase inhibitors target and inhibit the proliferation of myeloma cells. They might do this especially efficient because they (additionally) induce apoptosis in the presence of strained or deranged cell-cycle checkpoints present in primary myeloma cells and human myeloma cell lines.
Conclusion
Aurora-A is expressed at varying frequencies in CD138-purified myeloma cells of newly-diagnosed patients. Its expression significantly interrelates with proliferation, but not with a higher number of chromosomal aberrations (aneuploidy) or subclonal aberrations (genetic instability). Using gene expression profiling, Aurora-kinase inhibitors as promising therapeutic option for newly-diagnosed patients can be tailoredly given to patients expressing Aurora-A with adverse prognosis.
Supplementary Material
Acknowledgments
We thank Véronique Pantesco, Katrin Heimlich, Maria Dörner and Margit Happich for technical assistance. This work was supported in part by grants from the Hopp-Foundation, Germany, the University of Heidelberg, Germany, the National Centre for Tumor Diseases, Heidelberg, Germany, the Tumorzentrum Heidelberg/Mannheim, Germany, the European Myeloma Stem Cell Network (MSCNET) funded within the 6th framework program of the European Community, the Deutsche Krebshilfe, Bonn, Germany, the Ligue Nationale Contre Le Cancer (équipe labellisée), Paris, France. It is also part of a national program called “Carte d’Identité des Tumeurs” (CIT) funded by the Ligue Nationale Contre le Cancer, France.
Footnotes
Contribution: D.H. designed research, wrote the paper and participated in the microarray experiments; T. Meissner, A.B. and T.H. performed statistical analysis, A.S. performed experiments and participated in the writing of the paper; J.L., V.B., J.B. and J.Z. participated in the analysis of the data; J.F.R., U.B., J.H. and M.H. collected bone marrow samples and clinical data; B.B. and J.S. performed the total therapy 2-trial and J.S. the microarray experiments for the Arkansas-group; J.M., K.N. and A.K. participated in the writing and review of the paper; J.D.V participated in the microarray experiments; A.J. contributed in performing the interphase-FISH experiments; T.R., T. Möhler, B.K. and H.G. participated in the analyzing of the data and in the writing of the paper.
Conflict-of-interest disclosure: The authors declare no competing financial interests.
References
- 1.Kyle RA, Rajkumar SV. Multiple myeloma. N Engl J Med. 2004;351:1860–1873. doi: 10.1056/NEJMra041875. [DOI] [PubMed] [Google Scholar]
- 2.Witzig TE, Timm M, Larson D, Therneau T, Greipp PR. Measurement of apoptosis and proliferation of bone marrow plasma cells in patients with plasma cell proliferative disorders. Br J Haematol. 1999;104:131–137. doi: 10.1046/j.1365-2141.1999.01136.x. [DOI] [PubMed] [Google Scholar]
- 3.Boccadoro M, Gavarotti P, Fossati G, et al. Low plasma cell 3(H) thymidine incorporation in monoclonal gammopathy of undetermined significance (MGUS), smouldering myeloma and remission phase myeloma: a reliable indicator of patients not requiring therapy. Br J Haematol. 1984;58:689–696. doi: 10.1111/j.1365-2141.1984.tb06116.x. [DOI] [PubMed] [Google Scholar]
- 4.Greipp PR, Lust JA, O’Fallon WM, et al. Plasma cell labeling index and beta 2-microglobulin predict survival independent of thymidine kinase and C-reactive protein in multiple myeloma. Blood. 1993;81:3382–3387. [PubMed] [Google Scholar]
- 5.Cremer FW, Bila J, Buck I, et al. Delineation of distinct subgroups of multiple myeloma and a model for clonal evolution based on interphase cytogenetics. Genes Chromosomes Cancer. 2005;44:194–203. doi: 10.1002/gcc.20231. [DOI] [PubMed] [Google Scholar]
- 6.Fonseca R, Barlogie B, Bataille R, et al. Genetics and cytogenetics of multiple myeloma: a workshop report. Cancer Res. 2004;64:1546–1558. doi: 10.1158/0008-5472.can-03-2876. [DOI] [PubMed] [Google Scholar]
- 7.Andrews PD, Knatko E, Moore WJ, Swedlow JR. Mitotic mechanics: the auroras come into view. Curr Opin Cell Biol. 2003;15:672–683. doi: 10.1016/j.ceb.2003.10.013. [DOI] [PubMed] [Google Scholar]
- 8.Dutertre S, Descamps S, Prigent C. On the role of aurora-A in centrosome function. Oncogene. 2002;21:6175–6183. doi: 10.1038/sj.onc.1205775. [DOI] [PubMed] [Google Scholar]
- 9.Keen N, Taylor S. Aurora-kinase inhibitors as anticancer agents. Nat Rev Cancer. 2004;4:927–936. doi: 10.1038/nrc1502. [DOI] [PubMed] [Google Scholar]
- 10.Fu J, Bian M, Jiang Q, Zhang C. Roles of Aurora kinases in mitosis and tumorigenesis. Mol Cancer Res. 2007;5:1–10. doi: 10.1158/1541-7786.MCR-06-0208. [DOI] [PubMed] [Google Scholar]
- 11.Carmena M, Earnshaw WC. The cellular geography of aurora kinases. Nat Rev Mol Cell Biol. 2003;4:842–854. doi: 10.1038/nrm1245. [DOI] [PubMed] [Google Scholar]
- 12.Bischoff JR, Anderson L, Zhu Y, et al. A homologue of Drosophila aurora kinase is oncogenic and amplified in human colorectal cancers. EMBO J. 1998;17:3052–3065. doi: 10.1093/emboj/17.11.3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nadler Y, Camp RL, Schwartz C, et al. Expression of Aurora A (but Not Aurora B) Is Predictive of Survival in Breast Cancer. Clin Cancer Res. 2008;14:4455–4462. doi: 10.1158/1078-0432.CCR-07-5268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Giet R, Petretti C, Prigent C. Aurora kinases, aneuploidy and cancer, a coincidence or a real link? Trends Cell Biol. 2005;15:241–250. doi: 10.1016/j.tcb.2005.03.004. [DOI] [PubMed] [Google Scholar]
- 15.Kimmins S, Crosio C, Kotaja N, et al. Differential functions of the Aurora-B and Aurora-C kinases in mammalian spermatogenesis. Mol Endocrinol. 2007;21:726–739. doi: 10.1210/me.2006-0332. [DOI] [PubMed] [Google Scholar]
- 16.Andrews PD. Aurora kinases: shining lights on the therapeutic horizon? Oncogene. 2005;24:5005–5015. doi: 10.1038/sj.onc.1208752. [DOI] [PubMed] [Google Scholar]
- 17.Glover DM, Leibowitz MH, McLean DA, Parry H. Mutations in aurora prevent centrosome separation leading to the formation of monopolar spindles. Cell. 1995;81:95–105. doi: 10.1016/0092-8674(95)90374-7. [DOI] [PubMed] [Google Scholar]
- 18.Hannak E, Kirkham M, Hyman AA, Oegema K. Aurora-A kinase is required for centrosome maturation in Caenorhabditis elegans. J Cell Biol. 2001;155:1109–1116. doi: 10.1083/jcb.200108051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tyler RK, Shpiro N, Marquez R, Eyers PA. VX-680 inhibits Aurora A and Aurora B kinase activity in human cells. Cell Cycle. 2007;6:2846–2854. doi: 10.4161/cc.6.22.4940. [DOI] [PubMed] [Google Scholar]
- 20.Ditchfield C, Johnson VL, Tighe A, et al. Aurora B couples chromosome alignment with anaphase by targeting BubR1, Mad2, and Cenp-E to kinetochores. J Cell Biol. 2003;161:267–280. doi: 10.1083/jcb.200208091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hauf S, Cole RW, LaTerra S, et al. The small molecule Hesperadin reveals a role for Aurora B in correcting kinetochore-microtubule attachment and in maintaining the spindle assembly checkpoint. J Cell Biol. 2003;161:281–294. doi: 10.1083/jcb.200208092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Girdler F, Gascoigne KE, Eyers PA, et al. Validating Aurora B as an anti-cancer drug target. J Cell Sci. 2006;119:3664–3675. doi: 10.1242/jcs.03145. [DOI] [PubMed] [Google Scholar]
- 23.Evans R, Naber C, Steffler T, et al. Aurora A kinase RNAi and small molecule inhibition of Aurora kinases with VE-465 induce apoptotic death in multiple myeloma cells. Leuk Lymphoma. 2008;49:559–569. doi: 10.1080/10428190701824544. [DOI] [PubMed] [Google Scholar]
- 24.Evans RP, Naber C, Steffler T, et al. The selective Aurora B kinase inhibitor AZD1152 is a potential new treatment for multiple myeloma. Br J Haematol. 2008;140:295–302. doi: 10.1111/j.1365-2141.2007.06913.x. [DOI] [PubMed] [Google Scholar]
- 25.Shi Y, Reiman T, Li W, et al. Targeting aurora kinases as therapy in multiple myeloma. Blood. 2007;109:3915–3921. doi: 10.1182/blood-2006-07-037671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Greipp PR, San Miguel J, Durie BG, et al. International staging system for multiple myeloma. J Clin Oncol. 2005;23:3412–3420. doi: 10.1200/JCO.2005.04.242. [DOI] [PubMed] [Google Scholar]
- 27.Durie BG. Staging and cinetics of Multiple Myeloma. Semin Oncol. 1986;13:300–309. [PubMed] [Google Scholar]
- 28.Blade J, Samson D, Reece D, et al. Criteria for evaluating disease response and progression in patients with multiple myeloma treated by high-dose therapy and haemopoietic stem cell transplantation. Myeloma Subcommittee of the EBMT. European Group for Blood and Marrow Transplant. Br J Haematol. 1998;102:1115–1123. doi: 10.1046/j.1365-2141.1998.00930.x. [DOI] [PubMed] [Google Scholar]
- 29.Goldschmidt H, Sonneveld P, Cremer FW, et al. Joint HOVON-50/GMMG-HD3 randomized trial on the effect of thalidomide as part of a high-dose therapy regimen and as maintenance treatment for newly diagnosed myeloma patients. Ann Hematol. 2003;82:654–659. doi: 10.1007/s00277-003-0685-2. [DOI] [PubMed] [Google Scholar]
- 30.Barlogie B, Tricot G, Rasmussen E, et al. Total therapy 2 without thalidomide in comparison with total therapy 1: role of intensified induction and posttransplantation consolidation therapies. Blood. 2006;107:2633–2638. doi: 10.1182/blood-2005-10-4084. [DOI] [PubMed] [Google Scholar]
- 31.Mahtouk K, Hose D, Raynaud P, et al. Heparanase influences expression and shedding of syndecan-1, and its expression by the bone marrow environment is a bad prognostic factor in multiple myeloma. Blood. 2007;109:4914–4923. doi: 10.1182/blood-2006-08-043232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Moreaux J, Cremer FW, Reme T, et al. The level of TACI gene expression in myeloma cells is associated with a signature of microenvironment dependence versus a plasmablastic signature. Blood. 2005;106:1021–1030. doi: 10.1182/blood-2004-11-4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang XG, Gaillard JP, Robillard N, et al. Reproducible obtaining of human myeloma cell lines as a model for tumor stem cell study in human multiple myeloma. Blood. 1994;83:3654–3663. [PubMed] [Google Scholar]
- 34.Gu ZJ, De Vos J, Rebouissou C, et al. Agonist anti-gp130 transducer monoclonal antibodies are human myeloma cell survival and growth factors. Leukemia. 2000;14:188–197. doi: 10.1038/sj.leu.2401632. [DOI] [PubMed] [Google Scholar]
- 35.Rebouissou C, Wijdenes J, Autissier P, et al. A gp130 interleukin-6 transducer-dependent SCID model of human multiple myeloma. Blood. 1998;91:4727–4737. [PubMed] [Google Scholar]
- 36.Tarte K, De Vos J, Thykjaer T, et al. Generation of polyclonal plasmablasts from peripheral blood B cells: a normal counterpart of malignant plasmablasts. Blood. 2002;100:1113–1122. [PubMed] [Google Scholar]
- 37.Corre J, Mahtouk K, Attal M, et al. Bone marrow mesenchymal stem cells are abnormal in multiple myeloma. Leukemia. 2007;21:1079–1088. doi: 10.1038/sj.leu.2404621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hose D, Rossi JF, Ittrich C, et al. A New Molecular Classification of Multiple Myeloma Using Gene Expression Profiling and Fluorescence In Situ Hybridisation as Predictor for Event Free Survival. ASH Annual Meeting Abstracts. 2004;104:73. [Google Scholar]
- 39.Wuilleme S, Robillard N, Lode L, et al. Ploidy, as detected by fluorescence in situ hybridization, defines different subgroups in multiple myeloma. Leukemia. 2005;19:275–278. doi: 10.1038/sj.leu.2403586. [DOI] [PubMed] [Google Scholar]
- 40.Mahtouk K, Hose D, Reme T, et al. Expression of EGF-family receptors and amphiregulin in multiple myeloma. Amphiregulin is a growth factor for myeloma cells. Oncogene. 2005;24:3512–3524. doi: 10.1038/sj.onc.1208536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.De Vos J, Couderc G, Tarte K, et al. Identifying intercellular signaling genes expressed in malignant plasma cells by using complementary DNA arrays. Blood. 2001;98:771–780. doi: 10.1182/blood.v98.3.771. [DOI] [PubMed] [Google Scholar]
- 42.De Vos J, Jourdan M, Tarte K, Jasmin C, Klein B. JAK2 tyrosine kinase inhibitor tyrphostin AG490 downregulates the mitogen-activated protein kinase (MAPK) and signal transducer and activator of transcription (STAT) pathways and induces apoptosis in myeloma cells. Br J Haematol. 2000;109:823–828. doi: 10.1046/j.1365-2141.2000.02127.x. [DOI] [PubMed] [Google Scholar]
- 43.Ro TB, Holt RU, Brenne AT, et al. Bone morphogenetic protein-5, -6 and -7 inhibit growth and induce apoptosis in human myeloma cells. Oncogene. 2004;23:3024–3032. doi: 10.1038/sj.onc.1207386. [DOI] [PubMed] [Google Scholar]
- 44.Jourdan M, Ferlin M, Legouffe E, et al. The myeloma cell antigen syndecan-1 is lost by apoptotic myeloma cells. Br J Haematol. 1998;100:637–646. doi: 10.1046/j.1365-2141.1998.00623.x. [DOI] [PubMed] [Google Scholar]
- 45.Wu Z, Irizarry RA, Gentleman RC, Martinez-Murillo F, Spencer F. A Model-Based Background Adjustment for Oligonucleotide Expression Arrays. Journal of the American Statistical Association. 2004;99:909–917. [Google Scholar]
- 46.Taylor J, Tibshirani R, Efron B. The ‘miss rate’ for the analysis of gene expression data. Biostatistics. 2005;6:111–117. doi: 10.1093/biostatistics/kxh021. [DOI] [PubMed] [Google Scholar]
- 47.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat Appl Genet Mol Biol. 2004;3 doi: 10.2202/1544-6115.1027. Article3. [DOI] [PubMed] [Google Scholar]
- 48.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society Series B. 1995;57:289–300. [Google Scholar]
- 49.Chng WJ, Braggio E, Mulligan G, et al. The centrosome index is a powerful prognostic marker in myeloma and identifies a cohort of patients that might benefit from aurora kinase inhibition. Blood. 2008;111:1603–1609. doi: 10.1182/blood-2007-06-097774. [DOI] [PubMed] [Google Scholar]
- 50.R Development Core Team. R: A Language and Environment for Statistical Computing. Vienna, Austria: R Foundation for Statistical Computing; 2008. [Google Scholar]
- 51.Gentleman RC, Carey VJ, Bates DM, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5:R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hanamura I, Stewart JP, Huang Y, et al. Frequent gain of chromosome band 1q21 in plasma-cell dyscrasias detected by fluorescence in situ hybridization: incidence increases from MGUS to relapsed myeloma and is related to prognosis and disease progression following tandem stem-cell transplantation. Blood. 2006;108:1724–1732. doi: 10.1182/blood-2006-03-009910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Avet-Loiseau H, Facon T, Daviet A, et al. 14q32 translocations and monosomy 13 observed in monoclonal gammopathy of undetermined significance delineate a multistep process for the oncogenesis of multiple myeloma. Intergroupe Francophone du Myelome Cancer Res. 1999;59:4546–4550. [PubMed] [Google Scholar]
- 54.Shaughnessy JD, Jr, Zhan F, Burington BE, et al. A validated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes mapping to chromosome 1. Blood. 2007;109:2276–2284. doi: 10.1182/blood-2006-07-038430. [DOI] [PubMed] [Google Scholar]
- 55.Castedo M, Perfettini JL, Roumier T, et al. Cell death by mitotic catastrophe: a molecular definition. Oncogene. 2004;23:2825–2837. doi: 10.1038/sj.onc.1207528. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.