

Abstract
The molecular mechanisms by which the Abelson (Abl) or Abl-related gene (Arg) kinases interface with the actin polymerization machinery to promote cell edge protrusions during cell–matrix adhesion are unclear. In this study, we show that interactions between Arg and the Arp2/3 complex regulator cortactin are essential to mediate actin-based cell edge protrusion during fibroblast adhesion to fibronectin. Arg-deficient and cortactin knockdown fibroblasts exhibit similar defects in adhesion-dependent cell edge protrusion, which can be restored via reexpression of Arg and cortactin. Arg interacts with cortactin via both binding and catalytic events. The cortactin Src homology (SH) 3 domain binds to a Pro-rich motif in the Arg C terminus. Arg mediates adhesion-dependent phosphorylation of cortactin, creating an additional binding site for the Arg SH2 domain. Mutation of residues that mediate Arg–cortactin interactions abrogate the abilities of both proteins to support protrusions, and the Nck adapter, which binds phosphocortactin, is also required. These results demonstrate that interactions between Arg, cortactin, and Nck1 are critical to promote adhesion-dependent cell edge protrusions.
Introduction
Carefully controlled cell movement is essential for diverse biological events such as embryogenesis, wound healing, and proper brain development, whereas aberrant cell migration underlies numerous pathological states such as inflammatory diseases and cancer metastasis. Directed cell migration requires changes in cell shape powered by dynamic rearrangements of the actin cytoskeleton. Actin polymerization promotes protrusions at the cell edge (Mitchison and Cramer, 1996; Pollard and Borisy, 2003; Ponti et al., 2004), whereas actomyosin networks direct cellular contractility to provide traction force for cell body translocation (Jay et al., 1995; Lauffenburger and Horwitz, 1996; Mitchison and Cramer, 1996; Ridley et al., 2003; de Rooij et al., 2005; Gupton and Waterman-Storer, 2006). These rearrangements are guided locally by extracellular cues that bind cell surface receptors to activate signaling pathways that control the actin cytoskeletal machinery.
Abelson (Abl) family kinases, which include the vertebrate Abl/Abl1 and Abl-related gene (Arg)/Abl2 proteins, are critical mediators of cytoskeletal rearrangements in response to growth factor or adhesion receptor engagement (Plattner et al., 1999, 2003, 2004; Woodring et al., 2002, 2004; Hernandez et al., 2004; Miller et al., 2004; Sini et al., 2004; Moresco et al., 2005). Several studies indicate that Abl family kinases promote localized actin network assembly in response to cell–cell or cell–ECM adhesion. For example, Abl family kinases stimulate actin-based cell edge protrusions in fibroblasts (Woodring et al., 2002; Miller et al., 2004) and neurite branching in neurons (Woodring et al., 2002; Moresco et al., 2005) as they adhere and spread on ECM molecules. Abl family kinases also promote actin assembly during immune synapse formation between B and T lymphocytes (Huang et al., 2008) and strengthen F-actin networks that connect adherens junctions (Zandy et al., 2007). Abl family kinases can phosphorylate diverse cytoskeletal effector proteins including the Dok (downstream of the Tyr kinase) family adapters (Cong et al., 1999; Master et al., 2003; Woodring et al., 2004), Abl-interacting (Abi) family proteins (Dai and Pendergast, 1995; Shi et al., 1995; Biesova et al., 1997), Enabled/mammalian Enabled (Comer et al., 1998; Juang and Hoffmann, 1999; Tani et al., 2003), neural Wiskott-Aldrich syndrome protein (N-WASp; Burton et al., 2005), WAVE2 (Leng et al., 2005; Stuart et al., 2006), and cortactin (Boyle et al., 2007). The molecular mechanisms by which Abl family kinases act through these proteins to induce actin polymerization-dependent protrusions are largely unclear.
The formation of cell edge protrusions requires actin polymerization nucleated by the Arp2/3 complex or formins (Pollard, 2007). The Arp2/3 complex regulator cortactin localizes to and promotes dynamic actin-rich protrusions of the cell membrane, including circular dorsal ruffles, lamellipodia, and invadopodia (Weed et al., 1998, 2000; Bowden et al., 1999; McNiven et al., 2000; Head et al., 2003; Bryce et al., 2005; Boyle et al., 2007). An N-terminal acidic (NTA) region in cortactin binds the Arp3 subunit of the Arp2/3 complex and can weakly stimulate F-actin nucleation by this complex (Weaver et al., 2002). Cortactin synergizes with N-WASp to stimulate robust F-actin nucleation by the Arp2/3 complex (Uruno et al., 2001; Weaver et al., 2002; Martinez-Quiles et al., 2004; Kowalski et al., 2005). Cortactin can also stabilize Arp2/3-mediated F-actin branches in vitro (Weaver et al., 2001), and this activity may be critical for the stability of F-actin–rich cellular protrusions in vivo (Bryce et al., 2005).
We recently used an unbiased high throughput screen to identify cortactin as an Abl and Arg substrate (Boyle et al., 2007). Although other Tyr kinases (e.g., Src family kinases) can also phosphorylate cortactin (Wu et al., 1991; Thomas et al., 1995; Shah and Vincent, 2005), Abl and Arg are unique in that they also bind stably to cortactin and the cortactin homologue HS1 (Boyle et al., 2007; Huang et al., 2008). In the case of HS1, this interaction is at least partly mediated by binding of the Abl/Arg Src homology (SH) 2 domains to Tyr-phosphorylated HS1 (Huang et al., 2008). These observations do not resolve how Abl/Arg interface initially with cortactin/HS1 to mediate the phosphorylation required for this binding interaction or what role cortactin phosphorylation plays in actin-based protrusion.
In this study, we show that interactions between Arg, cortactin, and Nck1 are essential for efficient cell edge protrusion during fibroblast adhesion to fibronectin. We provide evidence that Arg interacts with cortactin via a series of binding and catalytic events. Mutations in Arg or cortactin that disrupt these interactions abrogate the ability of these proteins to support adhesion-dependent cell edge protrusion. Phosphorylation of cortactin reinforces Arg binding to cortactin and helps recruit the Nck1 adapter, which is required for protrusion. These results elucidate the molecular interactions between Arg, cortactin, and Nck1 that promote localized cell edge protrusion during cell–matrix adhesion.
Results
Arg and cortactin colocalize to adhesion-induced protrusive structures
We have previously reexpressed Arg-YFP at endogenous levels in arg−/− fibroblasts (arg−/− + Arg-YFP cells) to demonstrate that Arg localizes to and promotes dynamic cell edge protrusions during fibroblast adhesion and spreading on fibronectin (Miller et al., 2004; Peacock et al., 2007). In this study, we retrovirally expressed a functional cortactin-RFP fusion protein (Boyle et al., 2007) in arg−/− + Arg-YFP cells and observed the localization of both proteins in near simultaneous time-lapse imaging during fibroblast adhesion and spreading on glass coverslips coated with 10 µg/ml fibronectin. Arg-YFP and cortactin-RFP localize to protrusive lamellar structures at the cell periphery (Fig. 1, compare B with C [enlarged in E and F]; and Video 1). A portion of cortactin-RFP was also found associated with intracellular vesicular structures, which is consistent with a well-established role for cortactin in endocytosis and vesicle trafficking (Cao et al., 2003; Merrifield et al., 2005). Interestingly, both Arg-YFP and cortactin-RFP localize to the cell periphery (Fig. 1, E and F, arrowheads) just before the formation of a protrusion (Fig. 1, E and F, asterisks). We find that in 86% (12/14) of the cases in which we observed newly formed areas of Arg–cortactin colocalization (n = 4 cells), they were associated with subsequent protrusion. These colocalization experiments suggest that Arg and cortactin may interact to promote adhesion-dependent cell edge protrusions.
Figure 1.

Arg-YFP and cortactin-RFP colocalize to membrane protrusions in response to adhesion to fibronectin. Individual frames from time-lapse videos of arg−/− cells expressing Arg-YFP and cortactin-RFP (Video 1). (A–F) Phase-contrast (A and D), YFP channel (B and E), and RFP channel (C and F) images are shown. (D–F) Enlargements of the regions boxed in A, showing colocalization of Arg-YFP and cortactin-RFP (indicated by arrowheads) followed by the formation of a cell edge protrusion (indicated by asterisks). The elapsed time is shown in minutes:seconds. Bars, 10 µm.
Cortactin knockdown (KD) abrogates adhesion-dependent cell edge protrusion
Wild-type (WT) fibroblasts exhibit frequent dynamic cell edge protrusions as they adhere and spread on fibronectin-coated surfaces. These protrusions occur much less frequently in arg−/− fibroblasts, a defect which can be repaired by Arg-YFP reexpression (see Fig. 5, B–D; Miller et al., 2004). Similarly, we used kymography (Hinz et al., 1999) to determine whether cortactin is required for adhesion-dependent cell edge protrusion. During spreading on fibronectin, WT cells or WT cells expressing an empty short hairpin RNA vector (WT + vector) exhibit frequent dynamic protrusions and retractions of the cell edge, yielding kymographs that resemble “rolling hills” (Fig. 2, B, C, I, and J). In contrast, cortactin KD cells, which retain only 20% of endogenous cortactin (Fig. S1 E), exhibit a significantly reduced number of protrusions and retractions (Fig. 2 D), yielding flat, “prairie-like” kymographs. This deficiency was restored via reexpression of a functional RNAi-resistant cortactin monomeric RFP (cortactin-RFP) fusion (Fig. 2 E). These data indicate that cortactin, like Arg, is required for dynamic cell edge protrusion during fibroblast adhesion on fibronectin.
Figure 5.

A Pro-rich region in the Arg C terminus is required for cell edge dynamics. (A) Graphical depiction of Arg mutants used in this experiment. See Fig. 3 for a description of the domains. KI, KI (K317M) point mutation in the active site. Arg mutSH2 contains a point mutation (R176K) in the SH2 domain that abrogates binding to phospho-Tyr–containing binding partners. Arg 557-C PXXP mut1 and Arg 557-C PXXP mut23 contain point mutations in PXXP motif 1, and PXXP motifs 2 and 3, respectively. (B–J) Representative images of 10-min time-lapse videos of cells spreading on glass coverslips coated with fibronectin. The three rightmost panels are representative kymographs for each genotype. Kymographs were constructed as in Fig. 2. WT cells (n = 20 cells; B) and arg−/− cells expressing YFP (n = 22 cells; C), Arg-YFP (n = 34; D), KI Arg (n = 29 cells; E), Arg mutSH2 (n = 20 cells; F), Arg C terminus 557-C (n = 29 cells; G), Arg 688-C (n = 17 cells; H), Arg 557-C PXXP mut1 (n = 22 cells; I), and Arg 557-C PXXP mut23 (n = 16 cells; J) are shown. (K and L) Quantification of cell edge dynamics. Mean ± SEM. ANOVA between all cell types: protrusions, P < 0.0001; retractions, P < 0.0001. Fisher’s PLSD for all cells versus arg−/−: *, P < 0.01. Bars, 10 µm.
Figure 2.

Cortactin is required for adhesion-dependent cell edge protrusion. (A) Domain structure of RFP-tagged cortactin mutants used in this study. Cortactin has an NTA domain required for Arp2/3 binding followed by 6.5 cortactin repeats (shaded). A Pro-rich region (white box) contains three Tyr (Y) that can be phosphorylated by Arg and is followed by an SH3 domain. Cortactin 3F contains Tyr to Phe (F) point mutations in the phosphorylatable Tyr (Y421F, Y466F, and Y482F). The C-terminal SH3 domain is deleted in cortactinΔSH3 (aa 1–494). Cortactin W525A contains a mutation in a key Trp in the SH3 domain (W525A) that abrogates SH3 binding to ligands. (B–H) Representative frames from 10-min time-lapse videos of cells plated on 10 µg/ml fibronectin. For kymography analysis, a radial grid of eight lines was placed over the phase images (as shown in B), and kymographs were constructed for each of the indicated lines. WT (n = 24 cells; B), WT expressing empty pSuper (pS) RNAi vector (n = 23 cells; C), cortactin KD (n = 26 cells; D), cortactin KD expressing RNAi resistant FL cortactin-RFP (n = 20 cells; E), cortactin 3F–RFP (n = 20 cells; F), cortactinΔSH3-RFP (n = 17 cells; G), and cortactin WA–RFP (n = 23 cells; H) are shown. (I and J) Quantification of the number of protrusions (I) and retractions (J) per 10-min video. Mean ± SEM. ANOVA between all cell types: protrusions, P < 0.0001; retractions, P < 0.0001. Fisher’s protected least significant difference (PLSD) for all cells versus WT: *, P ≤ 0.0002. Bars, 10 µm.
The cortactin SH3 domain binds to one of several PXXP motifs in Arg
We used a bead affinity pull-down assay to characterize the interactions between Arg and cortactin. We incubated a constant concentration of cortactin-agarose beads with increasing Arg concentrations. After allowing binding to reach equilibrium, the amount of cortactin-bound Arg or Arg fragments was measured (Fig. 3 A). Arg binds to cortactin and was retained on the beads in a concentration-dependent manner, but only trace amounts of Arg were recovered on the control beads (Fig. 3 A).
Figure 3.

Cortactin uses its SH3 domain to bind to the first PXXP region of Arg. (A) Representative Coomassie blue–stained SDS-PAGE gel showing binding of the Arg C-terminal half (Arg 557-C) to cortactin beads but not to ethanolamine-blocked control beads. Arg 557-C binding to cortactin increases as a function of concentration. (B, D, and F) Plots of concentration (x axis) versus the amount bound (y axis) were fitted to a binding curve. Arg 557-C binding to cortactin (Kd = 0.85 ± 0.18 µM; n = 4; B), Arg 557-C binding to the cortactin SH3 domain (Kd = 0.44 ± 0.14 µM; n = 3; D), and Arg 557-C PXXP mut23 binding to the cortactin SH3 domain (Kd = 1.25 ± 0.25 µM; n = 2; F) are shown. (C, E, and G) Affinities of Arg and cortactin binding. Each binding reaction was tested at least twice, with combinations showing positive binding tested at least three times. (C) Affinity of Arg/Arg mutants for cortactin. Arg is composed of an N-terminal SH2 and SH3 domain followed by a kinase domain. This fragment does not bind to cortactin. Arg 557-C containing three conserved PXXP motifs (indicated by vertical lines), two F-actin–binding sites and a microtubule (MT)-binding site binds cortactin, whereas a shorter fragment lacking the Pro-rich stretch (Arg 688-C) does not. (E) Affinity of cortactin/cortactin fragments for Arg 557-C. See Fig. 2 for a description of the cortactin domains. The cortactin SH3 domain is necessary and sufficient to bind Arg 557-C. (G) Affinity of Arg 557-C PXXP mutants for the cortactin SH3 domain. Mutation of PXXP1 in Arg 557-C abrogates binding to the cortactin SH3 domain.
To identify the domains and motifs that mediate Arg binding to cortactin, we next measured binding affinities of various Arg truncation mutants for cortactin (Fig. 3 and Fig. S3). The Arg N-terminal half containing its SH3, SH2, and kinase domains (ArgΔC) does not bind cortactin (Fig. 3 C and Fig. S3 B). The Arg C-terminal half (Arg 557-C) contains a series of three PXXP motifs located in the linker region between the kinase domain and cytoskeletal-binding domains that could serve as potential binding sites for cortactin’s C-terminal SH3 domain. Arg 557-C binds to cortactin with submicromolar affinity (Kd = 0.85 ± 0.18 µM; Fig. 3, A–C). Although Arg 557-C binds tightly to cortactin, a slightly smaller Arg fragment (Arg 688-C) lacking the PXXP motifs does not bind cortactin (Fig. 3 C and Fig. S3 D). We could not obtain full-length (FL) Arg at sufficient concentrations in solution to achieve binding saturation, and, thus, we can only place a lower limit (Kd ≥ 1.1 µM) on this interaction (Fig. 3 C and Fig. S3 A). The reduced affinity of Arg for cortactin relative to Arg 557-C suggests that the Arg N-terminal half may restrict binding of cortactin to the Arg C-terminal half. Collectively, these data suggest that Arg residues 557–687 mediate cortactin binding.
Cortactin contains a C-terminal SH3 domain that might interact with the PXXP motifs in Arg. Purified GST-cortactin SH3 domain fusion protein binds Arg 557-C with a similar affinity as FL cortactin (Kd = 0.44 ± 0.14 µM), but mutation of a key binding residue in the cortactin SH3 domain (W525A) eliminates binding of cortactin to Arg 557-C (Fig. 3 E and Fig. S3, E and F). Mutation of either all three PXXP motifs or just PXXP1 in Arg 557-C (Arg 557-C mut1) disrupts its binding of the cortactin SH3 domain (Fig. 3 G and Fig. S3, G and H). It is important to note that the PXXP1 motif in Arg (residues 567–576) is a tandem array of PXXP motifs (PXXPXXPXXP). Only the central pair of Pro in this motif is conserved in Abl and other Abl family kinases. Mutation of all four Pro in this set (e.g., AXXAXXAXXA) was required to disrupt the ability of Arg 557-C to bind the cortactin SH3 domain or to support adhesion-dependent protrusions (Fig. 3 G and Fig. S3, G and H; see Fig. 5 I). However, mutation of both PXXP2 (P622A and P625A) and PXXP3 (P664A and P667A; Arg 557-C mut23) leads to only a modest reduction in binding of Arg 557-C to the cortactin SH3 domain (Kd = 1.25 ± 0.25 µM; Fig. 3 G and Fig. S3 I). Together, these data indicate that cortactin binds to the Arg PXXP1 motif via its SH3 domain.
Adhesion stimulates Arg-dependent cortactin phosphorylation and creates a binding site for the Arg SH2 domain
Arg phosphorylates cortactin on three sites (Tyr Y421, Y466, and Y482) after stimulation of fibroblasts with PDGF (Boyle et al., 2007). This observation led us to investigate whether Arg phosphorylates cortactin upon adhesion to fibronectin. Adhesion of serum-starved WT fibroblasts to fibronectin stimulates Tyr phosphorylation of cortactin threefold compared with cells held in suspension (Fig. 4 A). Arg is required for this phosphorylation, as adhesion to fibronectin does not stimulate cortactin phosphorylation in arg−/− cells (Fig. 4 B) or in WT cells treated with the Abl/Arg kinase inhibitor STI-571 (Fig. 4 C), even though they adhere with similar kinetics to fibronectin-coated plates (Bradley et al., 2006). Adhesion stimulates Tyr phosphorylation of cortactin-RFP expressed in cortactin KD cells 2.7-fold, which is a similar induction observed for endogenous cortactin in WT cells (Fig. 4, E and F). However, the cortactin SH3 domain mutant (W525A), which does not bind Arg, exhibited only minimal Tyr phosphorylation, and this was unaffected by adhesion (Fig. 4, E and F). Together with the data presented in Fig. 3, these data suggest that interactions between the cortactin SH3 domain and Arg PXXP1 are essential for adhesion-dependent phosphorylation of cortactin.
Figure 4.

Adhesion-dependent cortactin phosphorylation by Arg creates an additional Arg–cortactin-binding site. (A–D) Arg is required for adhesion-dependent phosphorylation of cortactin. (A–C) WT (n = 6 experiments; A), arg−/− cells (n = 3 experiments; B), or WT cells treated with STI-571 (n = 3 experiments; C) were plated on 10 µg/ml fibronectin, and cortactin was immunoprecipitated and immunoblotted with anti–phospho-Tyr (PY) or anti-cortactin antibodies. Phospho-Tyr content of cortactin was normalized to the amount of cortactin immunoprecipitated at each time point. (D) The fold increase at each time point indicates the normalized phospho-Tyr content compared with the 0 time point. Mean ± SEM; Post-hoc Fisher’s PLSD test for WT at 0 versus the 20-, 30-, or 40-min time point: *, P < 0.05. (E) Cortactin KD cells expressing FL cortactin-RFP or cortactin WA–RFP were plated and immunoprecipitated as in B and C. (F) The phospho-Tyr content of cortactin-RFP or cortactin WA was quantitated as in D (n = 2 experiments). Mean ± SEM; Post-hoc Fisher’s PLSD test for FL cortactin versus cortactin WA at the 30-min time point: *, P < 0.05. (G) Diagram showing the layout of the protein dots on the nitrocellulose panels. 0.4 µg dots of cortactin-P, a nonphosphorylatable cortactin mutant (3F), pervanadate-treated cell lysate positive control, or phosphatase-treated cell lysate negative control were dotted in duplicate. (H) Arg SH2 domain binds to cortactin-P. Horseradish peroxidase–conjugated GST-SH2 domains of Arg, Abl, and Grb2 or GST alone were incubated with the nitrocellulose membranes described in E and then exposed to film using chemiluminescent detection. Only the Arg SH2 domain showed binding to cortactin-P but not to the 3F mutant (n = 4). The asterisk indicates a smudge. (I–K) Representative SDS-PAGE gels and fitted binding curves showing binding of Arg and Arg fragments to Tyr-phosphorylated cortactin (cortactin-P). Each binding reaction was performed in triplicate. (I) Arg 557-C binds to cortactin-P (Kd = 0.57 ± 0.30 µM) with similar affinity to nonphosphorylated cortactin (Kd = 0.85 ± 0.18 µM; Fig. 3, C and E). (J) Although it does not bind nonphosphorylated cortactin (Fig. 3 C), ArgΔC binds well to cortactin-P (Kd = 0.37 ± 0.07 µM). (K) FL Arg binds cortactin-P with higher affinity (Kd = 0.21 ± 0.05 µM) than nonphosphorylated cortactin (Kd ≥ 1.1 µM; Fig. 3 C). This representative gel tore during destaining process but was reassembled on the scanner bed before rescanning. (I–K) Error bars represent SEM calculated from three replicate experiments. IP, immunoprecipitation.
SH2 domains bind to phospho-Tyr–containing sequences in target proteins (Cohen et al., 1995). We generated phosphorylated cortactin (cortactin-P) by coexpression of His-tagged cortactin with untagged Arg in insect cells. We next tested whether cortactin-P could bind the Arg SH2 domain. Membranes spotted with cortactin-P, a nonphosphorylatable cortactin mutant with Phe substitutions at its three phosphorylation sites (cortactin 3F), a positive control of pervanadate-treated cell extract, which is enriched for phospho-Tyr–containing proteins, and a negative control of phosphatase-treated cell extract (Fig. 4 G) were probed with a collection of GST-SH2 domain fusion proteins (Abl, Arg, and Grb2; Fig. 4 H). Each of the GST-SH2 domains binds to the pervanadate-treated cell lysate positive control, whereas none of the GST-SH2 domains binds to the phosphatase-treated cell lysate negative control (Fig. 4 H). The Arg SH2 domain binds to cortactin-P, but not to cortactin 3F.
We next measured the extent to which cortactin phosphorylation alters its affinity for Arg. Cortactin phosphorylation does not significantly alter its affinity for the Arg C-terminal half, Arg 557-C. Cortactin-P binds to Arg 557-C with an affinity (Kd = 0.57 ± 0.30 µM) quite similar to that of cortactin binding to Arg 557-C (Kd = 0.85 ± 0.18 µM; Figs. 3 C and 4 I). However, in marked contrast to cortactin, which does not bind the Arg N-terminal half (ArgΔC; Fig. 3 C), cortactin-P binds ArgΔC with high affinity (Kd = 0.37 ± 0.07 µM; Fig. 4 J). This interaction is mediated by the Arg SH2 domain, which binds cortactin with similar high affinity (Kd = 0.24 ± 0.07 µM; unpublished data). Most notably, cortactin-P exhibits a greater than fivefold enhanced binding affinity for FL Arg (Kd = 0.21 ± 0.05 µM) compared with cortactin (Kd > 1.1 µM; Figs. 3 C and 4 K). Together, these experiments indicate that adhesion to fibronectin stimulates Arg-dependent cortactin phosphorylation, which creates a high affinity binding surface for the Arg SH2 domain.
Disruption of SH3-mediated cortactin binding to Arg compromises adhesion-dependent cell edge protrusion
The cell edge protrusion defects of arg−/− or cortactin KD cells can be quantitatively restored via reexpression of Arg-YFP (Miller et al., 2004) or cortactin-RFP (Fig. 2 E), respectively. We used this complementation assay to explore whether Arg–cortactin interactions mediate adhesion-dependent cell edge protrusions in vivo. Each mutant was retrovirally expressed at levels similar to WT control cells (See Fig. S1 and Fig. S2 for quantification and Fig. S2 and Fig. S4 for localization of Arg-YFP and cortactin-RFP fusion proteins; Miller et al., 2004; Peacock et al., 2007). Although they localize normally to the cell periphery (Fig. S1, A–D), cortactin mutants lacking the C-terminal SH3 domain (ΔSH3) or containing an inactivating point mutation in the SH3 domain (WA) that abrogates SH3-mediated binding to Arg (Fig. 3 E) do not support adhesion-dependent cell edge protrusions (Fig. 2, G–J). These data show that cortactin requires its Arg-binding SH3 domain to support adhesion-dependent cell edge protrusion.
An Arg C-terminal half fragment (Arg 557-C–YFP) can restore adhesion-dependent cell edge dynamics to arg−/− cells to 70% of WT levels (Fig. 5 G; Miller et al., 2004). Interestingly, a C-terminal Arg fragment (Arg 688-C–YFP) lacking the three PXXP motifs did not rescue adhesion-dependent cell edge protrusion (Fig. 5 H), even though it undergoes normal adhesion-dependent localization to the cell periphery (Fig. S4, G and J; Wang et al., 2001). Similarly, an Arg 557-C–YFP mutant in which all Pro residues in the three PXXP motifs were mutated to Ala (Arg 557-C PXXP mut123-YFP) does not support adhesion-dependent cell edge protrusions (Fig. S5). Significantly, mutation of the first PXXP motif only (Arg 557-C PXXP mut1-YFP) was sufficient to reduce cell edge protrusions to levels observed in arg−/− + YFP cells (Fig. 5 I). The Arg 557-C mutant in which only PXXP1 was intact (Arg 557-C PXXP mut23-YFP) supported adhesion-dependent protrusions to similar levels to Arg 557-C–YFP (Fig. 5 J). Coupled with the binding experiments presented in Fig. 3, these data strongly suggest that Arg PXXP1 binding to the cortactin SH3 domain is required for adhesion-dependent cell edge dynamics.
Disruption of cortactin phosphorylation reduces cell edge dynamics
Having shown that Arg is required for cortactin phosphorylation, which creates a binding site for the Arg SH2 domain, we tested whether cortactin phosphorylation is required for adhesion-dependent cell edge protrusion. The cortactin 3F phosphorylation site mutant rescued cell edge dynamics to only ∼30% of the rescue level observed with WT cortactin (Fig. 2, F, I, and J). The rare protrusions that did form in these cells did not extend as far (2.0 ± 0.3 µM) as protrusions observed in WT cells (2.5 ± 0.2 µM). Similarly, an Arg SH2 domain–binding defective point mutant (R176K) could partially restore protrusion to arg−/− cells (Fig. 5, A, F, K, and L) but only to the intermediate protrusion levels similar to those observed with Arg 557-C–YFP. Together with the aforementioned binding experiments, these data demonstrate that cortactin phosphorylation and its reinforcement of Arg–cortactin binding interactions are required to support robust adhesion-dependent cell edge protrusion.
Abl can substitute for Arg kinase activity but not for a kinase-independent Arg scaffold function in supporting adhesion-dependent protrusions
Surprisingly, when introduced into arg−/− cells, a kinase-inactive (KI) catalytic point mutant of Arg (Arg KI-YFP) fully restored the protrusion and retraction rate to WT levels (Fig. 5 E). This observation seemed inconsistent with the requirements for Arg in adhesion-dependent cortactin phosphorylation (Fig. 4, A–C) and for cortactin phosphorylation for cell edge protrusion. We used serum-free conditions in adhesion-dependent phosphorylation assays to isolate the contribution of adhesion receptors. However, in contrast to the serum-free conditions of the adhesion-dependent phosphorylation assays, the cell edge protrusion assays must be performed in complete serum-containing medium to observe robust protrusion activity. We reasoned that in this context, some other kinase (e.g., Abl) may phosphorylate cortactin to allow cell edge protrusion. In support of this hypothesis, we find that the Abl/Arg inhibitor STI-571, which can block adhesion-dependent cortactin phosphorylation (Fig. 4 C), inhibits adhesion-dependent cell edge protrusion (Fig. 6, A, B, and D). We noted a similar low level of adhesion-dependent cells edge protrusions in abl−/−arg−/− double knockout (dko) mouse fibroblasts expressing YFP (Fig. 6, A, B, and E; Koleske et al., 1998). Expression of Arg-YFP in dko cells restored adhesion-dependent protrusions nearly to levels observed in WT cells, whereas Abl-YFP does not support adhesion-dependent protrusions in dko cells (Fig. 6, A, B, F, and H). Although Arg KI-YFP could support adhesion-dependent protrusions in arg−/− cells (Fig. 5, E, K, and L), it does not restore adhesion-dependent protrusions to dko cells (Fig. 6, A, B, and G). These data indicate that Arg provides two functions in supporting adhesion-dependent cell edge protrusion: a kinase activity that phosphorylates cortactin and a kinase-independent cortactin-binding scaffold function. Abl can substitute its kinase activity for that of Arg, but it is insufficient to supply the scaffold function.
Figure 6.

Either Abl or Arg kinase activity is sufficient for protrusions, but Arg uniquely provides a scaffold function required for cell edge protrusion. (A and B) Quantification of protrusion (A) and retraction (B) data for WT cells treated with DMSO vehicle or 10 µM STI-571 and Abl/Arg dko cells expressing YFP, Arg-YFP, KI Arg-YFP (Arg KI), or Abl-YFP. Mean ± SEM. Fisher’s PLSD of WT versus WT + STI-571: *, P < 0.0001. (C–H) Representative frames from 10-min time-lapse videos of cells plated on glass coverslips coated with 10 µg/ml fibronectin and treated with DMSO vehicle (n = 20 cells; C), 10 µM STI-571 (n = 20 cells; D), and dko cells expressing YFP (n = 20 cells; E), Arg-YFP (n = 16 cells; F), Arg KI-YFP (n = 20 cells; G), or Abl-YFP (n = 30; H) are shown. The three rightmost panels are representative kymographs for each genotype. Kymographs were constructed as in Fig. 2. Bars, 10 µm.
A cortactin-Arg fusion protein can rescue protrusion defects in both arg−/− and cortactin KD cells
Mutations that disrupt Arg–cortactin interactions compromise the abilities of the respective proteins to support adhesion-dependent protrusions. We reasoned that covalent fusion of otherwise noninteracting Arg and cortactin fragments might restore the ability of these proteins to support protrusions. Like FL Arg, the Arg 688-C fragment is targeted to the cell periphery via its interactions with F-actin and microtubules. However, because it lacks the PXXP motifs, Arg 688-C does not bind cortactin (Fig. 3 C, and Fig. S3 D) and therefore cannot support adhesion-dependent cell edge protrusion (Fig. 5, A, H, K, and L; Miller et al., 2004). Similarly, cortactinΔSH3 neither binds Arg nor supports protrusions (Fig. 2, A, G, I, and J). We tested whether fusion of these inert moieties could restore at least partial function to either arg−/− or cortactin KD cell types. A cortactinΔSH3–Arg 688-C–RFP fusion protein was expressed at levels similar to endogenous Arg (Fig. S2 F) and localizes to the cell periphery and intracellular vesicles similarly to cortactin (Fig. S2, G and H). Interestingly, the cortactinΔSH3–Arg 688-C–RFP fusion protein restored adhesion-dependent protrusions to both arg−/− and cortactin KD cells, even though these levels were only 40% or 45% of the levels supported by WT Arg or cortactin, respectively (Fig. 7, A, B, D, F, and G). Importantly, mutations of Tyr 421, 466, and 482 in the cortactin portion of the fusion protein (cortactin 3FΔSH3–Arg 688-C) abrogate its ability to rescue protrusions in either cell type (Fig. 7, A, C, and E–G). These data indicate that interactions between Arg and cortactin are necessary and at least partly sufficient to support protrusions in this context. These findings also further reinforce a model in which Arg acts as a scaffold to mediate downstream cortactin phosphorylation and function in the protrusion process.
Figure 7.

A cortactin/Arg fusion protein lacking the SH3–PXXP interaction regions partially rescues adhesion-dependent cell edge protrusion. (A) Graphical depiction of a cortactinΔSH3–Arg 688-C–RFP fusion protein (Fusion) or the same with Tyr 421, 466, and 482 in cortactin mutated to Phe (Fusion 3F). MT, microtubule. (B–E) representative images of 10-min time-lapse videos of cells spreading on glass coverslips covered with fibronectin. The three rightmost panes depict representative kymographs. Kymographs were constructed as in Fig. 2. arg−/− cells expressing the fusion protein (B) and the nonphosphorylatable fusion protein (Fusion 3F; C) and cortactin KD cells expressing the fusion protein (D), or the fusion 3F protein (E) are shown. (F and G) Quantitation of protrusions of either arg−/− cells (F) or cortactin KD cells (G) expressing the indicated constructs. Mean ± SEM; *, P < 0.05. Bars, 10 µm.
The cortactin- and Arg-binding protein Nck1 is critical for cell edge protrusions
Cortactin phosphorylation enhances Arp2/3 complex–mediated actin nucleation in a purified recombinant system by potentiating interactions between cortactin and the Nck1 adapter protein (Tehrani et al., 2007). Interestingly, we also identified Nck1 in a high throughput screen for cortactin-P–binding proteins (unpublished data). We tested whether Nck1 is required for adhesion-dependent protrusions. Nck1 KD cells, in which >80% KD of Nck1 was achieved (Fig. 8 G), exhibited significantly less adhesion-dependent protrusive activity than WT cells (Fig. 8, B, D, and E). Interestingly, protrusion levels in Nck1 KD cells are lower than those observed in arg−/− and cortactin KD cells. Reexpression of Nck-GFP in these cells restored protrusive activity to WT levels (Fig. 8, C–E). Importantly, Nck1-GFP localizes with cortactin-RFP to protrusive areas at the cell periphery (Fig. 8 F). These observations suggest that cortactin-P may recruit Nck1 to bridge interactions with the Arp2/3 complex during protrusion formation.
Figure 8.

Nck1 is required for cell edge protrusion. (A) Graphical depiction of the Nck1 adapter protein. Nck1 consists of three SH3 domains followed by a single SH2 domain. (B and C) Representative frames from the 10-min time-lapse videos of cells plated on 10 µg/ml fibronectin-coated coverslips. The three rightmost panels are representative kymographs constructed as in Fig. 2. Nck1 KD cells (n = 29 cells; B) and Nck1 KD cells expressing Nck-GFP (n = 19 cells; C) are shown. (D and E) Quantification of the number of protrusions (D) and retractions (E) per 10-min video. Mean ± SEM. ANOVA between all cell types: protrusions, P < 0.0001; retractions, P < 0.0001. Fisher’s PLSD for all cells versus WT: *, P ≤ 0.001. (F) Localization of Nck1-GFP and cortactin in Nck1 KD cells. Nck1 KD cells were infected with Nck1-GFP (a and f) and endogenous cortactin was visualized by immunostaining (b and g). Cells were plated on fibronectin-coated coverslips and fixed 45 min after plating. F-actin was visualized with Alexa Fluor 350 phalloidin (c and h). The merged images of Nck1-GFP and cortactin images (d and i) are shown. The merged images of Nck1, cortactin, and actin (e and j) are also shown. f–j are enlargements of the areas indicated in a. (G) Immunoblot of WT and Nck1 KD cell lysates for Nck1. The HSP-70 panel represents a loading control. Bars, 10 µm.
Discussion
Abl family kinases act downstream of adhesion receptors to promote actin-based protrusions in a variety of contexts, but the molecular mechanisms by which they interface with the actin polymerization machinery are largely unclear. We present evidence that Arg interacts functionally with the Arp2/3 complex activator cortactin to promote adhesion-dependent cell edge protrusions. The cortactin SH3 domain binds to a PXXP motif in the Arg C terminus, and both features are required for adhesion-dependent cell edge protrusion formation. Adhesion stimulates Arg to phosphorylate cortactin, creating a novel high affinity binding site for the Arg SH2 domain and allowing Nck1 recruitment. Disruption of cortactin phosphorylation or Arg binding to phosphocortactin significantly impairs adhesion-dependent cell edge protrusion. Our data indicate that Arg acts as both a cortactin-binding scaffold and a cortactin kinase in the protrusion process. Under appropriate conditions, Abl can substitute for Arg to phosphorylate cortactin, but it cannot subserve the Arg scaffold function. Together, these data indicate that Arg and cortactin interact via both binding and catalytic events to promote cell edge protrusion during fibroblast adhesion and spreading.
Abl/Arg–cortactin interactions mediate actin assembly in diverse biological contexts
We demonstrate that Arg–cortactin interactions promote adhesion-dependent cell edge protrusion. Abl family kinases appear to interact with cortactin to promote actin assembly in a growing number of cellular contexts. For example, we showed previously that Abl- or Arg-mediated cortactin phosphorylation was required for PDGF-induced F-actin–rich circular dorsal ruffles (Boyle et al., 2007). Huang et al. (2008) have shown that Abl interacts with the hematopoietic cortactin homologue HS1 to regulate actin polymerization and lamellipodial spreading at the T cell immunological synapse. Both Abl family kinases and cortactin localize to epithelial adherens junctions where they may similarly interact to promote actin assembly critical to their stability (Helwani et al., 2004; Zandy et al., 2007; Zandy and Pendergast, 2008). Finally, Abl family kinases and cortactin both promote cancer cell invasiveness (Kain et al., 2003; Bowden et al., 2006; Hill et al., 2006; Rothschild et al., 2006; Srinivasan and Plattner, 2006; Clark et al., 2007; Clark and Weaver, 2008; Srinivasan et al., 2008). In this context, Abl and Arg may potentiate cortactin’s known ability to promote formation of invadopodia, cellular protrusions that invasive tumor cells use to degrade ECM, and basement membrane surrounding the tumor (Clark et al., 2007; Ayala et al., 2008; Clark and Weaver, 2008).
Arg functions primarily as a scaffold to promote protrusion formation
Despite our finding that Arg is required for adhesion-dependent cortactin phosphorylation, several observations suggest that Arg functions primarily in a scaffolding role to support cell edge protrusion. Indeed, we find that a KI Arg point mutant restores normal adhesion-dependent cell edge dynamics to arg−/− cells. In fact, we find that the Arg C-terminal half, which lacks the SH3, SH2, and kinase domains, supports adhesion-dependent cell edge protrusions, albeit with reduced potency relative to the FL Arg. In addition to its cortactin-binding PXXP1 motif, the Arg 557-C fragment contains both F-actin and microtubule-binding domains that mediate its proper localization to the cell periphery (Wang et al., 2001; Miller et al., 2004). Thus, the Arg C-terminal half serves as both a physical and functional bridge between the F-actin and microtubule cytoskeletons and the actin polymerization machinery.
Does cortactin act as a scaffold for the assembly of actin regulatory complexes?
The cortactin N-terminal half contains the NTA and cortactin repeats that mediate interactions with the Arp2/3 complex and F-actin, respectively (Weaver et al., 2001; for review see Weed and Parsons, 2001). These cortactin features are sufficient to weakly activate the Arp2/3 complex and to stabilize the resulting F-actin branch points (Uruno et al., 2001; Weaver et al., 2001). Bryce et al. (2005) have shown that cortactin enhances lamellipodial persistence in human fibrosarcoma cells, and this function requires just the NTA and cortactin repeats. Together, these findings strongly suggest that cortactin’s role in F-actin branch formation/stabilization contributes to overall lamellipodial persistence.
The cortactin C-terminal half contains additional domains/motifs (e.g., phospho-Tyr residues, a Pro-rich region, and an SH3 domain) that can potentially mediate interactions with several cytoskeletal effectors, including the Arp2/3 activator N-WASp and the Wiskott-Aldrich syndrome protein–interacting protein (Mizutani et al., 2002; Kinley et al., 2003; for reviews see Weed and Parsons, 2001; Weaver et al., 2003; Daly, 2004; Cosen-Binker and Kapus, 2006). In this study, we demonstrate that the cortactin SH3 domain binds Arg. It is unclear how cortactin can use its SH3 domain to interact with both Arg and N-WASp or other proteins simultaneously. One possibility is that initial cortactin SH3 binding to Arg is remodeled to allow subsequent cortactin SH3 domain binding to N-WASp. Alternatively, Arg and N-WASp could bind simultaneously to SH3 domains on different cortactin molecules within the same Arp2/3 regulatory complex. A role for cortactin as an actin regulatory scaffold may explain the ability of the C-terminal cortactin fragment lacking the NTA and cortactin repeats to enhance migration of mammary epithelial cells (Kowalski et al., 2005).
Cortactin phosphorylation reinforces Arg–cortactin interactions and may promote recruitment of additional cytoskeletal regulators
We also show that the Arg SH2 domain can bind cortactin-P. This binding interaction significantly increases the overall affinity of Arg for cortactin by at least fivefold. Mutations that disrupt cortactin phosphorylation or Arg SH2 domain binding reduce protrusions to levels intermediate between WT and arg−/− or cortactin KD cells. Importantly, this protrusion level is similar to those supported by the Arg 557-C C-terminal fragment. These data suggest that a core Arg–cortactin complex is able to support low level protrusive activity but that Arg-mediated cortactin phosphorylation can significantly increase activity of this complex in vivo.
The binding of cortactin-P to the Arg SH2 domain would be expected to enhance Arg kinase activity by locking it in an active conformation (Hantschel et al., 2003). Thus, cortactin binding to Arg could serve as a positive feedback loop for the phosphorylation of additional cortactin molecules or other substrates. Our data and work by Tehrani et al. (2007) suggest that cortactin phosphorylation also recruits the SH2 domain–containing Nck1 adapter protein to regulate actin polymerization at protrusion sites. Tehrani et al. (2007) demonstrate that the stimulation of Arp2/3 complex–mediated actin nucleation in a purified reconstituted system requires Nck1. Having identified Nck1 as a cortactin-P–binding protein (unpublished data), in this study, we show that Nck1 colocalizes with cortactin at the cell periphery, where it is required for protrusions. Antibody-induced Nck1 clustering is sufficient to trigger localized actin polymerization (Rivera et al., 2004). Nck1 has also been shown to bind the third PXXP motif in Abl to promote filopodial protrusion in adhering fibroblasts, and this motif is conserved in Arg (Antoku et al., 2008). Together, these data strongly suggest that Nck1 promotes formation of adhesion-dependent protrusions by serving as a critical reinforcing bridge between Arg, cortactin-P, and other components of the actin polymerization apparatus, most likely N-WASp (Rohatgi et al., 2001).
Model
We propose a model (Fig. 9) for how Arg interacts with cortactin to promote adhesion-dependent cell edge protrusion. (1) Arg localizes to the cell periphery where it uses its PXXP1 motif to bind the cortactin SH3 domain. (2) Adhesion stimulates Arg kinase activity. Arg phosphorylates cortactin, creating an additional high affinity binding site for Arg and possibly other proteins. In the absence of Arg, Abl is also capable of mediating cortactin phosphorylation in response to soluble cues. (3a) Phosphorylation-dependent remodeling of the Arg–cortactin complex allows the SH3 domain of cortactin to interact with N-WASp, stimulating actin polymerization and leading to cell edge protrusion. (3b) As an alternative or in addition, cortactin phosphorylation recruits other SH2 domain–containing proteins such as Nck1. Nck1’s three SH3 domains bind and activate N-WASp to promote actin polymerization (Rohatgi et al., 2001; Rivera et al., 2004), leading to cell edge protrusions.
Figure 9.

Model for adhesion-dependent cell edge protrusion. (Step 1) Arg localizes to the cell periphery and binds the SH3 domain of cortactin via its PXXP1 motif. (Step 2) The kinase activity of Arg is activated upon adhesion. Arg phosphorylates cortactin, creating an additional binding site to the Arg SH2 domain. In the absence of Arg, Abl can phosphorylate cortactin in the presence of soluble growth factors. (Step 3a) Cortactin phosphorylation leads to a remodeling of the Arg–cortactin complex, allowing the cortactin SH3 domain to interact with N-WASp. These interactions lead to actin remodeling and protrusion formation. (Step 3b) Alternatively or in addition, cortactin phosphorylation recruits other SH2 domain–containing proteins such as Nck1. Nck1 contains three SH3 domains, which can interact with and activate N-WASp to promote actin polymerization. GBD, GTPase-binding domain; MT, microtubule; VCA, verprolin-cofilin-acidic domain.
Materials and methods
Molecular cloning and purification of recombinant protein
Murine Arg and Arg mutant cDNAs were cloned into pFastBac expression vectors (QIAGEN), expressed, and purified as described previously (Wang et al., 2001). Murine cortactin and cortactin mutants were expressed and purified as described previously (Boyle et al., 2007).
Cell culture and retroviral expression
WT, arg−/−, and cortactin KD cells were maintained as previously described (Miller et al., 2004; Boyle et al., 2007). Arg KI-YFP and Arg 557-C–YFP have been previously described (Miller et al., 2004). Arg 688-C–YFP was amplified by PCR from murine Arg using the amino acid numbering system for type IV myristoylated Arg and cloned into N1-EYFP (Clontech Laboratories, Inc.). The tagged sequence was then subcloned into the retroviral expression vector PK1 (a gift from A.M. Pendergast; Duke University, Durham, NC). Arg PXXP point mutants were introduced by PCR and site-directed mutagenesis. For PXXP mut123, we mutated P567A, P570A, P573A, and P576A (PXXP1), P622A and P625A (PXXP2), and P664A and P667A (PXXP3). The cortactinΔSH3–Arg 688-C–RFP fusion protein was constructed by inserting PCR-amplified cortactinΔSH3 sequence (aa 1–494) followed by a 5-aa linker (GSGGG) upstream of Arg 688-C into the monomeric RFP–N1 vector. This tagged fusion was subcloned into the PK1 expression vector. Expression levels of retrovirally expressed Arg or Arg mutants or the cortactinΔSH3–Arg 688-C–RFP fusions were determined by semiquantitative immunoblotting of 1.5 µg/ml puromycin-selected cells, and the signal was compared with standardized concentrations of either purified Arg or purified YFP.
RNAi-mediated cortactin KD and rescue
Cortactin KD cells and rescue constructs for FL cortactin and cortactin 3F were previously described (Boyle et al., 2007). Cortactin WA contains a W525A mutation. All mutants were subcloned into the PK1 retroviral expression system. Cortactin was knocked down by 80%, as determined by immunoblotting using pSuper empty vector cell extract as a control.
Nck1 was stably knocked down in WT fibroblasts using methods described previously (Boyle et al., 2007) using a 19-mer sequence (5′-GATTATGGCTCCTGGATGA-3′) to murine Nck1 (Simpson et al., 2008). Nck1 was knocked down by 92%, as determined by immunoblotting (Nck1 antibody; Cell Signaling Technology). Cells were rescued with a retrovirally expressed human Nck1-GFP fusion protein, which is RNAi resistant because of sequence differences in the targeted region.
Immunofluorescence microscopy
arg−/− + Arg-YFP cells and cortactin KD cells expressing Arg/Arg mutant–YFP fusions or cortactin/cortactin mutant–RFP fusions were plated on glass coverslips coated with 10 µg/ml fibronectin (Invitrogen), fixed, stained, and imaged as previously described (Miller et al., 2004).
Time-lapse microscopy and kymography
For colocalization experiments, arg−/− + Arg-YFP cells were selected in 1.5 µg/ml puromycin and subsequently infected with RFP-tagged cortactin. Cells were adapted to growth in microscopy media (DME + 10% FBS and 10 mM Hepes; Invitrogen) for ∼16 h and imaged between 20 min and 2 h after plating using a microscope (TE2000-S; Nikon) driven by Openlab software (PerkinElmer). Cells were maintained at 37°C during imaging with an in-line flow heater and a heated chamber (Warner Instruments). 40× NA 1.0 objective (Nikon) phase-contrast and YFP videos were 10-min long with frames taken every 10 s with a cooled mono 12-bit camera (Retiga; QImaging). For colocalization experiments, exposures of the cells in the phase, YFP, and RFP channels were taken sequentially every 30 s. Kymography was performed as described previously (Miller et al., 2004) using ImageJ software (National Institutes of Health). Images and kymographs were cropped in Photoshop (Adobe) and were adjusted using the auto-level function. Statistical analysis of the protrusion data was performed by using analysis of variance (ANOVA) using StatView software (SAS Institute).
Adhesion assays and immunoprecipitation
WT and arg−/− cells were serum-starved in DME containing 0.1% FBS for at least 1 h and kept in suspension for 15 min. An equal volume of cells was harvested by centrifugation (for the 0 time point) or plated on tissue culture dishes coated with 10 µg/ml fibronectin. Cells were harvested by scraping at 20, 30, or 40 min after plating and lysed in modified radio immunoprecipitation assay buffer (50 mM Tris, pH 7.2, 150 mM NaCl, 1% NP-40, 0.5% deoxycholate, 0.1% SDS, 1 mM EDTA, 2 mM NaF, 1 mM Na3VO4, and protease inhibitors). Total protein concentration was determined using a bicinchoninic acid protein assay kit (Thermo Fisher Scientific), and an equal amount of total protein per time point was subjected to immunoprecipitation with 2 µg anticortactin antibody (4F11; Millipore) and protein A/G beads (EMD). The immunoprecipitate was immunoblotted as described previously (Boyle et al., 2007). Phospho-Tyr and total cortactin levels were quantitated using ImageJ software. Treatment of WT cells with 10 µM STI-571 was conducted overnight, during serum starvation, and during plating of cells on fibronectin.
Measurements of cortactin–Arg binding
Recombinant cortactin protein was covalently coupled to Affigel 15 resin (at a concentration of 1 mg/ml of gel bed [16.3 µM]; Bio-Rad Laboratories) according to the manufacturer’s protocol. The cortactin beads were washed twice with ice-cold binding buffer (50 mM Hepes, pH 7.25, 125 mM NaCl, 0.01% NP-40, and 5% glycerol) and stored as a 50% slurry. Arg protein was diluted 1:3 in a serial fashion; 20 µl was saved as input sample, and 490 µl of each dilution was added to Eppendorf tubes containing 50 µl of cortactin bead slurry or 50 µl of blank bead slurry blocked with an excess of ethanolamine. The reaction was incubated at 4°C while rotating for 2 h. The supernatant was removed, and the beads were rapidly resuspended with 1 ml of ice-cold binding buffer. Bound material was recovered with 4× Laemmli sample buffer, analyzed on 8% SDS-PAGE gels, stained with Coomassie blue R-250, and scanned using a densitometer (ImageQuant 1; Bio-Rad Laboratories). Intensity levels were adjusted using the auto-adjust feature in Quantity One software (Bio-Rad Laboratories). Integrated density measurements were made of the band as well as an empty area of the cell immediately above the band. This background measurement was subtracted from the band density measurement. Binding curves were generated using GraphPad Prism software (Bio-Rad Laboratories) using the equation
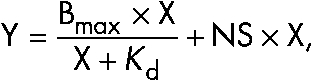 |
where Y equals the specific binding signal, X equals the concentration of ligand added to the cortactin beads, and NS equals the slope of the least-squares linear regression fit for the nonspecific binding as measured with the ethanolamine beads. For representative gels, the Photoshop auto-level function was used, and the gels were cropped to display the bands of interaction for each gel.
Recombinant cortactin-P was generated by coexpressing His-tagged cortactin with untagged Arg using baculoviral vectors in insect cells. Cell lysates were incubated with nickel beads, and three high salt washes (0.5 M, 1.25 M, 0.5 M KCl, respectively) were used to dislodge cortactin-bound Arg. Cortactin-P was eluted from the nickel column using 200 mM imidazole, and its phosphorylation status was verified via Western blotting with a cocktail of anti–phosphor-Tyr antibodies.
Recombinant SH2 domain binding was measured on cortactin affixed to nitrocellulose membranes as described previously (Nollau and Mayer, 2001; Machida et al., 2007). In brief, 0.4 µg in 1 µl dots of cortactin-P, cortactin 3F, pervanadate-treated cell lysate rich in phosphor-Tyr–containing proteins, or phosphatase-treated cell lysate was spotted in duplicate on a nitrocellulose membrane and allowed to dry overnight at room temperature in a 6-well culture dish. Membranes were soaked in transfer buffer (20% MeOH, 12.5 mM Tris-HCl, pH 8.0, and 100 mM Gly) for 30 min, rinsed twice with TBST (150 mM NaCl,10 mM Tris-HCl, pH 8.0, and 0.05% Tween 20), and blocked at room temperature for 1 h in TBST with 10% nonfat milk, 1 mM Na3VO4, and 1 mM EDTA. Purified recombinant GST-SH2 domains of Arg, Abl, and Grb2 conjugated to glutathione–horseradish peroxidase were added at a concentration of 1 µg/ml to separate chambers of the 6-well culture dish and allowed to incubate at room temperature for 1 h with constant orbital rotation. The membranes were then washed three times with TBST for 5 min each, dried, incubated with ECL, and exposed to film.
Online supplemental material
Fig. S1 shows the expression levels and localization of cortactin or cortactin mutants in cortactin KD cells. Fig. S2 shows the expression levels of Arg and Arg mutants in arg−/− cells. Fig. S3 shows that cortactin uses its SH3 domain to bind to the first PXXP region of Arg. Fig. S4 shows that Arg mutants colocalize with F-actin at the cell periphery. Fig. S5 shows that arg−/− cells expressing Arg 557-C PXXP mut123 have reduced protrusion dynamics. Video 1 shows that Arg-YFP and cortactin-RFP colocalize to membrane protrusions in response to adhesion to fibronectin. Online supplemental material is available at http://www.jcb.org/cgi/content/full/jcb.200809085/DC1.
Acknowledgments
We are grateful to Xianyun Ye for technical support. We thank Scott Boyle, Bill Bradley, Kevin Collins, Hava Henn, Michael Koelle, and Sloan Warren for stimulating discussions and critical feedback on this work. We thank Susan Keezer at Cell Signaling Technology for the Nck and Src antibodies.
This work was supported by U.S. Public Health Service grants NS39475 and MH77306 and a grant from the Elsa U. Pardee Foundation (to A.J. Koleske). C.C. Mader was supported by a National Science Foundation Predoctoral Fellowship. A.J. Koleske is an American Heart Association Established Investigator.
Footnotes
Abbreviations used in this paper: Abl, Abelson; ANOVA, analysis of variance; Arg, Abl-related gene; cortactin-P, phosphorylated cortactin; dko, double knockout; FL, full length; KD, knockdown; KI, kinase inactive; NTA, N-terminal acidic; N-WASp, neural Wiskott-Aldrich syndrome protein; PLSD, protected least significant difference; SH, Src homology; WT, wild type.
References
- Antoku S., Saksela K., Rivera G.M., Mayer B.J. 2008. A crucial role in cell spreading for the interaction of Abl PxxP motifs with Crk and Nck adaptors.J. Cell Sci. 121:3071–3082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala I., Baldassarre M., Giacchetti G., Caldieri G., Tete S., Luini A., Buccione R. 2008. Multiple regulatory inputs converge on cortactin to control invadopodia biogenesis and extracellular matrix degradation.J. Cell Sci. 121:369–378 [DOI] [PubMed] [Google Scholar]
- Biesova Z., Piccoli C., Wong W.T. 1997. Isolation and characterization of e3B1, an eps8 binding protein that regulates cell growth.Oncogene. 14:233–241 [DOI] [PubMed] [Google Scholar]
- Bowden E.T., Barth M., Thomas D., Glazer R.I., Mueller S.C. 1999. An invasion-related complex of cortactin, paxillin and PKCmu associates with invadopodia at sites of extracellular matrix degradation.Oncogene. 18:4440–4449 [DOI] [PubMed] [Google Scholar]
- Bowden E.T., Onikoyi E., Slack R., Myoui A., Yoneda T., Yamada K.M., Mueller S.C. 2006. Co-localization of cortactin and phosphotyrosine identifies active invadopodia in human breast cancer cells.Exp. Cell Res. 312:1240–1253 [DOI] [PubMed] [Google Scholar]
- Boyle S.N., Michaud G.A., Schweitzer B., Predki P.F., Koleske A.J. 2007. A critical role for cortactin phosphorylation by Abl-family kinases in PDGF-induced dorsal-wave formation.Curr. Biol. 17:445–451 [DOI] [PubMed] [Google Scholar]
- Bradley W.D., Hernandez S.E., Settleman J., Koleske A.J. 2006. Integrin signaling through Arg activates p190RhoGAP by promoting its binding to p120RasGAP and recruitment to the membrane.Mol. Biol. Cell. 17:4827–4836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryce N.S., Clark E.S., Leysath J.L., Currie J.D., Webb D.J., Weaver A.M. 2005. Cortactin promotes cell motility by enhancing lamellipodial persistence.Curr. Biol. 15:1276–1285 [DOI] [PubMed] [Google Scholar]
- Burton E.A., Oliver T.N., Pendergast A.M. 2005. Abl kinases regulate actin comet tail elongation via an N-WASP-dependent pathway.Mol. Cell. Biol. 25:8834–8843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao H., Orth J.D., Chen J., Weller S.G., Heuser J.E., McNiven M.A. 2003. Cortactin is a component of clathrin-coated pits and participates in receptor-mediated endocytosis.Mol. Cell. Biol. 23:2162–2170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark E.S., Weaver A.M. 2008. A new role for cortactin in invadopodia: regulation of protease secretion.Eur. J. Cell Biol. 87:581–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark E.S., Whigham A.S., Yarbrough W.G., Weaver A.M. 2007. Cortactin is an essential regulator of matrix metalloproteinase secretion and extracellular matrix degradation in invadopodia.Cancer Res. 67:4227–4235 [DOI] [PubMed] [Google Scholar]
- Cohen G.B., Ren R., Baltimore D. 1995. Modular binding domains in signal transduction proteins.Cell. 80:237–248 [DOI] [PubMed] [Google Scholar]
- Comer A.R., Ahern-Djamali S.M., Juang J.L., Jackson P.D., Hoffmann F.M. 1998. Phosphorylation of Enabled by the Drosophila Abelson tyrosine kinase regulates the in vivo function and protein-protein interactions of Enabled.Mol. Cell. Biol. 18:152–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong F., Yuan B., Goff S.P. 1999. Characterization of a novel member of the DOK family that binds and modulates Abl signaling.Mol. Cell. Biol. 19:8314–8325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosen-Binker L.I., Kapus A. 2006. Cortactin: the gray eminence of the cytoskeleton.Physiology (Bethesda). 21:352–361 [DOI] [PubMed] [Google Scholar]
- Dai Z., Pendergast A.M. 1995. Abi-2, a novel SH3-containing protein interacts with the c-Abl tyrosine kinase and modulates c-Abl transforming activity.Genes Dev. 9:2569–2582 [DOI] [PubMed] [Google Scholar]
- Daly R.J. 2004. Cortactin signalling and dynamic actin networks.Biochem. J. 382:13–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rooij J., Kerstens A., Danuser G., Schwartz M.A., Waterman-Storer C.M. 2005. Integrin-dependent actomyosin contraction regulates epithelial cell scattering.J. Cell Biol. 171:153–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupton S.L., Waterman-Storer C.M. 2006. Spatiotemporal feedback between actomyosin and focal-adhesion systems optimizes rapid cell migration.Cell. 125:1361–1374 [DOI] [PubMed] [Google Scholar]
- Hantschel O., Nagar B., Guettler S., Kretzschmar J., Dorey K., Kuriyan J., Superti-Furga G. 2003. A myristoyl/phosphotyrosine switch regulates c-Abl.Cell. 112:845–857 [DOI] [PubMed] [Google Scholar]
- Head J.A., Jiang D., Li M., Zorn L.J., Schaefer E.M., Parsons J.T., Weed S.A. 2003. Cortactin tyrosine phosphorylation requires Rac1 activity and association with the cortical actin cytoskeleton.Mol. Biol. Cell. 14:3216–3229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helwani F.M., Kovacs E.M., Paterson A.D., Verma S., Ali R.G., Fanning A.S., Weed S.A., Yap A.S. 2004. Cortactin is necessary for E-cadherin-mediated contact formation and actin reorganization.J. Cell Biol. 164:899–910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez S.E., Settleman J., Koleske A.J. 2004. Adhesion-dependent regulation of p190RhoGAP in the developing brain by the Abl-related gene tyrosine kinase.Curr. Biol. 14:691–696 [DOI] [PubMed] [Google Scholar]
- Hill A., McFarlane S., Mulligan K., Gillespie H., Draffin J.E., Trimble A., Ouhtit A., Johnston P.G., Harkin D.P., McCormick D., Waugh D.J. 2006. Cortactin underpins CD44-promoted invasion and adhesion of breast cancer cells to bone marrow endothelial cells.Oncogene. 25:6079–6091 [DOI] [PubMed] [Google Scholar]
- Hinz B., Alt W., Johnen C., Herzog V., Kaiser H.W. 1999. Quantifying lamella dynamics of cultured cells by SACED, a new computer-assisted motion analysis.Exp. Cell Res. 251:234–243 [DOI] [PubMed] [Google Scholar]
- Huang Y., Comiskey E.O., Dupree R.S., Li S., Koleske A.J., Burkhardt J.K. 2008. The c-Abl tyrosine kinase regulates actin remodeling at the immune synapse.Blood. 112:111–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jay P.Y., Pham P.A., Wong S.A., Elson E.L. 1995. A mechanical function of myosin II in cell motility.J. Cell Sci. 108:387–393 [DOI] [PubMed] [Google Scholar]
- Juang J.L., Hoffmann F.M. 1999. Drosophila abelson interacting protein (dAbi) is a positive regulator of abelson tyrosine kinase activity.Oncogene. 18:5138–5147 [DOI] [PubMed] [Google Scholar]
- Kain K.H., Gooch S., Klemke R.L. 2003. Cytoplasmic c-Abl provides a molecular ‘Rheostat’ controlling carcinoma cell survival and invasion.Oncogene. 22:6071–6080 [DOI] [PubMed] [Google Scholar]
- Kinley A.W., Weed S.A., Weaver A.M., Karginov A.V., Bissonette E., Cooper J.A., Parsons J.T. 2003. Cortactin interacts with WIP in regulating Arp2/3 activation and membrane protrusion.Curr. Biol. 13:384–393 [DOI] [PubMed] [Google Scholar]
- Koleske A.J., Gifford A.M., Scott M.L., Nee M., Bronson R.T., Miczek K.A., Baltimore D. 1998. Essential roles for the Abl and Arg tyrosine kinases in neurulation.Neuron. 21:1259–1272 [DOI] [PubMed] [Google Scholar]
- Kowalski J.R., Egile C., Gil S., Snapper S.B., Li R., Thomas S.M. 2005. Cortactin regulates cell migration through activation of N-WASP.J. Cell Sci. 118:79–87 [DOI] [PubMed] [Google Scholar]
- Lauffenburger D.A., Horwitz A.F. 1996. Cell migration: a physically integrated molecular process.Cell. 84:359–369 [DOI] [PubMed] [Google Scholar]
- Leng Y., Zhang J., Badour K., Arpaia E., Freeman S., Cheung P., Siu M., Siminovitch K. 2005. Abelson-interactor-1 promotes WAVE2 membrane translocation and Abelson-mediated tyrosine phosphorylation required for WAVE2 activation.Proc. Natl. Acad. Sci. USA. 102:1098–1103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machida K., Thompson C.M., Dierck K., Jablonowski K., Karkkainen S., Liu B., Zhang H., Nash P.D., Newman D.K., Nollau P., et al. 2007. High-throughput phosphotyrosine profiling using SH2 domains.Mol. Cell. 26:899–915 [DOI] [PubMed] [Google Scholar]
- Martinez-Quiles N., Ho H.Y., Kirschner M.W., Ramesh N., Geha R.S. 2004. Erk/Src phosphorylation of cortactin acts as a switch on-switch off mechanism that controls its ability to activate N-WASP.Mol. Cell. Biol. 24:5269–5280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Master Z., Tran J., Bishnoi A., Chen S.H., Ebos J.M., Van Slyke P., Kerbel R.S., Dumont D.J. 2003. Dok-R binds c-Abl and regulates Abl kinase activity and mediates cytoskeletal reorganization.J. Biol. Chem. 278:30170–30179 [DOI] [PubMed] [Google Scholar]
- McNiven M.A., Cao H., Pitts K.R., Yoon Y. 2000. The dynamin family of mechanoenzymes: pinching in new places.Trends Biochem. Sci. 25:115–120 [DOI] [PubMed] [Google Scholar]
- Merrifield C.J., Perrais D., Zenisek D. 2005. Coupling between clathrin-coated-pit invagination, cortactin recruitment, and membrane scission observed in live cells.Cell. 121:593–606 [DOI] [PubMed] [Google Scholar]
- Miller A.L., Wang Y., Mooseker M.S., Koleske A.J. 2004. The Abl-related gene (Arg) requires its F-actin-microtubule cross-linking activity to regulate lamellipodial dynamics during fibroblast adhesion.J. Cell Biol. 165:407–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchison T.J., Cramer L.P. 1996. Actin-based cell motility and cell locomotion.Cell. 84:371–379 [DOI] [PubMed] [Google Scholar]
- Mizutani K., Miki H., He H., Maruta H., Takenawa T. 2002. Essential role of neural Wiskott-Aldrich syndrome protein in podosome formation and degradation of extracellular matrix in src-transformed fibroblasts.Cancer Res. 62:669–674 [PubMed] [Google Scholar]
- Moresco E.M., Donaldson S., Williamson A., Koleske A.J. 2005. Integrin-mediated dendrite branch maintenance requires Abelson (Abl) family kinases.J. Neurosci. 25:6105–6118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nollau P., Mayer B.J. 2001. Profiling the global tyrosine phosphorylation state by Src homology 2 domain binding.Proc. Natl. Acad. Sci. USA. 98:13531–13536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peacock J.G., Miller A.L., Bradley W.D., Rodriguez O.C., Webb D.J., Koleske A.J. 2007. The Abl-related gene tyrosine kinase acts through p190RhoGAP to inhibit actomyosin contractility and regulate focal adhesion dynamics upon adhesion to fibronectin.Mol. Biol. Cell. 18:3860–3872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plattner R., Kadlec L., DeMali K.A., Kazlauskas A., Pendergast A.M. 1999. c-Abl is activated by growth factors and Src family kinases and has a role in the cellular response to PDGF.Genes Dev. 13:2400–2411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plattner R., Irvin B.J., Guo S., Blackburn K., Kazlauskas A., Abraham R.T., York J.D., Pendergast A.M. 2003. A new link between the c-Abl tyrosine kinase and phosphoinositide signalling through PLC-gamma1.Nat. Cell Biol. 5:309–319 [DOI] [PubMed] [Google Scholar]
- Plattner R., Koleske A.J., Kazlauskas A., Pendergast A.M. 2004. Bidirectional signaling links the Abelson kinases to the platelet-derived growth factor receptor.Mol. Cell. Biol. 24:2573–2583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard T.D. 2007. Regulation of actin filament assembly by Arp2/3 complex and formins.Annu. Rev. Biophys. Biomol. Struct. 36:451–477 [DOI] [PubMed] [Google Scholar]
- Pollard T.D., Borisy G.G. 2003. Cellular motility driven by assembly and disassembly of actin filaments.Cell. 112:453–465 [DOI] [PubMed] [Google Scholar]
- Ponti A., Machacek M., Gupton S.L., Waterman-Storer C.M., Danuser G. 2004. Two distinct actin networks drive the protrusion of migrating cells.Science. 305:1782–1786 [DOI] [PubMed] [Google Scholar]
- Ridley A.J., Schwartz M.A., Burridge K., Firtel R.A., Ginsberg M.H., Borisy G., Parsons J.T., Horwitz A.R. 2003. Cell migration: integrating signals from front to back.Science. 302:1704–1709 [DOI] [PubMed] [Google Scholar]
- Rivera G.M., Briceno C.A., Takeshima F., Snapper S.B., Mayer B.J. 2004. Inducible clustering of membrane-targeted SH3 domains of the adaptor protein Nck triggers localized actin polymerization.Curr. Biol. 14:11–22 [DOI] [PubMed] [Google Scholar]
- Rohatgi R., Nollau P., Ho H.Y., Kirschner M.W., Mayer B.J. 2001. Nck and phosphatidylinositol 4,5-bisphosphate synergistically activate actin polymerization through the N-WASP-Arp2/3 pathway.J. Biol. Chem. 276:26448–26452 [DOI] [PubMed] [Google Scholar]
- Rothschild B.L., Shim A.H., Ammer A.G., Kelley L.C., Irby K.B., Head J.A., Chen L., Varella-Garcia M., Sacks P.G., Frederick B., et al. 2006. Cortactin overexpression regulates actin-related protein 2/3 complex activity, motility, and invasion in carcinomas with chromosome 11q13 amplification.Cancer Res. 66:8017–8025 [DOI] [PubMed] [Google Scholar]
- Shah K., Vincent F. 2005. Divergent roles of c-Src in controlling platelet-derived growth factor-dependent signaling in fibroblasts.Mol. Biol. Cell. 16:5418–5432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y., Alin K., Goff S.P. 1995. Abl-interactor-1, a novel SH3 protein binding to the carboxy-terminal portion of the Abl protein, suppresses v-abl transforming activity.Genes Dev. 9:2583–2597 [DOI] [PubMed] [Google Scholar]
- Simpson J.P., Di Leo R., Dhanoa P.K., Allan W.L., Makhmoudova A., Clark S.M., Hoover G.J., Mullen R.T., Shelp B.J. 2008. Identification and characterization of a plastid-localized Arabidopsis glyoxylate reductase isoform: comparison with a cytosolic isoform and implications for cellular redox homeostasis and aldehyde detoxification.J. Exp. Bot. 59:2545–2554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sini P., Cannas A., Koleske A.J., Di Fiore P.P., Scita G. 2004. Abl-dependent tyrosine phosphorylation of Sos-1 mediates growth-factor-induced Rac activation.Nat. Cell Biol. 6:268–274 [DOI] [PubMed] [Google Scholar]
- Srinivasan D., Plattner R. 2006. Activation of Abl tyrosine kinases promotes invasion of aggressive breast cancer cells.Cancer Res. 66:5648–5655 [DOI] [PubMed] [Google Scholar]
- Srinivasan D., Sims J.T., Plattner R. 2008. Aggressive breast cancer cells are dependent on activated Abl kinases for proliferation, anchorage-independent growth and survival.Oncogene. 27:1095–1105 [DOI] [PubMed] [Google Scholar]
- Stuart J.R., Gonzalez F.H., Kawai H., Yuan Z.M. 2006. c-Abl interacts with the WAVE2 signaling complex to induce membrane ruffling and cell spreading.J. Biol. Chem. 281:31290–31297 [DOI] [PubMed] [Google Scholar]
- Tani K., Sato S., Sukezane T., Kojima H., Hirose H., Hanafusa H., Shishido T. 2003. Abl interactor 1 promotes tyrosine 296 phosphorylation of mammalian enabled (Mena) by c-Abl kinase.J. Biol. Chem. 278:21685–21692 [DOI] [PubMed] [Google Scholar]
- Tehrani S., Tomasevic N., Weed S., Sakowicz R., Cooper J.A. 2007. Src phosphorylation of cortactin enhances actin assembly.Proc. Natl. Acad. Sci. USA. 104:11933–11938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas S.M., Soriano P., Imamoto A. 1995. Specific and redundant roles of Src and Fyn in organizing the cytoskeleton.Nature. 376:267–271 [DOI] [PubMed] [Google Scholar]
- Uruno T., Liu J., Zhang P., Fan Y., Egile C., Li R., Mueller S.C., Zhan X. 2001. Activation of Arp2/3 complex-mediated actin polymerization by cortactin.Nat. Cell Biol. 3:259–266 [DOI] [PubMed] [Google Scholar]
- Wang Y., Miller A.L., Mooseker M.S., Koleske A.J. 2001. The Abl-related gene (Arg) nonreceptor tyrosine kinase uses two F-actin-binding domains to bundle F-actin.Proc. Natl. Acad. Sci. USA. 98:14865–14870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver A.M., Karginov A.V., Kinley A.W., Weed S.A., Li Y., Parsons J.T., Cooper J.A. 2001. Cortactin promotes and stabilizes Arp2/3-induced actin filament network formation.Curr. Biol. 11:370–374 [DOI] [PubMed] [Google Scholar]
- Weaver A.M., Heuser J.E., Karginov A.V., Lee W.L., Parsons J.T., Cooper J.A. 2002. Interaction of cortactin and N-WASp with Arp2/3 complex.Curr. Biol. 12:1270–1278 [DOI] [PubMed] [Google Scholar]
- Weaver A.M., Young M.E., Lee W.L., Cooper J.A. 2003. Integration of signals to the Arp2/3 complex.Curr. Opin. Cell Biol. 15:23–30 [DOI] [PubMed] [Google Scholar]
- Weed S.A., Parsons J.T. 2001. Cortactin: coupling membrane dynamics to cortical actin assembly.Oncogene. 20:6418–6434 [DOI] [PubMed] [Google Scholar]
- Weed S.A., Du Y., Parsons J.T. 1998. Translocation of cortactin to the cell periphery is mediated by the small GTPase Rac1.J. Cell Sci. 111:2433–2443 [DOI] [PubMed] [Google Scholar]
- Weed S.A., Karginov A.V., Schafer D.A., Weaver A.M., Kinley A.W., Cooper J.A., Parsons J.T. 2000. Cortactin localization to sites of actin assembly in lamellipodia requires interactions with F-actin and the Arp2/3 complex.J. Cell Biol. 151:29–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodring P.J., Litwack E.D., O’Leary D.D., Lucero G.R., Wang J.Y., Hunter T. 2002. Modulation of the F-actin cytoskeleton by c-Abl tyrosine kinase in cell spreading and neurite extension.J. Cell Biol. 156:879–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodring P.J., Meisenhelder J., Johnson S.A., Zhou G.L., Field J., Shah K., Bladt F., Pawson T., Niki M., Pandolfi P.P., et al. 2004. c-Abl phosphorylates Dok1 to promote filopodia during cell spreading.J. Cell Biol. 165:493–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H., Reynolds A.B., Kanner S.B., Vines R.R., Parsons J.T. 1991. Identification and characterization of a novel cytoskeleton-associated pp60src substrate.Mol. Cell. Biol. 11:5113–5124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zandy N.L., Pendergast A.M. 2008. Abl tyrosine kinases modulate cadherin-dependent adhesion upstream and downstream of Rho family GTPases.Cell Cycle. 7:444–448 [DOI] [PubMed] [Google Scholar]
- Zandy N.L., Playford M., Pendergast A.M. 2007. Abl tyrosine kinases regulate cell-cell adhesion through Rho GTPases.Proc. Natl. Acad. Sci. USA. 104:17686–17691 [DOI] [PMC free article] [PubMed] [Google Scholar]