

Abstract
Background
The correct folding and dimerization of tubulins, before their addition to the microtubular structure, needs a group of conserved proteins called cofactors A to E. The biochemical analysis of cofactors gave an insight to their general functions, however not much is known about the domain structure and detailed, molecular function of these proteins.
Results
Combining modelling and fold prediction tools, we present 3D models of all cofactors, including several previously unannotated domains of cofactors B-E. Apart from the new HEAT and Armadillo domains in cofactor D and an unusual spectrin-like domain in cofactor C, we have identified a new subfamily of ubiquitin-like domains in cofactors B and E. Together, these observations provide a reliable, molecular level model of cofactor complex.
Conclusion
Distant homology searches allowed the identification of unknown regions of cofactors as self-reliant domains and allow us to present a detailed hypothesis of how a cofactor complex performs its function.
Background
The cytoskeleton plays a major role in many cell processes. However, it is not a passive scaffold. It is an active system, continuously changing its characteristics, and in particular constantly building and destroying the opposite ends of polymers forming the scaffold. Microtubule metabolism performs one of the leading roles in regulating various features of the cytoskeleton. The α- and β-tubulin dimerization and polymerization processes leading to microtubule formation are controlled by several multi-domain proteins. After translation of both proteins a group of chaperones called chaperonines (e.g. CCT) capture the tubulins and begin the folding of tubulin monomers [1]. Chaperonines are sufficient to fold completely actin and γ-tubulin but another set of proteins is needed for the final folding and dimerization of α- and β-tubulins [2-5]. In the last ten years cofactors from different eukaryotic organisms were cloned and analyzed [4,6-18]. Apparently the set consists of five cofactors A, B, C, D and E and an ADP ribosylation factor-like protein 2, Arl2 that regulates the interaction of cofactor D with native tubulin.
The process of folding and dimerization of tubulins is complex. After release from chaperonin α-tubulin is seized by cofactorB while β-tubulin is captured by cofactor A. The next step is the replacement of cofactors B and A by cofactors E and D, respectively. The last stage of this process involves the formation of a super-complex consisting of cofactors C-E [2,4,10,19]. The release of tubulin α/β heterodimer is catalyzed in the presence of GTP [4,18]. In the last few years, the function of the cofactors has been described in increasing detail (Fig. 1) [8,19], but little is known about their domains and structural organization. As a result, we lack understanding of how the complex of cofactors may achieve its function.
Figure 1.

Simplified model depicting the reactions involved in the assembly of the tubulin heterodimer [18]. The colors depict the previously known regions of the cofactors and correspond to the colors ascribed to various domains in Figure 2A. The α-tubulin is gray colored and β-tubulin is black colored. For clarity, the involvement of ADP ribosylation factor-like protein 2 (Arl2) in modulating the interaction of cofactors with tubulins [18] was omitted.
In recent years, advances in sensitive sequence analysis tools and fold recognition algorithms have allowed us to recognize distant homologies, giving us important insights into the function and specific activities of many proteins. Here, we describe such analysis of the domain architecture of cofactors B-D. In particular, the application of fold recognition algorithms allowed us to predict three-dimensional structures of all but one domain in cofactors B, C, D and E, and provided us with critical information to link their structure with function (Fig. 2). Such predictions complement our understanding of how the cofactor complex might work in assembling tubulin dimers by providing structure-based understanding of function of individual domains and their possible interactions.
Figure 2.

Domain structure of cofactors B-E: Domain ranges and fold recognition algorithms used for function assignment are shown under each domain. Schematic 3D structures are shown when comparative modeling could be used with significant reliability. The same colors of the boxes depict regions belonging to the same domain family. Interestingly, in the cofactor B sequence the coiled-coil region overlaps with the CAP-Gly domain. Following sequences were used for analysis: cofactor B, Q99426; cofactor C, Q15814; cofactor D, O95458; cofactor E, Q15813. The structural models are available at http://ffas.ljcrf.edu/ffas/tubulin/.
Results
The Schizosaccharomyces pombe homologue of cofactor B, Alp11 was studied in detail in a few recent publications [8,9]. This analysis identified the CLIP-170 domain, described in PFAM as the cytoskeleton-associated domain (CAP-Gly domain, Pfam01302), required for efficient binding to α-tubulin. Our analysis suggests that this domain is about twice as long as that described in PFAM (about 80 amino acids). The structure of this domain cannot be predicted with confidence; however, the secondary structure prediction programs strongly suggest the presence of six beta strands. This domain is found in many other proteins (kinesin, dynactin, restin, dynein-associated protein). It is often repeated twice or three times in one protein suggesting a possibility of a beta barrel fold for this domain. The structure of this domain from the C. elegans homologue was solved recently by the SouthWest Structure Genomics Center [20], confirming this prediction and identifying the CAP-Gly domains as having an OB-fold of a beta barrel, formed by a twisted B-sheet. This fold was previously found to be involved in DNA binding and several enzymatic activities [21], but its presence in a cytoskeleton binding proteins comes as a complete surprise.
The center of cofactor B is occupied by a short helical domain. It was suggested to have a coiled-coil structure [9], although this is not confirmed by most of the coiled-coil prediction algorithms. This fragment was also characterized as necessary for a satisfactory maintenance of cellular α-tubulin level.
The N-terminal part of cofactor B (Fig. 2 and Fig. 3A) was shown to be essential for its function [9]. A homologous domain is present in the C-terminus of cofactor E (see later), and also in several proteins that do not contain the CLIP-170 domain, including some that bind other cytoskeletal proteins. Here we show, using fold recognition algorithms, that this domain has a structure similar to ubiquitin. Some sequence patterns characteristic for ubiquitin are strongly conserved, for instance the lysine residues important for linkage in multi-ubiquitin chains [22]. Others are not, for instance the lack of the carboxyl-terminal extension containing an exposed double glycine motif essential for conjugation to target proteins [23]. The new domain can be classified in a broad ubiquitin-like (UBL) family consisting of sequences from multi-domain proteins known to be involved in the proteasome or other protein-protein interactions [23,24]. As this domain is crucial for cofactor's function we hypothesize its involvement in α-tubulin binding might be to assist CAP-Gly domain in keeping the tubulin in the folded state.
Figure 3.

A. The alignment of representative subset of ubiquitin-like domain homologs. Abbreviations: cUBL-1, (Q07371) Caenorhabditis briggsae UBL-1; hFUBI, (P35544) human FUBI; hUCRP, (P05161) human UCRP domain 2; fyUSPP, (Q92353) fission yeast ubiquitin-specific processing protease; rtRNAgtg, (P40826) rabbit tRNA-guanine transglycosylase; fyAlp11, (Q10235) fission yeast Alp11; gUBL, (Q9PTE2) Carassius auratus ubiquitin-like protein; hcofE, (Q15813) human cofactor E; hBAG1, (Q99933) human BAG1; hcofB, (Q99426) human cofactor B.B. Sequence alignment of Leucine Rich Repeats in cofactor E. Consensus amino acids are red colored and a repeating motif is shown below.
Cofactor C is composed of two domains. The N-terminal part has a strong coiled-coil sequence signature, but fold recognition programs are able to narrow down the prediction to indicate a significant similarity to the spectrin repeat. Spectrin consists of three α-helices that fold to form a short triple α-helical unit [25-27]. Several variants of the spectrin domains were identified and assigned to different functions [28-32]. Unfortunately, the cofactor C spectrin domain seems to be equally distant from all known spectrin variants. Therefore, no conclusion can be drawn regarding its function. The most intriguing problem lies in the fact that cofactor C contains only a single spectrin-like domain (the length on the N-terminal fragment corresponds to the average length of a spectrin repeat) while in all other known examples there are always many spectrin repeats forming specific super-structures [33]. This may be an indication of a complex formation with other proteins with similar domains.
The C-terminal domain of cofactor C shares a significant similarity with a domain found in other cytoskeleton-related proteins. Distantly homologous domains were detected using FFAS algorithm in the distal part of adenylyl cyclase-associated protein (CAP) which is implicated in F-actin binding (13 % sequence identity) [34] as well as in the proximal part of the RP2 protein (29 % identity). Mutations in RP2 cause the X-linked retinitis pigmentosa, a severe retinal degeneration that leads to the loss of visual acuity and blindness [35]. Secondary structure predictions for this domain suggest an all-beta structure, but there is no consensus among the fold prediction programs indicating possibly a novel fold.
Cofactor D interacts directly with β-tubulin in vivo [18]. Recent sequence analysis revealed the presence of HEAT motives in cofactor D [8]. The HEAT-repeat motif is characterized by the presence of pairs of anti-parallel helices stacked in a consecutive array. The repeats are then stacked in parallel [36] and are involved in protein-protein interactions [37]. Our analysis suggests that in addition to the previously identified HEAT repeats, the N-terminal part of cofactor D contains several Armadillo repeats which are evolutionary related to HEAT repeats, but differ by the presence of the additional short (two turns) helix followed by two longer helices [36]. Armadillo repeats are also involved in mediating the protein-protein interaction [36-38]. Fold recognition programs however were not able to recognize whether the second domain belongs to the HEAT or Armadillo family. Therefore, we conclude this part of the protein can be classified as the ARM repeat superfamily member.
It is worth noting that the analysis of the close homologue of cofactor D, the Alp1 protein from S. pombe, revealed a significant difference in structure of both proteins. The 615–843 region from cofactor D sequence is not similar to the corresponding 571–766 region from Alp1. In fission yeast protein, the HEAT motives are interrupted by a coiled-coil region similar to the last coiled-coil domain from S. cerevisiae kinesin-related Cin8p (898–1017 region). Cin8p is essential for yeast spindle function opposing the force that draws separated poles back [39-41]. Its coiled-coil region might be involved in dimerization as it is in the case of kinesin [42]. This difference may be important since fission yeast Alp1 is able to bind microtubules [13] whereas mammalian cofactor D does not [43].
In the supercomplex α-tubulin is directly bound to cofactor E [5]. The central 300 amino acids of cofactor E contain ten leucine rich repeats (LRRs) which are usually involved in protein-protein interactions [44,45]. The most important positions for LRR function are conserved (forming the typical subfamily of LRRs) although a few of them seem to be lost (Fig. 3B). The changed repeats could more or less fit the RI-like consensus from animals 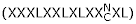 . But, according to Kajava [44], different LRR subfamilies never occur concomitantly within one LRR protein because the specific packing of repeats from one subfamily allows the formation of a specific hydrogen bond network between neighboring LRRs that could not be accomplished with LRRs from different subfamilies (Fig. 3B).
. But, according to Kajava [44], different LRR subfamilies never occur concomitantly within one LRR protein because the specific packing of repeats from one subfamily allows the formation of a specific hydrogen bond network between neighboring LRRs that could not be accomplished with LRRs from different subfamilies (Fig. 3B).
Two terminal domains of cofactor E are the same as in cofactor B, but in a reversed order. The CAP-Gly domain is on the N-terminus (in cofactor B it was on the C-terminus), and by simple analogy from the cofactor B function, it is probably involved in α-subunit binding as well, as suggested before [8]. The ubiquitin domain in cofactor E is in the C-terminal part (region 453–526). As discussed before, the function of this domain is still unknown.
Cofactor A is the first component of the β-tubulin folding pathway. It is discussed in the interest of integrity since its structure is known and it is not a subject of analysis here. It stabilizes the β-subunit and probably acts as a reservoir of tubulin folding intermediates for cofactor D [8]. It does not integrate into the super-complex but acts independently. Recently the crystal structure of the human cofactor A was described [46] and consists of a three-α-helix bundle. It interacts with β-tubulin via the helical regions.
Discussion
In the current study we have shown the complete domain composition of the cofactors involved in the final folding and dimerization of tubulin dimers. Our results narrow down the possible roles of previously unknown regions of these proteins.
The domain resemblance of cofactor B and E reflects their similar function in α-tubulin folding pathway, but the importance of the reverse order in which homologous domains appear in both proteins is unknown. CAP-Gly domain seems to be involved in α-subunit binding whereas the function of ubiquitin domain is unclear but most probably is involved in tubulin binding. It is interesting to note that both cofactors E and D contain extensive segments containing multiple domains involved in protein-protein interactions. The structure and length (1192 amino acids) of cofactor D predisposes this protein to bridge all the functions of the super-complex by means of spatial integration of all subunits (Fig. 4) [8].
Figure 4.

Model of domain interactions of the α/β-tubulin heterodimer folding pathway. The colors correspond to colors in Figure 2A. It is important to emphasize our hypothesis is most probable for the human system. As suggested by experiments, in fission yeast cofactors can act in a more linear manner with cofactor D homolog (Alp1) acting downstream from cofactors E and B [8].
There are still other known features of the cofactor super-complex, such as its GAP action manifested in stimulating the polymerization-independent hydrolysis of GTP by β-tubulin [19] that could not be explained by the analysis presented here. By the process of elimination, (i.e. the presence of protein-protein interaction motifs only in cofactor D and the lack of necessity for cofactor E in GAP activity) we can deduct that the GTPase-activating function possibly resides in cofactor C. The latest article by Bartolini and colleagues [47] presents evidence that the C-terminal part of cofactor C is indeed involved in this process. They also showed that mutation in an arginine residue, which they name "arginine finger", is responsible for the loss of function in both cofactor C and RP2 proteins [47].
To summarize we want to present a model for the interactions of cofactors in the final complex (Fig. 4). The goal of the cofactor complex is to facilitate the interaction of two tubulins. This is why they have to be brought together by cofactors D and E. After cofactor E binds β-tubulin both by CAP-Gly and ubiquitin-like domains it interacts with the repetitive region of cofactor D through the LRR region. The tubulin monomers are now brought together. After the process of complex formation is finished cofactor C joins it by binding with the spectrin-like domain to cofactor D or to both cofactor D and E. Then the cofactor C C-terminal domain stimulates the release of tubulin dimer ready to be incorporated in microtubule structures.
An interesting evolutionary association can be made by comparison of domains of proteins implicated in tubulin folding and chromosome segregation. The CAP-Gly domain was found in CLIP-170/restin and p150Glued proteins from the dynactin complex which is implicated as the dynein 'receptor' on organelles and kinetochores [14]. HEAT repeats were discovered in microtubule-associated protein family important for mitotic spindle assembly and chromosome movement [37]. LRRs are present in yeast topoisomerase II involved in chromosome segregation [48]. Finally ubiquitin is needed to separate sister chromatids [49]. It seems that the same bricks are used to build different houses on the same (microtubular) basis.
Conclusions
This analysis points to the importance of several newly recognized cofactor domains in the formation of the tubulin dimer. The discovery of ubiquitin-like domains (known to be essential for cofactor B function [9]) in cofactors B and E narrows down the possible functions of this domain, and suggests, together with other cofactor domains, the model of protein interactions in the complex.
Methods
Distant homology recognition PSI-BLAST [50], FFAS03 [51] and fold recognition Pcons [51-57] methods have been used to find possible structural templates for tubulin cofactor sequences. In addition, fold recognition Meta Server [58] (available at http://bioinfo.pl/) has been applied to cofactor C and D sequences. Table 1 summarizes the scores and templates used in this work. Domain boundaries were predicted by a combination of multiple alignment analysis, distant homology/fold recognition and modeling.
Table 1.
Confidence scores for cofactors domains. The best templates were used to build the theoretical model. In FFAS, all the scores below -9.000 are considered to be reliable, in Pcons – above 1.5.
| Protein | Domain | Score | Method | Best Template | Reference |
| Cofactor B | UBI-like | -12.600 | FFAS03 | 1J8C | [24] |
| CAP-Gly | -58.000 | FFAS03 | 1LPL | [20] | |
| Cofactor C | Spectrin-like | 1.604 | Pcons4 | 1CUN | [66] |
| RP2/CAP-like | -35.300 | FFAS03 | 1K4Z | [67] | |
| Cofactor D | Armadillo | 1.994 | Pcons4 | 1EJL | [68] |
| Armadillo HEAT | -32.500 -32.300 | FFAS03 FFAS03 | 1QBK 1B3U | [37,69] | |
| Cofactor E | CAP-Gly | -46.400 | FFAS03 | 1IXD | [70] |
| LRR | -44.100 | FFAS03 | 1H6T | [71] | |
| UBI-like | -13.000 | FFAS03 | 1J8C | [24] |
Best scoring alignments from FFAS03 [51] and Pcons [52] methods have been used for model building. FFAS03 and Pcons4 methods have consistently ranked among the best automated fold recognition methods over the last years both in LiveBench [59] and CAFASP competition [60]. Structural models based on these alignments have been obtained with WHATIF [61] modeling program with "slow" option (exhaustive side-chain packing optimization). The model visualization was done at the MICE [62] severs available at http://mice.sdsc.edu/.
The sequences of the ubiquitin family have been collected from PFAM domain database and subsequently supplemented with sequences from SWISS-PROT database [63] with help of literature sources. The resulting set was clustered at 40% sequence identity with CD-HIT algorithm [64]. The alignment has been obtained with the CLUSTALW algorithm [65].
Abbreviations
CCT = chaperonin containing TCP-1 complex
CAP-Gly = cytoskeleton-associated protein – glycine-rich
CLIP-170 = cytoplasmic linker protein-170
FFAS = Fold & Function Assignment System
RP2 = retinitis pigmentosa 2
LRR = Leucine-rich repeat
GAP = GTPase activating activity
PSI-BLAST = position-specific iterated BLAST
SECSG = Center for the Southeast Collaboratory on Structural Genomics
CD-HIT = Cluster Database at High Identity with Tolerance
Authors' contributions
MG conceived of the study, carried out the bioinformatics analysis, prepared the sequence alignment and drafted the manuscript. LJ prepared the structural models of the domains. AG provided general guidance in the project, coordinated the modeling and bioinformatics aspects of it and prepared the final version of the manuscript.
Acknowledgments
Acknowledgements
We thank Dr. Sally Lewis, Dr. Dorit Hanein, Dr. Andrzej Paszewski and Dr. Iddo Friedberg for critical reading of the manuscript, and Dr. Graham Thomas for helping with the spectrin repeat analysis. Part of the research described here was supported by NIH GM60049.
Contributor Information
Marcin Grynberg, Email: marcin@ljcrf.edu.
Lukasz Jaroszewski, Email: lukasz@sdsc.edu.
Adam Godzik, Email: adam@ljcrf.edu.
References
- Rommelaere H, Van Troys M, Gao Y, Melki R, Cowan NJ, Vandekerckhove J, Ampe C. Eukaryotic cytosolic chaperonin contains t-complex polypeptide 1 and seven related subunits. Proc Natl Acad Sci U S A. 1993;90:11975–11979. doi: 10.1073/pnas.90.24.11975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Vainberg IE, Chow RL, Cowan NJ. Two cofactors and cytoplasmic chaperonin are required for the folding of alpha- and beta-tubulin. Mol Cell Biol. 1993;13:2478–2485. doi: 10.1128/mcb.13.4.2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Melki R, Walden PD, Lewis SA, Ampe C, Rommelaere H, Vandekerckhove J, Cowan NJ. A novel cochaperonin that modulates the ATPase activity of cytoplasmic chaperonin. J Cell Biol. 1994;125:989–996. doi: 10.1083/jcb.125.5.989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian G, Huang Y, Rommelaere H, Vandekerckhove J, Ampe C, Cowan NJ. Pathway leading to correctly folded beta-tubulin. Cell. 1996;86:287–296. doi: 10.1016/s0092-8674(00)80100-2. [DOI] [PubMed] [Google Scholar]
- Lewis SA, Tian G, Cowan NJ. The a- and b-tubulin folding pathways. Trends Cell Biol. 1997;7:479–484. doi: 10.1016/S0962-8924(97)01168-9. [DOI] [PubMed] [Google Scholar]
- Radcliffe P, Hirata D, Childs D, Vardy L, Toda T. Identification of novel temperature-sensitive lethal alleles in essential beta-tubulin and nonessential alpha 2-tubulin genes as fission yeast polarity mutants. Mol Biol Cell. 1998;9:1757–1771. doi: 10.1091/mbc.9.7.1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radcliffe PA, Garcia MA, Toda T. The cofactor-dependent pathways for alpha- and beta-tubulins in microtubule biogenesis are functionally different in fission yeast. Genetics. 2000;156:93–103. doi: 10.1093/genetics/156.1.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radcliffe PA, Hirata D, Vardy L, Toda T. Functional dissection and hierarchy of tubulin-folding cofactor homologues in fission yeast. Mol Biol Cell. 1999;10:2987–3001. doi: 10.1091/mbc.10.9.2987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radcliffe PA, Toda T. Characterisation of fission yeast alp11 mutants defines three functional domains within tubulin-folding cofactor B. Mol Gen Genet. 2000;263:752–760. doi: 10.1007/s004380000252. [DOI] [PubMed] [Google Scholar]
- Tian G, Lewis SA, Feierbach B, Stearns T, Rommelaere H, Ampe C, Cowan NJ. Tubulin subunits exist in an activated conformational state generated and maintained by protein cofactors. J Cell Biol. 1997;138:821–832. doi: 10.1083/jcb.138.4.821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melki R, Rommelaere H, Leguy R, Vandekerckhove J, Ampe C. Cofactor A is a molecular chaperone required for beta-tubulin folding: functional and structural characterization. Biochemistry. 1996;35:10422–10435. doi: 10.1021/bi960788r. [DOI] [PubMed] [Google Scholar]
- Llosa M, Aloria K, Campo R, Padilla R, Avila J, Sanchez-Pulido L, Zabala JC. The beta-tubulin monomer release factor (p14) has homology with a region of the DnaJ protein. FEBS Lett. 1996;397:283–289. doi: 10.1016/S0014-5793(96)01198-2. [DOI] [PubMed] [Google Scholar]
- Hirata D, Masuda H, Eddison M, Toda T. Essential role of tubulin-folding cofactor D in microtubule assembly and its association with microtubules in fission yeast. Embo J. 1998;17:658–666. doi: 10.1093/emboj/17.3.658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grishchuk EL, McIntosh JR. Sto1p, a fission yeast protein similar to tubulin folding cofactor E, plays an essential role in mitotic microtubule assembly. J Cell Sci. 1999;112 ( Pt 12):1979–1988. doi: 10.1242/jcs.112.12.1979. [DOI] [PubMed] [Google Scholar]
- Stearns T, Hoyt MA, Botstein D. Yeast mutants sensitive to antimicrotubule drugs define three genes that affect microtubule function. Genetics. 1990;124:251–262. doi: 10.1093/genetics/124.2.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ursic D, Culbertson MR. The yeast homolog to mouse Tcp-1 affects microtubule-mediated processes. Mol Cell Biol. 1991;11:2629–2640. doi: 10.1128/mcb.11.5.2629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Archer JE, Vega LR, Solomon F. Rbl2p, a yeast protein that binds to beta-tubulin and participates in microtubule function in vivo. Cell. 1995;82:425–434. doi: 10.1016/0092-8674(95)90431-x. [DOI] [PubMed] [Google Scholar]
- Bhamidipati A, Lewis SA, Cowan NJ. ADP ribosylation factor-like protein 2 (Arl2) regulates the interaction of tubulin-folding cofactor D with native tubulin. J Cell Biol. 2000;149:1087–1096. doi: 10.1083/jcb.149.5.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian G, Bhamidipati A, Cowan NJ, Lewis SA. Tubulin folding cofactors as GTPase-activating proteins. GTP hydrolysis and the assembly of the alpha/beta-tubulin heterodimer. J Biol Chem. 1999;274:24054–24058. doi: 10.1074/jbc.274.34.24054. [DOI] [PubMed] [Google Scholar]
- Li S, Finley J, Liu ZJ, Qiu SH, Chen H, Luan CH, Carson M, Tsao J, Johnson D, Lin G, Zhao J, Thomas W, Nagy LA, Sha B, DeLucas LJ, Wang BC, Luo M. Crystal structure of the cytoskeleton-associated protein glycine-rich (CAP-Gly) domain. J Biol Chem. 2002;277:48596–48601. doi: 10.1074/jbc.M208512200. [DOI] [PubMed] [Google Scholar]
- Agrawal V, Kishan RK. Functional evolution of two subtly different (similar) folds. BMC Struct Biol. 2001;1:5. doi: 10.1186/1472-6807-1-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubiel W, Gordon C. Ubiquitin pathway: another link in the polyubiquitin chain? Curr Biol. 1999;9:R554–7. doi: 10.1016/S0960-9822(99)80353-4. [DOI] [PubMed] [Google Scholar]
- Buchberger A, Howard MJ, Proctor M, Bycroft M. The UBX domain: a widespread ubiquitin-like module. J Mol Biol. 2001;307:17–24. doi: 10.1006/jmbi.2000.4462. [DOI] [PubMed] [Google Scholar]
- Walters KJ, Kleijnen MF, Goh AM, Wagner G, Howley PM. Structural studies of the interaction between ubiquitin family proteins and proteasome subunit S5a. Biochemistry. 2002;41:1767–1777. doi: 10.1021/bi011892y. [DOI] [PubMed] [Google Scholar]
- Yan Y, Winograd E, Viel A, Cronin T, Harrison SC, Branton D. Crystal structure of the repetitive segments of spectrin. Science. 1993;262:2027–2030. doi: 10.1126/science.8266097. [DOI] [PubMed] [Google Scholar]
- Speicher DW, Marchesi VT. Erythrocyte spectrin is comprised of many homologous triple helical segments. Nature. 1984;311:177–180. doi: 10.1038/311177a0. [DOI] [PubMed] [Google Scholar]
- Parry DA, Dixon TW, Cohen C. Analysis of the three-alpha-helix motif in the spectrin superfamily of proteins. Biophys J. 1992;61:858–867. doi: 10.1016/S0006-3495(92)81893-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahana E, Flood G, Gratzer WB. Physical properties of dystrophin rod domain. Cell Motil Cytoskeleton. 1997;36:246–252. doi: 10.1002/(SICI)1097-0169(1997)36:3<246::AID-CM5>3.3.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Djinovic-Carugo K, Young P, Gautel M, Saraste M. Structure of the alpha-actinin rod: molecular basis for cross-linking of actin filaments. Cell. 1999;98:537–546. doi: 10.1016/s0092-8674(00)81981-9. [DOI] [PubMed] [Google Scholar]
- Kennedy SP, Warren SL, Forget BG, Morrow JS. Ankyrin binds to the 15th repetitive unit of erythroid and nonerythroid beta-spectrin. J Cell Biol. 1991;115:267–277. doi: 10.1083/jcb.115.1.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett V. Ankyrins. Adaptors between diverse plasma membrane proteins and the cytoplasm. J Biol Chem. 1992;267:8703–8706. [PubMed] [Google Scholar]
- Amann KJ, Renley BA, Ervasti JM. A cluster of basic repeats in the dystrophin rod domain binds F-actin through an electrostatic interaction. J Biol Chem. 1998;273:28419–28423. doi: 10.1074/jbc.273.43.28419. [DOI] [PubMed] [Google Scholar]
- Djinovic-Carugo K, Gautel M, Ylanne J, Young P. The spectrin repeat: a structural platform for cytoskeletal protein assemblies. FEBS Lett. 2002;513:119–123. doi: 10.1016/S0014-5793(01)03304-X. [DOI] [PubMed] [Google Scholar]
- Stevenson VA, Theurkauf WE. Actin cytoskeleton: putting a CAP on actin polymerization. Curr Biol. 2000;10:R695–7. doi: 10.1016/S0960-9822(00)00712-0. [DOI] [PubMed] [Google Scholar]
- Schwahn U, Lenzner S, Dong J, Feil S, Hinzmann B, van Duijnhoven G, Kirschner R, Hemberger M, Bergen AA, Rosenberg T, Pinckers AJ, Fundele R, Rosenthal A, Cremers FP, Ropers HH, Berger W. Positional cloning of the gene for X-linked retinitis pigmentosa 2. Nat Genet. 1998;19:327–332. doi: 10.1038/1214. [DOI] [PubMed] [Google Scholar]
- Groves MR, Barford D. Topological characteristics of helical repeat proteins. Curr Opin Struct Biol. 1999;9:383–389. doi: 10.1016/S0959-440X(99)80052-9. [DOI] [PubMed] [Google Scholar]
- Groves MR, Hanlon N, Turowski P, Hemmings BA, Barford D. The structure of the protein phosphatase 2A PR65/A subunit reveals the conformation of its 15 tandemly repeated HEAT motifs. Cell. 1999;96:99–110. doi: 10.1016/s0092-8674(00)80963-0. [DOI] [PubMed] [Google Scholar]
- Andrade MA, Bork P. HEAT repeats in the Huntington's disease protein. Nat Genet. 1995;11:115–116. doi: 10.1038/ng1095-115. [DOI] [PubMed] [Google Scholar]
- Saunders WS, Koshland D, Eshel D, Gibbons IR, Hoyt MA. Saccharomyces cerevisiae kinesin- and dynein-related proteins required for anaphase chromosome segregation. J Cell Biol. 1995;128:617–624. doi: 10.1083/jcb.128.4.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyt MA, He L, Loo KK, Saunders WS. Two Saccharomyces cerevisiae kinesin-related gene products required for mitotic spindle assembly. J Cell Biol. 1992;118:109–120. doi: 10.1083/jcb.118.1.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders WS, Hoyt MA. Kinesin-related proteins required for structural integrity of the mitotic spindle. Cell. 1992;70:451–458. doi: 10.1016/0092-8674(92)90169-d. [DOI] [PubMed] [Google Scholar]
- Kozielski F, Sack S, Marx A, Thormahlen M, Schonbrunn E, Biou V, Thompson A, Mandelkow EM, Mandelkow E. The crystal structure of dimeric kinesin and implications for microtubule-dependent motility. Cell. 1997;91:985–994. doi: 10.1016/s0092-8674(00)80489-4. [DOI] [PubMed] [Google Scholar]
- Martin L, Fanarraga ML, Aloria K, Zabala JC. Tubulin folding cofactor D is a microtubule destabilizing protein. FEBS Lett. 2000;470:93–95. doi: 10.1016/S0014-5793(00)01293-X. [DOI] [PubMed] [Google Scholar]
- Kajava AV. Structural diversity of leucine-rich repeat proteins. J Mol Biol. 1998;277:519–527. doi: 10.1006/jmbi.1998.1643. [DOI] [PubMed] [Google Scholar]
- Kobe B, Deisenhofer J. Proteins with leucine-rich repeats. Curr Opin Struct Biol. 1995;5:409–416. doi: 10.1016/0959-440X(95)80105-7. [DOI] [PubMed] [Google Scholar]
- Guasch A, Aloria K, Perez R, Avila J, Zabala JC, Coll M. Three-dimensional structure of human tubulin chaperone cofactor A. J Mol Biol. 2002;318:1139–1149. doi: 10.1016/S0022-2836(02)00185-7. [DOI] [PubMed] [Google Scholar]
- Bartolini F, Bhamidipati A, Thomas S, Schwahn U, Lewis SA, Cowan NJ. Functional overlap between retinitis pigmentosa 2 protein and the tubulin-specific chaperone cofactor C. J Biol Chem. 2002;277:14629–14634. doi: 10.1074/jbc.M200128200. [DOI] [PubMed] [Google Scholar]
- Wang X, Watt PM, Louis EJ, Borts RH, Hickson ID. Pat1: a topoisomerase II-associated protein required for faithful chromosome transmission in Saccharomyces cerevisiae. Nucleic Acids Res. 1996;24:4791–4797. doi: 10.1093/nar/24.23.4791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konforti B. Picture story. The great divide. Nat Struct Biol. 2001;8:488. doi: 10.1038/88540. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rychlewski L, Jaroszewski L, Li W, Godzik A. Comparison of sequence profiles. Strategies for structural predictions using sequence information. Protein Sci. 2000;9:232–241. doi: 10.1110/ps.9.2.232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundstrom J, Rychlewski L, Bujnicki J, Elofsson A. Pcons: a neural-network-based consensus predictor that improves fold recognition. Protein Sci. 2001;10:2354–2362. doi: 10.1110/ps.08501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer D. Hybrid fold recognition: combining sequence derived properties with evolutionary information. Pac Symp Biocomput. 2000:119–130. [PubMed] [Google Scholar]
- Jones DT. GenTHREADER: an efficient and reliable protein fold recognition method for genomic sequences. J Mol Biol. 1999;287:797–815. doi: 10.1006/jmbi.1999.2583. [DOI] [PubMed] [Google Scholar]
- Karplus K, Barrett C, Hughey R. Hidden Markov models for detecting remote protein homologies. Bioinformatics. 1998;14:846–856. doi: 10.1093/bioinformatics/14.10.846. [DOI] [PubMed] [Google Scholar]
- Kelley LA, MacCallum RM, Sternberg MJ. Enhanced genome annotation using structural profiles in the program 3D-PSSM. J Mol Biol. 2000;299:499–520. doi: 10.1006/jmbi.2000.3741. [DOI] [PubMed] [Google Scholar]
- Shi J, Blundell TL, Mizuguchi K. FUGUE: sequence-structure homology recognition using environment-specific substitution tables and structure-dependent gap penalties. J Mol Biol. 2001;310:243–257. doi: 10.1006/jmbi.2001.4762. [DOI] [PubMed] [Google Scholar]
- Bujnicki JM, Elofsson A, Fischer D, Rychlewski L. Structure prediction meta server. Bioinformatics. 2001;17:750–751. doi: 10.1093/bioinformatics/17.8.750. [DOI] [PubMed] [Google Scholar]
- Bujnicki JM, Elofsson A, Fischer D, Rychlewski L. LiveBench-2: large-scale automated evaluation of protein structure prediction servers. Proteins. 2001;Suppl 5:184–191. doi: 10.1002/prot.10039. [DOI] [PubMed] [Google Scholar]
- Fischer D, Rychlewski L. The 2002 olympic games of protein structure prediction. Protein Eng. 2003;16:157–160. doi: 10.1093/proeng/gzg022. [DOI] [PubMed] [Google Scholar]
- Vriend G. WHAT IF: a molecular modeling and drug design program. J Mol Graph. 1990;8:52–6, 29. doi: 10.1016/0263-7855(90)80070-V. [DOI] [PubMed] [Google Scholar]
- Tate JG, Moreland JL, Bourne PE. Design and implementation of a collaborative molecular graphics environment. J Mol Graph Model. 2001;19:280–7, 369-73. doi: 10.1016/S1093-3263(00)00055-3. [DOI] [PubMed] [Google Scholar]
- Gasteiger E, Jung E, Bairoch A. SWISS-PROT: connecting biomolecular knowledge via a protein database. Curr Issues Mol Biol. 2001;3:47–55. [PubMed] [Google Scholar]
- Li W, Jaroszewski L, Godzik A. Tolerating some redundancy significantly speeds up clustering of large protein databases. Bioinformatics. 2002;18:77–82. doi: 10.1093/bioinformatics/18.1.77. [DOI] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grum VL, Li D, MacDonald RI, Mondragon A. Structures of two repeats of spectrin suggest models of flexibility. Cell. 1999;98:523–535. doi: 10.1016/s0092-8674(00)81980-7. [DOI] [PubMed] [Google Scholar]
- Roswarski DA, Fedorov AA, Dodatko T, Almo SC. Crystal Structure of the Actin-Binding Domain of Cyclase Associated Protein (CAP) from Saccharomyces Cerevisiae. to be published. [DOI] [PubMed]
- Fontes MR, Teh T, Kobe B. Structural basis of recognition of monopartite and bipartite nuclear localization sequences by mammalian importin-alpha. J Mol Biol. 2000;297:1183–1194. doi: 10.1006/jmbi.2000.3642. [DOI] [PubMed] [Google Scholar]
- Chook YM, Blobel G. Structure of the nuclear transport complex karyopherin-beta2-Ran x GppNHp. Nature. 1999;399:230–237. doi: 10.1038/20375. [DOI] [PubMed] [Google Scholar]
- Saito K, Koshiba S, Kigawa T, Yokoyama S. Solution Structure of the CAP-Gly Domain from Human Cylindromatosis Tumour-Suppressor Cyld. to be published.
- Schubert WD, Gobel G, Diepholz M, Darji A, Kloer D, Hain T, Chakraborty T, Wehland J, Domann E, Heinz DW. Internalins from the human pathogen Listeria monocytogenes combine three distinct folds into a contiguous internalin domain. J Mol Biol. 2001;312:783–794. doi: 10.1006/jmbi.2001.4989. [DOI] [PubMed] [Google Scholar]