

Abstract
The excitotoxic conopeptide ι-RXIA induces repetitive action potentials in frog motor axons and seizures upon intracranial injection into mice. We recently discovered that ι-RXIA shifts the voltage-dependence of activation of voltage-gated sodium channel NaV1.6 to a more hyperpolarized level. Here, we performed voltage-clamp experiments to examine its activity against rodent NaV1.1 through NaV1.7 co-expressed with the β1 subunit in Xenopus oocytes and NaV1.8 in dissociated mouse DRG neurons. The order of sensitivity to ι-RXIA was NaV1.6 > 1.2 > 1.7, and the remaining subtypes were insensitive. The time course of ι-RXIA-activity on NaV1.6 during exposure to different peptide concentrations were well fit by single-exponential curves that provided kobs. The plot of kobs versus [ι-RXIA] was linear, consistent with a bimolecular reaction with a Kd of ~3 μM, close to the steady-state EC50 of ~2 μM. ι-RXIA has an unusual residue, D-Phe, and the analog with an L-Phe instead, ι-RXIA[L-Phe44], had a two-fold lower affinity and two-fold faster off-rate than ι-RXIA on NaV1.6 and furthermore was inactive on NaV1.2. ι-RXIA induced repetitive action potentials in mouse sciatic nerve with conduction velocities of both A- and C-fibers, consistent with the presence of NaV1.6 at nodes of Ranvier as well as in unmyelinated axons. Sixteen peptides homologous to ι-RXIA have been identified from a single species of Conus, so these peptides represent a rich family of novel sodium channel-targeting ligands.
Keywords: channel-activation, conopeptide, excitotoxin, iota-conotoxin RXIA, neurotoxin, voltage-gated sodium channel
1. Introduction
Voltage-gated sodium channels (NaVs) are important in signaling in the nervous system not only for their classical role in the generation and propagation of action potentials, but also for their involvement in amplifying excitatory postsynaptic potentials (e.g., [1]). Furthermore, NaVs are found in glia and Schwann cells where their functions are unclear (see review by [2]). There are nine isoforms of the main, pore-bearing α-subunit of NaVs in mammals, NaV1.1 through NaV1.9 [3], and neurotoxins have proven to be very useful as probes to analyze the molecular structure and function of NaVs (see review by [4]). Venom from the marine snails of the genus Conus has long served as a rich source of neuroactive peptides that target ion channels and receptors, including NaVs [5, 6].
Four families of conopeptides that target NaVs have been identified (Fig. 1A). μ-Conopeptides, the first conotoxins identified to target NaVs [7], compete with tetrodotoxin (TTX) [8] at neurotoxin receptor site 1 and block the pore (see review by [4]). μO-Conopeptides also block NaVs [9, 10]; however, they do not compete with TTX for binding to site 1 [11]. They bind instead to sites different from those of μ-conopeptides [12, 13] and are thought to block channel opening by interfering with the voltage sensor in domain II of the channel [12]. δ-Conopeptides, unlike the preceding conotoxins which produce flaccid paralysis, are excitotoxic and produce rigid paralysis by inhibiting the inactivation of NaVs [14, 15]; they act by interfering with the voltage sensor in domain IV, like α-scorpion toxins [16]. Recently, we discovered a new family of excitatory conotoxins, the iota-conopeptides [17–19]. Among the initially studied peptides in the I1 superfamily of iota-peptides is ι-RXIA (“iota-R eleven A”, referred to by its provisional name, r11a, in publications cited above), from the fish-hunting snail Conus radiatus (the “R” in ι-RXIA reflects the species name of its source). An unusual structural feature of ι-RXIA is that it has an unconventional residue, D-phenylalanine, near its C-terminus (Fig. 1B). The solution structure of ι-RXIA has been determined (Fig. 1C) [20]. ι-RXIA produces seizures when injected intracranially into mice and induces repetitive action potentials in motor axons of the frog [19]. We found recently that ι-RXIA modulated the activity of mouse NaV1.6 channels expressed in Xenopus oocytes by shifting the voltage-dependence of activation to a more hyperpolarized level [20]. In this regard, it behaves like β-scorpion toxins (see review by Cestele and Catterall (2000)).
Figure 1.

A, Four families of peptide conotoxins that target Na channels. Members in the upper two, μ- and μO-conotoxins, inhibit Na channels, whereas those in the lower two, ι- and δ-conotoxins, are agonist. The cysteine framework of each peptide family is illustrated. All cysteines are disulfide-bonded, rendering each peptide into a compact structure. B, The sequence of ι-RXIA, the iota-conotoxin studied in this report, where O is hydroxyproline, and the underlined F (residue 44) is the D-enantiomer of phenylalanine. The synthetic homologue, ι-RXIA[L-Phe44], is identical to ι-RXIA except residue 44 is an L-phenylalanine. The disulfide bridges between Cys residues are indicated. C, Ribbon structure of ι-RXIA determined by NMR spectroscopy [20]. Locations of the Cys residues and their disulfide linkages are indicated; also shown is the side chain of D-Phe44. This figure was prepared using PyMol (http://www.pymol.org).
In this investigation, we determined the specificity of ι-RXIA by testing it against seven cloned NaVs expressed in Xenopus oocytes, as well as TTX-resistant INa (largely due to NaV1.8) in dissociated DRG neurons from mouse. Furthermore, to examine the functional role of the atypical D-Phe residue, we compared the activities of ι-RXIA and its analog with an L- (instead of D-) phenylalanine, ι-RXIA[L-Phe44]. Finally, we examined the conduction velocities of the action potentials that were induced by ι-RXIA in mouse sciatic nerve to identify the fiber types in mammals that are susceptible to the peptide.
This is the first account of the specificity, affinity, and efficacy of a conopeptide that exerts its excitotoxic activity by shifting the voltage dependence of activation of Na channels to a more hyperpolarized level. The overall results provide a rational explanation for the effects of ι-RXIA on brain and peripheral nerve.
2. Methods and Materials
2.1. Voltage-gated Na channel clones
Clones for rat (r) NaV1.1 through 1.5, mouse (m) NaV1.6, and rat β1 were generously provided by Alan Goldin. Gail Mandel kindly contributed the rat NaV1.7 clone. Each plasmid was designated as the following: rNav1.1 (pNaI), rNav1.2 (pNa200, resistant to tetracycline), rNav1.3 (pNa3T, resistant to tetracycline), rNav1.4 (pNaμ1, resistant to ampicillin), rNav1.5 (pNaSkM2, resistant to ampicillin) and rNav1.7 (PN1, resistant to ampicillin). Approximately 20 ng of each cDNA was transformed into DH10B electrocompetent cells and plated on an agar plate with the appropriate antibiotic. The following day, 10 clones were picked and grown overnight in 5 ml of LB media with the appropriate antibiotic. Of the ten inoculates, the cDNA was isolated from five and screened by BamHI or HindIII (Nav1.7) digestion. For Nav1.1, two additional digestion screens were obtained from HindIII and AflIII. For each isoform, one clone that yielded the correct restriction digest fragments was grown in 50 ml of LB media with the appropriate antibiotic to obtain sufficient cDNA for sequencing. The cDNA for each isoform with the correct sequence was linearized with either NotI (Nav1.1, Nav1.2, Nav1.3, Nav1.4, Nav1.7) or AseI (Nav1.5) and transcribed with T7 RNA polymerase (Nav1.1, Nav1.2, Nav1.3, Nav1.4, Nav1.7) or SP6 (Nav1.5), using the mMessage mMachine RNA trancription kit from Ambion (Austin, TX) for generation of capped cRNA. The cRNA was purified using the Qiagen RNeasy kit (Qiagen, Valencia, CA).
2.2. Two-electrode voltage clamp recordings from Xenopus oocytes
Xenopus oocytes were prepared as described previously [21]. All animals were used following protocols approved by the University of Utah’s Institutional Animal Care and Use Committee and conform to the National Institutes of Health Guide for the Care and Use of Laboratory Animals. A given oocyte was injected with 30–50 nl of cRNA in distilled water for one of the followings sodium channel isoforms: rNaV1.1, rNaV1.2, rNaV1.3, rNaV1.4, rNaV1.5, mNaV1.6, or rNaV1.7 (3, 1.5, 15, 0.6, 3, 30, or 15 ng, respectively) along with an equal weight of rβ1 subunit cRNA. Oocytes were incubated at 16 °C in ND96 (composition in mM: 96 NaCl, 2 KCl, 1.8 CaCl2, 1 MgCl2, 5 HEPES, pH ~7.3) supplemented with Pen/Strep, Septra and Amikacin for 2 to 6 days. The recording chamber was a 4 mm diameter well (30 μl total volume) sunk in the silicone elastomer, Sylgard (Dow Corning, Midland, MI). Oocytes were voltage clamped with a Warner OC-725C amplifier (Warner Instruments, Hamden, CT) using 3 M KCl-filled microelectrodes (< 0.5 Megohm resistance). The chamber was perfu sed with ND96 containing 0.1 mg/ml bovine serum albumin. The membrane potential was held at −100 mV and channels activated by a 60 ms test pulse of fixed amplitude (for kinetic data), or varying amplitude (for activation data), or a test pulse of fixed amplitude (−20 mV) immediately preceded by a 0.5 or 1 s conditioning prepulse of varying amplitude (for inactivation data), applied every 20 s. Current signals were low-pass filtered at 2 kHz, digitized at a sampling frequency of 10 kHz, and leak-subtracted by a P/6 protocol using in-house software written in LabVIEW (National Instruments, Austin, Texas). Conopeptides were dissolved in ND96 containing 0.1 mg/ml bovine serum albumin, and oocytes were exposed to toxin by applying 3 μl of peptide solution (at ten times the final concentration) to a static bath with a pipettor and manually stirring the bath for a few seconds by gently aspirating and expelling a few μl of bath fluid several times with the pipettor.
2.3. Whole-cell voltage clamping of dissociated mouse DRG neurons
Adult mouse (Swiss Webster) DRGs were dissociated and used as described previously (Zhang, et al., 2006). Briefly, ganglia were treated with collagenase followed by trypsin. Cells were mechanically dissociated by trituration, washed, and suspended in L15 medium supplemented with 14 mM glucose, 1 mM CaCl2 and 10% fetal bovine serum supplemented with penicillin/streptomycin. Dissociated DRG neurons were kept in suspension at 4 °C up to 3 days.
The extracellular solution was (in mM) 140 NaCl, 3 KCl, 1 MgCl2, 1 CaCl2, 20 HEPES, pH 7.3. Recording pipettes had resistances of < 2 Megohms and contained (in mM): 140 CsF, 10 NaCl, 1 EGTA, 10 HEPES, pH 7.3. Currents produced by NaV1.8 were obtained by recording from small neurons in the presence of 1 μM TTX. After achieving whole cell clamp conditions, recordings were not initiated until the holding current had settled, which required >10 min; for mammalian neurons, the contribution of NaV1.8, relative to that of NaV1.9, to the TTX-resistant current is reported to be maximized by this wait [22]. An example of the distinctive kinetics of the TTX-resistant current can be seen in Fig. 3 of our recent report [23]. Conotoxins were dissolved in extracellular solution containing 0.1 mg/ml bovine serum albumin and applied to neurons under study by bath exchange, and toxin exposures were conducted in static baths, as described previously [15, 24]. A MultiClamp 700A amplifier (Axon Instruments, Union City, California) was used to hold the membrane potential at −80 mV, and Na channels were activated by a 50 ms test pulse to 0 mV, applied every 20 s. To obtain voltage dependence of activation or inactivation, pulse protocols similar to those described above for oocytes were used. Current signals were low-pass filtered at 3 kHz and digitally processed as described above for oocyte recordings.
2.4. Extracellular recording of action potentials from mouse sciatic nerve
Sciatic nerves were dissected from adult Swiss Webster mice of either sex, desheathed, and used within 2 h. The nerve was placed in a 4-well Vaseline-gap chamber as described previously (Zhang et al., 2006). All wells contained mammalian Ringers consisting of (in mM): NaCl, 140; KCl, 5; CaCl2, 2; MgCl2, 1.1; HEPES, 5; pH 7.4. The nerve was stimulated with electrodes in wells 1 and 2, while compound action potentials were recorded with a pair of extracellular electrodes in wells 3 and 4 and another pair in wells 4 and 5. Toxins were added to either well 1 or 2. All electrodes were stainless steel wires. Extracellular records were acquired with differential AC preamplifiers, band-pass filtered (1 Hz to 1 kHz), and sampled at 4 kHz using in-house software written in LabVIEW. A-compound action potentials (A-CAPs), which have fast conduction velocities, were rapidly and totally blocked by 1 μM TTX, whereas C-compound action potentials (C-CAPs), which have slower conduction velocities, were incompletely blocked and ~20% persisted in 10 μM TTX [25].
All experiments were performed at room temperature (~ 21 °C).
2.5. Contotoxins and other reagents
The conotoxins ι-RXIA and ι-RXIA[L-Phe44], synthesized as described previously [17], were dissolved in extracellular solution containing 0.1 mg/ml bovine serum albumin. Responses in the presence of peptide (as well as control responses in the absence of peptide) were acquired in static baths to conserve material. TTX was obtained from Alomone Labs (Jerusalem, Israel). Unless noted otherwise, all other reagents were from Sigma Chemical Co. (St. Louis, MO).
2.6. Data analysis
Conductance values were calculated using the formula GNa = INa/(Vstep − Vrev), where GNa is the conductance, Vstep is the test potential, INa is the peak current amplitude in response to the potential step, and Vrev the estimated reversal potential. Normalized activation and inactivation curves were fit to the Boltzmann equation of the form Y = 1/(1 + exp[(Vstep − V1/2)/k]), where Y is the normalized GNa or INa, Vstep is the test pulse (for activation curves) or the conditioning prepulse (for inactivation curves), V1/2 is the voltage at half-maximal activation or inactivation, and k is the slope factor.
The time course of toxin action, monitored as described below, identified when steady-state had been achieved following exposure to toxin. We quantified the effect of different concentrations of ι-RXIA at steady-state using: (i) the shift in V1/2 of the Boltzmann relationship for the voltage dependence of activation, and (ii) the increase in peak INa specified by the ratio I′Na/IoNa, where I′Na and IoNa are the peak current values for a fixed Vstep to −30 mV in the presence and absence of toxin, respectively. We assumed, for simplicity, that a channel has two states, either unmodified or toxin-modified; thus, the G-V relationship for activation can be described by the double-Boltzmann function, Y = a(1/(1 + exp[(Vstep − V1/2)/k])) + (1 − a)(1/(1 + exp[(Vstep − V1/2)/k])), where a represents the fraction of channels that are unmodified, and the non-underlined and underlined constants correspond to the non-modified and toxin-modified channels, respectively [26]. It should be noted that the I′Na/IoNa ratio varies linearly with a; in contrast, the value of the voltage at half-maximal activation in the G-V relationship varies non-linearly with a, with the degree of non-linearity being dependent on the difference between V1/2 and V1/2; thus, I′Na/IoNa was used as the preferred measure of toxin activity.
For kinetic measurements, the I′Na/IoNa ratio, or simply the peak INa, in response to a fixed Vstep to −30 mV was plotted as a function of time during toxin-exposure and toxin-washout. The observed rate constant for the time course during exposure to toxin, kobs, and the rate constant for the return of INa to control values following toxin-washout, koff, were determined from single-exponential fits of their respective time courses. The on-rate constant, kon, and the extrapolated koff value were obtained, respectively, from the slope and Y-intercept of the kobs versus [toxin] plot, assuming the equation for a pseudo first-order reaction kobs = kon[toxin] + koff [27].
Data were analyzed and curve fit using Graphpad Prism (GraphPad Software, San Diego, CA). Numerical data are presented as means ± SE, and error bars on all graphs represent SE. EC50 values are expressed as means with 95% confidence intervals (CI’s). Statistical significance was assessed using Student’s t-test.
3. RESULTS
3.1. ι-RXIA modulates NaV1.6/β1 expressed in Xenopus oocytes by shifting the voltage dependence of activation with minimal effects on the voltage dependence of inactivation or on the kinetics of Na currents
Oocytes expressing NaV1.6 along with the β1 subunit were two-electrode voltage clamped, as described in Methods, and the effects of ι-RXIA on INa were examined after the effects of the toxin had achieved steady-state, which was <10 min when a saturating concentration (≥ 5 μM) of ι-RXIA was used (see below for kinetic analyses and dose-response curves). Figure 2A and B (and insets in C) show that, for a given Vstep, INa was markedly increased following exposure to 10 μM ι-RXIA; however, when their peaks are normalized, the traces largely overlap, showing that the time course of INa (and therefore channel kinetics) was minimally affected by ι-RXIA (Fig. 2C). The conductance-voltage curve in Fig. 2D shows that ι-RXIA shifted the voltage sensitivity of activation in the hyperpolarizing direction. In contrast, the voltage sensitivity of inactivation was minimally affected by ι-RXIA (Fig. 2E).
Figure 2.

ι-RXIA modulates the activity of NaV1.6 coexpressed with β1 in Xenopus oocytes by shifting its voltage-dependence of activation. Oocytes were two-electrode clamped at a holding potential of −100 mV, and Vm was stepped in 10 mV increments as described in Methods. Representative current records for voltage steps ranging from −60 to 10 mV in the absence (A) and presence (B) of 10 μM ι-RXIA. C, Normalized current traces from A and B in response to the voltage step to −30 mV (left) and −20 mV (right), in the absence (dots) and presence (solid line) of 10 μM ι-RXIA; each pair of responses to a given voltage step largely overlap. Insets show corresponding traces non-normalized. Voltage sensitivity of activation (D) and inactivation (E) obtained in the absence (open circles) and presence (closed circles) of ι-RXIA. The solid lines are best-fit curves to the Boltzmann equation. V1/2 values for activation in D were −22.38 ± 0.48 (control) and −38.29 ± 0.47 mV (10 μM ι-RXIA) with respective slope factors of −6.08 ± 0.42 and −7.53 ± 0.41 mV. V1/2 values for inactivation in E were −58.71 ± 1.03 (control) and −55.73 ± 0.75 mV (5 μM ι-RXIA) with respective slope factors of 5.98 ± 0.90 and 5.37 ± 0.61 mV. Each data point represents mean ± SE (N = 3 oocytes).
3.2. ι-RXIA modulates not only NaV1.6, but also NaV1.2 and 1.7, albeit at higher concentrations
To examine the channel subtype-selectivity of ι-RXIA, we compared its effect on oocytes expressing NaV1.1 through 1.7, each with the β1 subunit. To test the ι-RXIA-susceptibility of NaV1.8, TTX-resistant INa in whole cell voltage clamped mouse DRG neurons were examined as described in Methods. Of these eight channel subtypes, only NaV1.2/β1 and 1.6/β1 were significantly affected by ι-RXIA at concentrations of 10 and 50 μM, as judged by the shift in the V1/2 of activation (Fig. 3A). When the more direct measure of the I′Na/IoNa ratio for responses to a fixed depolarizing voltage step to −30 mV was used, the effect of 50μM ι-RXIA on NaV1.2 and 1.6 was very pronounced, and the susceptibility of NaV1.7/β1 could also be discerned (Fig. 3B). However, the remaining channel subtypes, including the TTX-resistant INa in DRG neurons (NaV1.8), were insensitive to 50 μM ι-RXIA, the highest concentration tested, by this measure (Fig. 3B).
Figure 3.
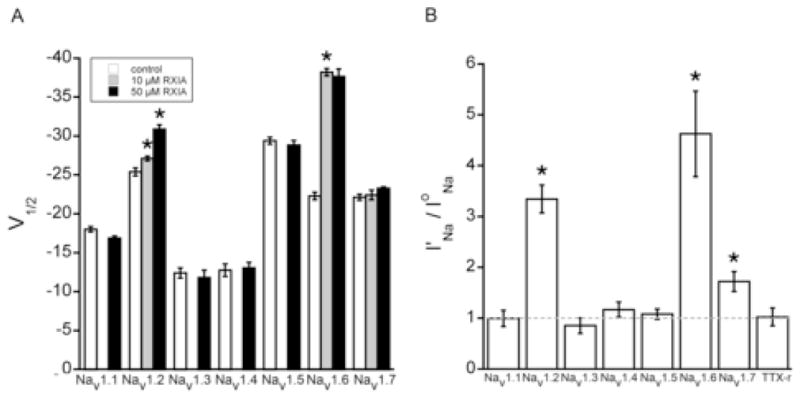
Effect of ι-RXIA on NaV1.1 through NaV1.7 coexpressed with β1 in Xenopus oocytes and TTX-resistant Na channels in dissociated neurons from mouse DRG. Oocytes were used as in Fig. 2, and neurons were whole-cell clamped and TTX-resistant INa were recorded as described in Methods. A, V1/2 of the voltage-dependence of activation in controls (white bars) and in the presence of 10 μM (gray bars) or 50 μM (black bars) ι-RXIA. In 10 μM ι-RXIA, V1/2‘s of only NaV1.2/β1 and 1.6/β1 differed significantly from control. In 50 μM ι-RXIA, V1/2 of NaV1.6/β1 remained unchanged, that of NaV1.2/β1 changed further, and those of NaV1.1/β1, 1.3/β1, 1.4/β1, 1.5/β1, and 1.7/β1 remained insignificantly different from controls. B, Ratio of I′Na/IoNa for Vstep to −30 mV, where I′Na and IoNa are peak INa in 50 μM ι-RXIA and control, respectively. The ratio measurement shows that in addition to NaV1.2/β1 and 1.6/β1, NaV1.7/β1 was also significantly up-modulated by 50 μM ι-RXIA. I′Na/IoNa value of none of the other Na channels differed significantly from 1, indicating they were not affected even at this high toxin concentration. Bars represent mean ± SE (N = 3). *p < .05, compared to bar immediately to its left (A) or compared to value of 1 (B).
The effect of ι-RXIA is reminiscent of that of β-scorpion toxins, which also shift the voltage dependence of activation in the hyperpolarizing direction, but the activity of the latter can be strongly potentiated by depolarizing conditioning prepulses [28]. To examine this possibility, the depolarizing conditioning pulse employed by Cestele and coworkers [28], consisting of a 50 mV, 3 ms pulse applied 70 ms before the test pulse, was used, but it did not affect the effect of ι-RXIA on any of the NaV1 subtypes (not illustrated). Furthermore, ι-RXIA‘s effect on NaV1.6 was also examined using the pulse protocol of Tsushima and colleagues [26], where a 50 ms pulse to −10 mV was applied with a 20 ms interval between its termination and the start of the test pulse. Here too, the conditioning pulse had no effect (not illustrated).
3.3. Dose-dependency of the kinetics of modulation of NaV1.6/β1 by ι-RXIA
The I′Na/IoNa ratio proved to be a facile means of quantifying the extent of the modulation induced by the toxin, and we used this measure to monitor the time courses of the development and washout of the effect of ι-RXIA. Both were well fit by single exponential curves and provided the rate constants kobs and koff, as exemplified in Fig 4A. Time courses were obtained at several different concentrations of ι-RXIA; the development of modulation was toxin-concentration dependent (Fig. 4B), but that of washout was concentration-independent (Fig. 4C). When the rate constants of the development of modulation, kobs, were plotted as a function of ι-RXIA concentration, a straight line was observed (Fig. 4D). For a pseudo first order reaction, the slope of the kobs plot in Fig. 4D corresponds to kon and the Y-intercept, the extrapolated koff (see Methods); the respective values were 0.137 μM−1•min−1 and 0.394 min−1. The Kd, given by the ratio of koff/kon, was 2.88 μM. The rate constant for ι-RXIA washout, which provides an independent measure of koff, remained essentially the same regardless of starting toxin-concentration (dashed line in Fig. 4D) with an average value of 0.229 min−1, which is ~0.6-times the extrapolated koff in Fig. 4D. Using kon and this lower koff value yields a Kd of 1.67 μM.
Figure 4.
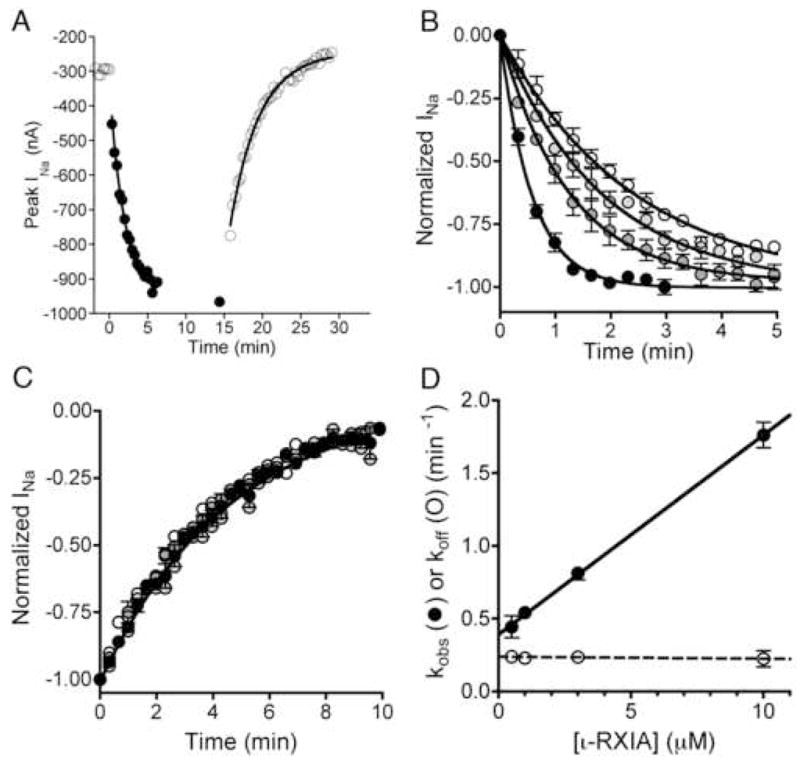
Kinetics of the modulation of NaV1.6/β1 by ι-RXIA. Oocytes were used essentially as described in Fig. 2, and the peak INa in response to a Vstep to −30 mV from a holding potential of −100 mV was measured at different times during the exposure to and washout of ι-RXIA. A, Representative time course of INa during exposure to 3 μM ι-RXIA (closed circles) and its subsequent washout (open circles). Absence of points for 5 min in middle of plot reflects period when I-V data were being acquired. Single-exponential best-fits of the data provided kobs (the observed rate constant for the toxin-induced increase in INa) and koff (the rate constant of recovery following washout). Values of kobs and koff for illustrated curves were 0.65 min−1 and 0.24 min−1, respectively. B, Time course of normalized peak INa upon exposure to 0.5, 1, 3, and 10 μM ι-RXIA (from white to progressively darker circles, respectively). C, Time course of normalized peak INa following washout of ι-RXIA (symbols as in B). The data points largely overlap, and the best fit single-exponential curve for all of them is shown, yielding an aggregate koff of 0.227 ± 0.007. D, Plot of kobs (closed circles) and koff (open circles) versus [ι-RXIA], from data shown in B and C, respectively. Values for kobs increased linearly with peptide concentration, best-fit linear regression line (solid curve) has slope of 0.137 ± 0.002 μM−1•min−1 and Y-intercept of 0.394 ± 0.011 min−1. In contrast, koff values remained invariant, with average value of 0.229 ± 0.021 min−1. Each data point represents mean ± S.E. (N = 3 oocytes).
3.4. Dose-dependency of the modulation of NaV1.6/β1 and 1.2/β1 by ι-RXIA at steady-state
To complement the kinetic data for NaV1.6/β1, the steady-state I′Na/IoNa ratio over a range of ι-RXIA concentrations was obtained. The plot of I′Na/IoNa as function of [ι-RXIA] yielded an EC50 of 1.80 μM (95% CI: 0.93 – 3.60 μM) with a Hill coefficient, nH, of 1.04 ± 0.26 (Fig. 5, circles). This EC50 value is in agreement with the Kd of 1.67 to 2.88 μM obtained in the kinetic analysis above.
Figure 5.
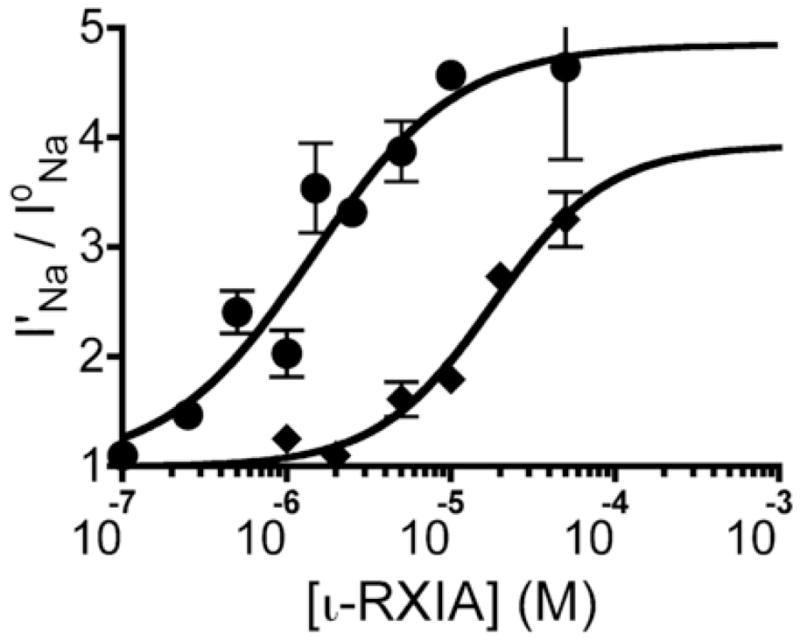
Modulation NaV1.6/β1 and 1.2/β1 as a function of ι-RXIA concentration at steady-state. Current measurements in response to a Vstep to −30 mV were made as in Fig. 4, and modulation is expressed as a ratio of peak currents, I′Na/IoNa, where I′Na and IoNa were obtained in toxin and control solution, respectively. Effect of ι-RXIA on NaV1.6/β1 (circles) and NaV1.2/β1 (diamonds) measured at steady-state, solid line represents data fit to the equation, Y = (Top −1)/(1 + 10^((LogEC50 − Log[ι-RXIA]) × nH)), where nH is the Hill coefficient. Respective EC50 values for NaV1.6/β1 and 1.2/β1 were 1.80 μM (95% CI: 0.93 –3.60 μM) and 17.78 μM (95% CI: 4.70 – 67.21 μM) and nH values were 1.04 ± 0.26 and 1.24 ± 0.44. Each point represents mean ± S.E. (N = 3 oocytes).
The steady-state I′Na/IoNa ratio was also obtained for NaV1.2/β1 challenged with different concentrations of ι-RXIA. The resulting EC50 was 17.78 μM (95% CI: 4.70 – 67.21 μM) with an nH of 1.24 ± 0.44 (Fig. 5, triangles). Thus, ι-RXIA had a significantly lower EC50 for NaV1.6/β1 than 1.2/β1.
3.5. Effects of the L-isomer analog, ι-RXIA[L-Phe44], on NaV1.6/β1 and 1.2/β1
A notable feature of ι-RXIA is the presence of an unusual amino acid, D-Phe, near its carboxy terminus (Fig. 1B). When ι-RXIA[L-Phe44] was tested on oocytes expressing NaV1.6/β1, both its affinity and efficacy were found to be less than that of ι-RXIA. Fig. 6A shows that 50 μM ι-RXIA[L-Phe44] (a saturating concentration, see below) shifts the V1/2 for activation by only 4 mV (versus 16 mV for ι-RXIA at saturation, see Fig 2A). The kinetics of modulation by ι-RXIA[L-Phe44] were also examined, The plot in Fig. 6B shows that, although its kon was essentially the same as that of ι-RXIA, its koff was two-fold faster, yielding a Kd of 5.52 μM. This result is in agreement with its steady-state EC50 of 5.45 μM (95% CI: 2.03 – 14.63 μM), as shown in the dose-response curve in Fig. 6C. Thus, the two-fold lower affinity of ι-RXIA[L-Phe44], relative to that of ι-RXIA, can be explained by its two-fold faster off-rate. Also evident in Fig. 6C is that the maximum increase in I′Na/IoNa, obtained at saturating concentrations of peptide, for ι-RXIA[L-Phe44] is half of that for ι-RXIA, indicating that the efficacy of ι-RXIA is twice that of its analog.
Figure 6.

Comparison of the modulation of NaV1.6/β1 and NaV1.2/β1 by ι-RXIA[L-Phe44] and ι-RXIA. Oocytes were tested essentially as described in Figs. 2, 3, and 4. A, Voltage dependence of activation of NaV1.6/β1 in the absence (open squares) and presence of 50 μM ι-RXIA[L-Phe44] (closed squares); respective V1/2 was −21.66 ± 0. 61 and −25.30 ± 0.27 mV and slope factor was 5.75 ± 0.54 and 5.58 ± 0.23 mV. For comparison, the curve for ι-RXIA from Fig. 2D is shown in gray. B, Plot of kobs vs. concentration of ι-RXIA[L-Phe44] for NaV1.6/β1 (black squares). Slope (or kon) was 0.152 ± 0.008 μM−1•min−1, and Y intercept (or extrapolated koff) was 0.841 ± 0.145 min-1, yielding a Kd of 5.52 μM. For comparison, the curve for ι-RXIA (from Fig. 4D) is shown in gray. C, Modulation of NaV1.6/β1 as a function of ι-RXIA[L-Phe44] concentration (black squares). EC50 was 5.45 μM (95% CI: 2.03 – 14.63 μM) and nH was 1.13 ± 0.39. For comparison, the curve for ι-RXIA (from Fig. 5) is shown in gray; note that at or near saturating [peptide] its efficacy is about twice that of ι-RXIA[L-Phe44]. D, Modulation of NaV1.2/β1 as a function of ι-RXIA[L-Phe44] concentration (black triangles) yields a flat line, showing that the peptide had no effect on NaV1.2/β1. For comparison, the curve for ι-RXIA (from Fig. 5) is shown in gray.
In contrast to its effects on NaV1.6/β1, ι-RXIA[L-Phe44] was found to be inactive on NaV1.2/β1 up to the highest concentration tested (50 μM, Fig. 6D.). To see whether ι-RXIA[L-Phe44] could still bind to NaV1.2/β1 (i.e., to examine the possibility that it had a finite affinity despite a zero efficacy), its ability to interfere with the action of ι-RXIA on NaV1.2/β1 was tested. Oocytes expressing NaV1.2/β1 were preincubated with 50 μM ι-RXIA[L-Phe44], then exposed to ι-RXIA while ι-RXIA[L-Phe44] was still present. The activity of ι-RXIA was unaltered in this experiment (not illustrated), indicating that ι-RXIA[L-Phe44] did not seem to even bind to NaV1.2/β1, much less affect its function.
3.6. ι-RXIA induces action potentials in both A- and C-fibers in mouse sciatic nerve
ι-RXIA induces repetitive activity not only in frog motor axons [19], but also in axons of frog sciatic nerve with a wide range of conduction velocities (not illustrated). This experiment was repeated with mouse sciatic nerve preparations. Conduction velocities were assessed by simultaneously recording extracellularly from two sites along the nerve. As illustrated in Fig. 7A, compound action potentials (CAPs) with two different conduction velocities could be readily resolved following electrical stimulation: A-CAPs and C-CAPs, with conduction velocities of ~12 m/s and <1 m/s, respectively. When a location near one end of the nerve was exposed to 0.05 μM ι-RXIA, spontaneous units of two sorts were observed: brief waveforms with conduction velocities >10 m/s and longer lasting waveforms with conduction velocities < 1 m/s corresponding to action potentials in A- and C-fibers, respectively (Fig. 7B).
Figure 7.
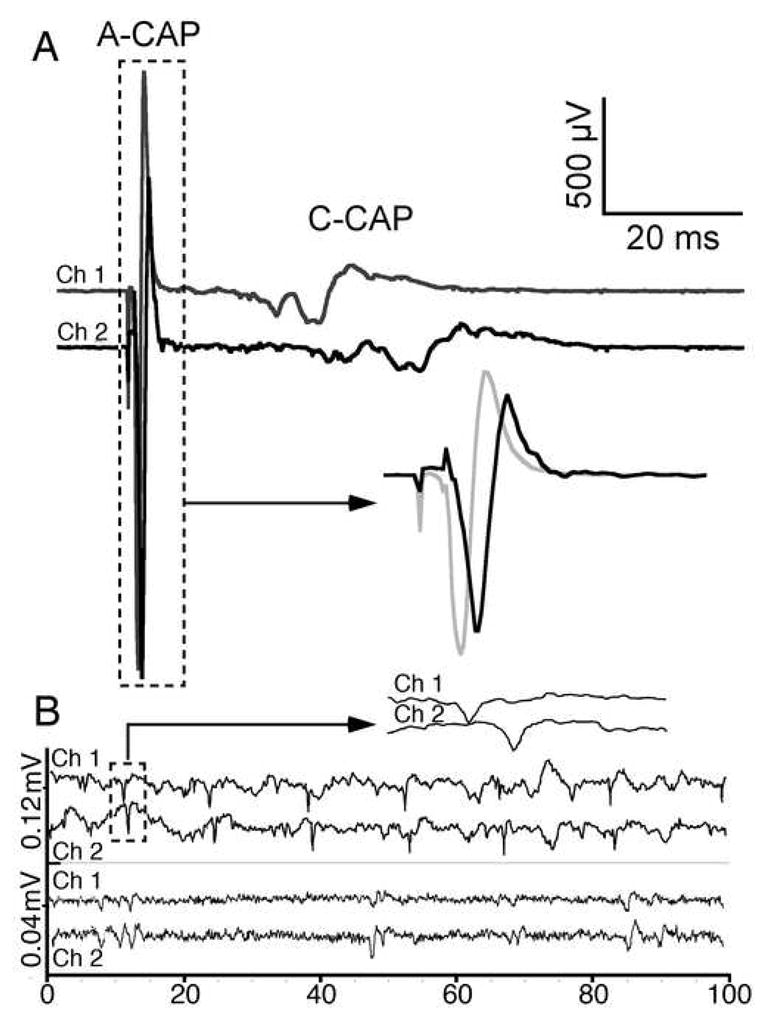
ι-RXIA induces action potentials in A- and C-fibers in mouse sciatic nerve. Extracellular action potentials were recorded simultaneously from two locations along the nerve as described in Methods, 7 mm apart. In each pair of traces, the upper (Channel 1) and lower (Channel 2) traces were recorded from a proximal versus distal location, respectively, relative to the site of electrical stimulus (A) or toxin-application (B). A, Stimulus-evoked compound action potentials (CAPs) in control solution showing fast A-CAP with conduction velocity of 11.8 m/s, and slower C-CAP with conduction velocity in the range of 0.72 to 0.48 m/s. Region of A-CAPs enclosed by (10 ms wide) dashed box is shown in inset with baselines superimposed and reveals a ~ 0.6 ms temporal offset; note 1 ms stimulus artifact near start of traces. B, “Spontaneous” action potentials induced by exposure to 0.05 μM ι-RXIA; representative recordings of single-units with conduction velocities of about 10 (top pair) and 0.7 (bottom pair) m/s. Region of top pair of traces enclosed by (5 ms wide) dashed box is shown in inset, revealing a ~ 0.7 ms temporal offset of this unit corresponding to that of an A-fiber. Lower trace in bottom pair of traces has been shifted leftward by 9.5 ms and reveals near coincidence of units corresponding to those of C-fibers.
4. DISCUSSION
4.1. Properties of ι-RXIA
At saturating concentrations of ι-RXIA (≥ 5 μM), the activation V1/2 of NaV1.6/β1 was shifted by about −16 mV (Fig. 2D). In previous experiments on NaV1.6 without β1, 5 μM ι-RXIA shifted the V1/2 of activation by about −12 mV[20]. Coexpression with β subunits is known to shift NaV1.6’s voltage sensitivity of activation in the hyperpolarized direction and accelerate the time course of fast inactivation [29, 30]. Despite these effects of β1 on NaV1.6, ι-RXIA modulated NaV1.6 whether β1 was present or not.
The interaction of ι-RXIA with NaV1.6/β1 was measured kinetically as well as at steady-state, yielding a Kd between 1.7 and 2.9 μM (Fig. 4) and an EC50 of 1.8 μM (Fig. 5). Furthermore, the Hill coefficient of the steady-state dose-response curve was near 1. Taken together, these results are consistent with a single ι-RXIA-binding site on the channel.
The EC50 of ι-RXIA for NaV1.2/β1 was 17.8 μM (Fig. 5), which is ~10-fold higher than that for NaV1.6/β1. In contrast, ι-RXIA had no measurable effect on NaV1.7/β1 at 10 μM and only a small effect at 50 μM, the maximum concentration tested (Fig. 3). Thus, the order of sensitivity to ι-RXIA was NaV1.6/β1 > NaV1.2/β1 > NaV1.7/β1, and none of the remaining isoforms tested, by coexpression with β1 in oocytes (NaV1.1, 1.3, 1.4, or 1.5) or in mouse DRG neurons (NaV1.8), was sensitive to 50 μM ι-RXIA (Fig. 3).
4.2. Properties of ι-RXIA[LPhe44]
The analog, ι-RXIA [LPhe44], affected NaV1.6/β1 much like ι-RXIA itself did insofar as it negatively shifted the V1/2 for activation, albeit at to a lesser extent (Fig. 6A). In comparison to the native peptide, it had a ~2-fold higher Kd (5.52 μM, Fig. 6B) and EC50 (5.45 μM, Fig. 6C). It had a similar kon and a two-fold faster koff, which account for its two-fold lower affinity.
In addition to its lower affinity, the analog had a lower efficacy, as reflected by the observation that at saturating peptide concentrations it increased the I′Na/IoNa ratio only half as much as ι-RXIA could. This suggests that binding and efficacy may be partially uncoupled, as discussed further below.
Another notable feature of ι-RXIA[LPhe44] is that it was inactive against NaV1.2. Given the aforementioned observations, it seemed possible that the peptide could bind to the channel but be incapable of functionally affecting the channel’s activity. However, this is unlikely since the presence of high concentrations of ι-RXIA[LPhe44] did not inhibit ι-RXIA‘s activity; in other words, ι-RXIA[LPhe44] was unable to shield NaV1.2/β1 against ι-RXIA, as would be expected if the analog could bind to the channel.
In previous studies with mice, involving intracranial administration of conopeptides, we observed that ι-RXIA[L-Phe44] was 10- to 20-fold less active than ι-RXIA in inducing seizures [17]. Both NaV1.2 and 1.6 are present in brain, and the lower potency of ι-RXIA[L-Phe44] in inducing seizures is consistent with its inactivity against NaV1.2 as well as its attenuated activity against NaV1.6.
4.3. Comparison with delta-conopeptides
δ-Conotoxins are excitotoxic (Fig. 1A), and those such as δ-PVIA and δ-SVIE, like ι-RXIA, can shift the voltage sensitivity of activation in the hyperpolarizing direction (West et al., 2005). However, these δ-conopeptides also shift the voltage sensitivity of inactivation, and most saliently, greatly decrease the level of fast inactivation (West et al., 2005). Although RXIA exhibits little tendency to do either, close examination of normalized current traces in the presence and absence of ι-RXIA suggests that the level of inactivation may be slightly decreased by ι-RXIA, but by no more than a few percent (e.g., Fig. 2C).
4.4. Comparison with β-scorpion toxins
Among the NaV1 channels tested, ι-RXIA has highest affinity for NaV1.6. In this regard, it is of interest to note the behavior of β-scorpion toxin Cn2, which was shown to be specific for (human) NaV1.6 among NaV1.1 through 1.7 expressed in transfected HEK cells [31]. Cn2 acts by a voltage-sensor trapping mechanism like that first described for the β-scorpion toxin CssIV, where the leftward shift in the voltage dependence of activation requires a depolarizing priming (or conditioning) pulse [28]. β-Scorpion toxins such as TdVIII [26] and Tz1 [32] can shift the activation curve to some extent without any priming prepulse, but significantly less than when priming is used. In contrast, ι-RXIA did not require priming to be effective, nor have any depolarizing conditioning pulses tested so far enhanced its effect. Furthermore, unlike the β-scorpion toxins mentioned above, ι-RXIA did not significantly shift the voltage dependence of inactivation.
As mentioned previously, the behavior of ι-RXIA[L-Phe44] compared to that of ι-RXIA is consistent with partial uncoupling between binding and efficacy of modulation. Complete uncoupling of binding and modulation has been reported for the insect-specific scorpion β-toxin Bj-xtrIT by replacing its Glu15 with an Arg [33]. Relevant here is the converse experiment by Catterall and colleagues who observed that a G845N mutation in NaV1.2 lowered the channel’s affinity for β-scorpion toxin Css IV by about 10 fold (as measured by radiolabeled toxin-binding) but its voltage dependence of activation (which was unaltered by the mutation) was no longer susceptible to modulation by the toxin [28].
Domain II has been identified as a major binding site of β-scorpion toxins TiTxγ [34] and Css IV [28], specifically the S3-S4 loop, where the critical G845 mentioned above, is located. In addition, the C-terminal loop (SS2) of domain III was also implicated in the interaction of Css IV with the channel [28], and more recent work indicates that this region is a major determinant of β-scorpion toxin Tz1 [32], leading the latter investigators to conclude that the β-scorpion toxin binding site spans domains II and III. It remains to be shown whether this site is also targeted by ι-RXIA.
4.5. The structure of ι-RXIA
The solution structure of ι-RXIA has been determined [20] and can be viewed as composed of a well structured inhibitory cystine knot [35, 36] and C-terminal “tail” of around ten residues which is disordered in solution (Fig. 1C). The D-Phe44 location in the disordered tail implicates this region of the toxin as being involved in modulation. In the parlance of Olivera and colleagues [37], one might consider the knot as interacting with one microsite on the channel, and the tail with another.
A significant difference between ι-RXIA and β-scorpion toxins such as CssIV is that the latter have much faster on- and off-kinetics of modulation [28]. In terms of a voltage sensor trapping mechanism, ι-RXIA may be slow to modulate because it is unable to access the voltage sensor as readily as a toxin that can exploit the unmasking of interaction sites on the voltage sensor provided by a depolarizing conditioning pulse. Moreover, the unstructured tail of the peptide may take time to assume the correct conformation (perhaps via an induced-fit mechanism) to interact with the modulation site.
4.6. Susceptibility of peripheral axons to ι-RXIA
ι-RXIA induces “spontaneous” action potentials in mouse sciatic nerve with shapes and conduction velocities corresponding to A- and C-fibers (Fig. 7). Immunolabeling studies reveal that NaV1.6 is present at nodes of Ranvier of both sensory and motor axons in sciatic nerve [38–40]. In contrast, NaV1.2 and 1.7 are not immunodetected at PNS nodes [38]. Thus, we conclude that ι-RXIA induces action potentials in A-fibers by targeting nodal NaV1.6.
Likewise, the observation that C-fibers were susceptible to ι-RXIA is consistent with NaV1.6-immunoreactivity of unmyelinated peripheral axons [41, 42]. Small sensory neurons, whose axons are C-fibers, also express NaV1.7 and 1.9, and both channel isoforms are immunodetectable in C-fibers [38, 43]. Thus, it is possible that NaV1.7 might participate in the induction of C-fiber activity by ι-RXIA, although this seems unlikely at the low concentration of peptide used (0.05 μM). Finally, we cannot rule out a role for NaV1.9 at this time, since its susceptibility to ι-RXIA is not known.
It should be noted that NaV1.6 has also been identified in non-neuronal cells such as Schwann cells [44] and microglia [45]. ι-RXIA may be useful in helping to identify the role NaV1.6 plays in the functioning of cells such as these, just as the β-CssIV scorpion toxin was exploited to explore the functional involvement of TTX-sensitive channels in cardiac myocytes [46]. Furthermore, at least 17 isoforms of iota-conopeptides homologous to ι-RXIA are produced by Conus radiatus, of which five have been tested and shown to have excitotoxic activity [19]. Comparison of their specificities, affinities, and efficacies should shed light on the structure-activity relationships of both the conopeptides in this remarkably large family of excitotoxins as well as their Na channel targets.
Acknowledgments
This work was supported by National Institute of General Medical Sciences Grant GM 48677. We thank Alan Goldin for the clones for NaV1.1 through 1.6 and β1, Gail Mandel for the NaV1.7 clone, Pradip Bandyopadhyay for help with NaV1.7 cRNA synthesis, and Alex Morrison for help with peptide syntheses.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gonzalez-Burgos G, Barrionuevo G. Voltage-gated sodium channels shape subthreshold EPSPs in layer 5 pyramidal neurons from rat prefrontal cortex. J Neurophysiol. 2001;86:1671–84. doi: 10.1152/jn.2001.86.4.1671. [DOI] [PubMed] [Google Scholar]
- 2.Sontheimer H, Black JA, Waxman SG. Voltage-gated Na+ channels in glia: properties and possible functions. Trends Neurosci. 1996;19:325–31. doi: 10.1016/0166-2236(96)10039-4. [DOI] [PubMed] [Google Scholar]
- 3.Catterall WA, Goldin AL, Waxman SG. International Union of Pharmacology. XLVII. Nomenclature and structure-function relationships of voltage-gated sodium channels. Pharmacol Rev. 2005;57:397–409. doi: 10.1124/pr.57.4.4. [DOI] [PubMed] [Google Scholar]
- 4.Cestele S, Catterall WA. Molecular mechanisms of neurotoxin action on voltage-gated sodium channels. Biochimie. 2000;82:883–92. doi: 10.1016/s0300-9084(00)01174-3. [DOI] [PubMed] [Google Scholar]
- 5.Norton RS, Olivera BM. Conotoxins down under. Toxicon. 2006;48:780–98. doi: 10.1016/j.toxicon.2006.07.022. [DOI] [PubMed] [Google Scholar]
- 6.Terlau H, Olivera BM. Conus venoms: a rich source of novel ion channel-targeted peptides. Physiological reviews. 2004;84:41–68. doi: 10.1152/physrev.00020.2003. [DOI] [PubMed] [Google Scholar]
- 7.Cruz LJ, Gray WR, Olivera BM, Zeikus RD, Kerr L, Yoshikami D, et al. Conus geographus toxins that discriminate between neuronal and muscle sodium channels. The Journal of biological chemistry. 1985;260:9280–8. [PubMed] [Google Scholar]
- 8.Moczydlowski E, Olivera BM, Gray WR, Strichartz GR. Discrimination of muscle and neuronal Na-channel subtypes by binding competition between [3H]saxitoxin and mu-conotoxins. Proc Natl Acad Sci U S A. 1986;83:5321–5. doi: 10.1073/pnas.83.14.5321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.McIntosh JM, Hasson A, Spira ME, Gray WR, Li W, Marsh M, et al. A new family of conotoxins that blocks voltage-gated sodium channels. The Journal of biological chemistry. 1995;270:16796–802. doi: 10.1074/jbc.270.28.16796. [DOI] [PubMed] [Google Scholar]
- 10.Fainzilber M, van der Schors R, Lodder JC, Li KW, Geraerts WP, Kits KS. New sodium channel-blocking conotoxins also affect calcium currents in Lymnaea neurons. Biochemistry. 1995;34:5364–71. doi: 10.1021/bi00016a007. [DOI] [PubMed] [Google Scholar]
- 11.Terlau H, Stocker M, Shon KJ, McIntosh JM, Olivera BM. MicroO-conotoxin MrVIA inhibits mammalian sodium channels, but not through site I. J Neurophysiol. 1996;76:1423–9. doi: 10.1152/jn.1996.76.3.1423. [DOI] [PubMed] [Google Scholar]
- 12.Leipold E, de Bie H, Zorn S, Adolfo B, Olivera BM, Terlau H, et al. μO-Conotoxins Inhibit NaV Channels by Interfering with their Voltage Sensors in Domain-2. Channels. 2007;1:253–62. doi: 10.4161/chan.4847. [DOI] [PubMed] [Google Scholar]
- 13.Zorn S, Leipold E, Hansel A, Bulaj G, Olivera BM, Terlau H, et al. The muO-conotoxin MrVIA inhibits voltage-gated sodium channels by associating with domain-3. FEBS letters. 2006;580:1360–4. doi: 10.1016/j.febslet.2006.01.057. [DOI] [PubMed] [Google Scholar]
- 14.Terlau H, Shon KJ, Grilley M, Stocker M, Stuhmer W, Olivera BM. Strategy for rapid immobilization of prey by a fish-hunting marine snail. Nature. 1996;381:148–51. doi: 10.1038/381148a0. [DOI] [PubMed] [Google Scholar]
- 15.West PJ, Bulaj G, Yoshikami D. Effects of delta-conotoxins PVIA and SVIE on sodium channels in the amphibian sympathetic nervous system. J Neurophysiol. 2005;94:3916–24. doi: 10.1152/jn.01304.2004. [DOI] [PubMed] [Google Scholar]
- 16.Leipold E, Hansel A, Olivera BM, Terlau H, Heinemann SH. Molecular interaction of delta-conotoxins with voltage-gated sodium channels. FEBS letters. 2005;579:3881–4. doi: 10.1016/j.febslet.2005.05.077. [DOI] [PubMed] [Google Scholar]
- 17.Buczek O, Yoshikami D, Bulaj G, Jimenez EC, Olivera BM. Post-translational amino acid isomerization: a functionally important D-amino acid in an excitatory peptide. The Journal of biological chemistry. 2005;280:4247–53. doi: 10.1074/jbc.M405835200. [DOI] [PubMed] [Google Scholar]
- 18.Buczek O, Yoshikami D, Watkins M, Bulaj G, Jimenez EC, Olivera BM. Characterization of D-amino-acid-containing excitatory conotoxins and redefinition of the I-conotoxin superfamily. Febs J. 2005;272:4178–88. doi: 10.1111/j.1742-4658.2005.04830.x. [DOI] [PubMed] [Google Scholar]
- 19.Jimenez EC, Shetty RP, Lirazan M, Rivier J, Walker C, Abogadie FC, et al. Novel excitatory Conus peptides define a new conotoxin superfamily. J Neurochem. 2003;85:610–21. doi: 10.1046/j.1471-4159.2003.01685.x. [DOI] [PubMed] [Google Scholar]
- 20.Buczek O, Wei D, Babon JJ, Yang X, Fiedler B, Chen P, et al. Structure and sodium channel activity of an excitatory I1-superfamily conotoxin. Biochemistry. 2007;46:9929–40. doi: 10.1021/bi700797f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cartier GE, Yoshikami D, Gray WR, Luo S, Olivera BM, McIntosh JM. A new alpha-conotoxin which targets alpha3beta2 nicotinic acetylcholine receptors. The Journal of biological chemistry. 1996;271:7522–8. doi: 10.1074/jbc.271.13.7522. [DOI] [PubMed] [Google Scholar]
- 22.Choi JS, Hudmon A, Waxman SG, Dib-Hajj SD. Calmodulin regulates current density and frequency-dependent inhibition of sodium channel Nav1.8 in DRG neurons. J Neurophysiol. 2006;96:97–108. doi: 10.1152/jn.00854.2005. [DOI] [PubMed] [Google Scholar]
- 23.Zhang MM, Green BR, Catlin P, Fiedler B, Azam L, Chadwick A, et al. Structure/function characterization of micro-conotoxin KIIIA, an analgesic, nearly irreversible blocker of mammalian neuronal sodium channels. The Journal of biological chemistry. 2007;282:30699–706. doi: 10.1074/jbc.M704616200. [DOI] [PubMed] [Google Scholar]
- 24.Zhang MM, Fiedler B, Green BR, Catlin P, Watkins M, Garrett JE, et al. Structural and functional diversities among mu-conotoxins targeting TTX-resistant sodium channels. Biochemistry. 2006;45:3723–32. doi: 10.1021/bi052162j. [DOI] [PubMed] [Google Scholar]
- 25.Fiedler B, Zhang M, Olivera BM, Bulaj G, Yoshikami D. Tetrodotoxin-resistant action potentials in C-fibers of rodent and amphibian sciatic nerve. Soc Neurosci Abstr. 2006:130.13. [Google Scholar]
- 26.Tsushima RG, Borges A, Backx PH. Inactivated state dependence of sodium channel modulation by beta-scorpion toxin. Pflugers Arch. 1999;437:661–8. doi: 10.1007/s004240050830. [DOI] [PubMed] [Google Scholar]
- 27.West PJ, Bulaj G, Garrett JE, Olivera BM, Yoshikami D. Mu-conotoxin SmIIIA, a potent inhibitor of tetrodotoxin-resistant sodium channels in amphibian sympathetic and sensory neurons. Biochemistry. 2002;41:15388–93. doi: 10.1021/bi0265628. [DOI] [PubMed] [Google Scholar]
- 28.Cestele S, Qu Y, Rogers JC, Rochat H, Scheuer T, Catterall WA. Voltage sensor-trapping: enhanced activation of sodium channels by beta-scorpion toxin bound to the S3-S4 loop in domain II. Neuron. 1998;21:919–31. doi: 10.1016/s0896-6273(00)80606-6. [DOI] [PubMed] [Google Scholar]
- 29.Dietrich PS, McGivern JG, Delgado SG, Koch BD, Eglen RM, Hunter JC, et al. Functional analysis of a voltage-gated sodium channel and its splice variant from rat dorsal root ganglia. J Neurochem. 1998;70:2262–72. doi: 10.1046/j.1471-4159.1998.70062262.x. [DOI] [PubMed] [Google Scholar]
- 30.Smith MR, Smith RD, Plummer NW, Meisler MH, Goldin AL. Functional analysis of the mouse Scn8a sodium channel. J Neurosci. 1998;18:6093–102. doi: 10.1523/JNEUROSCI.18-16-06093.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schiavon E, Sacco T, Cassulini RR, Gurrola G, Tempia F, Possani LD, et al. Resurgent current and voltage sensor trapping enhanced activation by a beta-scorpion toxin solely in Nav1.6 channel.Significance in mice Purkinje neurons. The Journal of biological chemistry. 2006;281:20326–37. doi: 10.1074/jbc.M600565200. [DOI] [PubMed] [Google Scholar]
- 32.Leipold E, Hansel A, Borges A, Heinemann SH. Subtype specificity of scorpion beta-toxin Tz1 interaction with voltage-gated sodium channels is determined by the pore loop of domain 3. Molecular pharmacology. 2006;70:340–7. doi: 10.1124/mol.106.024034. [DOI] [PubMed] [Google Scholar]
- 33.Karbat I, Cohen L, Gilles N, Gordon D, Gurevitz M. Conversion of a scorpion toxin agonist into an antagonist highlights an acidic residue involved in voltage sensor trapping during activation of neuronal Na+ channels. Faseb J. 2004;18:683–9. doi: 10.1096/fj.03-0733com. [DOI] [PubMed] [Google Scholar]
- 34.Marcotte P, Chen LQ, Kallen RG, Chahine M. Effects of Tityus serrulatus scorpion toxin gamma on voltage-gated Na+ channels. Circulation research. 1997;80:363–9. doi: 10.1161/01.res.80.3.363. [DOI] [PubMed] [Google Scholar]
- 35.Norton RS, Pallaghy PK. The cystine knot structure of ion channel toxins and related polypeptides. Toxicon. 1998;36:1573–83. doi: 10.1016/s0041-0101(98)00149-4. [DOI] [PubMed] [Google Scholar]
- 36.Pallaghy PK, Nielsen KJ, Craik DJ, Norton RS. A common structural motif incorporating a cystine knot and a triple-stranded beta-sheet in toxic and inhibitory polypeptides. Protein Sci. 1994;3:1833–9. doi: 10.1002/pro.5560031022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Olivera BM, Rivier J, Scott JK, Hillyard DR, Cruz LJ. Conotoxins. The Journal of biological chemistry. 1991;266:22067–70. [PubMed] [Google Scholar]
- 38.Caldwell JH, Schaller KL, Lasher RS, Peles E, Levinson SR. Sodium channel Na(v)1.6 is localized at nodes of ranvier, dendrites, and synapses. Proc Natl Acad Sci U S A. 2000;97:5616–20. doi: 10.1073/pnas.090034797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Krzemien DM, Schaller KL, Levinson SR, Caldwell JH. Immunolocalization of sodium channel isoform NaCh6 in the nervous system. The Journal of comparative neurology. 2000;420:70–83. [PubMed] [Google Scholar]
- 40.Tzoumaka E, Tischler AC, Sangameswaran L, Eglen RM, Hunter JC, Novakovic SD. Differential distribution of the tetrodotoxin-sensitive rPN4/NaCh6/Scn8a sodium channel in the nervous system. Journal of neuroscience research. 2000;60:37–44. doi: 10.1002/(SICI)1097-4547(20000401)60:1<37::AID-JNR4>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 41.Black JA, Renganathan M, Waxman SG. Sodium channel Na(v)1.6 is expressed along nonmyelinated axons and it contributes to conduction. Brain research. 2002;105:19–28. doi: 10.1016/s0169-328x(02)00385-6. [DOI] [PubMed] [Google Scholar]
- 42.Rush AM, Wittmack EK, Tyrrell L, Black JA, Dib-Hajj SD, Waxman SG. Differential modulation of sodium channel Na(v)1.6 by two members of the fibroblast growth factor homologous factor 2 subfamily. The European journal of neuroscience. 2006;23:2551–62. doi: 10.1111/j.1460-9568.2006.04789.x. [DOI] [PubMed] [Google Scholar]
- 43.Fjell J, Hjelmstrom P, Hormuzdiar W, Milenkovic M, Aglieco F, Tyrrell L, et al. Localization of the tetrodotoxin-resistant sodium channel NaN in nociceptors. Neuroreport. 2000;11:199–202. doi: 10.1097/00001756-200001170-00039. [DOI] [PubMed] [Google Scholar]
- 44.Musarella M, Alcaraz G, Caillol G, Boudier JL, Couraud F, Autillo-Touati A. Expression of Nav1.6 sodium channels by Schwann cells at neuromuscular junctions: role in the motor endplate disease phenotype. Glia. 2006;53:13–23. doi: 10.1002/glia.20252. [DOI] [PubMed] [Google Scholar]
- 45.Craner MJ, Damarjian TG, Liu S, Hains BC, Lo AC, Black JA, et al. Sodium channels contribute to microglia/macrophage activation and function in EAE and MS. Glia. 2005;49:220–9. doi: 10.1002/glia.20112. [DOI] [PubMed] [Google Scholar]
- 46.Maier SK, Westenbroek RE, Schenkman KA, Feigl EO, Scheuer T, Catterall WA. An unexpected role for brain-type sodium channels in coupling of cell surface depolarization to contraction in the heart. Proc Natl Acad Sci U S A. 2002;99:4073–8. doi: 10.1073/pnas.261705699. [DOI] [PMC free article] [PubMed] [Google Scholar]