

Abstract
Ir(I)-catalyzed intermolecular allylic amidation of ethyl allylic carbonates with soft nitrogen nucleophiles under completely “salt-free” conditions is described. A combination of [Ir(COD)Cl]2, a chiral phosphoramidite ligand L*, and DBU as a base in THF effects the reaction. The reaction appears to be quite general, accommodating a wide variety of R-groups and soft nitrogen nucleophiles, and proceeds with excellent regio- and enantioselectivities to afford the branched N-protected allylic amines. The developed reaction was conveniently utilized in the asymmetric synthesis of Boc protected α- and β-amino acids as well as (−)-cytoxazone.
Keywords: iridium, phosphoramidite ligand, asymmetric amidation, α-amino acid, β-amino acid, cytoxazone
Ir(I)-catalyzed stereoselective allylic amination reactions have quickly become a method of choice for the preparation of optically enriched allylic amines due to their mild reaction conditions, as well as high regio- and enantioselectivity.1–5 As an alternative and complementary approach, we recently reported that a catalytic system involving [Ir(COD)Cl]2, a chiral phosphoramidite ligand L*, DBU, and proton sponge (PS) in THF enabled highly regio- and enantioselective decarboxylative allylic amidation of substituted allyl benzyl imidodicarboxylates 1 to the branched N-Cbz protected allylic amines 2, and the catalytic reaction was likely to proceed through the Ir-π-allyl complex (eq 1 in Figure 1).6,7 Three possible pathways are conceivable for the carbon-nitrogen bond formation step in the reaction (eq 2 in Figure 1). Oxidative addition of 1 to the Ir(I) catalyst gives rise to the intermediate I, which after proton shift from the nitrogen to the oxygen attacks the Ir-π-allyl complex to form the carbon-nitrogen bond. Alternatively, the intermediate I can undergo decarboxylation to generate the intermediate II, where the anion of benzyl carbamate acts as a nucleophile toward the Ir-π-allyl complex. The third possibility is an intermolecular reaction between the Ir-π-allyl complex and the anion of 1 generated in the reaction.
Figure 1.

Ir(I)-catalyzed decarboxylative allylic amidation reaction of allyl benzyl imidodicarbonates and three possible reaction pathways
We were interested in developing the intermolecular version of the above decarboxylative allylic amidation reaction, and envisioned that pathways I and III could be mimicked by reactions between ethyl allyl carbonates 3 and di-benzyl imidodicarboxylate (or other soft nitrogen nucleophiles) in the presence of a base and the iridium catalyst, whereas the reaction between 3 and benzyl carbamate under similar conditions would follow pathway II (Figure 2).
Figure 2.

Ir(I)-catalyzed intermolecular allylic amidation
Herein, we disclose that the Ir(I) catalyzed intermolecular enantioselective allylic amidation reaction occurs between ethyl allyl carbonates 3 and di-benzyl imidodicarboxylate (or other soft nitrogen nucleophiles) in the presence of DBU to give the corresponding branched N-Bis-Cbz protected allylic amine 4 (or N-Bis-Boc/N-Ac, N-Boc protected allylic amines) in good to excellent yield and stereoselectivity (Figure 2 and Table 1). So far, to the best of our knowledge, there have been only one literature report on the Ir(I)-catalyzed enantioselective allylic amidation reaction, which can give Ac, Cbz, and Boc (arguably three most popular N-protection groups) protected chiral allylic amines.8 While we were preparing the present paper, Helmchen’s group reported a similar work on the enantioselective allylic amidation. Based on all data reported to date, our catalytic systems involving Ir(I), L*, and DBU appears to be more general than the Helmchen’s, accommodating a variety of alkyl/aryl groups and soft nitrogen nucleophiles such as (Ac)(Boc)NH, (Boc)2NH, (Cbz)(Boc)NH, and (Cbz)2NH, especially under “salt-free” conditions.8
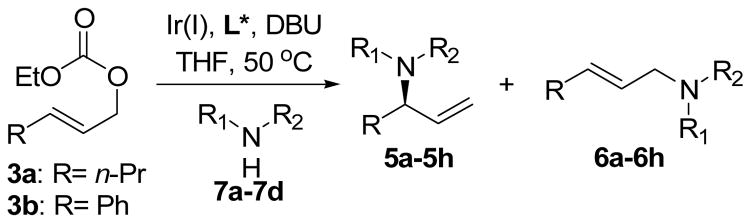
Table 1.
Allylic amidation of ethyl allyl carbonates with different imidodicarboxylates.a
 | |||||||
|---|---|---|---|---|---|---|---|
| entry | carbonate | nucleophile | product | time (h) | yieldb (%) | 5/6c | % ee of 5d |
| 1 | 3a |
7a: R1= Ac- R2= Boc |
5a | 0.5 | 95 | 97:03 | 96 |
| 2 | 3b | 5b | 1.0 | 94 | >99:01 | >99 | |
| 3 | 3a |
7b: R1= Boc- R2= Boc |
5c | 0.5 | 93 | 98:02 | 96 |
| 4 | 3b | 5d | 1.0 | 90 | >99:01 | >99 | |
| 5 | 3a |
7c: R1= Cbz- R2= Boc |
5e | 0.5 | 94 | 98:02 | 96 |
| 6 | 3b | 5f | 1.0 | 92 | >99:01 | >99 | |
| 7 | 3a |
7d: R1= Cbz- R2= Cbz- |
5g | 3.0e | 90 | 99:01 | 96 |
| 8 | 3b | 5h | f | f | f | f | |
All reactions were performed at 0.2 mmol scale.
Isolated yields.
Ratios of regioisomers were determined by 1H-NMR of crude reaction mixtures.
Enantiomeric excesses (ee’s) were determined by converting 5 to their mono N-Cbz/Boc derivatives and using chiral HPLC (Chiracel OD-H).
The reaction was conducted at room temperature.
see the text part.
An allylic amidation reaction was initially performed on ethyl trans-hex-2-enyl carbonate (3a) using [Ir(COD)Cl]2 (2 mol %), a chiral phosphoramidite ligand L* (4 mol %), the anion of benzyl carbamate (generated in situ from benzyl carbamate and n-BuLi at 0 °C) as a nucleophile (1.2 equiv), and DBU as a base (1 equiv) in THF (0.5 mL). Not even a trace amount of the desired product was observed, and the starting materials were recovered. All efforts using other counterions for the anion of benzyl carbamate, other solvents, and halide anions proved to be fruitless.9
However, when N-acetyl tert-butyl carbamate (7a)10 was used as a nucleophile, the corresponding branched allylation product 5a formed in 60% yield after 5 h with excellent regio- and enantioselectivities. Increasing the reaction temperature to 55 °C improved the reaction yield to 95% and reduced the reaction time to 30 min without affecting regio- and enantioselectivities. Subsequently, the amount of DBU was adjusted to 20 mol % without any detrimental effects on reaction yield as well as regio- and enantioselectivities (entry 1 of Table 1). The determined optimal conditions were applied to ethyl cinnamyl carbonate (3b) (the corresponding cinnamyl benzyl imidodicarboxylate was a slower and more difficult substrate in the decarboxylative allylic amidation reaction under similar conditions), and the allylic amidation occurred equally well to give the branched product 5b (entry 2).
Other soft nitrogen nucleophiles such as di-tert-butyl imidodicarboxylate (7b), benzyl tert-butyl imidodicarboxylate (7c),11 and di-benzyl imidodicarboxylate (7d)11 were examined with both ethyl trans-2-hexenyl carbonate and ethyl cinnamyl carbonate. All these reactions proceeded well to give the corresponding branched allylation products in high yields and excellent regio- and enantioselectivities (entries 3–6). In the case using 3a and 7d, a lower reaction temperature (room temperature) was necessary to suppress the transesterification reaction between the branched allylation product 5g and ethoxide anion generated in the reaction (entry 7). The reaction of 3b with 7d was sluggish at room temperature, and gave a mixture of the transesterification product and the usual allylation product (entry 8)
Selective/mono deprotections of the different nitrogen protecting groups of 5a–5g were studied (Scheme 1). Selective deprotection of the Boc group of 5a–5b was effected by TFA in CH2Cl2, while selective deprotection of the acetyl group was achieved by K2CO3 in MeOH. Mono deprotection of the Boc groups of 5c–5d was accomplished by Zn in refluxing MeOH.12 Similarly, treatment of either K2CO3 in MeOH or TFA in CH2Cl2 could be used for selective/mono deprotection of the Cbz/Boc groups of 5e–5g.
Scheme 1.

Selective/mono deprotections of the different nitrogen protection groups
Having established the reaction conditions for the allylic amidation and deprotection reactions, we pursued a one-pot operation of two reactions. The allylation reaction between 7a and 3a in THF at 55 °C followed by the addition of K2CO3 and MeOH at room temperature furnished the corresponding N-Boc protected allylic amine 8a in reaction yield and stereoselectivities comparable to those from the corresponding two step reactions.
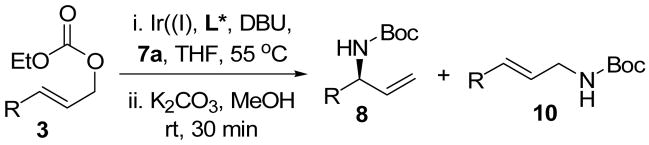
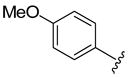
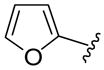
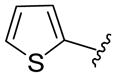
The generality of the one-pot allylic amidation and deprotection sequence was probed with 7a as a nitrogen nucleophile and a variety of different ethyl allyl carbonates, and the results were summarized in Table 2. According to the data, the Ir(I)-catalyzed allylic amidation and selective deprotection sequence appears to be quite general, tolerating a wide variety of substituents such as linear and branched alkyl (entries 1–2), conjugated alkenyl (entry 3), heteroatom functionalized alkyl (entry 4), aryl (entries 5–6), and heteroaryl groups (entries 7–8). In all cases, the N-Boc protected branched allylic amines 5 were obtained in high yields and excellent regio- and enantioselectivities.
Table 2.
One-pot allylic amidation and deprotection using methyl tert-butyl imidodicarboxylate as a nitrogen nucleophile.a
 | |||||
|---|---|---|---|---|---|
| entry | R= | time (h) | yieldb (%) | 8/10c | ee of 8 (%) |
| 1 |
|
0.5 | 95 | 97:03 | 97d |
| 2 |
|
1.5 | 85 | 99:01 | >99e |
| 3 |
|
0.5 | 92 | 98:02 | 96e |
| 4 |

|
0.5 | 94 | 99:01 | 99e |
| 5 |
|
1.0 | 94 | >99:01 | >99d |
| 6 |
|
1.0 | 92 | >99:01 | >99d |
| 7 |
|
1.0 | 85 | >99:01 | 98.5d |
| 8 |
|
1.0 | 80 | 97:03 | ndf |
All reactions were performed at 0.2 mmol scale.
Isolated yields.
Ratios of regioisomers were determined by 1H-NMR of crude reaction mixtures.
Enantiomeric excesses were determined using chiral HPLC.
Enantiomeric excesses were determined after converting to the respective Cbz derivatives.
The recemic compound could not be separated on HPLC using chiral columns OD-H, OJ-H and OB-H in repeated attempts.
To explore a synthetic utility of N-Boc protected chiral allylic amines and to ascertain their stereochemistry, the compound 11 (from entry 5 of Table 2) was converted to the known α- and β-amino acids 12 and 14 as depicted in Scheme 2. The oxidative cleavage of 11 by RuCl3 and NaIO413 produced N-Boc protected (R)-(−)-phenylglycine (12).14 On the other hand, the hydroboration-oxidation15 of 11 followed by oxidation of the resulting alcohol 13 by RuCl3 and NaIO4 gave N-Boc protected β-amino acid 14.16
Scheme 2.

Synthesis of N-Boc protected α- and β-amino acids
As shown in Scheme 3, combined with the Sharpless dihydroxylation reaction,17 the developed allylic amidation reaction could also provide a convenient route for the asymmetric synthesis of (−)-cytoxazone,18 a novel cytokinin modulator isolated from Streptomyces sp.19 The dihydroxylation reaction of the N-Boc protected allylic amine 15 (from entry 6 of Table 2) generated an inseparable mixture of syn- and anti-diols 16 and 17 in the ratio of 3:7. The use of the (DHQD)2-PHAL/(DHQ)2-PHAL ligand in the dihydroxylation reaction did not affect the syn-/anti- ratio in any meaningful way. Thus, the diol mixture was converted to the corresponding TBDPS ethers 18 and 19, which were separated by column chromatography. Upon treatment with NaH in THF, 19 cyclized to form the O-TBDPS protected (−)-cytoxazone, and the final deprotection of the TBDPS group by TBAF provided (−)-cytoxazone (20) in 53% overall yield in five steps starting from the corresponding achiral ethyl allylic carbonate.
Scheme 3.

Asymmetric Synthesis of (−)-cytoxazone
In summary, a highly regio- and enantioselective intermolecular allylic amidation reaction has been developed, which employs [Ir(COD)Cl]2, a chiral phosphoramidite ligand L*, and DBU in THF. The reaction operates under completely “salt-free” conditions, and proves to be quite general, accommodating a wide variety of allylic substrates and nitrogen nucleophiles. Also shown is the possibility of one pot operation of the allylic amidation reaction and subsequent selective/mono deprotection reaction. Since the developed allylic amidation reaction can provide the N-Ac, N-Boc, and N-Cbz (three popular nitrogen protection groups) protected chiral allylic amines, it is nicely complementary to the stereoselective allylic aminations developed by other research groups2–5 and should be of particular value in the asymmetric synthesis of nitrogen-containing chiral compounds.
Acknowledgments
Financial support from National Institute of Health (GM 08194) and The Welch Foundation (AX-1534) is gratefully acknowledged. We are also grateful to Dr. K. Ditrich, BASF, Germany for a generous gift of a chiral amine for the synthesis of ligand L*
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.For reviews: Takeuchi R, Kezuka S. Synthesis. 2006:3349–3366.Helmchen G, Dahnz A, Duebon P, Schelwise M, Weihofen R. Chem Commun. 2007:675–691. doi: 10.1039/b614169b.
- 2.Takeuchi R, Ue N, Tanabe K, Yamashita K, Shiga N. J Am Chem Soc. 2001;123:9525–9534. doi: 10.1021/ja0112036. [DOI] [PubMed] [Google Scholar]
- 3.(a) Yamashita Y, Gopalarathnam A, Hartwig JF. J Am Chem Soc. 2007;129:7508–7509. doi: 10.1021/ja0730718. [DOI] [PubMed] [Google Scholar]; (b) Leitner A, Shekhar S, Pouy MJ, Hartwig JF. J Am Chem Soc. 2005;127:15506–15514. doi: 10.1021/ja054331t. [DOI] [PubMed] [Google Scholar]; (c) Leitner A, Shu C, Hartwig JF. Org Lett. 2005;7:1093–1096. doi: 10.1021/ol050029d. [DOI] [PubMed] [Google Scholar]; (d) Kiener CA, Shu C, Incarvito C, Hartwig JF. J Am Chem Soc. 2003;125:14272–14273. doi: 10.1021/ja038319h. [DOI] [PubMed] [Google Scholar]; (e) Ohmura T, Hartwig JF. J Am Chem Soc. 2002;124:15614–15615. doi: 10.1021/ja028614m. [DOI] [PubMed] [Google Scholar]; (f) Shu C, Leitner A, Hartwig JF. Angew Chem Int Ed. 2004;43:4797–4800. doi: 10.1002/anie.200460276. [DOI] [PubMed] [Google Scholar]
- 4.(a) Polet D, Alexakis A, Tissot-Croset K, Corminboeuf C, Ditrich K. Chem Eur J. 2006;12:3596–3609. doi: 10.1002/chem.200501180. [DOI] [PubMed] [Google Scholar]; (b) Polet D, Alexakis A. Org Lett. 2005;7:1621–1624. doi: 10.1021/ol050350w. [DOI] [PubMed] [Google Scholar]; (c) Tissot-Croset K, Polet D, Alexakis A. Angew Chem Int Ed. 2004;43:2426–2428. doi: 10.1002/anie.200353744. [DOI] [PubMed] [Google Scholar]
- 5.(a) Weihofen R, Dahnz A, Brunner B, Streiff S, Duebon P, Helmchen G. Org Lett. 2005;7:1239–1242. doi: 10.1021/ol047351t. [DOI] [PubMed] [Google Scholar]; (b) Welter C, Koch O, Lipowsky G, Helmchen G. Chem Commun. 2004:896–897. doi: 10.1039/b400023d. [DOI] [PubMed] [Google Scholar]
- Lipowsky G, Helmchen G. Chem Commun. 2004:116–117. doi: 10.1039/b311502j. [DOI] [PubMed] [Google Scholar]
- 6.Singh OV, Han H. J Am Chem Soc. 2007;129:774–775. doi: 10.1021/ja067966g. [DOI] [PubMed] [Google Scholar]
- 7.For recent reviews for the decarboxylative allylation: Tunge JA, Burger EC. Eur J Org Chem. 2005:1715–1726.Tsuji J. Proc Jpn Acad, Ser B. 2004;80:349–358.For recent references on decarboxylative amination/amidation: Wang C, Tunge JA. Org Lett. 2006;8:3211–3214. doi: 10.1021/ol0610744.Mellegaard-Waetzig SR, Rayabarapu DK, Tunge JA. Synlett. 2005:2759–2762.
- 8.Weihofen R, Tverskoy O, Helmchen G. Angew Chem, Int Ed. 2006;45:5546–5549. doi: 10.1002/anie.200601472.. Although the reported catalytic conditions showed some generality for the intermolecular allylic amidation, di-tert-butyl imidodicarboxylate could not be used as a nitrogen nucleophile under the “salt-free” conditions. Furthermore, only two allylic substrates were studied. Weihofen R, Dahnz A, Tverskoy O, Helmchen G. Chem Commun. 2005:3541–3543. doi: 10.1039/b505197e. sulfonamides were used as nucleophiles.
- 9.(a) Shu C, Hartwig JF. Angew Chem, Int Ed. 2004;43:4794–4797. doi: 10.1002/anie.200460214. [DOI] [PubMed] [Google Scholar]; (b) Evans PA, Leahy DK. J Am Chem Soc. 2002;124:7882–7883. doi: 10.1021/ja026337d. [DOI] [PubMed] [Google Scholar]; (c) Kim H, Lee C. Org Lett. 2002;4:4369–4371. doi: 10.1021/ol027104u. [DOI] [PubMed] [Google Scholar]; (d) Fagnou K, Lautens M. Angew Chem Int, Ed. 2002;41:26–47. doi: 10.1002/1521-3773(20020104)41:1<26::aid-anie26>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 10.Tanaka K, Yoshifuji S, Nitta Y. Chem Pharm Bull. 1988;36:3125–3129. [Google Scholar]
- 11.Grehn L, Lurdes M, Almeida S, Ragnarsson U. Synthesis. 1988:992–994. doi: 10.3891/acta.chem.scand.43-0990. [DOI] [PubMed] [Google Scholar]
- 12.Yadav JS, Reddy BVS, Reddy KS, Reddy KB. Tetrahedron Lett. 2002;43:1548–1551. [Google Scholar]
- 13.Carlsen PHJ, Katsuki T, Martin VS, Sharpless KB. J Org Chem. 1981;46:3936–3938. [Google Scholar]
- 14.Dolence EK, Minnick AA, Lin CE, Miller MJ. J Med Chem. 1991;34:968–978. doi: 10.1021/jm00107a014. [DOI] [PubMed] [Google Scholar]
- 15.Alcon M, Canas M, Poch M, Moyano A, Pericas MA, Riera A. Tetrahedron Lett. 1994;35:1589–1592. [Google Scholar]
- 16.Laschat S, Kunz H. J Org Chem. 1991;56:5883–5889. [Google Scholar]
- 17.Becker H, Sharpless KB. In: Asymmetric Oxidation Reactions. Katsuki T, editor. Oxford University Press; Oxford: 2001. pp. 81–104. [Google Scholar]; (b) Zaitsev AB, Adolfsson H. Synthesis. 2006:1725–1756. [Google Scholar]
- 18.For leading references on the synthesis of cytoxazone: Kim IS, Kim JD, Ryu CB, Zee OP, Jung YH. Tetrahedron. 2006;62:9349–9358.Rozwadowska MD. Tetrahedron: Asymmetry. 2006;17:1749–1753.Birmna VB, Jiang H, Li X, Guo L, Uffman EW. J Am Chem Soc. 2006;128:6536–6537. doi: 10.1021/ja061560m.Sohtome Y, Hashimoto Y, Nagasawa K. Eur J Org Chem. 2006:2894–2897.Tosaki SY, Tsuji R, Ohshima T, Shibasaki M. J Am Chem Soc. 2005;127:2147–2155. doi: 10.1021/ja043770+.Boruwa J, Borah JC, Kalita B, Barua NC. Tetrahedron Lett. 2004;45:7355–7358.Sugiyama S, Arai S, Ishii K. Tetrahedron: Asymmetry. 2004;15:3149–3153.Milicevic S, Matovic R, Saicic RN. Tetrahedron Lett. 2004;45:955–957.Davies SG, Hughes DG, Nicholson RL, Smith AD, Wright A. J Org Biomol Chem. 2004;2:1549–1553. doi: 10.1039/b402437k.Carter PH, LaPorte JR, Scherle PA, Decicco CP. Bioorg Med Chem Lett. 2003;13:1237–1239. doi: 10.1016/s0960-894x(03)00131-8.Carda M, Gonzalez F, Sanchez R, Marco JA. Tetrahedron: Asymmetry. 2002;13:1005–1010.Madham A, Kumar AR, Rao BV. Tetrahedron: Asymmetry. 2001;12:2009–2011.Park JN, Ko SY, Koh HY. Tetrahedron Lett. 2000;41:5553–5556.
- 19.Kakeya H, Morishita M, Kobinata K, Osono M, Osada H. J Antibiot. 1998;51:1126–1128. doi: 10.7164/antibiotics.51.1126. [DOI] [PubMed] [Google Scholar]