

Abstract
In continuation of our structure-activity relationship studies on anti-HCV activity of the title imidazo[4,5-e][1,3]diazepine ring system, we report here the synthesis and effect on biological activity of introducing hydrophobic substituents at the 2-position of the heterocycle. Our results suggest that there is no particular advantage to that end as the observed antiviral activity of the test compounds was lower than the unmodified 2-bromo derivative used for comparison. The activity/toxicity profile of all target compounds, however, was still better than the reference compound ribavirin used in the antiviral assay, but not as good as that of interferon-α, the other reference compound used in the assay.
Keywords: HCV; Inhibition; Ring-expanded nucleobases; Imidazo[4,5-e][1,3]diazepines
With nearly three percent of the world population infected with, and at risk of developing liver cancer from, hepatitis C virus (HCV) is one of the most dreadful viruses currently threatening the global health.1-8 While a new T-cell HCV genetic vaccine capable of protecting chimpanzees from acute hepatitis virus challenge has recently been reported,2 there still appears to be no prospect of an effective human vaccine on the horizon.3 The existing therapeutic treatment options are limited, and include a combination therapy with interferon-α and a non-selective and toxic drug ribavirin.9 While a few drugs are currently in clinical trials,4,6,8,10,11 none has yet been approved by FDA. Therefore, the search must continue for an efficacious and non-toxic therapeutic as well as for an effective vaccine to combat this deadly virus.
A number of ring-expanded (“fat”) nucleoside analogues containing the title ring system I, recently synthesized in this laboratory, have exhibited potent in vitro inhibitory activity against viral NTPase/ helicase, a crucial enzyme involved in the replication of not only HCV, but also of other notorious viruses belonging to the same Flaviviridae family such as the West Nile virus (WNV) and the Japanese encephalitis virus (JEV).12,13 We also fortuitously discovered that the sugar moiety in these nucleoside analogues is not always necessary for activity, provided that the N-1 position in I is substituted by an appropriate aralkyl group. A compound identified as ZP-33 (I; R= C8H17, R′=Br, R″=p-methoxybenzyl) showed a promising in vitro anti-HCV activity in a human (Huh7 ET) cell line, using an HCV RNA replicon assay with a stable luciferase (LUC) reporter.14 The present work is an attempt to enhance the antiviral potency of this lead heterocyclic compound through further structure-activity relationship studies.
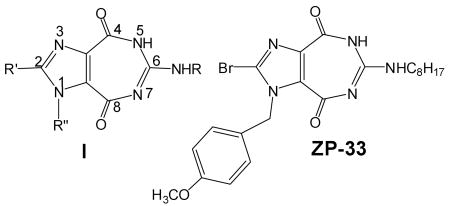
Most of our previous studies were directed at exploring the 1- and 6-positions of I by substitutions, for example, with various deoxy/oxy sugar moieties and α- or β-configurations at position-1 or with various alkyl or aralkyl chains at position-6, while position-2 remained largely unexplored. In this regard, the bromo group at position-2 of ZP-33 provides an excellent opportunity for substitution of both hydrophobic and hydrophilic substituents. In the current preliminary study, we focused on three compounds, including two with hydrophobic (phenyl and p-methoxyphenyl) and one with semi-hydrophobic/hydrophilic (alkynol) substituents. The choice of an ω-alkynol group was based on the reported antiviral activities of ampipathic oligo- and polyribonucleotides.15 The molecular modeling studies have suggested that the distance between the C-2 and the OH of the alkynol group in imidazole nucleosides is approximately the same as that between the C-2 and the 5′-OH of purine nucleosides.16 Therefore, the OH group of an alkynol moiety may play the role of a 5′-OH in nucleosides.
Synthesis of the target compounds (Scheme 1) is straightforward, and started with a precursor to ZP-33, namely ZP-74, reported earlier.14 The Suzuki coupling reaction17 was employed for conversion of the bromo group of ZP-33 to the desired phenyl derivatives ZP-91 and ZP-100, while the Sonogashira alkyne synthesis16,18,19 provided the required alkynol precursor ZP-92. The final step involved the condensation of the above precursors with octylguanidine, which in turn was prepared by the reaction of 3,5-dimethylpyrazole-1-carboxamidinium nitrate with n-octylamine in methanol at reflux, using the procedure of Scott, et al.20 The target 5:7 fused products ZP-94, ZP-101, and ZP-95 were isolated and fully characterized by spectroscopic and microanalytical data.21,22
Scheme 1.

The target compounds were screened against HCV through contractual arrangements with the National Institute of Allergy and Infectious Diseases (NIAID), employing standard protocols, published on NIAID-AACF website.23 Anti-HCV activity and toxicity were assessed by the HCV RNA Replicon assay.24 The results are collected in Table 1. All three compounds were found to be less active and had lower selectivity index (SI) than ZP-33. Nevertheless, the SI values of all four compounds are still better than ribavirin, one of the reference compounds used, although not as good as that of interferon-α, the other reference compound employed in the assay.
Table 1.
Anti-HCV Activity of Ring-Expanded Heterocycles In Vitroψ
| Compd ID | Antiviral Activity HCV RNAa % Control | Toxicity β-Actin RNAb % Control | Selectivity Index (SI)c Toxicity/Antiviral Activity |
|---|---|---|---|
| ZP-94 | 88±9 | 82±4 | 0.932 |
| ZP-101 | 54±2 | 79±1 | 1.463 |
| ZP-95 | 90±6 | 52±6 | 0.578 |
| ZP-33 | 32±13 | 69±3 | 2.155 |
| Interferon-α (10 IU/mL) | 10±1 | 108±4 | 11.3 |
| Ribavirin | 89±10 | 12±1 | 0.42 |
The antiviral activity is based on a primary assay employing 10 μM concentrations of the test compound for determination of both antiviral activity and toxicity. The assay was performed using an Huh7 ET cell line, which contains the HCV RNA replicon with a stable luciferase (LUC) reporter.
HCV RNA-derived LUC activity is used as an indirect measure of HCV RNA levels.
β-Actin RNA level is used as a positive control for cellular RNA in order to compute cytotoxicity.
Selectivity index (SI) is represented as a ratio of the levels of β-Actin RNA/HCV RNA.
In conclusion, there appears to be no specific advantage in replacing the bromo group at the 2-position of ZP-33 with a hydrophobic substituent. It remains to be seen if a hydrophilic substituent at the same position or a smaller hydrophobic group than phenyl, such as alkyl substituents, would enhance the antiviral activity, and the work is currently in progress to that end. Studies on the mechanism of action of ZP-33 are also underway, which are anticipated to further assist in accelerating and properly steering the undertaken SAR studies.
Supplementary Material
Acknowledgments
The research was supported in part by grants (# 5 RO1 AI55452 & #1 R21 AI071802) from the National Institute of Allergy and Infectious Diseases (NIAID) of the National Institutes of Health, Bethesda, Maryland, and an unrestricted grant from Nabi Biopharmaceuticals, Rockville, Maryland. We sincerely thank Dr. Christopher Tseng, the Program Officer of Antiviral Research and Antimicrobial Chemistry of the Virology Branch of the National Institute of Allergy and Infectious Diseases (NIAID), Bethesda, Maryland for his support and encouragement throughout the course of this work. We also acknowledge the continual assistance provided by Dr. Cecil Kwong, the coordinator of NIAID's Antimicrobial Acquisition and Coordinating Facility (AACF) at the Southern Research Institute, Birmingham, Alabama.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Dufour DR. Molecular Diagnostics. (2nd) 2006:461. [Google Scholar]
- 2.Ferrari C. J Hepatol. 2006;45:163. doi: 10.1016/j.jhep.2006.04.005. [DOI] [PubMed] [Google Scholar]
- 3.Houghton M, Abrignani S. Nature. 2005;436:961. doi: 10.1038/nature04081. [DOI] [PubMed] [Google Scholar]
- 4.Huang Z, Murray MG, Secrist JA. Antiviral Res. 2006;71:351. doi: 10.1016/j.antiviral.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 5.MacDonald A, Harris M. Liver Dis. 2006;2:439. [Google Scholar]
- 6.Neyts J. Antiviral Res. 2006;71:363. doi: 10.1016/j.antiviral.2006.06.006. [DOI] [PubMed] [Google Scholar]
- 7.Pol S, Mallet VO. Expert Opin Biol Ther. 2006;6:923. doi: 10.1517/14712598.6.9.923. [DOI] [PubMed] [Google Scholar]
- 8.Toniutto P, Fabris C, Pirisi M. Expert Opin Pharmacother. 2006;7:2025. doi: 10.1517/14656566.7.15.2025. [DOI] [PubMed] [Google Scholar]
- 9.Feld JJ, Hoofnagle JH. Nature. 2005;436:967. doi: 10.1038/nature04082. [DOI] [PubMed] [Google Scholar]
- 10.Zhang R, Durkin JP, Windsor WT. Biorg Med Chem Lett. 2002;12:1005. doi: 10.1016/s0960-894x(02)00102-6. [DOI] [PubMed] [Google Scholar]
- 11.De Francesco R, Migliaccio G. Nature. 2005;436:953. doi: 10.1038/nature04080. [DOI] [PubMed] [Google Scholar]
- 12.Zhang N, Chen HM, Koch V, Schmitz H, Liao CL, Bretner M, Bhadti VS, Fattom AI, Naso RB, Hosmane RS, Borowski P. J Med Chem. 2003;46:4149. doi: 10.1021/jm030842j. [DOI] [PubMed] [Google Scholar]
- 13.Zhang N, Chen HM, Koch V, Schmitz H, Minczuk M, Stepien P, Fattom AI, Naso RB, Kalicharran K, Borowski P, Hosmane RS. J Med Chem. 2003;46:4776. doi: 10.1021/jm030277k. [DOI] [PubMed] [Google Scholar]
- 14.Zhang P, Zhang N, Korba BE, Hosmane RS. Bioorg Med Chem Lett. 2005;15:5397. doi: 10.1016/j.bmcl.2005.09.015. [DOI] [PubMed] [Google Scholar]
- 15.Hyde RM, Broom AD, Buckheit RW. J Med Chem. 2003;46:1878. doi: 10.1021/jm0203276. [DOI] [PubMed] [Google Scholar]
- 16.Lang P, Magnin G, Mathis G, Burger A, Biellmann JF. J Org Chem. 2000;65:7825. doi: 10.1021/jo000841o. [DOI] [PubMed] [Google Scholar]
- 17.Schomaker JM, Delia TJ. J Org Chem. 2001;66:7125. doi: 10.1021/jo010573+. [DOI] [PubMed] [Google Scholar]
- 18.Li P, Wang L. Synlett. 2006:2261. [Google Scholar]
- 19.Firth AG, Fairlamb IJS, Darley K, Baumann CG. Tetrahedron Lett. 2006;47:3529. [Google Scholar]
- 20.Scott FL, O'Donovan DG, Reilly J. J Am Chem Soc. 1953;75:4053. [Google Scholar]
- 21.Experimental: Butyl 1-p-Methoxybenzyl-2-phenyl imidazole-4,5-dicarboxylate (ZP-91). A 25 mL round-bottom flask was charged with 0.58 g of ZP-74 (1.24 mmol), 0.15 g (1.24 mmol) of phenylboronic acid, 0.39 g of sodium carbonate (3.75 mmol dissolved in 1.8 mL water), 3.6 mL of benzene and 0.36 mL of ethanol. The flask was covered with aluminum foil and a trace amount of tetrakis(triphenyl phosphine)palladium was added. The reaction was heated to reflux for 60 h and the solvent was evaporated. The residue was dissolved in 50 mL of dichloromethane and washed with water (3×50 mL). The organic layer was dried over anhy. Na2SO4, filtered and evaporated to give a brown foam, which was purified by column chromatography, eluting with chloroform. The appropriate fractions (Rf = 0.23, chloroform) were pooled and evaporated to give a syrup, which was dried in vacuo overnight. Yield: 0.37 g (63.5%), 1H NMR (CDCl3) δ 7.58, 7.34, 6.94, 6.75 (4 m, 9H, Ar-H), 5.40 (s, 2H, NCH2Ph), 4.33 (t, J=6.6 Hz, 2H, OCH2 of Butyl), 4.17 (t, J=7.0 Hz, 2H, OCH2 of Butyl), 3.77 (s, 3H, OCH3), 1.74, 1.57, 1.44, 1.27 (4 m, 8H, (CH2)2 of 2 Butyl), 0.95, 0.88 (2 t, J=7.0 Hz, 6H, CH3 of Butyl). Anal. (C27H32N2O5 . H2O) C, H, N.Butyl 2-(But-3-yn-1-ol)-1-p-methoxybenzylimidazole-4,5-di carboxylate (ZP-92). To a solution of ZP-74 (0.66 g, 1.41 mmol) in dry THF (20 mL), diisopropylamine (0.24 mL, 1.7 mmol), CuI (0.06 g), trans-dichlorobis(triphenyl phosphine)palladium(II) (0.03 g) and 3-butyn-1-ol (0.11 mL, 1.41 mmol) was added successively under N2 protection. The reaction system was degassed several times whenever necessary. The obtained mixture was heated at 45 °C for 36 h, cooled down and concentrated to dryness. The residue was purified by silica gel flash chromatography, eluting with a mixture of chloroform: methanol (100:1). The appropriate fractions (Rf =0.39, chloroform:Methanol = 30:1) were collected and evaporated to give 0.2 g of brown liquid (31%). 1H NMR (CDCl3) δ 7.13, 6.82 (2 m, 4H, Ar-H), 5.43 (s, 2H, NCH2Ph), 4.28 (t, J=6.6 Hz, 2H, OCH2 of Butyl), 4.20 (t, J=7.6 Hz, 2H, OCH2 of Butyl), 3.82 (m, 2H, CH2OH), 3.78 (s, 3H, OCH3), 2.72 (t, J=6.2 Hz, 2H, CH2) 2.22 (br, 1H, OH, exchangeable with D2O), 1.71, 1.58, 1.42, 1.30 (4 m, 8H, (CH2)2 of 2 Butyl), 0.93, 0.89 (2 t, J=7.3 Hz, 6H, CH3 of Butyl). Anal. (C25H32N2O6 . ½ H2O) C, H, N.General Method for Ring Closure Condensation Reactions of Diesters with Substituted Guanidine to Synthesize ZP-94, ZP-95, and ZP-101. Hemisulfate or nitrate salt of octylguanidine (4 mmol) was suspended in anhydrous methanol (6.0 mL) and cooled to 0 °C. A solution of sodium methoxide (25 wt%, 2.1 mL, 9.2 mmol) was added. The resulting mixture was stirred in an ice bath for 30 min. The precipitated sodium chloride was removed by filtration, and the filtrate was poured into a methanolic solution (20 mL) of the appropriate diester precursor (1 mmol) (ZP-91, ZP-92 or ZP-100). The mixture was stirred at room temperature for 16-72 h, and was monitored by frequent TLC analysis to check for the completion of reaction. The reaction mixture was filtered if necessary and the clear solution was evaporated to dryness. The residue was purified by flash chromatography on a silica gel column. The appropriate fractions were combined and evaporated to obtain the product. The latter was recrystallized from an appropriate solvent when necessary. The spectral and analytical data, along with solvent of recrystallization and/or solvent of elution for chromatography are collected as below.7,8-Dihydro-4H-1-(p-methoxy benzyl)-6-N-octylamino-2-phenylimidazo[4,5-e][1,3]diazepine-4,8-dione (ZP-94). The precursor ZP-91 was condensed with octylguanidine hemisulfate using the General Procedure given above. Yield 69%, Rf = 0.21 (chloroform:methanol (30:1)), purified by silica gel flash chromatography, eluting with chloroform/methanol (30:1). 1H NMR (CDCl3) δ 10.67, 7.92 (2 br, 2H, NH, exchangeable with D2O), 7.46, 7.45, 6.82, 6.70 (4 m, 9H, Ar-H), 5.69 (s, 2H, CH2), 3.73 (s, 3H, OCH3), 3.19 (m, 2H, NHCH2), 1.49-1.78 (2 m, 12 H, C6H12), 0.85 (t, J=7.3 Hz, 3H, CH3). Anal. (C28H33N5O3. 5/4 H2O) C, H, N.2-(But-3-yn-1-ol)-7,8-dihydro-4H-1-(p-methoxy benzyl)-6-N-octylaminoimidazo[4,5-e][1,3]diazepine-4,8-dione (ZP-95). The precursor ZP-92 was condensed with octylguanidine hemisulfate using the General Procedure given above. Yield: 30%, Rf = 0.06 (chloroform: methanol (30:1)), mp 212 °C, purified by silica gel flash chromatography, eluting with a mixture of chloroform: methanol (30:1). 1H NMR (DMSO-d6) δ 10.50, 7.10 (2 br, 2H, NH, exchangeable with D2O), 7.19, 6.86, (2 m, 4H, Ar-H), 5.61 (s, 2H, CH2), 5.06 (1H, OH, exchangeable with D2O), 3.71 (s, 3H, OCH3), 3.60 (q, J=5.8 Hz, 2H, CH2OH) 3.20 (t, J=5.5 Hz, 2H, NHCH2), 2.68 (t, J=5.9 Hz, 2H, CH2) 1.45, 1.25 (2 m, 12 H, C6H12), 0.85 (t, J=6.5 Hz, 3H, CH3). 13C NMR (75 MHz, DMSO-d6) δ 162.7, 159.2, 158.7, 155.1, 151.2, 147.7, 143.4, 137.6, 129.0, 129.0, 123.5, 113.9, 60.2, 58.9, 50.0, 40.6, 31.1, 28.6, 28.5, 26.2, 23.1, 22.0, 13.8; Anal. (C26H33N5O4) C, H, N.Butyl 1-p-Methoxybenzyl-2-(p-methoxy phenyl)imidazole-4,5-dicarboxylate (ZP-100). Experimental procedure is the same as that given for ZP-91 except that p-methoxyphenyl boronic acid is used in place of phenylboronic acid. Yield: 74%, Rf = 0.21 (chloroform), purified by silica gel flash chromatography, eluting with a mixture of chloroform:methanol (100:1). 1H NMR (CDCl3) δ 7.48, 6.92, 6.82 (3 m, 8H, Ar-H), 5.39 (s, 2H, NCH2Ph), 4.33 (t, J=7.3 Hz, 2H, OCH2 of Butyl), 4.16 (t, J=6.6 Hz, 2H, OCH2 of Butyl), 3.83, 3.77 (2 s, 6H, OCH3), 1.74, 1.55, 1.44, 1.27 (4 m, 8H, (CH2)2 of 2 Butyl), 0.95 (t, J=7.7 Hz, 3H, CH3 of Butyl), 0.88 (t, J=7.7 Hz, 3H, CH3 of Butyl). Anal. (C28H34N2O6) C, H, N.7,8-Dihydro-4H-1-(p-methoxybenzyl)-2-(p-methoxy phenyl)-6-N-octylamino imidazo[4,5-e][1,3]diazepine-4,8-dione (ZP-101). The precursor: ZP-100 was condensed with octylguanidine hemisulfate. Yield: 53%, Rf = 0.16 (chloroform:methanol (30:1)), purified by silica gel flash chromatography, eluting with a mixture of chloroform: methanol (30:1). 1H NMR (CDCl3) δ 10.96, 8.02 (2 br, 2H, NH, exchangeable with D2O), 7.41, 7.38, 6.94, 6.86, 6.72 (5 m, 8H, Ar-H), 5.68 (s, 2H, CH2), 3.85, 3.73 (2 s, 6H, OCH3), 3.20 (m, 2H, NHCH2), 1.42-0.8 (m, 15 H, C7H15). 13C NMR (75 MHz, CDCl3) δ 162.8, 161.4, 159.2, 153.3, 147.9, 134.4, 132.4, 131.2, 129.2, 128.8, 128.4, 121.2, 114.5, 114.3, 55.5, 55.4, 49.1, 42.2, 32.0, 29.5, 29.4, 29.1, 27.0, 22.8, 14.2. Anal. (C29H35N5O4 .H2O) C, H, N.
- 22.The observed C, H & N microanalyses were within 0.4% of the theoretical values (see Suppl. Data).
- 23.HCV: see http://www.niaid-aacf.org/protocols/HCV.htm.
- 24.Okuse C, Rinaudo JA, Farrar K, W F, Korba BE. Antivir Res. 2005;65:23. doi: 10.1016/j.antiviral.2004.09.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.