

Abstract
Emotional-affective and cognitive dimensions of pain are less well understood than nociceptive and nocifensive components, but the forebrain is believed to play an important role. Recent evidence suggests subcortical and cortical brain areas outside the traditional pain processing network contribute critically to emotional-affective responses and cognitive deficits related to pain. These brain areas include different nuclei of the amygdala and certain prefrontal cortical areas. Their roles in various aspects of pain will be discussed. Biomarkers of cortical dysfunction are being identified that may evolve into therapeutic targets to modulate pain experience and improve pain-related cognitive impairment. Supporting data from preclinical studies in neuropathic pain models will be presented. Neuroimaging analysis provides evidence for plastic changes in the pain processing brain network. Results of clinical studies in neuropathic pain patients suggest that neuroimaging may help determine mechanisms of altered brain functions in pain as well as monitor the effects of pharmacologic interventions to optimize treatment in individual patients. Recent progress in the analysis of higher brain functions emphasizes the concept of pain as a multidimensional experience and the need for integrative approaches to determine the full spectrum of harmful or protective neurobiological changes in pain.
Keywords: Pain, emotion, cognition, decision-making, amygdala, prefrontal cortex
1. Introduction
A multidimensional experience pain not only includes nociceptive and nocifensive but also emotional-affective and cognitive components (Figure 1). This article will draw attention to some brain areas and functions that have been implicated in the well-documented reciprocal interactions between pain, affect and cognition (Rhudy and Meagher, 2001; Gallagher and Verma, 2004; Flor and Turk, 2006; Seminowicz and Davis, 2007; Tracey and Mantyh, 2007; Baliki et al., 2008). Pain hurts. Pain can lead to anxiety and depression. Conversely, patients suffering from anxiety and depression experience pain more strongly and are more likely to develop chronic pain. Pain also impairs cortical functions such as our ability to think clearly and make advantageous decisions. In turn, cognitive processes can modulate pain perception (deCharms et al., 2005; Mayer et al., 2006; Seminowicz and Davis, 2007; Rhudy et al., 2008). Understanding these complex interactions is required for improved assessment and management of pain.
Figure 1. Pain as a multidimensional experience.
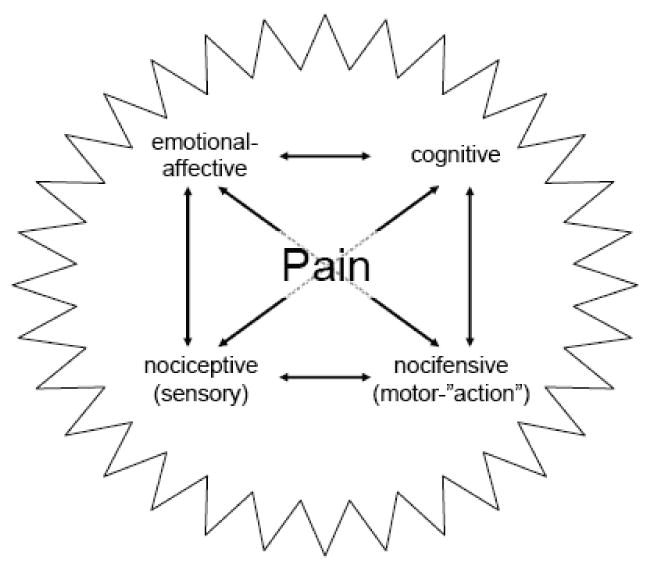
Different components interact to form the complex nature of pain. Their reciprocal relationships are indicated by arrows.
Neurobiological mechanisms of the different aspects of pain are only beginning to emerge. A network of brain structures that process pain-related information has emerged based on neuroimaging studies (Casey, 1999; Seminowicz et al., 2004; Mackey and Maeda, 2004; Apkarian et al., 2005; Mayer et al., 2006; Tracey and Mantyh, 2007; Zhuo, 2008). This so-called “pain matrix” or “homeostatic afferent processing network” consistently includes primary (S1) and secondary (S2) somatosensory cortices, insular cortex, anterior cingulate cortex (ACC), and thalamic nuclei. S1 cortex is generally associated with sensory-discriminative aspects (but see Craig, 2003), S2 likely has additional affective/cognitive functions, while the insula and ACC are important for affective-motivational and certain cognitive aspects of pain, including anticipation, attention and evaluation (Seminowicz et al., 2004; Apkarian et al., 2005; Ohara et al., 2005; Zhuo, 2008).
It is clear now that prefrontal cortical areas other than ACC and subcortical areas such as the amygdala are also part of the brain network for pain (Neugebauer et al., 2004; Apkarian et al., 2005; Ochsner et al., 2006; Neugebauer, 2006; Baliki et al., 2006; Mayer et al., 2006; Kulkarni et al., 2007; Tracey and Mantyh, 2007). These brain areas may play a role in “secondary pain affect”, which includes the conscious awareness and cognitive evaluation of pain (Price, 2000). Pain-related changes in these brain areas may contribute to the emotional-affective and emotion-based cognitive consequences of pain. Conversely, pain can be modulated by emotional (fear and anxiety) and cognitive (attention, expectation, or memory) factors (deCharms et al., 2005; Mayer et al., 2006; Seminowicz and Davis, 2007; Rhudy et al., 2008). The neurobiology of these top-down processes remains to be determined and will not be discussed here. It should be noted, however, that activation of the ACC (Calejesan et al., 2000; Zhuo, 2008) or disinhibition of the insula (Jasmin et al., 2003) can facilitate nocifensive reflexes whereas stimulation of the medial PFC inhibited nocifensive responses and mixed results have been reported for orbitofrontal cortex activation (see Ohara et al., 2005).
2. Emotional-affective aspects of pain and amygdala dysfunction
The amygdala has long been known for its important role in emotions and affective disorders (Pare et al., 2004; Maren, 2005; Phelps and Ledoux, 2005). An increasing body of evidence from anatomical, neurochemical, electrophysiological and behavioral studies strongly supports the concept of the amygdala as an important player in the emotional-affective dimension of pain (Heinricher and McGaraughty, 1999; Rhudy and Meagher, 2001; Gauriau and Bernard, 2002; Neugebauer et al., 2004; Neugebauer, 2006; Pedersen et al., 2007; Ikeda et al., 2007; Carrasquillo and Gereau, 2007; Ji et al., 2007).
2.1 Amygdala circuitry
The amygdala includes several nuclei (Figure 2). The lateral (LA), basolateral (BLA) and central (CeA) nuclei are of particular importance for sensory processing. The amygdala receives nociceptive information through anatomically and functionally distinct lines of input (Neugebauer et al., 2004; Braz et al., 2005; Neugebauer, 2006). Purely nociceptive information reaches the latero-capsular division of the CeA (CeLC) directly from spinal cord and brainstem (parabrachial area, PB), thus bypassing the thalamus (Bernard and Besson, 1990; Cliffer et al., 1991; Gauriau and Bernard, 2004). Polymodal sensory, including nociceptive, inputs from thalamus (posterior areas) and cortex (insula and association cortices) target the LA, the initial site of sensory convergence and integration in the amygdala (Shi and Davis, 1999; Pare et al., 2004; Phelps and Ledoux, 2005). Associative processing in the LA-BLA network is believed to attach emotional significance to sensory information and play an important role in fear and anxiety (Pare et al., 2004; Maren, 2005; Phelps and Ledoux, 2005). Highly processed affect-related information is then transmitted to the CeA, which can modulate pain behavior through projections to descending pain control centers in the brainstem (Heinricher and McGaraughty, 1999; Fields, 2000; Mason, 2005; Neugebauer, 2006; Tracey and Mantyh, 2007).
Figure 2. Amygdala function in pain.
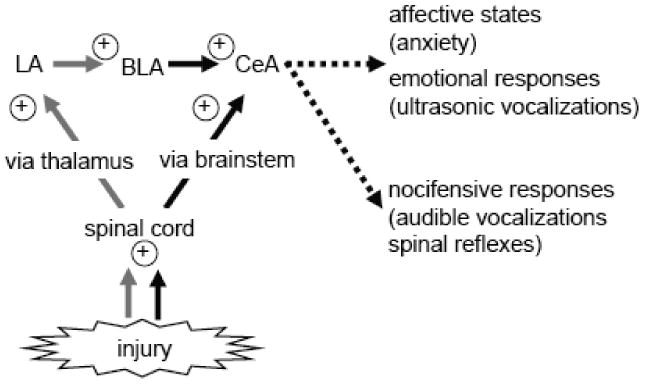
The amygdala receives nociceptive information through pathways that either do or do not involve the thalamus. A serial arrangement of different amygdala nuclei contributes to emotional-affective aspects of pain and modulates nocifensive responses. Thalamic inputs to the LA-BLA circuitry are indicated by gray lines. Black lines show inputs to the CeA that involve direct extra-thalamic pathways. Plus symbols indicate injury-induced increased excitability. LA, BLA, CeA: lateral, basolateral, central nuclei of the amygdala.
The amygdala is also closely interconnected with cortical areas. The BLA is a major source of input to certain prefrontal cortical (PFC) areas (Porrino et al., 1981; McDonald, 1991; Bacon et al., 1996; Price, 2003; Ishikawa and Nakamura, 2003; Orozco-Cabal et al., 2006; Gabbott et al., 2006). A neural circuit that involves medial and orbital PFC and amygdala (BLA) is of critical importance for cognitive functions such as decision-making based on reward expectancy, risk anticipation and punishment avoidance (Bechara et al., 1999; Bechara et al., 2003; Holland and Gallagher, 2004; Fukui et al., 2005; Stalnaker et al., 2007; Hampton et al., 2007; Floresco and Ghods-Sharifi, 2007). Therefore, it is tempting to speculate that the amygdala contributes not only to emotional-affective but also certain cognitive aspects of pain (see section 3).
2.2 Pain-related amygdala plasticity
Pain is associated with increased responsiveness of CeLC neurons to peripheral stimuli (see Neugebauer et al., 2004; Neugebauer, 2006). The so-called “central sensitization” includes enhanced neurotransmission at the BLA-CeLC and the PB-CeLC synapses (“synaptic plasticity”) as well as increased membrane excitability of CeLC neurons and has been shown in models of arthritic (Neugebauer et al., 2003), visceral (Han and Neugebauer, 2004) and neuropathic (Ikeda et al., 2007) pain. Synaptic plasticity in the CeLC was mimicked by tetanic stimulation of presumed nociceptive inputs from the parabrachial area (Lopez de Armentia and Sah, 2007). Recent imaging data also showed increased amygdala activation related to knee pain in patients with osteoarthritis (Kulkarni et al., 2007). Pain-related plasticity in the CeLC is believed to be the consequence of converging affect-related input from the BLA with nociceptive input from spinal cord and brainstem (reviewed in Neugebauer et al., 2004; Neugebauer, 2006; see Figure 1). Preliminary data (Fu and Neugebauer, unpublished observations) suggest that the BLA undergoes pain-related plastic changes as well.
Arthritis pain-related synaptic plasticity and central sensitization in the CeLC require the upregulation of presynaptic metabotropic glutamate receptors (mGluRs) of the group I type to enhance excitatory neurotransmission (Neugebauer et al., 2003; Li and Neugebauer, 2004b). Western blot analysis showed increased levels of mGluR1 and mGluR5 protein in the CeA of arthritic rats compared with controls. In contrast, presynaptic group II and III mGluRs have inhibitory effects on synaptic transmission and pain-related plasticity in the CeLC (Han et al., 2004; Li and Neugebauer, 2006; Han et al., 2006). On the postsynaptic site, NMDA receptor function is increased in the arthritis pain model through a mechanism that involves PKA-dependent NR1 phosphorylation (Li and Neugebauer, 2004a; Bird et al., 2005), but synaptic plasticity in a neuropathic pain model appears to be NMDA receptor-independent (Ikeda et al., 2007). Neuropeptides such as calcitonin gene-related peptide (CGRP) and corticotropin releasing factor (CRF), high levels of which are present in the amygdala, typically couple to the activation of cAMP through CGRP1 and CRF1 receptors, respectively. CGRP1 receptors (Han et al., 2005) and CRF1 receptors (Ji and Neugebauer, 2007; Ji and Neugebauer, 2008) contribute to pain-related plasticity in the CeLC through a PKA-dependent increase of NMDA receptor function. In addition to PKA, ERK enhances NMDA receptor function in the CeLC in the arthritis pain model (Fu et al., 2008). Pain-related ERK activation was directly measured in the CeLC (Carrasquillo and Gereau, 2007), but the underlying mechanisms are not known yet.
2.3 Amygdala-mediated emotional-affective pain behavior
A consequence of pain-related plasticity in the amygdala is increased pain behavior. Pharmacologic deactivation of the CeA by compounds with demonstrated inhibitory effects in this brain area decreased nocifensive and affective pain behaviors in arthritic animals. Antagonists for group I mGluR (Han and Neugebauer, 2005), CGRP1 (Han et al., 2005) and CRF1 (Fu and Neugebauer, 2008) receptors, PKA and ERK inhibitors (Fu et al., 2008), and group III mGluR8 agonist (Palazzo et al., 2008a) all inhibited spinal withdrawal reflexes and supraspinally organized audible and ultrasonic vocalizations that were increased in the arthritis pain model. A CRF1 receptor antagonist (Ji et al., 2007) and a mGluR8 agonist (Palazzo et al., 2008b) reversed pain-related anxiety-like behavior in arthritic rats. A CRF receptor antagonist also reduced opiate withdrawal-associated hyperalgesia (McNally and Akil, 2002). Inhibition of ERK activation in the CeA decreased formalin-induced mechanical, but not thermal, hypersensitivity and had no effect on “spontaneous” nocifensive responses (Carrasquillo and Gereau, 2007). Excitotoxic amygdala lesions abolished conditioned place aversion, a measure of negative affective-motivational behavior, in the formalin test and in a visceral pain model, but had no effect on “spontaneous” nocifensive behavior (Tanimoto et al., 2003). Deactivation of the CeA with a GABAA receptor agonist attenuated escape/avoidance behavior and reversed hindpaw mechanical hypersensitivity in neuropathic rats (Pedersen et al., 2007).
Conversely, activation of the CeA with corticosterone implants produced anxiety-like behavior (Greenwood-Van Meerveld et al., 2001; Myers et al., 2005; Myers and Greenwood-Van, 2007), increased visceromotor responses to colorectal distension (Greenwood-Van Meerveld et al., 2001; Qin et al., 2003b; Myers et al., 2005; Myers et al., 2007; Myers and Greenwood-Van, 2007), produced mechanical but not thermal hypersensitivity (Myers et al., 2007), and sensitized viscerosensitive spinal neurons (Qin et al., 2003a; Qin et al., 2003b; Qin et al., 2003c).
These results clearly show a positive correlation of amygdala activity with pain behavior and affective states such as anxiety. However, the amygdala is also important for pain inhibition, particularly in the context of conditioned and stress- or morphine-induced analgesia (Helmstetter, 1992; Watkins et al., 1993; Oliveira and Prado, 1994; Manning and Mayer, 1995; Pavlovic and Bodnar, 1998; McGaraughty and Heinricher, 2002; McGaraughty et al., 2004; Shin and Helmstetter, 2005). Amygdala-driven pain inhibition may involve projections to descending pain control centers in the brainstem (Heinricher and McGaraughty, 1999; Fields, 2000; Mason, 2005; Neugebauer, 2006; Tracey and Mantyh, 2007). It remains to be determined if pain inhibition results from decreased amygdala activity or increased amygdala output to inhibitory pain control centers or from the integration of both.
3. Cognitive impairment and cortical dysfunction in pain
Cognitive impairment, the inability to think clearly and make advantageous decisions, is one of the consequences of persistent pain but the underlying neural mechanisms are not known. A neural circuit that involves prefrontal cortical (PFC) areas and the amygdala appears to be of critical importance for cognitive functions such as decision-making.
3.1 Cortical mechanisms
A crucial cognitive process that depends on prefrontal areas is decision-making guided by the ability to learn from the outcome of a given choice in order to direct future action when facing a similar situation (Gold and Shadlen, 2007). The neural mechanisms of decision-making learning are more intricate than those involved in hippocampal-dependent forms of declarative memory. Learning from mistakes (or successes) depends on the correct formulation of an expected value for each possible outcome. More importantly, it requires continuous updates of expectations and values so that future options may be properly weighed (Rangel et al., 2008). The PFC plays an important role in higher order cognitive functions such as planning, decision making, reward expectancy, avoidance of risky choices, and goal-directed behaviors in animals and humans (Goldman-Rakic, 1996; Bechara et al., 2000a; Windmann et al., 2006; Vertes, 2006).
Since decision-making relies on frontal brain areas anatomically separate from the thalamocortical somatosensory system, it might be expected to remain largely unaffected in pain patients. However, two lines of evidence suggest that this is not the case. First, chronic pain shows significant comorbidity with clinical depression (Fishbain et al., 1997; Dersh et al., 2002) while depressive states are known to alter performance in decision-making tasks (Must et al., 2006). Second, brain areas of the so-called reward-aversion circuitry that are important for decision-making are also implicated in pain processing, because they respond to noxious stimuli and their activation or inhibition modulates the level of perceived pain (Becerra et al., 2001; Ploghaus et al., 2003).
Recent studies have detailed the participation of brain areas outside the somatosensory system in the perception of noxious stimuli (Borsook et al., 2007; Leknes and Tracey, 2008). Hypoactivity in the PFC, yet hyperactivity in the amygdala and insula, were observed in patients with somatoform pain disorder (Gundel et al., 2008). Patients with prolonged surgical dental pain (Derbyshire et al., 1999) or persistent pain in rheumatoid arthritis (Jones and Derbyshire, 1997) and ulcerative colitis (Mayer et al., 2005) also showed inhibition or reduced activation of PFC areas. In contrast to PFC deactivation in persistent pain, acute noxious stimuli in normal subjects appear to be associated with PFC activation. PFC activation was found during painful thermal stimulation, pain rating or pain expectancy (Casey et al., 1996; Derbyshire et al., 1997; Porro et al., 2002; Lorenz and Casey, 2005; Kong et al., 2006). Attention to a painful laser stimulus was associated with increased synchrony between medial PFC and primary somatosensory cortex in humans (Ohara et al., 2006). Single- unit recordings in anesthetized rats showed that medial PFC neurons are activated by noxious mechanical stimuli (Zhang et al., 2004).
The involvement of primarily “non-sensory” reward-aversion brain areas in pain perception is consistent with the concept that these largely “limbic” areas play an important role in pain modulation, including endogenous analgesia through opioidergic mechanisms and descending antinociceptive control (Fields, 2000; Millan, 2002; Rainville, 2002). Beyond the simple recruitment of antinociceptive systems, these recent studies view analgesia as a rewarding process (Seymour et al., 2005) and conversely raise the possibility that prolonged pain conditions constitute an aversive overload to the reward circuitry, which disrupts the functional balance and may cause long-term perturbation in these brain areas (Chapman et al., 2008). If this hypothesis is correct, pain-induced aversive overload likely would impair the performance in cognitive tasks that depend on proper functioning of the reward-aversion circuitry such as the generation of subjective valuation. Importantly, the PFC with its major input from limbic structures such as the amygdala is believed to play a key role in value-based decision-making that guides goal-directed behavior in humans (Bechara et al., 1998; Holland and Gallagher, 2004; Daw et al., 2006) and animals (Gisquet-Verrier and Delatour, 2006; Vertes, 2006), because PFC activation in humans correlates with the expectation of reward value and prediction error in neuroimaging studies (Breiter et al., 2001; Daw et al., 2006; Hampton et al., 2006; Windmann et al., 2006; Taylor et al., 2006).
3.2 Impaired decision-making in humans
A substantial amount of information about subjective valuation comes from studies of decision-making based on gambling tasks such as the Iowa Gambling Task (IGT) (Damasio et al, 1994; (Bechara et al., 1994; Damasio et al., 1994; Damasio, 1996; Bechara et al., 2005). IGT is a laboratory task that models real-life decision-making involving an emotion-based valuation (Bechara et al., 1999; Bechara et al., 2005; Sevy et al., 2006). Subjects choose from different decks of cards that yield either high immediate monetary reward but lead to long-term losses or relatively low immediate reward but lead to long-term gain. They are instructed to make choices that maximize their gain. The net score of advantageous versus disadvantageous choices and the total amount of money won assess the subject's decision-making strategy (Bechara et al., 2005; Sevy et al., 2006).
A number of seminal studies showed that strategic planning in the IGT is critically dependent on normal activity of PFC and amygdala (Bechara et al., 1997; Bechara et al., 1999; Bechara et al., 2003). Specifically, patients with damage to ventromedial PFC or amygdala performed poorly on the IGT (Bechara et al., 1999; Bechara et al., 2000b; Bechara et al., 2001). These patients perseverated in making disadvantageous choices that yield high immediate monetary gains at the risk of higher future losses. Accordingly, electrophysiological activity recorded in the ventromedial PFC of humans performing the IGT increased before card selection (Bechara et al., 2001) and correlated with reward prediction error, which encodes the discrepancy between predicted and actual outcome (Oya et al., 2005). Performance on the IGT is not explained by intelligence (IQ) or memory since patients with abnormally low IQ or dementia tend to choose randomly on this task (Bechara et al., 2001). Impaired decision-making is also not necessarily associated with structural brain damage but can be observed in substance abusers and compulsive gamers in the absence of discrete brain lesion (Bolla et al., 2003; Goudriaan et al., 2006; Noel et al., 2007).
Impaired decision-making in the IGT and dysfunction of the ventromedial PFC was only recently shown in pain patients with chronic back pain or complex regional pain syndrome (Apkarian et al., 2004a). These patients continued to choose immediate reward at the risk of incurring future negative consequences; this disadvantageous decision results in a lower net score and less profitable outcome (Apkarian et al., 2004a). Importantly, other cognitive abilities that do not depend entirely on the ventromedial PFC, such as attention, short-term memory, and general intelligence, tested normal in these patients. Further, performance on the IGT did not correlate with anxiety or depression in these patients (Apkarian et al., 2004a). Chronic pain was shown to induce a decrease in gray matter tissue in prefrontal and thalamic areas in a time-dependent manner (Apkarian et al., 2004b) and alter the connectivity between brain areas of the “Default Mode Network”, i.e., regions that are active during resting state (Baliki et al., 2008). Subsequently, pain-induced changes in brain volume (decreased grey matter) were detected in cortical and subcortical areas, including reward-related brain areas such as orbitofrontal cortex and striatum (Schmidt-Wilcke et al., 2006; Schmidt-Wilcke et al., 2007; Kuchinad et al., 2007). Understanding the mechanisms by which chronic pain causes brain dysfunctions requires the use of animal models.
3.3 Impaired decision-making in animals
Suitable decision-making tasks for animal studies were not available until recently. We developed a novel behavioral task for rodents based on the IGT. This rodent gambling task (RGT) requires the animal to make consecutive choices between two levers associated with rewards of different value and probability (Pais-Vieira et al., 2007). Results of the RGT showed that rats are capable of complex emotional decision-making performance. The RGT retains key features of the human IGT. Like the IGT, the RGT is sensitive to changes in amygdalar (Neugebauer et al., 2007) and orbital PFC functioning (Pais-Vieira et al., 2007). We used this task to test if rats with chronic pain would also show impaired performance in the RGT.
The RGT consists of one probe session of 90 consecutive trials, in which the animal enters a partially divided chamber and chooses between going to the left or the right area of that chamber to press the corresponding lever and retrieve a food reward. One of the levers is designated as “low-risk” choice, rewarding the animal with one chocolate-flavored food pellet in 8 out of every 10 visits. The other lever represents the “high-risk” choice, providing 3 pellets at a time but in only 3 out of 10 visits. Only one lever choice is allowed per entry, and 10 seconds after pressing the lever the animal is placed back in the adjacent connecting chamber to await the next trial. Levers are counterbalanced between animals and the reward pattern follows a pseudorandom sequence for each lever, which is kept the same for every animal. Before the probe session with levers set to different values, each animal received 10 days of training while both levers were set to give rewards at the “low-risk” setting. The number of trials in each training session varied pseudo-randomly from 30 to 120 so that the animals could not formulate an expectation about the duration of each session. After the 10 training sessions the animals were split between sham and pain groups. Complete Freund's adjuvant (CFA)-induced arthritis (Butler et al., 1992) and spared nerve injury (SNI)-induced neuropathy (Decosterd and Woolf, 2000) were used as pain models. Daily training resumed and five days later the animals were tested in the probe session. Therefore, each animal started the probe session naïve to the fact that the reward probability of one lever was changed. The test monitors the way the animal develops its choice of preferred lever using outcome experience gained throughout the 90 trials of the probe session.
During the training phase the animals did not show any laterality in their choices (left-right ratio of visits). In the first 30 trials of the probe session both control and chronic pain animals developed a tendency for visiting the high-risk lever, but as the task progresses the performance of each group became very distinct (Figure 3). Control animals began to avoid the high-risk lever, whereas both groups of chronic pain animals increased their preference for the high-risk lever (Pais-Vieira et al., 2007; and unpublished observations). The decision-making behavior of the pain groups was very similar to that of animals with lesions of the orbital PFC (Figure 3). Preliminary data show that animals with deactivated amygdala (BLA) also failed to switch from high-risk to low-risk behavior, indicating impaired decision-making (Neugebauer et al., 2007). Evidence from these studies strongly suggests that persistent pain may profoundly change ventromedial PFC-amygdala functioning.
Figure 3. Cognitive performance of animals in a decision-making task.
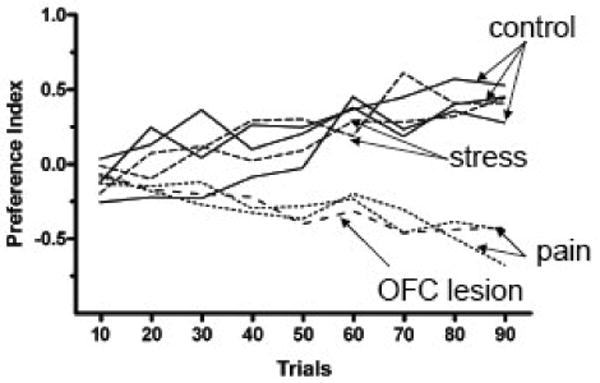
Rodent Gambling Task. Results of the so-called Rodent Gambling Task are shown for different experimental groups as the preference index in 90 trials. Normal animals and animals subjected to mild stress developed a positive preference index whereas animals with inflammatory or neuropathic pain and animals with orbitofrontal cortex (OFC) lesions had a negative preference index. Preference Index is defined as in the Iowa Gambling Task (IGT, Bechara et al, 1994) as the number of low-risk choices minus the number of high-risk choices, divided by the number of completed trials. The index is calculated for each group of 10 consecutive trials. Solid lines, control groups; dotted lines, pain groups; narrow-spaced dashed lines, stress groups; wide-spaced dashed line, OFC lesion group. Original data presented in Pais-Vieira et al, 2007, 2008. Error bars are omitted for clarity.
Impaired decision-making in the RGT could also result from elevated stress levels associated with chronic pain. To test this possibility we used two animal models of mild stress in the RGT, repeated restraint or daily injections of ACTH for 5 days. Individual stress responses (anxiety-like behavior) were measured in the elevated plus maze test before the first training session and again before the probe session. Both stress groups showed significantly different open-arm preference compared to control animals and to their own pre-stress baseline behavior. Somewhat surprisingly, neither stress group showed any decision-making impairment in the RGT, behaving instead similar to control animals (Figure 3). This may be the first behavioral task in which chronic pain produces a time-dependent pattern of mild stress-related behavior. One explanation is that stress does not gain access to the mechanisms of pain-induced decision-making impairment. Another possibility is that non-painful stressors require more time to induce decision-making impairment. The results of our experiments showing that pharmacological blockade of CRF1 receptors in the amygdala restores normal performance in the RGT (Neugebauer et al., 2007) appears to favor the latter explanation.
3.4 Neurochemistry of cortical dysfunction
The absence of any gross structural damage in these pain models led us to hypothesize that cognitive dysfunction was linked to changes of neurotransmitter levels in brain areas relevant for decision-making mechanisms. Chronic pain can be modulated by tonic levels of serotonin and dopamine (Diatchenko et al., 2006; Zhao et al., 2007), while stress has been reported to alter neurotransmitter levels (Inglis and Moghaddam, 1999; Mizoguchi et al., 2000). We used high-performance liquid chromatography (HPLC) to analyze dopaminergic and serotonergic content in the orbital PFC (OFC) and amygdala, in control and in animals with chronic inflammatory pain (CFA model; 5, 21 and 50 days after injection). The analysis showed that pain induced a significant decrease in the levels of both dopamine and serotonin in the OFC and a small increase of both molecules in the amygdala (Pais-Vieira, Mendes-Pinto, Lima, Galhardo, unpublished observations). Despite the large variability of the individual levels of neurotransmitters in all experimental groups, we found a striking correlation between individual levels of dopamine and serotonin in OFC with those in the amygdala. The OFC/amygdala ratio for both neurotransmitters showed an inverse correlation in control animals as well as 50 days after CFA induction but not at 5 or 15 days.
Pain-related decreases of dopamine and serotonin levels in the OFC support the notion that hypoactivity of prefrontal areas lead to poor performance in gambling tasks (Knoch et al., 2006; Sevy et al., 2006). Similarly, increased levels of both neurotransmitters in the amygdala are consistent with the effect of increased functions of corticotropin releasing factor (CRF) in the amygdala in models of stress (Rainnie et al., 2004; Funk et al., 2006) and pain (Ji and Neugebauer, 2007; Fu and Neugebauer, 2008). As a consequence, pharmacological blockade of amygdalar CRF1 receptors restores normal performance in the RGT (Neugebauer et al., 2007). Importantly, patients with chronic back pain showed abnormal brain chemistry, including decreased levels of excitatory neurotransmitters in the PFC consistent with PFC dysfunction (Grachev et al., 2001) and decreased gray matter density suggestive of PFC atrophy (Apkarian et al., 2004b). Evidence described in section 3.1 suggests decreased activation or inhibition in persistent and pathological pain states (prolonged dental pain, rheumatoid arthritis, ulcerative colitis, and somatoform pain disorder) but (increased) activation by acute noxious stimuli in normal subjects (brief thermal or mechanical stimulation). The potentially different roles of the PFC in acute physiological and chronic pathological pain remain to be determined, but it appears that normal cognitive evaluative functions of the PFC are impaired in persistent pain states. Recent advances in the understanding of chemical changes associated with cortical dysfunction will be discussed in section 4 (Biomarkers of cortical dysfunction in neuropathic pain).
3.5 Reward mechanisms and expectation in pain
Understanding reward-aversion brain areas in pain perception is clinically important since pain-induced depressive states can result from “role” disability, which refers to the degradation of social interactions in pain patients who are no longer able to fulfill their previous social roles (Von Korff et al., 2005; Eccleston et al., 2008). However, if the alteration in reward-aversion brain areas results directly from the pain stimulus rather than indirectly from the pain-induced disability, assessment and prevention of depressive states are of critical importance in the early stages of pain management. More research is needed for further understanding the affective and mood disorders that are comorbid with pain. Evidence from recent studies, including our own, suggests that altered expectation plays a preeminent role and may be more important than affective change such as anxiety itself in pain prediction, severity and disability (Boersma & Linton, 2006; Brown et al, 2008).
4. Biomarkers of cortical dysfunction in neuropathic pain
Chronic pain is associated with functional and morphological changes in subcortical and cortical brain areas that lead to cognitive impairment. However, objective markers for pain and related dysfunctions are generally missing. Neuropathic pain resulting from peripheral nerve injury is a chronic pain condition that is difficult to manage and seriously affects quality of life. Enhanced transmission of nociceptive messages at peripheral and spinal levels occurs. Functional and morphological plastic changes such as grey matter atrophy can also take place in brain areas participating in pain processing. Changes in cortical caspases and specific cytokine release may represent an index of cell degeneration processes that contribute to neuropathic pain and cognitive impairment. It is in these conditions that cannabinoid receptor agonists, which reduce particular caspase and cytokine levels, could become novel analgesics to attenuate neuropathic pain syndromes.
4.1 Functional changes in chronic pain
Neuropathic pain is a devastating and difficult to manage consequence of peripheral nerve injury, the main aspect of which is enhanced and altered transmission of nociceptive information (Bonica, 1970; Zhuo, 2007). In this condition, noxious stimuli are perceived as more painful (hyperalgesia), and normally innocuous stimuli may elicit pain (allodynia). Neuropathic pain may be considered a progressive nervous system disease that results from poorly-defined neurophysiological and neurochemical changes. Support for this hypothesis comes from animal models of chronic pain, in which a long-lasting increase in excitability (central sensitization) of pain-integrating neurons is observed. More recently, multidisciplinary research approaches have shown that chronic pain may be a consequence of long-term plastic changes along the entire pain matrix (Zhuo, 2007). Apart from peripheral nociceptors and the spinal cord, morphological and functional plastic changes also take place in subcortical and cortical areas that participate in pain processing. Indeed, growing evidence now suggests that long term plastic modifications in cortical networks may represent a possible basic mechanism underlying chronic pain.
Among cortical regions, S1 and S2 cortices, ACC, insular cortex, and PFC areas consistently respond to acute “physiological” pain stimuli in healthy subjects Importantly, depending on the stimulus applied (i.e., somatosensory or visceral stimuli), specific cortical network activation or deactivation has been found in human brain imaging studies (Dunckley et al., 2005; Iannetti et al., 2005). The reduction of chronic pain-related deactivation of specific cortical areas with anti-neuropathic drugs (Iannetti et al., 2005; Governo et al., 2008) further suggests that deactivation per se might contribute to allodynia and hyperalgesia. The majority of pain imaging studies, however, have been obtained from healthy subjects. Less is known about changes in cortical network functioning or responsiveness during chronic pain. It is not entirely clear if chronic pain involves the same or different brain areas than those associated with acute physiological pain. Activity changes in the thalamus and medial (emotional) pain system are more consistently observed in spontaneous neuropathic pain, whereas data on the involvement of cortical areas in allodynia appear more variable (Moisset and Bouhassira, 2007). Different “sources” of allodynia may induce distinct patterns of brain activity, reflecting different pathophysiological mechanisms and suggesting that specific “allodynia networks” in the cortex may exist (Apkarian et al., 2005; Schweinhardt et al., 2006).
4.2 Morphological changes in chronic pain
In addition to functional changes, morphological alterations at spinal and supraspinal levels have been reported in chronic pain. Neuropathic pain is accompanied by apoptosis of spinal cord cells (Whiteside and Munglani, 2001; Moore et al., 2002; de Novellis et al., 2004), sprouting of nerve terminals in somatosensory cortex (Flor et al., 2006), grey matter density decrease in PFC associated with reduced cognitive abilities (Apkarian et al., 2004a,b), and thalamic atrophy (Apkarian et al., 2004b). Morphometric analysis showed that chronic back pain particularly in patients with a neuropathic pain component is associated with 5-10% of brain grey matter atrophy in prefrontal cortex and thalamus (Apkarian et al., 2004b). A decrease in grey matter was also found in brainstem, temporal lobe and somatosensory cortex in addition to PFC in patients with chronic back pain; cortical changes were more pronounced in the right hemisphere (Schmidt-Wilcke et al., 2006). It remains to be determined if reduced grey matter density is related exclusively or predominantly to a specific cell population (projection neurons, inhibitory interneurons, and microglia) or if different cell types are affected equally. Nerve injury-induced apoptosis in the spinal dorsal horn caused a loss of GABAergic inhibitory interneurons and a decrease in inhibitory synaptic transmission (Scholz et al., 2005). Microglia was hyperactivated at the spinal level after nerve injury (Tsouda et al., 2003; see Scholz and Woolf, 2007) but possibly inhibited in cortical areas in chronic back pain (Apkarian et al., 2004b).
The majority of currently available data that correlate cortical damage and chronic pain come from brain imaging studies in humans, which do not allow the detailed analysis of the cell type(s) involved and the source of neuronal activity changes, namely excitatory vs. inhibitory neurons and circuits. Therefore, studies in animal models are critical to determine basic mechanisms of chronic pain.
4.3 Biochemical changes in chronic pain
Changes in cortical caspase levels may represent an index of cell degenerative processes leading to cognitive deficits in chronic pain states (Thornberry and Lazebnik, 1998). In particular, caspase-1 plays a pivotal role in controlling the release of pro-inflammatory cytokines such as IL-1beta, which appears to be upregulated in the prefrontal cortex of rats with neuropathic pain (Apkarian et al., 2006; Martinon and Tschopp, 2007). Cytokines and chemokines contribute directly to the pathophysiology of neuropathic pain at peripheral (Watkins et al., 1994; Wagner et al., 1998) spinal and supraspinal levels (Watkins et al., 1997; Clark et al., 2007).
A preliminary study from our laboratory (Maione et al., unpublished observations; see Figure 4) shows a significant increase in gene expression of caspase-1, caspase-12, and caspase-8 in the PFC of neuropathic mice. Concomitantly, staining of IL-1beta, which correlates with astrocyte activation, and the active form of caspase-3 in microglia increased in the PFC following spared nerve injury in mice. The occurrence of PFC caspase-3 activation may suggest microglial cell death and secondary cortical dysfunction or cortical damage induced by peripheral nerve injury and subsequent development of neuropathic pain.
Figure 4. Biochemical PFC changes in neuropathic pain.
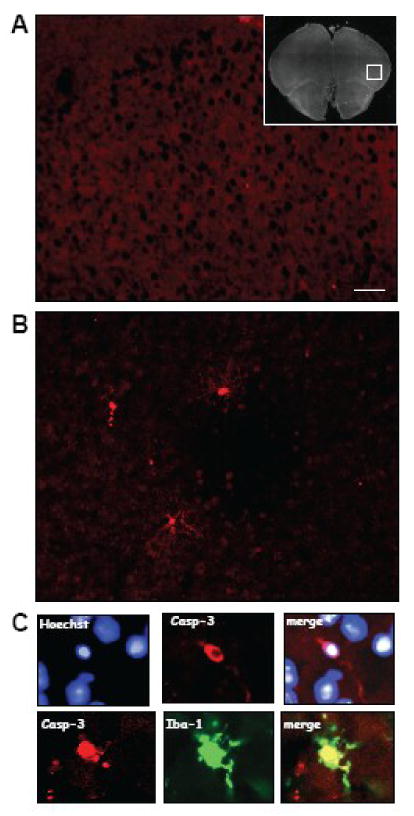
Caspase-3 immunostaining in prefrontal cortex (PFC) of sham controls (A) and mice with spared nerve injury (B). (C) Double staining for caspase-3 (casp-3) and nuclei (Hoechst) (upper panel) and double staining for caspase-3 and Iba-1 (microglial marker) (bottom panel). The microphotographs show an increased immunoreactivity for caspase-3 10 days after spared nerve injury compared to sham mice. Double staining shows microglial localization of caspase 3 as revealed by Iba-1, a typical microglial marker. Scale bar: 20 micrometer.
Activation of cannabinoid receptor subtype 2 (CB2) attenuates the induction and maintenance of inflammatory and neuropathic pain (Valenzano et al., 2005; Beltramo et al., 2006; Cheng and Hitchcock, 2007; Jhaveri et al., 2007) by inhibiting the release of pro-inflammatory cytokines and chemokines (Malan et al., 2002; Elmes et al., 2005). CB2 receptors are expressed primarily on immune cells and tissues (see Cheng and Hitchcock, 2007). CB2 receptors may therefore represent a new target for chronic pain management. A preliminary study from our laboratory (Maione et al., unpublished observations) shows that prolonged systemic administration of a selective CB2 receptor agonist (GW405833) reduced mechanical allodynia (Figure 4) and mRNA levels of pro-inflammatory caspase-1, caspase 8 and caspase-12 (Figure 5) that were increased in the PFC of neuropathic mice.
Figure 5. Behavioral effects of a CB2 agonist.
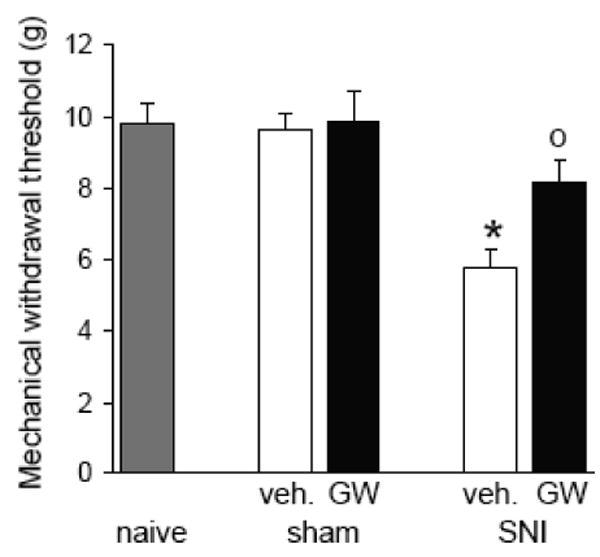
Chronic treatment with a selective CB2 receptor agonist (GW405833, GW; 5 mg/kg i.p., for 10 days) prevented the appearance of mechanical allodynia in neuropathic mice but had no effect in sham controls. Bar histograms show effects of a 10-day treatment with GW405833 or vehicle (veh., 20% DMSO in saline i.p.) on mechanical withdrawal threshold of sham mice or neuropathic mice (10 days after spared nerve injury, SNI). Data represent the mean ± SEM (n = 5) of paw withdrawal thresholds (PWT) in grams. SNI mice treated with vehicle showed mechanical allodynia (decreased thresholds) 10 days post-surgery. GW405833 partially reversed the allodynia. * P < 0.05 (vehicle-treated SNI compared to vehicle-treated sham mice), 0 P < 0.05 (CB2 agonist-treated SNI mice compared to vehicle-treated SNI mice), one-way ANOVA followed by Student's t-test.
In conclusion, changes in microglia/astrocyte activation (through different expression of caspases and specific cytokines) may serve as possible biomarkers of neural damage during chronic pain states. Drugs such as CB2 agonists that can change the activation of caspases and the release of particular cytokines may represent a tool to modulate chronic pain and avoid cortical impairment.
5. Imaging cortical plasticity in clinical neuropathic pain
Until recently our understanding of pain generation, mechanisms, and transmission from the spinal cord to the brain has been based primarily on animal studies, which have yielded a wealth of knowledge about nociceptive processing, analgesia, and plasticity. Animal models often permit rapid advances in understanding many underlying disease mechanisms — understanding that can be translated to human models. However, with neuropathic pain (NP) translating what has been learned in animals to humans can require invasive procedures.
5.1 Brain network for pain in humans
With recently introduced noninvasive neuroimaging technologies we can directly examine and compare the human central nervous system (CNS) in both healthy subjects and chronic pain patients and translate knowledge gleaned from animal research more confidently to humans. Many human neuroimaging studies have focused on defining brain regions that respond to various experimental noxious stimuli and on conceptualizing a “pain matrix” — a matrix of brain regions mapped to components of pain perception (sensory-discriminative, affective, reward, motivational, attentional and evaluative) (Melzack, 1999; Price, 2000). Some brain regions and their surmised pain components include anterior cingulate cortex (affective, attentional), insula (affective, attentional), amygdala (affect, reward), nucleus accumbens and striatum (reward), prefrontal cortex (motivational-affective, cognitive), and primary and secondary somatosensory cortices (S1, S2; sensory-discriminative, cognitive) (Melzack, 1999; Price, 2000; Mackey and Maeda, 2004; Apkarian et al., 2005). Additional regions include periaqueductal gray, premotor cortex, parietal cortex, and hypothalamus.
Recent research suggests that the “pain matrix” concept may be outdated. Newer studies reveal subtleties in the pain experience and its associated brain-activation patterns, suggesting that assigning specific pain components to specific brain regions is overly simplistic. For instance, the insular cortex (IC) is now thought to play a variety of roles. For example, the posterior IC is somatotopically organized (Brooks et al., 2005); stimulating it in awake humans causes well-localized pain (Ostrowsky et al., 2002). Additionally, activation of the posterior IC has been reported in numerous acute and chronic pain studies (see Apkarian et al., 2005), using graded noxious heat or cold stimuli (Brooks et al., 2002) and allodynic stimulation in neuropathic pain patients (Schweinhardt et al., 2006). Homeostatic afferent input is thought to project to the mid-posterior insula, where it is re-represented in the ipsilateral anterior insula, a region that may mediate interoception (the subjective evaluation of the internal physiological state of the body) (Craig et al., 2002). These more recent data suggest that the IC and other brain structures participate in the multidimensional experience of pain. As investigators refine their neuroimaging tools and methods, it is expected that we will learn that brain structures formerly thought to have simple roles in pain, will turn out to be quite complex.
Neuroimaging has also helped elucidate many neural correlates of factors well-known to modulate the experience of pain — attention (Petrovic et al., 2000), anticipation (Koyama et al., 2005), placebo (Wager et al., 2004), empathy (Singer et al., 2004; Zaki et al., 2007), fear/anxiety (Ochsner et al., 2006) and direct control (deCharms et al., 2005) — as well as the neural correlates of individual differences in the experience of pain (Coghill et al., 2003; Ochsner et al., 2006; Henderson et al., 2008). Based on these results we hypothesize that there is not simply a direct link between the degree of nociception and the overall experience of pain and furthermore, voluntary brain mechanisms can modulate that pain experience.
5.2 Functional changes in chronic pain
Changes in brain activity associated with neuropathic pain (NP) conditions are less thoroughly investigated than the brain's response to acutely evoked noxious stimuli (see Apkarian et al., 2005). NP is caused by lesion or dysfunction of the peripheral or central nervous systems (see 4.1). NP is challenging to treat, often responding poorly to traditional analgesic medications (Dworkin et al., 2007), but it affects well over three million individuals in the US and can become continuous, lifelong, and disabling. Recent research in animal and human subjects has yielded several possible mechanisms of NP, although there is still a considerable amount to be learned. One critical challenge to understanding NP is determining to what degree the CNS, particularly the brain, contributes to NP's generation and maintenance. Neuroimaging has recently provided important clues to these challenges.
Earlier positron emission tomography (PET) neuroimaging work demonstrated that the “pain matrix” (see section 5.1) is involved in chronic NP (Hsieh et al., 1995; Willoch et al., 2000). PET and other functional magnetic resonance imaging (fMRI) studies show that NP leads to activity changes in the normal pain-processing network. The brain regions of NP patients react differently depending on whether the stimulation is evoked on the affected or unaffected side (Becerra et al., 2006). Stimulation to unaffected regions in NP patients seems to evoke a normal brain response (Peyron et al., 2004). The various NP conditions are associated with abnormal central processing of pain and the chronicity of these conditions may be related to central plasticity and changes over time (Iadarola et al., 1995; Ducreux et al., 2006). For example, investigators using neuroimaging tools to investigate spontaneous pain in NP patients have consistently found evidence of reduced thalamic activity on the side contralateral to the affected limb (Hsieh et al., 1995). Brain sites of abnormal activity may vary depending on the specific NP syndrome and the type of stimulus applied (e.g., heat, cold, and mechanical) (Becerra et al., 2006). In summary, NP may result in plasticity within structures associated with acute evoked pain, leading to abnormalities in the processing and perception of pain.
Neuroimaging studies of phantom-limb pain have helped us understand the mechanisms of NP. Such pain often follows traumatic injury to a limb; the pain has a clear, dramatic peripheral origin. Animal research showed that after limb amputation, central plasticity changes occur despite the fact that the original peripheral etiology has healed. Microelectrode recordings in the somatosensory cortex after an animal's appendage had been amputated revealed a rapid reorganization of somatosensory cortical maps (Merzenich et al., 1984; see also Zhuo, 2007). Subsequent studies using magnetoencephalography (MEG) and functional magnetic resonance imaging (fMRI) techniques showed that a similar reorganization occurred in humans (Ramachandran, 1993; Lotze et al., 2001). These cortical plastic changes differed in phantom-limb patients who experienced pain from those who did not (Lotze et al., 2001). Similar patterns of cortical reorganization were observed in patients with complex regional pain syndrome (CRPS) (Maihöfner et al., 2003). A recent neuroimaging study suggests that cortical reorganization (in S1 and S2) and pain perception in CRPS can be reversed with behavioral treatment consisting of graded sensorimotor retuning therapy (Pleger et al., 2005). Functional neuroimaging has helped translate the knowledge gained from basic science work into the better understanding of clinical NP conditions.
5.3 Biochemical and structural changes in chronic pain
Functional imaging has been the primary tool to investigate brain activity in NP patients and healthy volunteers. Recently, a number of novel brain imaging techniques have been developed and applied to pain research, including magnetic resonance spectroscopy (MRS) and voxel based morphometry (VBM).
Imaging of the human CNS's biochemical environment has helped reveal abnormalities in chronic pain (see also section 4.3). Investigators used MRS to characterize the biochemical differences in brain regions of healthy individuals and those with chronic pain. Specifically, investigators observed decreased N-acetylaspartate (NAA) in the dorsolateral prefrontal cortex of patients with chronic low back pain (Grachev et al., 2000) and CRPS type I (Grachev et al., 2002) as well as decreases in NAA in the thalami of NP patients (Fukui et al., 2006). Investigators have proposed NAA, a marker of neuronal activity found only in mature neurons (Miller, 1991), as a possible biomarker of neuronal health and function that can be assessed non-invasively using MRS (Grachev et al., 2000, 2002; Fukui et al., 2006). Investigators are using this information to generate and confirm hypotheses about the mechanisms of clinical NP. This field is expected to advance rapidly with the introduction of methods to imaging a variety of neurotransmitters such as glutamate, glycine and GABA (Ross and Bluml, 2001).
VBM is a structural imaging technique that measures differences in local concentrations of brain tissue through a voxel-wise comparison of multiple brain images (Ashburner and Friston, 2000). It has been applied sparsely to pain research. One of the earliest studies found a 5-11% reduction of global cortical gray matter density in patients with low back pain compared with aged and gender matched controls (Apkarian et al., 2004b). This gray matter reduction correlated with the duration of pain. A more recent study compared phantom limb patients to matched controls and noted a reduction in gray matter in the posterolateral thalamus contralateral to the side of the amputation (Draganski et al., 2006). This gray matter reduction was positively correlated with the duration of time since the amputation, but not the severity or frequency of pain. These VBM studies need to be expanded and replicated. Additionally, the question regarding the cellular basis for the morphometric variations remains unanswered and may require the direct comparison to histological data. Combining these methods may ultimately yield useful information about the mechanisms of the maladaptive plasticity underlying NP.
5.4 Cortical plasticity – cause or consequence of chronic pain?
Many questions remain regarding the role of brain dysregulation in chronic pain such as NP. The causal relationship of changes noted in NP patient studies has yet to be determined. Does the abnormal brain activity contribute to the NP or does the abnormal brain cause the NP? How can we characterize individual differences in NP patients? Do these differences correlate with specific brain changes? For example, recent studies have identified considerable individual variability in pain sensitivity, even within subclasses of neuropathic disorders such as postherpetic neuralgia (Pappagallo et al., 2000). Chronic NP may therefore reflect varying degrees of central dysregulation, ranging from purely abnormal peripheral nociceptive input to pain that is mediated solely through dysregulated central mechanisms. Central sensitization is likely to depend on a combination of the specific neuropathic condition and individual trait factors.
Although we do not yet know the central cause-effect relationships underlying NP, neuroimaging can help advance our understanding of both pharmacologic and non-pharmacologic treatments. Despite the variety of available analgesics, many chronic NP patients receive inadequate relief, and physicians treating them face daunting obstacles. To optimize compliance, minimize side-effects, and maximize analgesia, each anti-neuropathic medication needs to be started at a low dose and titrated to effect over many weeks, a “labor-intensive” process (Collins et al., 2000). Unfortunately, each anti-neuropathic medication produces pain relief for only a minority of NP patients (Katz et al., 2008). Not surprisingly, few physicians have the patience to persist in a strategy involving months of frustrating, side-effect-laden medication trials. Pharmacologic neuroimaging may be a novel method to optimize the matching of a particular drug to a particular NP patient. It may also provide important information related to both the drug's activity and the underlying mechanism of NP. For example, recently investigators studied the effect of topical lidocaine patch therapy (Lidoderm) in postherpetic neuralgia patients and noted reward-related brain regions (amygdala and ventral striatum) that changed only after long-term treatment. These brain regions also best reflected changes in pain with treatment (Geha et al., 2007).
Neuroimaging will play an important role in characterizing the central correlates of different types of neuropathic pain, helping to separate aberrant nociceptive processing in the periphery and spine from the influences of central amplification of pain. This discrimination will help clinicians rapidly diagnose individual cases of NP and more effectively treat the peripheral and central dysregulation elements of pain.
6. Conclusions
It is clear now that the brain network for pain also includes primarily “non-sensory” forebrain areas such as PFC and amygdala that are concerned with emotional-affective and emotion-based cognitive aspects of pain. As part of the reward-aversion circuitry these brain areas play a key role in value-based decision-making that guides goal-directed behavior. Well-documented pain-related cognitive deficits may be attributed to abnormal processing in these structures.
Activation versus deactivation of a particular brain area depends on a number of factors and may be different for acute painful stimuli under normal conditions and prolonged pathophysiological pain states. Functional studies are needed to determine the interactions of different brain areas. The inverse relationship that is emerging between some areas such as amygdala and PFC may suggest that amygdala-cortical interactions are important for the emotional-affective modulation of cognitive function in pain. A detailed analysis of neuronal mechanisms and circuits involved in connectivity changes and cortical dysfunctions in pain can be expected from electrophysiological animal studies. Behavioral studies using new tasks for complex decision-making assessment will link changes in brain function to cognitive deficits in pain.
In addition to functional changes, biochemical and structural brain changes are observed in prolonged pain states. Cortical dysfunction is associated with neurochemical changes. The analysis of underlying molecules and mechanisms may yield biomarkers of neural impairment in persistent pain. Biomarkers associated with biochemical changes such as microglia/astrocyte activation may serve as useful diagnostic tools but also provide an opportunity for the development of novel therapeutic strategies that target these biomarkers. CB2 receptor agonists can modulate the pain-related expression of caspases and release of cytokines; they may be useful tools to modulate persistent pain and avoid cortical impairment.
Pain impairs cortical function but in turn is modulated by cortical processes. Are cortical changes the consequence or cause of persistent pain? This intriguing question awaits an answer. Imaging studies argue against a simple direct link between the degree of nociception and the overall experience of pain and suggest that voluntary brain mechanisms can modulate that pain experience. Neuroimaging will likely continue to play an important role in the diagnosis of pain-related dysfunctions and assessment of mechanism and efficacy of various treatments.
The significance of the results presented in this article is that they emphasize the need for a concerted multidisciplinary effort to provide basic science information that helps explain and improve therapeutic strategies, including cognitive therapies and psychotherapy, and unravel the mysteries of the multidimensional disorder pain.
Figure 6. Biochemical effects of a CB2 agonist.
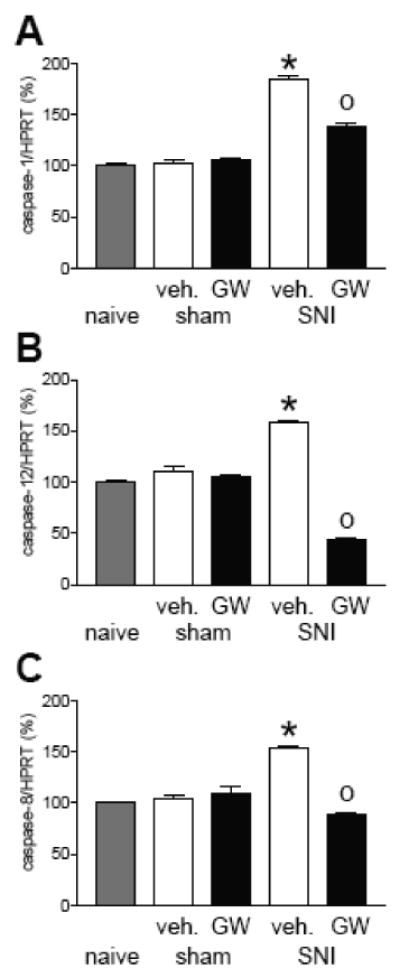
Chronic treatment with a selective CB2 receptor agonist (GW405833, GW; 5 mg/kg i.p., for 10 days) prevented the increase of caspase expression in neuropathic mice. Bar histograms show effects of a 10-day treatment with GW405833 or vehicle (veh., 20% DMSO in saline i.p.) on the levels of caspase mRNA normalized to HPRT (housekeeping gene) and expressed as percent (mean ± SEM) of control levels in naïve mice (set to 100 %)(n = 5). (A) Caspase-1, (B) caspase-12, (C) caspase 8 mRNA expression in naïve, sham or neuropathic mice (10 days after spared nerve injury, SNI). SNI mice treated with vehicle showed an increase of caspases 10 days post-surgery. GW405833 significantly prevented caspase hyperexpression. * P < 0.05 (vehicle-treated SNI compared to vehicle-treated sham mice), 0 P < 0.05 (CB2 agonist-treated SNI mice compared to vehicle-treated SNI mice), one-way ANOVA followed by Student's Neuman–Keuls test.
Acknowledgments
Work in the authors' laboratories was supported by National Institutes of Health (NIH) grants NS38261 and NS11255 (to V.N.), Fundação para a Ciência e Tecnologia (FCT) grants POCI/SAU-NEU/63034/2004 and POCI/SAU-NEU/55811/2004 and Bial Foundation grant 84/04 (to V.G.), Ministero dell'Università e della Ricerca Scientifica (MIUR) grant PRIN-2005 (to S.M.), and NIH grant NS053961 and the John and Dodie Rosekranz Endowment (to S.C.M.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Apkarian AV, Bushnell MC, Treede RD, Zubieta JK. Human brain mechanisms of pain perception and regulation in health and disease. Eur J Pain. 2005;9:463–484. doi: 10.1016/j.ejpain.2004.11.001. [DOI] [PubMed] [Google Scholar]
- Apkarian AV, Sosa Y, Krauss BR, Thomas PS, Fredrickson BE, Levy RE, Harden RN, Chialvo DR. Chronic pain patients are impaired on an emotional decision-making task. Pain. 2004a;108:129–136. doi: 10.1016/j.pain.2003.12.015. [DOI] [PubMed] [Google Scholar]
- Apkarian AV, Sosa Y, Sonty S, Levy RM, Harden RN, Parrish TB, Gitelman DR. Chronic Back Pain Is Associated with Decreased Prefrontal and Thalamic Gray Matter Density. J Neurosci. 2004b;24:10410–10415. doi: 10.1523/JNEUROSCI.2541-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacon SJ, Headlam AJ, Gabbott PL, Smith AD. Amygdala input to medial prefrontal cortex (mPFC) in the rat: a light and electron microscope study. Brain Res. 1996;720:211–219. doi: 10.1016/0006-8993(96)00155-2. [DOI] [PubMed] [Google Scholar]
- Baliki MN, Geha PY, Apkarian AV, Chialvo DR. Beyond feeling: chronic pain hurts the brain, disrupting the default-mode network dynamics. J Neurosci. 2008;28:1398–1403. doi: 10.1523/JNEUROSCI.4123-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baliki MN, Chialvo DR, Geha PY, Levy RM, Harden RN, Parrish TB, Apkarian AV. Chronic Pain and the Emotional Brain: Specific Brain Activity Associated with Spontaneous Fluctuations of Intensity of Chronic Back Pain. J Neurosci. 2006;26:12165–12173. doi: 10.1523/JNEUROSCI.3576-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becerra L, Breiter HC, Wise R, Gonzalez RG, Borsook D. Reward circuitry activation by noxious thermal stimuli. Neuron. 2001;32:927–946. doi: 10.1016/s0896-6273(01)00533-5. [DOI] [PubMed] [Google Scholar]
- Bechara A, Damasio AR, Damasio H, Anderson SW. Insensitivity to future consequences following damage to human prefrontal cortex. Cognition. 1994;50:7–15. doi: 10.1016/0010-0277(94)90018-3. [DOI] [PubMed] [Google Scholar]
- Bechara A, Damasio H, Damasio AR. Emotion, decision making and the orbitofrontal cortex. Cereb Cortex. 2000a;10:295–307. doi: 10.1093/cercor/10.3.295. [DOI] [PubMed] [Google Scholar]
- Bechara A, Damasio H, Damasio AR. Role of the Amygdala in Decision-Making. Annals of the New York Academy of Sciences. 2003;985:356–369. doi: 10.1111/j.1749-6632.2003.tb07094.x. [DOI] [PubMed] [Google Scholar]
- Bechara A, Damasio H, Tranel D, Anderson SW. Dissociation of working memory from decision making within the human prefrontal cortex. J Neurosci. 1998;18:428–437. doi: 10.1523/JNEUROSCI.18-01-00428.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bechara A, Damasio H, Tranel D, Damasio AR. Deciding advantageously before knowing the advantageous strategy. Science. 1997;275:1293–1295. doi: 10.1126/science.275.5304.1293. [DOI] [PubMed] [Google Scholar]
- Bechara A, Damasio H, Tranel D, Damasio AR. The Iowa Gambling Task and the somatic marker hypothesis: some questions and answers. Trends Cogn Sci. 2005;9:159–162. doi: 10.1016/j.tics.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Bechara A, Dolan S, Denburg N, Hindes A, Anderson SW, Nathan PE. Decision-making deficits, linked to a dysfunctional ventromedial prefrontal cortex, revealed in alcohol and stimulant abusers. Neuropsychologia. 2001;39:376–389. doi: 10.1016/s0028-3932(00)00136-6. [DOI] [PubMed] [Google Scholar]
- Bechara A, Tranel D, Damasio H. Characterization of the decision-making deficit of patients with ventromedial prefrontal cortex lesions. Brain. 2000b;123(Pt 11):2189–2202. doi: 10.1093/brain/123.11.2189. [DOI] [PubMed] [Google Scholar]
- Bechara A, Damasio H, Damasio AR, Lee GP. Different Contributions of the Human Amygdala and Ventromedial Prefrontal Cortex to Decision-Making. J Neurosci. 1999;19:5473–5481. doi: 10.1523/JNEUROSCI.19-13-05473.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard JF, Besson JM. The spino(trigemino)pontoamygdaloid pathway: electrophysiological evidence for an involvement in pain processes. J Neurophysiol. 1990;63:473–490. doi: 10.1152/jn.1990.63.3.473. [DOI] [PubMed] [Google Scholar]
- Bird GC, Lash LL, Han JS, Zou X, Willis WD, Neugebauer V. Protein kinase A-dependent enhanced NMDA receptor function in pain-related synaptic plasticity in rat amygdala neurones. J Physiol. 2005;564:907–921. doi: 10.1113/jphysiol.2005.084780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolla KI, Eldreth DA, London ED, Kiehl KA, Mouratidis M, Contoreggi C, Matochik JA, Kurian V, Cadet JL, Kimes AS, Funderburk FR, Ernst M. Orbitofrontal cortex dysfunction in abstinent cocaine abusers performing a decision-making task. Neuroimage. 2003;19:1085–1094. doi: 10.1016/s1053-8119(03)00113-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borsook D, Becerra L, Carlezon WA, Jr, Shaw M, Renshaw P, Elman I, Levine J. Reward-aversion circuitry in analgesia and pain: implications for psychiatric disorders. Eur J Pain. 2007;11:7–20. doi: 10.1016/j.ejpain.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Braz JM, Nassar MA, Wood JN, Basbaum AI. Parallel “pain” pathways arise from subpopulations of primary afferent nociceptor. Neuron. 2005;47:787–793. doi: 10.1016/j.neuron.2005.08.015. [DOI] [PubMed] [Google Scholar]
- Breiter HC, Aharon I, Kahneman D, Dale A, Shizgal P. Functional imaging of neural responses to expectancy and experience of monetary gains and losses. Neuron. 2001;30:619–639. doi: 10.1016/s0896-6273(01)00303-8. [DOI] [PubMed] [Google Scholar]
- Butler SH, Godefroy F, Besson JM, Weil-Fugazza J. A limited arthritic model for chronic pain studies in the rat. Pain. 1992;48:73–81. doi: 10.1016/0304-3959(92)90133-V. [DOI] [PubMed] [Google Scholar]
- Calejesan AA, Kim SJ, Zhuo M. Descending facilitatory modulation of a behavioral nociceptive response by stimulation in the adult rat anterior cingulate cortex. Eur J Pain. 2000;4:83–96. doi: 10.1053/eujp.1999.0158. [DOI] [PubMed] [Google Scholar]
- Carrasquillo Y, Gereau RW. Activation of the extracellular signal-regulated kinase in the amygdala modulates pain perception. J Neurosci. 2007;27:1543–1551. doi: 10.1523/JNEUROSCI.3536-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey KL. Forebrain mechanisms of nociception and pain: analysis through imaging. Proc Natl Acad Sci U S A. 1999;96:7668–7674. doi: 10.1073/pnas.96.14.7668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casey KL, Minoshima S, Morrow TJ, Koeppe RA. Comparison of human cerebral activation pattern during cutaneous warmth, heat pain, and deep cold pain. J Neurophysiol. 1996;76:571–581. doi: 10.1152/jn.1996.76.1.571. [DOI] [PubMed] [Google Scholar]
- Chapman CR, Tuckett RP, Song CW. Pain and stress in a systems perspective: reciprocal neural, endocrine, and immune interactions. J Pain. 2008;9:122–145. doi: 10.1016/j.jpain.2007.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliffer KD, Burstein R, Giesler GJ., Jr Distributions of spinothalamic, spinohypothalamic, and spinotelencephalic fibers revealed by anterograde transport of PHA-L in rats. J Neurosci. 1991;11:852–868. doi: 10.1523/JNEUROSCI.11-03-00852.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Craig AD. Pain mechanisms: labeled lines versus convergence in central processing. Annu Rev Neurosci. 2003;26:1–30. doi: 10.1146/annurev.neuro.26.041002.131022. [DOI] [PubMed] [Google Scholar]
- Damasio AR. The somatic marker hypothesis and the possible functions of the prefrontal cortex. Philos Trans R Soc Lond B Biol Sci. 1996;351:1413–1420. doi: 10.1098/rstb.1996.0125. [DOI] [PubMed] [Google Scholar]
- Damasio H, Grabowski T, Frank R, Galaburda AM, Damasio AR. The return of Phineas Gage: clues about the brain from the skull of a famous patient. Science. 1994;264:1102–1105. doi: 10.1126/science.8178168. [DOI] [PubMed] [Google Scholar]
- Daw ND, O'Doherty JP, Dayan P, Seymour B, Dolan RJ. Cortical substrates for exploratory decisions in humans. Nature. 2006;441:876–879. doi: 10.1038/nature04766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- deCharms RC, Maeda F, Glover GH, Ludlow D, Pauly JM, Soneji D, Gabrieli JD, Mackey SC. Control over brain activation and pain learned by using real-time functional MRI. Proc Natl Acad Sci U S A. 2005;102:18626–18631. doi: 10.1073/pnas.0505210102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decosterd I, Woolf CJ. Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain. 2000;87:149–158. doi: 10.1016/S0304-3959(00)00276-1. [DOI] [PubMed] [Google Scholar]
- Derbyshire SW, Jones AK, Collins M, Feinmann C, Harris M. Cerebral responses to pain in patients suffering acute post-dental extraction pain measured by positron emission tomography (PET) Eur J Pain. 1999;3:103–113. doi: 10.1053/eujp.1998.0102. [DOI] [PubMed] [Google Scholar]
- Derbyshire SWG, Jones AKP, Gyulai F, Clark S, Townsend D, Firestone LL. Pain processing during three levels of noxious stimulation produces differential patterns of central activity. Pain. 1997;73:431–445. doi: 10.1016/S0304-3959(97)00138-3. [DOI] [PubMed] [Google Scholar]
- Dersh J, Polatin PB, Gatchel RJ. Chronic pain and psychopathology: research findings and theoretical considerations. Psychosom Med. 2002;64:773–786. doi: 10.1097/01.psy.0000024232.11538.54. [DOI] [PubMed] [Google Scholar]
- Diatchenko L, Nackley AG, Slade GD, Bhalang K, Belfer I, Max MB, Goldman D, Maixner W. Catechol-O-methyltransferase gene polymorphisms are associated with multiple pain-evoking stimuli. Pain. 2006;125:216–224. doi: 10.1016/j.pain.2006.05.024. [DOI] [PubMed] [Google Scholar]
- Fields HL. Pain modulation: expectation, opioid analgesia and virtual pain. Prog Brain Res. 2000;122:245–253. doi: 10.1016/s0079-6123(08)62143-3. [DOI] [PubMed] [Google Scholar]
- Fishbain DA, Cutler R, Rosomoff HL, Rosomoff RS. Chronic pain-associated depression: antecedent or consequence of chronic pain? A review Clin J Pain. 1997;13:116–137. doi: 10.1097/00002508-199706000-00006. [DOI] [PubMed] [Google Scholar]
- Flor H, Turk DC. Cognitive and learning aspects. In: McMahon SB, Koltzenburg M, editors. Textbook of Pain. Elsevier; Amsterdam: 2006. pp. 241–258. [Google Scholar]
- Floresco SB, Ghods-Sharifi S. Amygdala-prefrontal cortical circuitry regulates effort-based decision making. Cereb Cortex. 2007;17:251–260. doi: 10.1093/cercor/bhj143. [DOI] [PubMed] [Google Scholar]
- Fu Y, Han J, Ishola T, Scerbo M, Adwanikar H, Ramsey C, Neugebauer V. PKA and ERK, but not PKC, in the amygdala contribute to pain-related synaptic plasticity and behavior. Mol Pain. 2008;4:26–46. doi: 10.1186/1744-8069-4-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Neugebauer V. Differential mechanisms of CRF1 and CRF2 receptor functions in the amygdala in pain-related synaptic facilitation and behavior. J Neurosci. 2008;28:3861–3876. doi: 10.1523/JNEUROSCI.0227-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukui H, Murai T, Fukuyama H, Hayashi T, Hanakawa T. Functional activity related to risk anticipation during performance of the Iowa Gambling Task. Neuroimage. 2005;24:253–259. doi: 10.1016/j.neuroimage.2004.08.028. [DOI] [PubMed] [Google Scholar]
- Gabbott PLA, Warner TA, Busby SJ. Amygdala input monosynaptically innervates parvalbumin immunoreactive local circuit neurons in rat medial prefrontal cortex. Neuroscience. 2006;139:1039–1048. doi: 10.1016/j.neuroscience.2006.01.026. [DOI] [PubMed] [Google Scholar]
- Gallagher RM, Verma S. Mood and anxiety disorders in chronic pain. Prog Pain Res Management. 2004;27:139–178. [Google Scholar]
- Gauriau C, Bernard JF. Pain pathways and parabrachial circuits in the rat. Exp Physiol. 2002;87:251–258. doi: 10.1113/eph8702357. [DOI] [PubMed] [Google Scholar]
- Gauriau C, Bernard JF. A comparative reappraisal of projections from the superficial laminae of the dorsal horn in the rat: the forebrain. J Comp Neurol. 2004;468:24–56. doi: 10.1002/cne.10873. [DOI] [PubMed] [Google Scholar]
- Gisquet-Verrier P, Delatour B. The role of the rat prelimbic/infralimbic cortex in working memory: Not involved in the short-term maintenance but in monitoring and processing functions. Neuroscience. 2006;141:585–596. doi: 10.1016/j.neuroscience.2006.04.009. [DOI] [PubMed] [Google Scholar]
- Gold JI, Shadlen MN. The neural basis of decision making. Annu Rev Neurosci. 2007;30:535–574. doi: 10.1146/annurev.neuro.29.051605.113038. [DOI] [PubMed] [Google Scholar]
- Goldman-Rakic PS. The prefrontal landscape: implications of functional architecture for understanding human mentation and the central executive. Philos Trans R Soc Lond B Biol Sci. 1996;351:1445–1453. doi: 10.1098/rstb.1996.0129. [DOI] [PubMed] [Google Scholar]
- Goudriaan AE, Oosterlaan J, De BE, Van Den BW. Psychophysiological determinants and concomitants of deficient decision making in pathological gamblers. Drug Alcohol Depend. 2006;84:231–239. doi: 10.1016/j.drugalcdep.2006.02.007. [DOI] [PubMed] [Google Scholar]
- Grachev ID, Fredickson BE, Apkarian AV. Dissociating anxiety from pain: mapping the neuronal marker N-acetyl aspartate to perception distinguishes closely interrelated characteristics of chronic pain. Mol Psychiatry. 2001;6:256–258. doi: 10.1038/sj.mp.4000834. [DOI] [PubMed] [Google Scholar]
- Greenwood-Van Meerveld B, Gibson M, Gunder W, Shepard J, Foreman R, Myers D. Stereotaxic delivery of corticosterone to the amygdala modulates colonic sensitivity in rats. Brain Res. 2001;893:135–142. doi: 10.1016/s0006-8993(00)03305-9. [DOI] [PubMed] [Google Scholar]
- Hampton AN, Adolphs R, Tyszka JM, O'Doherty JP. Contributions of the Amygdala to Reward Expectancy and Choice Signals in Human Prefrontal Cortex. Neuron. 2007;55:545–555. doi: 10.1016/j.neuron.2007.07.022. [DOI] [PubMed] [Google Scholar]
- Hampton AN, Bossaerts P, O'Doherty JP. The Role of the Ventromedial Prefrontal Cortex in Abstract State-Based Inference during Decision Making in Humans. J Neurosci. 2006;26:8360–8367. doi: 10.1523/JNEUROSCI.1010-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JS, Bird GC, Neugebauer V. Enhanced group III mGluR-mediated inhibition of pain-related synaptic plasticity in the amygdala. Neuropharmacology. 2004;46:918–926. doi: 10.1016/j.neuropharm.2004.01.006. [DOI] [PubMed] [Google Scholar]
- Han JS, Fu Y, Bird GC, Neugebauer V. Enhanced group II mGluR-mediated inhibition of pain-related synaptic plasticity in the amygdala. Mol Pain. 2006;2:18–29. doi: 10.1186/1744-8069-2-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JS, Li W, Neugebauer V. Critical role of calcitonin gene-related peptide 1 receptors in the amygdala in synaptic plasticity and pain behavior. J Neurosci. 2005;25:10717–10728. doi: 10.1523/JNEUROSCI.4112-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JS, Neugebauer V. Synaptic plasticity in the amygdala in a visceral pain model in rats. Neuroscience Letters. 2004;361:254–257. doi: 10.1016/j.neulet.2003.12.027. [DOI] [PubMed] [Google Scholar]
- Han JS, Neugebauer V. mGluR1 and mGluR5 antagonists in the amygdala inhibit different components of audible and ultrasonic vocalizations in a model of arthritic pain. Pain. 2005;113:211–222. doi: 10.1016/j.pain.2004.10.022. [DOI] [PubMed] [Google Scholar]
- Heinricher MM, McGaraughty S. Pain-modulating neurons and behavioral state. In: Lydic R, Baghdoyan HA, editors. Handbook of Behavioral State Control. CRC Press; New York: 1999. pp. 487–503. [Google Scholar]
- Helmstetter FJ. The amygdala is essential for the expression of conditional hypoalgesia. Behav Neurosci. 1992;106:518–528. doi: 10.1037//0735-7044.106.3.518. [DOI] [PubMed] [Google Scholar]
- Holland PC, Gallagher M. Amygdala-frontal interactions and reward expectancy. Curr Opin Neurobiol. 2004;14:148–155. doi: 10.1016/j.conb.2004.03.007. [DOI] [PubMed] [Google Scholar]
- Ikeda R, Takahashi Y, Inoue K, Kato F. NMDA receptor-independent synaptic plasticity in the central amygdala in the rat model of neuropathic pain. Pain. 2007;127:161–172. doi: 10.1016/j.pain.2006.09.003. [DOI] [PubMed] [Google Scholar]
- Ishikawa A, Nakamura S. Convergence and Interaction of Hippocampal and Amygdalar Projections within the Prefrontal Cortex in the Rat. J Neurosci. 2003;23:9987–9995. doi: 10.1523/JNEUROSCI.23-31-09987.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jasmin L, Rabkin SD, Granato A, Boudah A, Ohara PT. Analgesia and hyperalgesia from GABA-mediated modulation of the cerebral cortex. Nature. 2003;424:316–320. doi: 10.1038/nature01808. [DOI] [PubMed] [Google Scholar]
- Ji G, Fu Y, Ruppert KA, Neugebauer V. Pain-related anxiety-like behavior requires CRF1 receptors in the amygdala. Mol Pain. 2007;3:13–17. doi: 10.1186/1744-8069-3-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji G, Neugebauer V. Differential effects of CRF1 and CRF2 receptor antagonists on pain-related sensitization of neurons in the central nucleus of the amygdala. J Neurophysiol. 2007;97:3893–3904. doi: 10.1152/jn.00135.2007. [DOI] [PubMed] [Google Scholar]
- Ji G, Neugebauer V. Pro- and Anti-Nociceptive Effects of Corticotropin-Releasing Factor (CRF) in Central Amygdala Neurons Are Mediated Through Different Receptors. J Neurophysiol. 2008;99:1201–1212. doi: 10.1152/jn.01148.2007. [DOI] [PubMed] [Google Scholar]
- Jones AK, Derbyshire SW. Reduced cortical responses to noxious heat in patients with rheumatoid arthritis. Ann Rheum Dis. 1997;56:601–607. doi: 10.1136/ard.56.10.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong J, White NS, Kwong KK, Vangel MG, Rosman IS, Gracely RH, Gollub RL. Using fMRI to dissociate sensory encoding from cognitive evaluation of heat pain intensity. Hum Brain Mapp. 2006;27:715–721. doi: 10.1002/hbm.20213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuchinad A, Schweinhardt P, Seminowicz DA, Wood PB, Chizh BA, Bushnell MC. Accelerated brain gray matter loss in fibromyalgia patients: premature aging of the brain? J Neurosci. 2007;27:4004–4007. doi: 10.1523/JNEUROSCI.0098-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni B, Bentley DE, Elliott R, Julyan PJ, Boger E, Watson A, Boyle Y, El-Deredy W, Jones AK. Arthritic pain is processed in brain areas concerned with emotions and fear. Arthritis Rheum. 2007;56:1345–1354. doi: 10.1002/art.22460. [DOI] [PubMed] [Google Scholar]
- Leknes S, Tracey I. A common neurobiology for pain and pleasure. Nat Rev Neurosci. 2008;9:314–320. doi: 10.1038/nrn2333. [DOI] [PubMed] [Google Scholar]
- Li W, Neugebauer V. Block of NMDA and non-NMDA receptor activation results in reduced background and evoked activity of central amygdala neurons in a model of arthritic pain. Pain. 2004a;110:112–122. doi: 10.1016/j.pain.2004.03.015. [DOI] [PubMed] [Google Scholar]
- Li W, Neugebauer V. Differential roles of mGluR1 and mGluR5 in brief and prolonged nociceptive processing in central amygdala neurons. J Neurophysiol. 2004b;91:13–24. doi: 10.1152/jn.00485.2003. [DOI] [PubMed] [Google Scholar]
- Li W, Neugebauer V. Differential changes of group II and group III mGluR function in central amygdala neurons in a model of arthritic pain. J Neurophysiol. 2006;96:1803–1815. doi: 10.1152/jn.00495.2006. [DOI] [PubMed] [Google Scholar]
- Lopez de Armentia M, Sah P. Bidirectional synaptic plasticity at nociceptive afferents in the rat central amygdala. J Physiol. 2007;581:961–970. doi: 10.1113/jphysiol.2006.121822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenz J, Casey KL. Imaging of acute versus pathological pain in humans. Eur J Pain. 2005;9:163–165. doi: 10.1016/j.ejpain.2004.07.009. [DOI] [PubMed] [Google Scholar]
- Mackey SC, Maeda F. Functional imaging and the neural systems of chronic pain. Neurosurg Clin N Am. 2004;15:269–288. doi: 10.1016/j.nec.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Manning BH, Mayer DJ. The central nucleus of the amygdala contributes to the production of morphine antinociception in the formalin test. Pain. 1995;63:141–152. doi: 10.1016/0304-3959(95)00027-P. [DOI] [PubMed] [Google Scholar]
- Maren S. Synaptic Mechanisms of Associative Memory in the Amygdala. Neuron. 2005;47:783–786. doi: 10.1016/j.neuron.2005.08.009. [DOI] [PubMed] [Google Scholar]
- Mason P. Deconstructing endogenous pain modulations. J Neurophysiol. 2005;94:1659–1663. doi: 10.1152/jn.00249.2005. [DOI] [PubMed] [Google Scholar]
- Mayer EA, Naliboff BD, Craig AD. Neuroimaging of the brain-gut axis: from basic understanding to treatment of functional GI disorders. Gastroenterology. 2006;131:1925–1942. doi: 10.1053/j.gastro.2006.10.026. [DOI] [PubMed] [Google Scholar]
- Mayer EA, Berman S, Suyenobu B, Labus J, Mandelkern MA, Naliboff BD, Chang L. Differences in brain responses to visceral pain between patients with irritable bowel syndrome and ulcerative colitis. Pain. 2005;115:398–409. doi: 10.1016/j.pain.2005.03.023. [DOI] [PubMed] [Google Scholar]
- McDonald AJ. Organization of amygdaloid projections to the prefrontal cortex and associated striatum in the rat. Neuroscience. 1991;44:1–14. doi: 10.1016/0306-4522(91)90247-l. [DOI] [PubMed] [Google Scholar]
- McGaraughty S, Heinricher MM. Microinjection of morphine into various amygdaloid nuclei differentially affects nociceptive responsiveness and RVM neuronal activity. Pain. 2002;96:153–162. doi: 10.1016/s0304-3959(01)00440-7. [DOI] [PubMed] [Google Scholar]
- McGaraughty S, Farr DA, Heinricher MM. Lesions of the periaqueductal gray disrupt input to the rostral ventromedial medulla following microinjections of morphine into the medial or basolateral nuclei of the amygdala. Brain Research. 2004;1009:223–227. doi: 10.1016/j.brainres.2004.02.048. [DOI] [PubMed] [Google Scholar]
- McNally GP, Akil H. Role of corticotropin-releasing hormone in the amygdala and bed nucleus of the stria terminalis in the behavioral, pain modulatory, and endocrine consequences of opiate withdrawal. Neuroscience. 2002;112:605–617. doi: 10.1016/s0306-4522(02)00105-7. [DOI] [PubMed] [Google Scholar]
- Millan MJ. Descending control of pain. Prog Neurobiol. 2002;66:355–474. doi: 10.1016/s0301-0082(02)00009-6. [DOI] [PubMed] [Google Scholar]
- Must A, Szabo Z, Bodi N, Szasz A, Janka Z, Keri S. Sensitivity to reward and punishment and the prefrontal cortex in major depression. J Affect Disord. 2006;90:209–215. doi: 10.1016/j.jad.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Myers B, Dittmeyer K, Greenwood-Van MB. Involvement of amygdaloid corticosterone in altered visceral and somatic sensation. Behav Brain Res. 2007;181:163–167. doi: 10.1016/j.bbr.2007.03.031. [DOI] [PubMed] [Google Scholar]
- Myers B, Greenwood-Van MB. Corticosteroid receptor-mediated mechanisms in the amygdala regulate anxiety and colonic sensitivity. Am J Physiol Gastrointest Liver Physiol. 2007;292:G1622–G1629. doi: 10.1152/ajpgi.00080.2007. [DOI] [PubMed] [Google Scholar]
- Myers DA, Gibson M, Schulkin J, Greenwood Van-Meerveld B. Corticosterone implants to the amygdala and type 1 CRH receptor regulation: effects on behavior and colonic sensitivity. Behav Brain Res. 2005;161:39–44. doi: 10.1016/j.bbr.2005.03.001. [DOI] [PubMed] [Google Scholar]
- Neugebauer V. Subcortical processing of nociceptive information: basal ganglia and amygdala. In: Cervero F, Jensen TS, editors. Pain. Elsevier; Amsterdam: 2006. pp. 141–158. [DOI] [PubMed] [Google Scholar]
- Neugebauer V, Fu Y, Ji G, Galhardo V. CRF1 receptors in the basolateral amygdala contribute to pain-related decision-making deficits. Soc Neurosci Abstracts. 2007;37:723–3. [Google Scholar]
- Neugebauer V, Li W, Bird GC, Bhave G, Gereau RW. Synaptic plasticity in the amygdala in a model of arthritic pain: differential roles of metabotropic glutamate receptors 1 and 5. J Neurosci. 2003;23:52–63. doi: 10.1523/JNEUROSCI.23-01-00052.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neugebauer V, Li W, Bird GC, Han JS. The amygdala and persistent pain. Neuroscientist. 2004;10:221–234. doi: 10.1177/1073858403261077. [DOI] [PubMed] [Google Scholar]
- Noel X, Bechara A, Dan B, Hanak C, Verbanck P. Response inhibition deficit is involved in poor decision making under risk in nonamnesic individuals with alcoholism. Neuropsychology. 2007;21:778–786. doi: 10.1037/0894-4105.21.6.778. [DOI] [PubMed] [Google Scholar]
- Ochsner KN, Ludlow DH, Knierim K, Hanelin J, Ramachandran T, Glover GC, Mackey SC. Neural correlates of individual differences in pain-related fear and anxiety. Pain. 2006;120:69–77. doi: 10.1016/j.pain.2005.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohara PT, Vit JP, Jasmin L. Cortical modulation of pain. Cell Mol Life Sci. 2005;62:44–52. doi: 10.1007/s00018-004-4283-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohara S, Crone NE, Weiss N, Lenz FA. Analysis of synchrony demonstrates 'pain networks' defined by rapidly switching, task-specific, functional connectivity between pain-related cortical structures. Pain. 2006;123:244–253. doi: 10.1016/j.pain.2006.02.012. [DOI] [PubMed] [Google Scholar]
- Oliveira MA, Prado WA. Antinociception and behavioral manifestations induced by intracerebroventricular or intra-amygdaloid administration of cholinergic agonists in the rat. Pain. 1994;57:383–391. doi: 10.1016/0304-3959(94)90014-0. [DOI] [PubMed] [Google Scholar]
- Orozco-Cabal L, Pollandt S, Liu J, Vergara L, Shinnick-Gallagher P, Gallagher JP. A novel rat medial prefrontal cortical slice preparation to investigate synaptic transmission from amygdala to layer V prelimbic pyramidal neurons. Journal of Neuroscience Methods. 2006;151:148–158. doi: 10.1016/j.jneumeth.2005.07.002. [DOI] [PubMed] [Google Scholar]
- Oya H, Adolphs R, Kawasaki H, Bechara A, Damasio A, Howard MA., III Electrophysiological correlates of reward prediction error recorded in the human prefrontal cortex. Proc Natl Acad Sci U S A. 2005;102:8351–8356. doi: 10.1073/pnas.0500899102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pais-Vieira M, Lima D, Galhardo V. Orbitofrontal cortex lesions disrupt risk assessment in a novel serial decision-making task for rats. Neuroscience. 2007;145:225–231. doi: 10.1016/j.neuroscience.2006.11.058. [DOI] [PubMed] [Google Scholar]
- Palazzo E, Fu Y, Ji G, Maione S, Neugebauer V. Group III mGluR7 and mGluR8 in the amygdala differentially modulate nocifensive and affective pain behaviors. Neuropharmacology. 2008a doi: 10.1016/j.neuropharm.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palazzo E, Fu Y, Ji G, Maione S, Neugebauer V. Group III mGluR7 and mGluR8 in the amygdala differentially modulate nocifensive and affective pain behaviors. Neuropharmacology. 2008b doi: 10.1016/j.neuropharm.2008.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pare D, Quirk GJ, Ledoux JE. New Vistas on Amygdala Networks in Conditioned Fear. J Neurophysiol. 2004;92:1–9. doi: 10.1152/jn.00153.2004. [DOI] [PubMed] [Google Scholar]
- Pavlovic ZW, Bodnar RJ. Opioid supraspinal analgesic synergy between the amygdala and periaqueductal gray in rats. Brain Res. 1998;779:158–169. doi: 10.1016/s0006-8993(97)01115-3. [DOI] [PubMed] [Google Scholar]
- Pedersen LH, Scheel-Kruger J, Blackburn-Munro G. Amygdala GABA-A receptor involvement in mediating sensory-discriminative and affective-motivational pain responses in a rat model of peripheral nerve injury. Pain. 2007;127:17–26. doi: 10.1016/j.pain.2006.06.036. [DOI] [PubMed] [Google Scholar]
- Phelps EA, Ledoux JE. Contributions of the Amygdala to Emotion Processing: From Animal Models to Human Behavior. Neuron. 2005;48:175–187. doi: 10.1016/j.neuron.2005.09.025. [DOI] [PubMed] [Google Scholar]
- Ploghaus A, Becerra L, Borras C, Borsook D. Neural circuitry underlying pain modulation: expectation, hypnosis, placebo. Trends Cogn Sci. 2003;7:197–200. doi: 10.1016/s1364-6613(03)00061-5. [DOI] [PubMed] [Google Scholar]
- Porrino LJ, Crane AM, Goldman-Rakic PS. Direct and indirect pathways from the amygdala to the frontal lobe in rhesus monkeys. J Comp Neurol. 1981;198:121–136. doi: 10.1002/cne.901980111. [DOI] [PubMed] [Google Scholar]
- Porro CA, Baraldi P, Pagnoni G, Serafini M, Facchin P, Maieron M, Nichelli P. Does anticipation of pain affect cortical nociceptive systems? J Neurosci. 2002;22:3206–3214. doi: 10.1523/JNEUROSCI.22-08-03206.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price DD. Psychological and neural mechanisms of the affective dimension of pain. Science. 2000;288:1769–1772. doi: 10.1126/science.288.5472.1769. [DOI] [PubMed] [Google Scholar]
- Price JL. Comparative aspects of amygdala connectivity. Ann N Y Acad Sci. 2003;985:50–58. doi: 10.1111/j.1749-6632.2003.tb07070.x. [DOI] [PubMed] [Google Scholar]
- Qin C, Greenwood-Van Meerveld B, Foreman RD. Spinal Neuronal Responses to Urinary Bladder Stimulation in Rats With Corticosterone or Aldosterone Onto the Amygdala. J Neurophysiol. 2003a;90:2180–2189. doi: 10.1152/jn.00298.2003. [DOI] [PubMed] [Google Scholar]
- Qin C, Greenwood-Van Meerveld B, Foreman RD. Visceromotor and spinal neuronal responses to colorectal distension in rats with aldosterone onto the amygdala. J Neurophysiol. 2003b;90:2–11. doi: 10.1152/jn.00023.2003. [DOI] [PubMed] [Google Scholar]
- Qin C, Greenwood-Van Meerveld B, Myers DA, Foreman RD. Corticosterone Acts Directly at the Amygdala to Alter Spinal Neuronal Activity in Response to Colorectal Distension. J Neurophysiol. 2003c;89:1343–1352. doi: 10.1152/jn.00834.2002. [DOI] [PubMed] [Google Scholar]
- Rainville P. Brain mechanisms of pain affect and pain modulation. Curr Opin Neurobiol. 2002;12:195–204. doi: 10.1016/s0959-4388(02)00313-6. [DOI] [PubMed] [Google Scholar]
- Rangel A, Camerer C, Montague PR. A framework for studying the neurobiology of value-based decision making. Nat Rev Neurosci. 2008;9:545–556. doi: 10.1038/nrn2357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhudy JL, Meagher MW. The role of emotion in pain modulation. Curr Opin Psychiatry. 2001;14:241–245. [Google Scholar]
- Rhudy JL, Williams AE, McCabe KM, Russell JL, Maynard LJ. Emotional control of nociceptive reactions (ECON): do affective valence and arousal play a role? Pain. 2008;136:250–261. doi: 10.1016/j.pain.2007.06.031. [DOI] [PubMed] [Google Scholar]
- Schmidt-Wilcke T, Leinisch E, bauer S, Draganski B, Bogdahn U, Altmeppen J, May A. Affective components and intensity of pain correlate with structural differences in gray matter in chronic back pain patients. Pain. 2006;125:89–97. doi: 10.1016/j.pain.2006.05.004. [DOI] [PubMed] [Google Scholar]
- Schmidt-Wilcke T, Luerding R, Weigand T, Jurgens T, Schuierer G, Leinisch E, Bogdahn U. Striatal grey matter increase in patients suffering from fibromyalgia--a voxel-based morphometry study. Pain. 2007;132 1:S109–S116. doi: 10.1016/j.pain.2007.05.010. [DOI] [PubMed] [Google Scholar]
- Seminowicz DA, Mikulis DJ, Davis KD. Cognitive modulation of pain-related brain responses depends on behavioral strategy. Pain. 2004;112:48–58. doi: 10.1016/j.pain.2004.07.027. [DOI] [PubMed] [Google Scholar]
- Seminowicz DA, Davis KD. A re-examination of pain-cognition interactions: Implications for neuroimaging. Pain. 2007;130:8–13. doi: 10.1016/j.pain.2007.03.036. [DOI] [PubMed] [Google Scholar]
- Sevy S, Hassoun Y, Bechara A, Yechiam E, Napolitano B, Burdick K, Delman H, Malhotra A. Emotion-based decision-making in healthy subjects: short-term effects of reducing dopamine levels. Psychopharmacology (Berl) 2006;188:228–235. doi: 10.1007/s00213-006-0450-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seymour B, O'Doherty JP, Koltzenburg M, Wiech K, Frackowiak R, Friston K, Dolan R. Opponent appetitive-aversive neural processes underlie predictive learning of pain relief. Nat Neurosci. 2005;8:1234–1240. doi: 10.1038/nn1527. [DOI] [PubMed] [Google Scholar]
- Shi C, Davis M. Pain pathways involved in fear conditioning measured with fear-potentiated startle: lesion studies. J Neurosci. 1999;19:420–430. doi: 10.1523/JNEUROSCI.19-01-00420.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin MS, Helmstetter FJ. Antinociception following application of DAMGO to the basolateral amygdala results from a direct interaction of DAMGO with Mu opioid receptors in the amygdala. Brain Res. 2005;1064:56–65. doi: 10.1016/j.brainres.2005.09.065. [DOI] [PubMed] [Google Scholar]
- Stalnaker TA, Franz TM, Singh T, Schoenbaum G. Basolateral Amygdala Lesions Abolish Orbitofrontal-Dependent Reversal Impairments. Neuron. 2007;54:51–58. doi: 10.1016/j.neuron.2007.02.014. [DOI] [PubMed] [Google Scholar]
- Tanimoto S, Nakagawa T, Yamauchi Y, Minami M, Satoh M. Differential contributions of the basolateral and central nuclei of the amygdala in the negative affective component of chemical somatic and visceral pains in rats. Eur J Neurosci. 2003;18:2343–2350. doi: 10.1046/j.1460-9568.2003.02952.x. [DOI] [PubMed] [Google Scholar]
- Taylor SF, Martis B, Fitzgerald KD, Welsh RC, Abelson JL, Liberzon I, Himle JA, Gehring WJ. Medial frontal cortex activity and loss-related responses to errors. J Neurosci. 2006;26:4063–4070. doi: 10.1523/JNEUROSCI.4709-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tracey I, Mantyh PW. The Cerebral Signature for Pain Perception and Its Modulation. Neuron. 2007;55:377–391. doi: 10.1016/j.neuron.2007.07.012. [DOI] [PubMed] [Google Scholar]
- Vertes RP. Interactions among the medial prefrontal cortex, hippocampus and midline thalamus in emotional and cognitive processing in the rat. Neuroscience. 2006;142:1–20. doi: 10.1016/j.neuroscience.2006.06.027. [DOI] [PubMed] [Google Scholar]
- Watkins LR, Wiertelak EP, Maier SF. The amygdala is necessary for the expression of conditioned but not unconditioned analgesia. Behav Neurosci. 1993;107:402–405. doi: 10.1037//0735-7044.107.2.402. [DOI] [PubMed] [Google Scholar]
- Windmann S, Kirsch P, Mier D, Stark R, Walter B, Gunturkun O, Vaitl D. On framing effects in decision making: linking lateral versus medial orbitofrontal cortex activation to choice outcome processing. J Cogn Neurosci. 2006;18:1198–1211. doi: 10.1162/jocn.2006.18.7.1198. [DOI] [PubMed] [Google Scholar]
- Zhang R, Tomida M, Katayama Y, Kawakami Y. Response durations encode nociceptive stimulus intensity in the rat medial prefrontal cortex. Neuroscience. 2004;125:777–785. doi: 10.1016/j.neuroscience.2004.01.055. [DOI] [PubMed] [Google Scholar]
- Zhao ZQ, Chiechio S, Sun YG, Zhang KH, Zhao CS, Scott M, Johnson RL, Deneris ES, Renner KJ, Gereau RW, Chen ZF. Mice lacking central serotonergic neurons show enhanced inflammatory pain and an impaired analgesic response to antidepressant drugs. J Neurosci. 2007;27:6045–6053. doi: 10.1523/JNEUROSCI.1623-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo M. Cortical excitation and chronic pain. Trends Neurosci. 2008;31:199–207. doi: 10.1016/j.tins.2008.01.003. [DOI] [PubMed] [Google Scholar]