

Abstract
Akt is a highly regulated serine/threonine kinase involved in stress response and cell survival. Stress response pathways must cope with increasing chronic stress susceptibility with age. We found an age-related lesion in Akt activity via loss of phosphorylation on Ser473. In hepatocytes from old rats, basal phospho-Ser473 Akt is 30% lower when compared to young, but basal phospho-Thr308 Akt is unchanged. (R)-α-lipoic acid (LA), a dithiol compound with antioxidant properties, is effective against age-related increases in oxidative stress and has been used to improve glucose utilization through insulin receptor (IR) pathway-mediated Akt phosphorylation. Treatment with physiologically relevant doses of LA (50 μM) provided a 30% increase in phospho-Ser473. Furthermore, two phosphatases that antagonize Akt, PTEN and PP2A, were both partially inhibited by LA. Thus, LA may be a nutritive agent that can remediate loss of function in the Akt pathway and aid in the survival of liver cells.
Keywords: PKB/Akt, PI3 kinase, Aging, Alpha-lipoic acid, Phosphatase, Oxidative stress
Introduction
Attenuation of cellular stress response mechanisms is a major contributing factor to cell and tissue decline with age. Susceptibility versus resistance to exogenous and endogenous stresses is now recognized as a key determinant of successful aging (Finkel and Holbrook 2000). Many genes linked to longevity mediate stress resistance. In particular, the insulin/PI3 kinase/Akt signaling network, which confers resistance to a variety of stress stimuli, also strongly influences longevity in both invertebrate and vertebrate species. For example, mutations in the InR and Daf-2 signaling pathways (homologs to the insulin receptor in Drosophila and C. elegans, respectively) result in increased lifespan, but this longevity gain comes at the price of reductions in body size, fertility, and resistance to exogenous stresses (Clancy et al. 2001; Tatar et al. 2001). Similar trade-offs are evident for mammals. Mice made deficient for insulin-like growth factor (IGF-1) reactivity through homozygous knockouts of the Prop-1 and Pit-1 genes show dwarfism but also increased lifespan (Flurkey et al. 2001). Prop-1 knockouts (Ames dwarf mice) display reduced PI3 kinase and Akt activities in response to insulin in skeletal muscle (Dominici et al. 2003) but an increase in phospho-Akt in liver (Dominici et al. 2002), whereas Pit-1 knockouts (Snell dwarf mice) show decreased hepatic Akt phosphorylation compared to normal counterparts (Hsieh and Papaconstantinou 2004). Thus, this signal transduction pathway may be necessary for growth and fertility, but reduced activity leads to longer lifespan of a given species. While the mechanisms associated with how insulin/IGF-1 limits lifespan in mammalian systems are not completely known, it can be hypothesized that the insulin/IGF-1 signaling axis may mediate some of their lifespan-enhancing properties through maintaining resistance to oxidative stresses.
In this regard, Akt (also known as protein kinase B) is an important serine/threonine kinase downstream of the insulin/IGF-1 receptors and in turn, confers resistance to a variety of cellular stresses through phosphorylating a diverse array of targets (Manning and Cantley 2007). Despite its recognized importance as a mediator of cell survival, very little is known about how the aging process affects Akt expression or its activity, or how Akt influences or mediates IGF-1-directed changes in lifespan. This is surprising, given the significant scrutiny afforded this enzyme for its involvement in carcinogenesis and cardiovascular disorders. Of greatest significance, it is not known whether transient or chronic low-grade induction of Akt activity may be beneficial to cell survival in aged tissues undergoing oxidative stress.
Akt has critical serine and threonine residues, which if phosphorylated, significantly induce enzyme activity (Alessi et al. 1996). It is well established that phosphoinositide-dependent kinase-1 (PDK1) governs phosphorylation of threonine 308 (Thr308), while it is still controversial as to the particular enzyme that mediates phosphorylation at the serine 473 (Ser473) residue. Ser473 may be targeted by mTOR (Jacinto et al. 2006), protein kinase C (Kawakami et al. 2004), DNA-protein kinase (Feng et al. 2004), and/or a putative “PDK2”. Regardless of the mediator, Akt phosphorylation on these two residues works synergistically. Akt activity is significantly attenuated when only one of these two sites is phosphorylated (Alessi et al. 1996), and it is possible for Ser473 to be phosphorylated independently of Thr308 (Alessi et al. 1996; Kawakami et al. 2004). Thus, overall steady-state phosphorylation levels largely mediate Akt activity and its dephosphorylation is known to cause membrane dissociation and lower cell survival in response to pro-apoptotic challenges.
In addition to the activity of kinases, further control of steady-state Akt phosphorylation status is mediated by cellular phosphatases. Previous studies have highlighted the effects of phosphatase and tensin homolog (PTEN) on Akt. PTEN is a protein tyrosine phosphatase that dephosphorylates PtdIns-3,4,5-P3, converting it back to PtdIns-4, 5-P2 (Maehama and Dixon 1998; Wu et al. 1998). This limits membrane colocalization of PDK1 and Akt, and lowers PDK1-dependent phosphorylation of Akt (Alessi et al. 1997). In addition to prevention of Akt phosphorylation through PTEN, phosphate groups may also be directly removed from active Akt via action of PP2A, a tripartite phosphatase (Millward et al. 1999).
Considering the generally limited characterization of age-associated changes to Akt, the primary goal of this study is to define general Akt phosphorylation state and its activity using cells from aged versus young rats. We further hypothesized that potential age-related changes in Akt activity adversely affect the cellular response to oxidative insults. To meet these goals, we used both liver tissue and hepatocytes isolated from young (2-4 months) and old (26-28 months) F344 rats placed in primary culture to examine Akt in a cellular context that preserves the aging phenotype (Shenvi et al. 2008). Herein we show that Akt is less active at a basal level in cells from aged animals, and we provide evidence that the redox active dithiol compound, (R)-α-lipoic acid (LA), reverses the age-related loss of Akt phosphorylation and its attenuated activity in hepatocytes from old rats, at least in part through suppressing phosphatase activities.
Materials and methods
Animal models and isolation of primary hepatocytes
All animal procedures were in accordance with the Oregon State University Guidelines for animal experimentation. Rats (male Fischer 344), both young (2-4 months) and old (24-28 months) were used throughout the study. This is a well-characterized rat strain that is approved for aging studies by the National Institutes on Aging (NIH/NIA). They were maintained on a standard chow diet, with food and water given ad libitum. Hepatocytes were isolated by collagenase perfusion as previously described (Smith et al. 2004). Briefly, animals were anesthetized with diethyl ether, a midline incision was made in the abdomen, and heparin solution was injected into the iliac vein. The portal vein was perfused with Hanks’ balanced salt solution (HBSS), then an HBSS buffer containing EDTA and BSA to loosen the basement membrane. The liver was perfused with an HBSS buffer containing collagenase to disperse the liver into single cells. Isolated cells were filtered through gauze to remove large debris and washed with Krebs-Henseleit buffer, pH 7.4, supplemented with 2 mM glucose and 7 mM l-glutamine. Following hepatocyte dispersal, cells were washed three times with Krebs-Henseleit buffer, pH 7.4, supplemented with 2 mM glucose and 7 mM l-glutamine before plating.
Gavage and liver tissue
Rats (male Fischer 344), both young (2-4 months) and old (24-28 months) were maintained as above, then given a bolus of 120 mg/kg LA by gavage at various times before sacrifice. Livers were excised and longitudinal slices were frozen at -80°C until use. Tissue was thawed on ice and homogenized in extraction buffer (50 mM Tris-HCl, (pH 7.5), 1% Igepal, 2 mM EDTA, 100 mM NaCl, 1 mM orthovanadate) plus protease and phosphatase inhibitors (Sigma-Aldrich). Protein concentrations were determined by spectrophotometry and the Bradford protein assay (Bio-Rad).
Primary hepatocyte culture
Isolated hepatocytes were plated at a density of 1 × 106 cells per well in 6-well collagen I-coated plates. Cells were maintained in Williams E medium containing 5% FBS, 1 μM dexamethasone, 10 μg/l insulin, 2 mM l-glutamine, and antibiotics. All cells were maintained in a 37°C incubator with 5% CO2. Medium was changed at the end of each day, at least 12 h before an experiment, to eliminate artifactual stimulation from fresh serum or insulin. All experiments took place between 24 and 72 h after plating. For glucose/glucose oxidase experiments, medium was replaced with medium containing 25 mM glucose, and cells were treated with 100 mU/ml glucose oxidase.
Whole cell lysates and immunoblotting
Cells were harvested by scraping, washed in PBS, and sonicated in PBS containing protease and phosphatase inhibitors (Sigma-Aldrich). Protein concentrations were determined by spectrophotometry and the Bradford protein assay (Bio-Rad). Proteins were solubilized in Laemmli loading buffer containing SDS and samples were heated for 5 min at 100°C. Normalized amounts of protein (30 μg) were then run on SDS-PAGE and transferred to PVDF membranes. Membranes were blocked in PBS containing 1% Tween-20 and 5% nonfat dry milk, incubated with primary antibodies for 2 h at room temperature, washed with PBS-Tween, and incubated with secondary antibodies for 1 h at room temperature. Following an additional wash, the membranes were incubated with chemiluminescence reagents (Pierce Pico West), exposed to film, and developed. Antibodies made to the following proteins were used at 1:1,000 dilutions: Akt, phosphoserine 473 Akt, phosphothreonine 308 Akt, PTEN, phosphoserine 380 PTEN, procaspase-3, GSK3α/β, and phosphoserine 21/9 GSK3α/β (Cell Signaling Technology), PP2A-C (Upstate), phosphotyrosine 307 PP2A (Epitomics), and actin (Sigma-Aldrich). Blots were densitometrically analyzed with ImageJ from NIH.
LA and chemical inhibitors
LA was a kind gift from Dr. Hans Tritschler (Asta Medica). A 50 mM solution was made in DMF vehicle, and LA was added to cell culture medium at 1:1,000 for a final concentration of 50 μM. Vehicle treatment was used as a control. The Akt inhibitor VIII (“Akti1/2”, Calbiochem, 124018, IC50 = 58 nM for Akt1, 210 nM for Akt2, and 2.12 μM for Akt3 (Lindsley et al. 2005)) was used as a direct inhibitor of Akt. It selectively inhibits Akt via the pleckstrin homology domain. LY294002 (Calbiochem) was used as a PI3 kinase inhibitor. Both were solubilized in DMF vehicle. Akti1/2 (10 μM) and LY294002 (50 μM) (Calbiochem) were supplied to cells at least 2 h prior to LA treatment.
Akt activity assay
Snap-frozen cells (6 × 106 per reaction) were lysed in buffer (50 mM Tris-HCl, pH 7.5, 100 mM NaCl, 1% Igepal, 2 mM EDTA, and 1 mM sodium orthovanadate, plus protease and phosphatase inhibitors (P8340, P2850, P5726, Sigma-Aldrich) and cleared by centrifugation at 13,000 rpm for 30 min at 4°C. Akt was immunoprecipitated with the antibody described above, plus protein A agarose beads (Pierce) for 2 h at 4°C, then washed 4× in buffer and used in an activity assay with 1 μg GST-GSK3α/β (Cell Signaling Technology) as the substrate. The reaction buffer (15 mM MgCl2, 150 mM NaCl, 20 mM HEPES, pH 7.5, 100 μM ATP, 1 mM DTT) was supplemented with the following inhibitors (Biosource): PKC inhibitor peptide, PKA inhibitor peptide, GF109203X, and calmidazolium. The reaction was incubated at 30°C for 10 min, then 6× Laemmli sample buffer was added, and samples were prepared for SDS-PAGE and immunoblotting with antibodies to phospho- and total GSK3α/β.
Statistics
Statistical analysis was performed with GraphPad Prism 4.0 software (GraphPad, San Diego). At least 3 replicates (animals) were performed for each experiment. Student’s t-test, Tukey’s Multiple Comparisons test, or ANOVA was used to determine statistical significance, using Tukey’s post-hoc test where appropriate. Results were considered significant if the P value was <0.05.
Results
Basal phosphorylation of Akt declines in hepatocytes from young and old rats
Hepatocytes from young and old rats placed in primary culture were the model used throughout this study. We previously showed that this culture system resulted in maintenance of the aging phenotype for at least 5 days without significant changes in gene profiles or activities of stress response proteins (Shenvi et al. 2008). Initial studies sought to characterize potential age-dependent changes in Akt activity by monitoring ratios of phospho-to-total Akt levels in hepatocytes from young and old rats. As shown in Fig. 1a, there was no significant difference in phosphorylation of Akt at the Thr308 site with age. In contrast, basal phosphorylation status at the Ser473 residue was significantly lower (P = 0.03), where a 30% loss with respect to age was observed. To pursue the physiological relevance of this observation, livers from young and old rats were used to measure Akt phosphorylation in vivo. Figure 1b shows that the Ser473 phosphorylation level was 67% lower in livers from old versus young animals (P = 0.01). These results confirm that the culture model largely conforms to the aging profile of Akt phosphorylation seen in vivo and also suggests that Akt activity declines significantly with age.
Fig. 1.
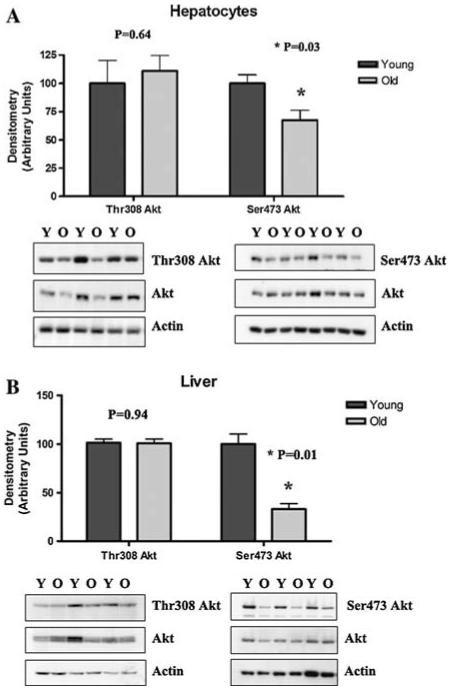
Basal phosphorylation of Akt in hepatocytes and liver from young and old rats. Basal phosphorylation status of Akt at the Thr308 and Ser473 residues in (a) hepatocytes from young (Y) and old (O) rats in primary culture and (b) homogenized liver tissue from young and old rats. Western blotting was performed for phospho-Ser473, phospho-Thr308 Akt, and total Akt. Actin was used as a loading control. Bands were scanned densitometrically (arbitrary units), and phospho-to-total Akt was graphed using the mean ± SE (N = 3-4 animals for each). Statistical analyses were performed using Student’s t-test a P = 0.64 for Thr308, *P = 0.03 for Ser473; b P = 0.94 for Thr308, *P = 0.01 for Ser473)
Insulin-induced phosphorylation of Thr308 and Ser473 Akt still occurs in hepatocytes from old rats
The above results suggest that the lower Ser473 Akt phosphorylation state in hepatocytes from old rats may be due to an age-related change in signaling pathways upstream of Akt. As Akt activity is regulated in large part by signals through the insulin receptor (IR), the ability of Akt in hepatocytes from old rats to become phosphorylated via insulin dependent signaling was determined. Insulin was withdrawn from cultured hepatocytes, and after a 4 h acclimation, a time course of insulin repletion was performed. Culture of hepatocytes in insulin-free medium for this short time did not appreciably alter cell viability (data not shown). Under insulin withdrawal (time 0), Akt phosphorylation was barely detectable by western blot. The readdition of insulin (10 ng/ml) to hepatocytes resulted, however, in significant phosphorylation to both Thr308 and Ser473 within 5 min (Fig. 2a, b). These results indicate that insulin is still able to stimulate Akt phosphorylation in hepatocytes from old rats in spite of the observed decline in phospho-Ser473. While there was no significant difference in Thr308 phosphorylation between cells from young and old animals (P > 0.05), Ser473 phosphorylation was more pronounced at an earlier time in cells from young animals (P < 0.05). This is in accord with our data showing lower steady-state phosphorylation of Akt on Ser473 in cells from old versus young animals.
Fig. 2.
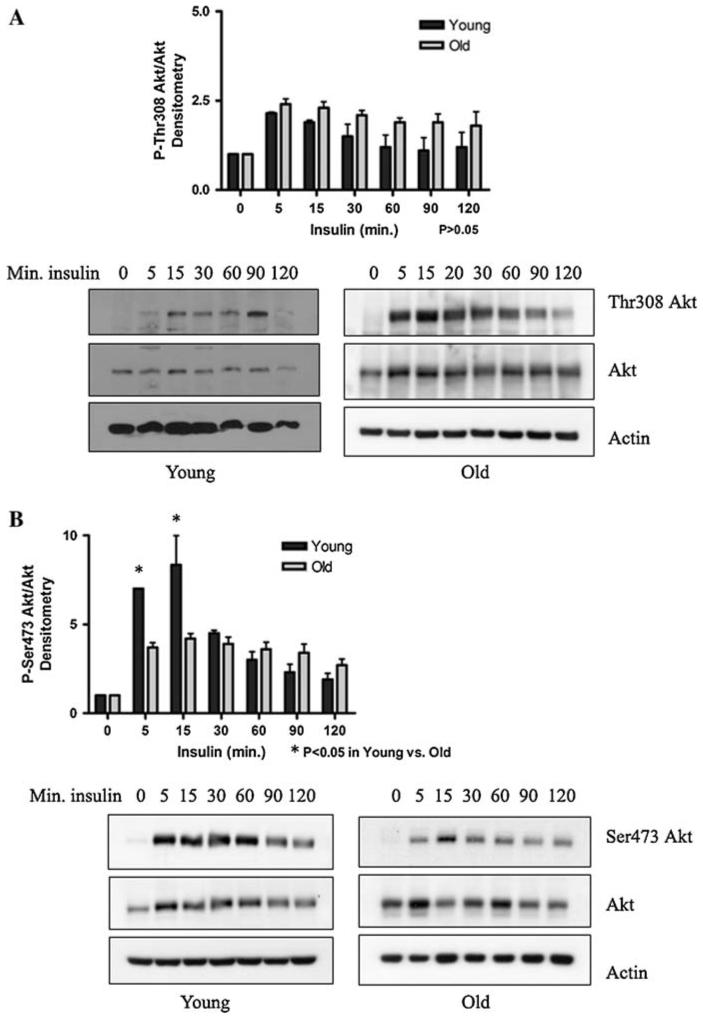
Insulin-induced phosphorylation of Thr308 and Ser473 Akt in hepatocytes from young and old rats. Insulin was withdrawn from cultured hepatocytes from young and old rats by washing the cells and replacing the media with insulin-free media. After a 4 h acclimation, a time course of insulin repletion was performed (10 ng/ml). Representative western blots (N = 3 animals) and graphs of (a) phospho-Thr308 Akt-tototal Akt, and (b) phospho-Ser473 Akt-to-total Akt. Actin was used as a loading control. Bands were scanned and densitometry was graphed (arbitrary units) using the mean ± SE. Statistical analyses were performed by two-way ANOVA (a P = 0.04 for time, P = 0.10 for age; b P < 0.0001 for time, P = 0.12 for age), with Bonferroni’s post-hoc test for young versus old (a P > 0.05 at all time points; b *P < 0.05 at 5 and 15 min)
Increased vulnerability to oxidative stress through loss of Akt activity is ameliorated by LA
The age-related decrement in Akt activity suggests that hepatocytes from old rats are more susceptible to a variety of insults to which the enzyme would typically confer resistance. Previous work has shown cell death increases with age in response to H2O2, particularly when the PI3K pathway is blocked (Ikeyama et al. 2002). We have previously shown in our culture model that there is equal viability for hepatocytes from young and old rats (Shenvi et al. 2008), but viability may be compromised by the addition of an external insult. We thus hypothesized that under oxidative stress, cells from old rats would be more susceptible to apoptosis than cells from young animals. To test this assertion, a glucose-glucose oxidase system (Gox) was used to generate H2O2 at a steady rate, and the cleavage of procaspase-3 as a marker of an early apoptotic event was quantified by western blot. Results in Fig. 3 show that cells from old animals were markedly more vulnerable to H2O2 than those from young rats; significant procaspase-3 cleavage (P < 0.001) was observed after 24 h for hepatocytes from old rats but not in cells from young animals. Control experiments where Akt was inhibited by pretreating hepatocytes with the specific Akt inhibitor Akti1/2 sensitized hepatocytes from old rats to oxidative stress. These results suggest that the age-specific loss of Akt activity results in significant susceptibility to oxidative stress.
Fig. 3.
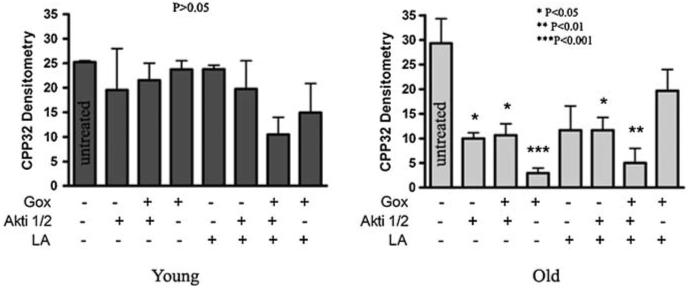
Increased vulnerability to oxidative stress through loss of Akt activity is ameliorated by LA. Hepatocytes from young and old rats were treated with 25 mM glucose and 100 mU/ml glucose oxidase (system designated as Gox), and/or 10 μM Akt inhibitor 1/2 (Akti1/2). Graphic representation of uncleaved procaspase-3 cleavage in cells treated with Gox and/or Akti1/2, with or without 50 μM LA pretreatment. Actin was used as a loading control. Bands were scanned and densitometry was graphed (arbitrary units) using the mean ± SE (N = 3 animals). One-way ANOVA was performed (P = 0.40 for young and P = 0.001 for old), and Tukey’s multiple comparison test was performed to determine statistical significance (P < 0.05) in treated versus untreated cells (Young: P > 0.05 for all treatments versus untreated; Old: *P < 0.05 for Akti1/2, Gox + Akti1/2, and LA + Akti1/2 versus untreated, **P < 0.01 for Gox + LA + Akti1/2 versus untreated, and ***P < 0.001 for Gox versus untreated)
LA has been shown to improve resistance to oxidative stress (Packer et al. 1995; Smith et al. 2004). The addition of LA + Gox to Akti1/2-treated cells did not significantly change the amount of procaspase-3 cleavage compared to Gox + Akti1/2 alone, indicating that LA could not rescue H2O2 damaged cells from either age group without Akt (Fig. 3). However, when cells from old animals were not pretreated with Akti1/2, LA prevented H2O2-induced procaspase-3 cleavage. LA alone did not confer further resistance to cells from young rats, suggesting that LA worked by directly affecting the lesion that limited Akt-mediated resistance, likely Ser473 phosphorylation. These results indicate that the high susceptibility of hepatocytes from aging animals to apoptosis by oxidative stress can be ameliorated by LA, and that this beneficial event occurs partly through an Akt survival pathway.
LA induces transient phosphorylation of Akt at Ser473 and increases Akt activity
LA has been used to improve glucose uptake in type II diabetes, and mediates this effect partly through stimulating the insulin pathway (Konrad 2005). As insulin dependent signaling directly affects Akt Ser473 phosphorylation, and the above results suggest that LA prevents oxidative stress-induced apoptosis in part through Akt, we tested whether LA ameliorates loss of Akt Ser473 phosphorylation in insulin-replete cells. To determine whether LA induces Akt phosphorylation in cultured hepatocytes, particularly from old animals, cells were treated with 50 μM LA and Akt phosphorylation status was monitored over time. Akt phosphorylation at the Thr308 site was not significantly affected by LA treatment in cells from either young or old rats (data not shown). However, using site-specific antibodies for phosphorylation at the Ser473 epitope revealed that LA strongly induced phosphorylation at this locus in an age-dependent manner (Fig. 4a). Treating hepatocytes from old rats with LA resulted in a time-dependent increase in the level of Ser473 phosphorylation, which approached levels seen in young rats approximately 90 min after LA addition. LA also enhanced Akt activity. Akt was immunoprecipitated from lysates of hepatocytes from young and old rats, with or without a 90 min pretreatment with 50 μM LA. The activity assay was performed using a GST-GSK3α/β fusion protein as the substrate. Figure 4b shows Akt activity was deficient in cells from old rats compared to young, but in LA-treated hepatocytes from old rats, Akt activity approached the steadystate activity levels observed in young rats. These results thus show that low LA doses likely to be achieved in plasma following oral intake results in an age-specific improvement in Akt activity. Interestingly, in cells from young rats, little effect on Ser473 phosphorylation and Akt activity was observed, at least at the provided LA dose.
Fig. 4.
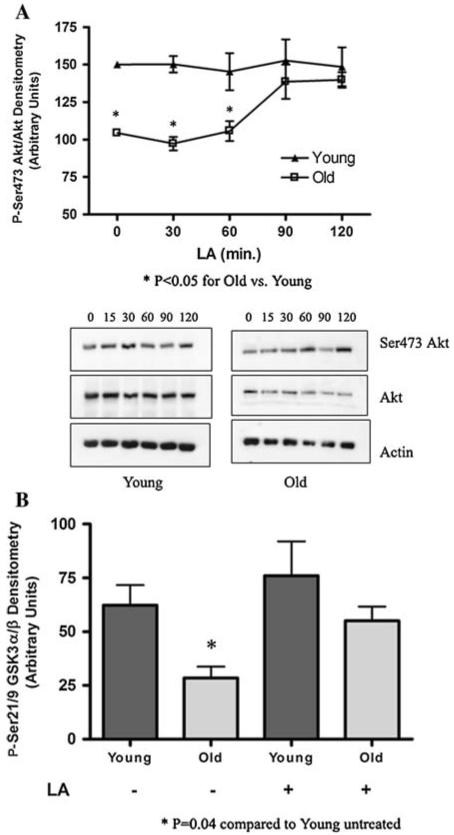
LA induces phosphorylation of Akt at Ser473 and increases overall activity. (a) Ser473 Akt phosphorylation status in cultured hepatocytes from young and old rats treated with 50 μM LA. Western blotting was performed for phospho-Ser473, total Akt, and actin. Bands were scanned densitometrically; data represents the mean ± SE (N = 7 animals) of phospho-to-total Akt. Statistical analyses were performed by two-way ANOVA (P = 0.04 for time, and P < 0.0001 for age), with Bonferroni’s post-hoc test for young versus old (*P < 0.05 at 0, 30, and 60 min). (b) Akt activity was tested by the phosphorylation of a GST-GSK3α/β fusion protein, using extracts of hepatocytes from young and old rats, with and without a 90 min. 50 μM LA pretreatment. Phospho-Ser21/9 GSK3α/β was determined by western blotting, bands were scanned densitometrically, and data represents the mean ± SE (N = 3 animals). Statistical analyses were performed using Student’s t-test. Young versus old *P = 0.04, young versus young + LA: P = 0.50, young versus old + LA: P = 0.56
To test whether LA induces Akt phosphorylation on Ser473 in vivo, young and old rats were gavaged with 120 mg/kg LA. Western blots of liver tissue from old rats showed a marked increase in Ser473 Akt phosphorylation in response to LA (Fig. 5). The phosphorylation level also increased in livers from young rats, but to a lesser degree. These data are in agreement with the hypothesis that Ser473 Akt becomes activated in the presence of LA, and extends the finding to include oral intake.
Fig. 5.
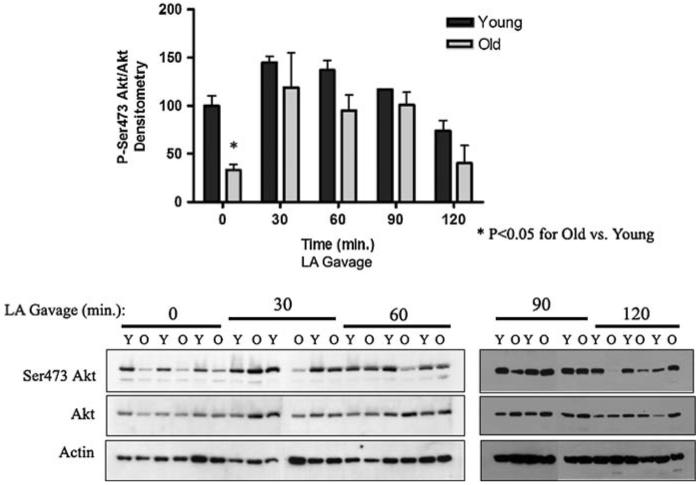
LA induces phosphorylation of Akt at Ser473 in liver tissue in young and old rats. Ser473 Akt phosphorylation status in liver tissue from young (Y) and old (O) rats given LA by gavage (120 mg/kg). Western blotting was performed for phospho-Ser473, total Akt, and Actin. Bands were scanned densitometrically; data represents the mean ± SE (N = 3 animals) for phospho-to-total Akt. Statistical analyses were performed by two-way ANOVA (P = 0.0003 for time, P = 0.001 for age), with Bonferroni’s post-hoc test for young versus old (*P < 0.05 at 0 min)
LA stimulates Akt phosphorylation in the absence of insulin
To ascertain whether LA-induced Akt phosphorylation state and activity via its action on the insulin signaling pathway, insulin was withdrawn from isolated hepatocytes in culture, as described above. Phospho-Thr308 Akt and phospho-Ser473 Akt levels were very low before treatment, due to the insulin withdrawal. When 50 μM LA, instead of insulin, was added to insulin-deprived cells from young and old rats, both Thr308 and Ser473 Akt phosphorylation significantly increased within 5 min in cells from old rats (Fig. 6a, b) (P < 0.05). Hepatocytes from young rats demonstrated increased Akt phosphorylation at the Ser473 site within 30 min, but Thr308 phosphorylation state was not significantly affected by LA. As Thr308 is phosphorylated by the PI3K/PDK1 pathway, a lack of insulin in the medium may account for this minor response. Overall, our data suggest that LA may stimulate the Akt pathway in cells from old animals, but not as a true insulin mimetic.
Fig. 6.
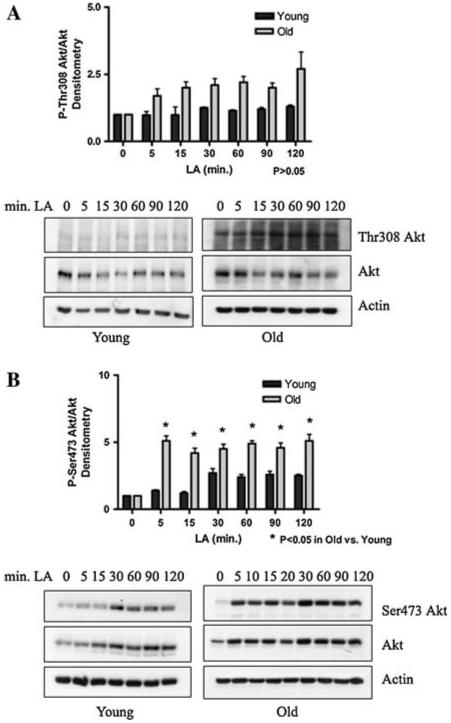
LA induces phosphorylation of Akt in the absence of insulin. Insulin was withdrawn from cultured hepatocytes from young and old rats by washing the cells and replacing the media with insulin-free media. After a 4 h acclimation, a time course of LA treatment was performed. Representative western blots and graphic representation (N = 3) of (a) phospho-Thr308 Akt-to-total Akt and (b) phospho-Ser473 Akt-to-total Akt. Actin was used as a loading control. Bands were scanned and densitometry was graphed (arbitrary units) using the mean ± SE. Statistical analyses were performed by two-way ANOVA (a P = 0.35 for time, P = 0.0007 for age; b P < 0.0001 for time, P < 0.0001 for age), with Bonferroni’s post-hoc test for young versus old (a P > 0.05 at all time points; b *P < 0.05 at 5, 15, 60, 90, and 120 min)
Ser473 Akt does not become phosphorylated in response to LA under PI3K inhibition
PI3 kinase is a key enzyme positioned “downstream” of the IR and “upstream” of Akt in the insulin signaling cascade. PI3K is known to phosphorylate PtdIns-4,5-P2, and its action is antagonized by PTEN. When PI3K is active, Akt becomes phosphorylated on Thr308, an event that precedes Ser473 phosphorylation. We thus sought to determine whether pharmacological inhibition of PI3K also antagonized LA-mediated stimulation of Ser473 phosphorylation, using the well-known PI3K inhibitor, LY294002 (Okano et al. 2003). Cultured hepatocytes from young and old rats were given LY294002 2 h prior to treatment of the cells with 50 μM LA. Treatment with LY294002 prevented LA-stimulated phosphorylation of Akt on Ser473 in cells from young and old animals (Fig. 7). Thus, phosphorylation of Akt on Ser473 in response to LA does not circumvent PI3K.
Fig. 7.
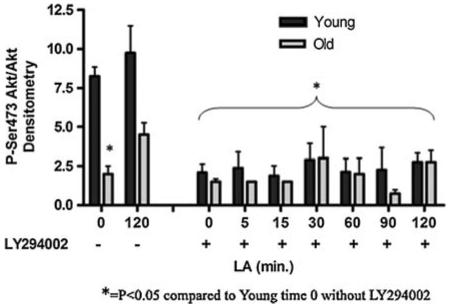
LA-induced Ser473 Akt phosphorylation is dependent upon PI3K. Hepatocytes from young and old rats were cultured in insulin-replete media, then pretreated with 50 μM LY294002 2 h prior to administration of LA. Phospho-Ser473 Akt-to-total Akt levels were measured by western blot (N = 3) and bands measured densitometrically. Actin was used as a loading control. Data was graphed (arbitrary units) using the mean ± SE. P = 0.009 for age and P < 0.0001 for time by two-way ANOVA. *P < 0.05 for young versus old at 0 min LA in the absence of inhibitor, as well as for all LY294002-treated samples, by Bonferroni’s post-hoc test
LA suppresses the activity of both PTEN and PP2A
To ascertain the origin of LA’s effect on Akt phosphorylation at Ser473, it is important to consider how the overall phosphorylation state of Akt is governed. Phosphorylation of Akt can be prevented by the action of PTEN, a phosphatase that converts PtdIns-3,4,5-P3 back to PtdIns-4,5-P2 through dephosphorylation and, in turn, prevents Akt recruitment to the membrane to be phosphorylated. In addition, phosphatases (e.g., PP2A) may directly remove phosphate groups from Akt once it has been activated. In either event, Akt activity declines and renders cells susceptible to apoptosis. As shown in Fig. 4a, LA treatment required a full 90 min in insulin-replete cells to reverse the age-related loss of Akt phosphorylation at the Ser473 site. Because signaling through the IR/PI3K pathway is generally quite rapid, we hypothesized that LA may also act indirectly to reverse Akt phosphorylation by inhibiting phosphatase activities. Phosphorylation status of Tyr307 on the holoenzyme’s C subunit, a site known to inhibit overall PP2A activity (Chen et al. 1992) was monitored along with total PP2A-C, as were the levels of PTEN and Ser380 PTEN (Odriozola et al. 2007). While there were no basal, age-related differences in either PTEN or PP2A (Fig. 8a, b, P > 0.05), nevertheless we tested whether treatment with LA had a suppressive effect on the phosphatases. LA treatment increased PP2A-C phosphorylation at Tyr307 (P < 0.05), indicating that it did indeed suppress the activity of this phosphatase (Fig. 8c). In addition, Fig. 8d shows that LA treatment caused a decrease in phosphorylation of PTEN on Ser380 (P < 0.05), which decreases its activity (Odriozola et al. 2007). Together, these results show that LA acts to increase Akt phosphorylation both by increasing its ability to become phosphorylated through lowered PTEN activity, as well as by preventing the removal of phosphate groups through lowered PP2A activity.
Fig. 8.
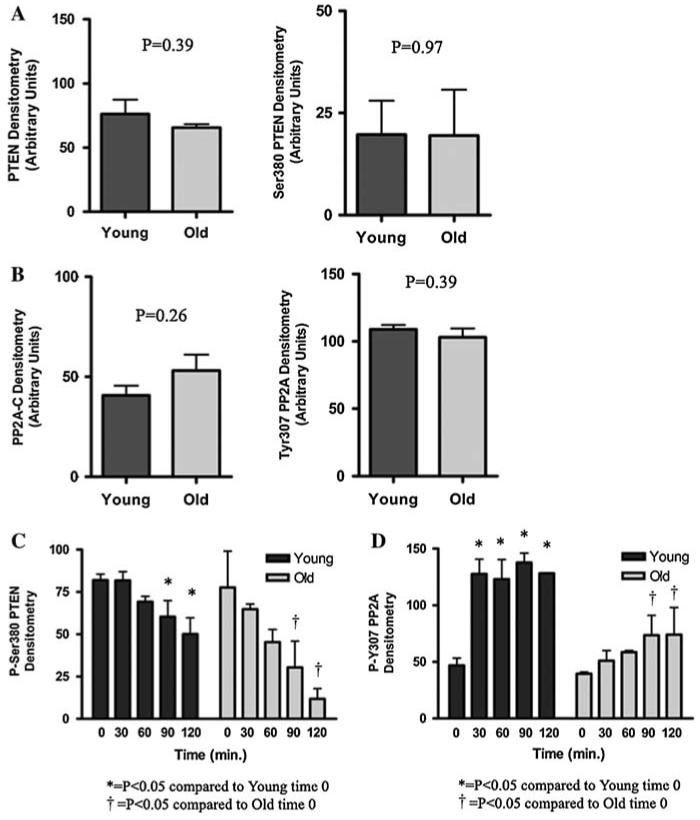
LA suppresses the activity of the phosphatases PTEN and PP2A. Graphic representation of western blot results for (a) total PTEN (P = 0.39) and phospho-Ser380 PTEN (P = 0.97), and (b) total PP2A-C (P = 0.26) and phospho-Tyr307 PP2A-C (P = 0.39) in homogenized liver tissue from young and old rats (N = 6 for each, statistical significance determined by Student’s t-test). Graphic representation of time course by western blot for (c) PTEN phosphorylation at Ser380 (P = 0.0009 for time and P = 0.03 for age), and (d) PP2A-C phosphorylation at Tyr307 (P = 0.0009 for time and P = 0.01 for age) in cultured hepatocytes from young and old rats, treated with 50 μM LA (N = 3 for each, statistical significance determined by ANOVA). Actin was used as a loading control. Data represent the mean ± SE. Bonferroni’s post-hoc test was used to determine statistical significance for treated versus untreated cells (c Young: *P < 0.05 for 0 vs. 90 min, *P < 0.01 for 0 vs. 120 min; Old: P < 0.05 for 0 vs. 90 and 120 min; d Young: *P < 0.01 for 0 vs. 30, 60, 90, and 120 min; Old: P < 0.05 for 0 vs. 90 and 120 min)
Discussion
The most significant results of this study suggest that a profound loss of constitutive Akt activity occurs in aging hepatocytes. Akt phosphorylation state, which is largely reflective of Akt activity, appears to be the primary lesion responsible for this marked decline. Taken to its logical extent, the data presented in this study further suggests that there is a link between Akt activity, age, and perhaps also oxidative stress resistance. These changes would be expected to render hepatocytes susceptible to a variety of stresses, which otherwise would be successfully repulsed. Thus, our data extends the concept of Akt as a significant regulator of cellular stress response to show that its loss of function with age may portend a decline in the capacity to resist a variety of oxidative, xenobiotic, and environmental insults.
The systematic loss of Akt activity is somewhat surprising, given that Akt activation regulates permissiveness toward apoptosis, which could therefore lead to depletion of cells especially in post-mitotic organs. However, downregulation of Akt activity may be balanced by greater FOXO-mediated transcription that would eventually lead to lower metabolism and upregulation of cellular antioxidants (Morris 2005), even in cells that are not undergoing continuous mitosis. This period of cell cycle arrest may have effects similar to caloric restriction (currently the best-known method of achieving longevity), and the dauer hibernation-like state that takes place in C. elegans (Kenyon et al. 1993). Thus, Akt may have a dual role in stress response and aging. Lower Akt activity may predispose the cell to death from oxidative or toxic stress, but in so doing, may also limit other more onerous risks to survival. For example, the prevalence of cancer increases with the 4th power of age (Ames et al. 1993), suggesting that lowered Akt activity could balance cell death with prevention of neoplasia, a compromise that is imperative for survival of the individual. Under advantageous conditions, cells from the aged animal survive, but under stress, apoptosis may prevail.
Regardless of the consequences, the underlying mechanism(s) responsible for diminished Akt activity with age lies in its clearly lower constitutive phosphorylation state. Phosphorylation loss had a very age-specific pattern. There was no age difference in Thr308 phosphorylation, but phosphorylation at the Ser473 residue in hepatocytes from old animals was significantly lower than that seen in cells from young rats. This is in accord with Li et al. (2003), who showed an age-related decrease in Akt phosphorylation in mouse skeletal muscle. Further analysis of our data revealed that the decline we observed was not due to hyper-activation of either of two relevant protein phosphatases, as there were no age-related differences in either PP2A or PTEN activities in hepatocytes. Because phosphorylation at the Thr308 site was not affected with age, we argue that the aging lesion must reside in signaling processes that ultimately phosphorylate the Ser473 site of Akt. However, the precise mechanism involved was not explored in the present study, as multiple kinases and signaling pathways recognize and contribute to Ser473 phosphorylation. Despite the often conflicting and controversial literature on the governance of Akt phosphorylation at Ser473, it must be recognized that phosphorylation at this site can be blocked by LY294002, a PI3K inhibitor, and so it was in our study. Thus, any signals generated through IR-mediated signal transduction pathways or those that trigger PI3K phosphorylation, may contribute to age-specific loss of Akt phosphorylation. The precise signaling mechanism(s) associated with loss of Ser473 phosphorylation represent a significant future focus in identifying the loss of Akt-dependent survival mechanisms with age.
In relation to the general control that PI3K affords to Akt phosphorylation, we found that LA reintroduces Akt phosphorylation and its activity specifically through the PI3K pathway (Fig. 7). Indeed, phosphorylation of Akt on Ser473 in hepatocytes from old rats is the same as observed in young animals after treatment with relatively small concentrations of LA (50 μM) (Fig. 5). These results are in accord with our previous work that showed a similar improvement in Akt activity in endothelial cells (Smith and Hagen 2003), and also work done in isolated monocytes (Zhang et al. 2007). Though the insulin pathway that stimulates Akt phosphorylation on both sites appears to be intact in hepatocytes from both young and old animals (Fig. 2), LA did not act as an insulin mimetic in the absence of insulin (Fig. 6). Interestingly, LA alone stimulated Akt phosphorylation to a greater degree in cells from old animals, suggesting that there is indeed an age-related lesion that is remediated by LA. However, none of these studies revealed the precise mechanism involved in LA-dependent Akt activation.
In this regard, it is interesting to note that the age-related improvement in Akt activity also agrees with a larger body of work showing orally supplied LA stimulates insulin receptor (IR) activation, thus initiating insulin dependent signal transduction pathways and cell survival systems (Yaworsky et al. 2000). In fact, LA has been used as a diabetes therapy in Germany for 50 years because of its insulin mimetic effects on the insulin/IGF-1 pathway and ability to improve glucose transport (Evans and Goldfine 2000). Recently, one group has shown a putative binding pocket for LA on the IR (Diesel et al. 2007), the importance of which is yet to be elucidated. Thus, LA may work in part, via a direct action on upstream kinases from Akt.
In addition to its activation of PI3K-mediated Akt phosphorylation, there is increasing evidence that LA inhibits specific phosphatases, including those that have direct relevance to Akt Ser473 phosphorylation. Cho et al. showed that LA inactivates the tyrosine phosphatase, PTP1B, in vitro (Cho et al. 2003), which halts insulin-generated signals by dephosphorylating proteins of the insulin pathway. We previously showed that LA lowers PP2A activity in isolated aortic endothelial cells by inhibiting neutral sphing-omyelinases (nSMase) and subsequent ceramide-induced activation of this particular phosphatase (Smith et al. 2006). It has also been shown that expression of wild type PTEN in rhabdomyosarcoma and myoblast cell lines reduces Akt phosphorylation at Ser473 but not Thr308 (Wan and Helman 2003), and the authors suggest a role for regulation of the putative PDK2 proteins by PTEN, which would target Ser473 Akt specifically. Overall, this prior evidence fits well with our present results showing that LA improves Akt phosphorylation state and activity by simultaneously increasing PI3K-dependent signaling and inhibiting PTEN and PP2A phosphatase action (Fig. 8). In fact, the data showing LA-induced gain of PP2A phosphorylation on Tyr307 and loss of phosphorylation of PTEN on Ser380, both of which are inhibitory, indicates that the observed phosphorylation of Ser473 Akt in response to LA may be in part a function of its action on phosphatases. The relatively slow timing of LA-induced Akt phosphorylation in insulin-replete medium (Fig. 5) also supports this contention.
The marked improvement of Akt activity and cell survival by LA treatment in hepatocytes from aged rats adds weight to the growing body of evidence that LA supplementation may be a useful adjunct to maintaining stress resistance mechanisms in aging. Showing the involvement of Akt in LA-mediated cell survival and reduced susceptibility to oxidative stress helps to establish a mechanism for this phenomenon. It is notable that LA does not induce Akt phosphorylation in isolated cells from young animals at the low concentration used, which is a level likely to be achieved in the blood plasma following oral supplementation (Petersen Shay et al. 2008). However, there is a noticeable increase in Ser473 Akt phosphorylation in liver tissue from young animals (Fig. 5) and the difference may be that the isolated cells are cultured in insulin-replete medium. We have also found that substantially higher and potentially supraphysiological levels of LA do stimulate Akt activity in vitro for cultured cells from young animals (data not shown). Thus, LA appears to induce a compensation for the constitutive decline in Akt activity, preserving this enzyme’s critical function which otherwise declines with age.
The reversible rather than inducible nature of LA action is an important concept in two ways, namely, (i) LA action must stimulate pathways distinct from the aging lesion, thereby compensating for the age-related loss of Akt phosphorylation, i.e., PTEN and PP2A; and (ii) LA improves resistance to Akt-driven stress response mechanisms without associated problems that may arise from chronic overstimulation of Akt activity. Thus, the risk for neoplasia that is associated with Akt overexpression would be minimized. The net effect of LA on Akt phosphorylation in cells from old animals is a reversal of loss, and is age-specific. This study thus presents a rationale for future work to examine the benefits of LA and other allied micronutrients to maintain Akt-driven stress response mechanisms in the elderly.
Acknowledgments
This work was supported by grants from the National Institutes of Health R01 2AG17141 and P01 AT002034-01. KPS was supported by T32 AT002688-01.
Contributor Information
Kate Petersen Shay, Linus Pauling Institute, Oregon State University, 571 Weniger Hall, Corvallis, OR 97331-6512, USA.
Tory M. Hagen, Linus Pauling Institute, Oregon State University, 571 Weniger Hall, Corvallis, OR 97331-6512, USA & Department of Biochemistry and Biophysics, Oregon State University, Corvallis, OR 97331, USA
References
- Alessi DR, Andjelkovic M, Caudwell B, Cron P, Morrice N, Cohen P, Hemmings BA. Mechanism of activation of protein kinase B by insulin and IGF-1. EMBO J. 1996;15:6541–6551. [PMC free article] [PubMed] [Google Scholar]
- Alessi DR, James SR, Downes CP, Holmes AB, Gaffney PR, Reese CB, Cohen P. Characterization of a 3-phosphoinositide-dependent protein kinase which phosphorylates and activates protein kinase B alpha. Curr Biol. 1997;7:261–269. doi: 10.1016/s0960-9822(06)00122-9. [DOI] [PubMed] [Google Scholar]
- Ames BN, Shigenaga MK, Hagen TM. Oxidants, antioxidants, and the degenerative diseases of aging. Proc Natl Acad Sci USA. 1993;90:7915–7922. doi: 10.1073/pnas.90.17.7915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Martin BL, Brautigan DL. Regulation of protein serine-threonine phosphatase type-2A by tyrosine phosphorylation. Science. 1992;257:1261–1264. doi: 10.1126/science.1325671. [DOI] [PubMed] [Google Scholar]
- Cho KJ, Moini H, Shon HK, Chung AS, Packer L. Alpha-lipoic acid decreases thiol reactivity of the insulin receptor and protein tyrosine phosphatase 1B in 3T3-L1 adipocytes. Biochem Pharmacol. 2003;66:849–858. doi: 10.1016/s0006-2952(03)00395-2. [DOI] [PubMed] [Google Scholar]
- Clancy DJ, Gems D, Harshman LG, Oldham S, Stocker H, Hafen E, Leevers SJ, Partridge L. Extension of life-span by loss of CHICO, a Drosophila insulin receptor substrate protein. Science. 2001;292:104–106. doi: 10.1126/science.1057991. [DOI] [PubMed] [Google Scholar]
- Diesel B, Kulhanek-Heinze S, Holtje M, Brandt B, Holtje HD, Vollmar AM, Kiemer AK. Alpha-lipoic acid as a directly binding activator of the insulin receptor: protection from hepatocyte apoptosis. Biochemistry. 2007;46:2146–2155. doi: 10.1021/bi602547m. [DOI] [PubMed] [Google Scholar]
- Dominici FP, Hauck S, Argentino DP, Bartke A, Turyn D. Increased insulin sensitivity and upregulation of insulin receptor, insulin receptor substrate (IRS)-1 and IRS-2 in liver of Ames dwarf mice. J Endocrinol. 2002;173:81–94. doi: 10.1677/joe.0.1730081. [DOI] [PubMed] [Google Scholar]
- Dominici FP, Argentino DP, Bartke A, Turyn D. The dwarf mutation decreases high dose insulin responses in skeletal muscle, the opposite of effects in liver. Mech Ageing Dev. 2003;124:819–827. doi: 10.1016/s0047-6374(03)00136-2. [DOI] [PubMed] [Google Scholar]
- Evans JL, Goldfine ID. Alpha-lipoic acid: a multifunctional antioxidant that improves insulin sensitivity in patients with type 2 diabetes. Diabetes Technol Ther. 2000;2:401–413. doi: 10.1089/15209150050194279. [DOI] [PubMed] [Google Scholar]
- Feng J, Park J, Cron P, Hess D, Hemmings BA. Identification of a PKB/Akt hydrophobic motif Ser-473 kinase as DNA-dependent protein kinase. J Biol Chem. 2004;279:41189–41196. doi: 10.1074/jbc.M406731200. [DOI] [PubMed] [Google Scholar]
- Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature. 2000;408:239–247. doi: 10.1038/35041687. [DOI] [PubMed] [Google Scholar]
- Flurkey K, Papaconstantinou J, Miller RA, Harrison DE. Lifespan extension and delayed immune and collagen aging in mutant mice with defects in growth hormone production. Proc Natl Acad Sci USA. 2001;98:6736–6741. doi: 10.1073/pnas.111158898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh CC, Papaconstantinou J. Akt/PKB and p38 MAPK signaling, translational initiation and longevity in Snell dwarf mouse livers. Mech Ageing Dev. 2004;125:785–798. doi: 10.1016/j.mad.2004.07.008. [DOI] [PubMed] [Google Scholar]
- Ikeyama S, Kokkonen G, Shack S, Wang XT, Holbrook NJ. Loss in oxidative stress tolerance with aging linked to reduced extracellular signal-regulated kinase and Akt kinase activities. FASEB J. 2002;16:114–116. doi: 10.1096/fj.01-0409fje. [DOI] [PubMed] [Google Scholar]
- Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, Huang Q, Qin J, Su B. SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell. 2006;127:125–137. doi: 10.1016/j.cell.2006.08.033. [DOI] [PubMed] [Google Scholar]
- Kawakami Y, Nishimoto H, Kitaura J, Maeda-Yamamoto M, Kato RM, Littman DR, Leitges M, Rawlings DJ, Kawakami T. Protein kinase C beta II regulates Akt phosphorylation on Ser-473 in a cell type- and stimulus-specific fashion. J Biol Chem. 2004;279:47720–47725. doi: 10.1074/jbc.M408797200. [DOI] [PubMed] [Google Scholar]
- Kenyon C, Chang J, Gensch E, Rudner A, Tabtiang R. A C. elegans mutant that lives twice as long as wild type. Nature. 1993;366:461–464. doi: 10.1038/366461a0. [DOI] [PubMed] [Google Scholar]
- Konrad D. Utilization of the insulin-signaling network in the metabolic actions of alpha-lipoic acid-reduction or oxidation? Antioxid Redox Signal. 2005;7:1032–1039. doi: 10.1089/ars.2005.7.1032. [DOI] [PubMed] [Google Scholar]
- Li M, Li C, Parkhouse WS. Age-related differences in the des IGF-I-mediated activation of Akt-1 and p70 S6K in mouse skeletal muscle. Mech Ageing Dev. 2003;124:771–778. doi: 10.1016/s0047-6374(03)00124-6. [DOI] [PubMed] [Google Scholar]
- Lindsley CW, Zhao Z, Leister WH, Robinson RG, Barnett SF, Defeo-Jones D, Jones RE, Hartman GD, Huff JR, Huber HE, Duggan ME. Allosteric Akt (PKB) inhibitors: discovery and SAR of isozyme selective inhibitors. Bioorg Med Chem Lett. 2005;15:761–764. doi: 10.1016/j.bmcl.2004.11.011. [DOI] [PubMed] [Google Scholar]
- Maehama T, Dixon JE. The tumor suppressor, PTEN/MMAC1, dephosphorylates the lipid second messenger, phosphatidylinositol 3,4,5-trisphosphate. J Biol Chem. 1998;273:13375–13378. doi: 10.1074/jbc.273.22.13375. [DOI] [PubMed] [Google Scholar]
- Manning BD, Cantley LC. AKT/PKB signaling: navigating downstream. Cell. 2007;129:1261–1274. doi: 10.1016/j.cell.2007.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millward TA, Zolnierowicz S, Hemmings BA. Regulation of protein kinase cascades by protein phosphatase 2A. Trends Biochem Sci. 1999;24:186–191. doi: 10.1016/s0968-0004(99)01375-4. [DOI] [PubMed] [Google Scholar]
- Morris BJ. A forkhead in the road to longevity: the molecular basis of lifespan becomes clearer. J Hypertens. 2005;23:1285–1309. doi: 10.1097/01.hjh.0000173509.45363.dd. [DOI] [PubMed] [Google Scholar]
- Odriozola L, Singh G, Hoang T, Chan AM. Regulation of PTEN activity by its carboxyl-terminal autoinhibitory domain. J Biol Chem. 2007;282:23306–23315. doi: 10.1074/jbc.M611240200. [DOI] [PubMed] [Google Scholar]
- Okano J, Shiota G, Matsumoto K, Yasui S, Kurimasa A, Hisatome I, Steinberg P, Murawaki Y. Hepatocyte growth factor exerts a proliferative effect on oval cells through the PI3K/AKT signaling pathway. Biochem Biophys Res Commun. 2003;309:298–304. doi: 10.1016/j.bbrc.2003.04.002. [DOI] [PubMed] [Google Scholar]
- Packer L, Witt EH, Tritschler HJ. Alpha-lipoic acid as a biological antioxidant. Free Radic Biol Med. 1995;19:227–250. doi: 10.1016/0891-5849(95)00017-r. [DOI] [PubMed] [Google Scholar]
- Petersen Shay K, Moreau RF, Smith EJ, Hagen TM. Is alpha-lipoic acid a scavenger of reactive oxygen species in vivo? Evidence for its initiation of stress signaling pathways that promote endogenous antioxidant capacity. IUBMB Life. 2008;60:362–367. doi: 10.1002/iub.40. [DOI] [PubMed] [Google Scholar]
- Shenvi SV, Dixon BD, Shay KP, Hagen TM. A rat primary hepatocyte culture model for aging studies. Curr Protoc Toxicol. 2008 doi: 10.1002/0471140856.tx1407s37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith AR, Hagen TM. Vascular endothelial dysfunction in aging: loss of Akt-dependent endothelial nitric oxide synthase phosphorylation and partial restoration by (R)-alpha-lipoic acid. Biochem Soc Trans. 2003;31:1447–1449. doi: 10.1042/bst0311447. [DOI] [PubMed] [Google Scholar]
- Smith AR, Shenvi SV, Widlansky M, Suh JH, Hagen TM. Lipoic acid as a potential therapy for chronic diseases associated with oxidative stress. Curr Med Chem. 2004;11:1135–1146. doi: 10.2174/0929867043365387. [DOI] [PubMed] [Google Scholar]
- Smith AR, Visioli F, Frei B, Hagen TM. Age-related changes in endothelial nitric oxide synthase phosphorylation and nitric oxide dependent vasodilation: evidence for a novel mechanism involving sphingomyelinase and ceramide-activated phosphatase 2A. Aging Cell. 2006;5:391–400. doi: 10.1111/j.1474-9726.2006.00232.x. [DOI] [PubMed] [Google Scholar]
- Tatar M, Kopelman A, Epstein D, Tu MP, Yin CM, Garofalo RS. A mutant Drosophila insulin receptor homolog that extends life-span and impairs neuroendocrine function. Science. 2001;292:107–110. doi: 10.1126/science.1057987. [DOI] [PubMed] [Google Scholar]
- Wan X, Helman LJ. Levels of PTEN protein modulate Akt on serine 473, but not on threonine 308, in IGF-II-overexpressing rhabdomyosarcoma cells. Oncogene. 2003;22:8205–8211. doi: 10.1038/sj.onc.1206878. [DOI] [PubMed] [Google Scholar]
- Wu X, Senechal K, Neshat MS, Whang YE, Sawyers CL. The PTEN/MMAC1 tumor suppressor phosphatase functions as a negative regulator of the phosphoinositide 3-kinase/Akt pathway. Proc Natl Acad Sci USA. 1998;95:15587–15591. doi: 10.1073/pnas.95.26.15587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaworsky K, Somwar R, Ramlal T, Tritschler HJ, Klip A. Engagement of the insulin-sensitive pathway in the stimulation of glucose transport by alpha-lipoic acid in 3T3-L1 adipocytes. Diabetologia. 2000;43:294–303. doi: 10.1007/s001250050047. [DOI] [PubMed] [Google Scholar]
- Zhang WJ, Wei H, Hagen T, Frei B. Alpha-lipoic acid attenuates LPS-induced inflammatory responses by activating the phosphoinositide 3-kinase/Akt signaling pathway. Proc Natl Acad Sci USA. 2007;104:4077–4082. doi: 10.1073/pnas.0700305104. [DOI] [PMC free article] [PubMed] [Google Scholar]