

Abstract
Background
Subtractive hybridization methods are valuable tools for identifying differentially regulated genes in a given tissue avoiding redundant sequencing of clones representing the same expressed genes, maximizing detection of low abundant transcripts and thus, affecting the efficiency and cost effectiveness of small scale cDNA sequencing projects aimed to the specific identification of useful genes for breeding purposes. The objective of this work is to evaluate alternative strategies to high-throughput sequencing projects for the identification of novel genes differentially expressed in sunflower as a source of organ-specific genetic markers that can be functionally associated to important traits.
Results
Differential organ-specific ESTs were generated from leaf, stem, root and flower bud at two developmental stages (R1 and R4). The use of different sources of RNA as tester and driver cDNA for the construction of differential libraries was evaluated as a tool for detection of rare or low abundant transcripts. Organ-specificity ranged from 75 to 100% of non-redundant sequences in the different cDNA libraries. Sequence redundancy varied according to the target and driver cDNA used in each case. The R4 flower cDNA library was the less redundant library with 62% of unique sequences. Out of a total of 919 sequences that were edited and annotated, 318 were non-redundant sequences. Comparison against sequences in public databases showed that 60% of non-redundant sequences showed significant similarity to known sequences. The number of predicted novel genes varied among the different cDNA libraries, ranging from 56% in the R4 flower to 16 % in the R1 flower bud library. Comparison with sunflower ESTs on public databases showed that 197 of non-redundant sequences (60%) did not exhibit significant similarity to previously reported sunflower ESTs. This approach helped to successfully isolate a significant number of new reported sequences putatively related to responses to important agronomic traits and key regulatory and physiological genes.
Conclusions
The application of suppressed subtracted hybridization technology not only enabled the cost effective isolation of differentially expressed sequences but it also allowed the identification of novel sequences in sunflower from a relative small number of analyzed sequences when compared to major sequencing projects.
Keywords: Helianthus annuus, sunflower, EST, SSH, organ-specific transcripts
Background
Cultivated sunflower (Helianthus annuus L.) is one of the most important sources of vegetable oil worldwide. During the last decade, rapid advances in applied genetics and genomic technologies have led to the development of saturated sunflower genetic maps based on different molecular markers including RFLP, AFLP and SSR [1-10]. More recently, large-scale cDNA sequencing projects have identified expressed sequence tags (ESTs) in different plant species. Today, more than 100 plant species are represented in the EST division (dbEST) of GenBank http://www.ncbi.nlm.nih.gov/dbEST/dbEST_summary.html with a total of 2,063,406 entries. However, Arabidopsis, rice, maize, tomato and soybean ESTs projects gather more than 50% of the total entries. The Compositae is represented by 113,149 entries, of which 44,961 correspond to sunflower ESTs. These projects allow the characterization of full sets of transcribed genes in the target organisms and provide, at the same time, a source of genetic markers that can be functionally associated to important agronomical traits reinforcing and complementing the use of anonymous markers. Thus, the use of EST-based markers could lead to genetic mapping of a gene that directly affects the trait or a specific sequence could be target due to its predicted function based on sequence comparison [11]. ESTs generated from cDNA libraries should represent, ideally, all expressed genes in a target organ/tissue, at a specific developmental stage and/ or in a specific environment. However, differences in expression level among genes in a given tissue yield mRNAs that differ in abundance, making it difficult to capture rare mRNA in cDNA libraries. This problem also leads to redundant sequencing of clones representing the same expressed genes, affecting the efficiency and cost effectiveness of the EST approach [12] which hinders research laboratories with small budgets to perform EST characterization studies. To avoid this problem, different strategies based on normalized cDNA libraries have been reported in many different organisms [12-14] including plants [15-17]. In this study, we report for the first time in sunflower, the isolation and characterization of ESTs from organ-specific cDNA libraries constructed by suppressed subtractive hybridization [18] as an alternative to identify differentially expressed sunflower transcripts. We analyzed the efficiency of the subtraction and enrichment methods for each cDNA library generated and present the differential level of representation for functional EST groups based on Gene Ontology annotation [19], as well as a comprehensive description of individual non-redundant sequences generated.
Results and Discussion
Construction of organ-specific cDNA libraries
Different cDNA libraries were constructed after subtractive hybridization. Firstly, a reciprocal experiment was designed to determine the efficiency of the subtraction procedure to clone differentially expressed genes in two different plant organs. Poly (A+) RNAs from R4 flower and from leaf were used to generate tester and driver cDNAs, respectively, for the flower library and vice versa for the leaf library. Clearly distinctive patterns of differential transcript abundance could be observed when these two cDNA libraries were compared. Sequence comparison among the two generated cDNA showed that as much as 92% (209 out of 227 sequences) and 62.5% (80 out of 128 sequences) of the analyzed sequences were unique to R4 flower and leaf libraries, respectively (Table 1). These results indicate the high efficiency of this technique to isolate organ specific transcripts compared to other reports on organ derived cDNA libraries [20]. Endo et al. [20] reported that 64.8% of ESTs sequences isolated from Lotus japonicum flower bud had not been found in EST sequences of the whole plant. Other reports indicated that only 12% of ESTs of an equalized cDNA library constructed from different developmental stages of inflorescence in Arabidopsis thaliana were unique to inflorescence tissue [21]. This high percent of organ-specific sequences for the flower and leaf libraries encouraged us to construct additional cDNA libraries. Stem and root cDNA libraries were subtracted with leaf cDNA in order to avoid high redundancy of photosynthesis related sequences. The R1 flower bud cDNA library was subtracted with R4 flower cDNA with the aim to identify specific gene induction during early stages of development.
Table 1.
Number of isolated, analyzed, differential and non redundant sequences by organ-specific cDNA library.
| Total ESTs | R1 flower (1) | R4 Flower (2) | Leaf (3) | Stem (4) | Root (5) | Total |
| Isolated | 504 | 384 | 268 | 400 | 115 | 1671 |
| Sequenced | 261 | 269 | 159 | 312 | 72 | 1073 |
| Analyzed | 245 | 227 | 128 | 282 | 55 | 919 |
| Differential sequences between (2) and (3) a,b | 209 | 80 | ||||
| Non-redundant | 32 | 140 | 63 | 79 | 4 | 318 |
| Non-redundant percentage b | 13 | 62 | 49 | 28 | 7 | 36 |
| Differential sequences c | 31 | 131 | 42 | 81 | 4 | 289 |
| Average ORF c | 269 | 220 | 257 | 265 | 225 | 247 |
| Average insert size (bp)b | 495 | 365 | 443 | 463 | 370 | 441 |
a Sequences detected exclusively either in the flower or leaf cDNA libraries. b Out of analyzed sequences. c Sequences detected exclusively in the indicated cDNA library out of non-redundant sequences.
Sequence analysis
Table 1 summarizes the total number of isolated, sequenced and analyzed clones, differential and non-redundant sequences and the average insert size and the average ORF per cDNA library. A total of 1073 randomly selected non-directionally clones from the different cDNA libraries were sequenced from which, after removing low quality and contaminant ribosomal RNA sequences, 919 readable sequences were generated, edited and annotated as described in experimental procedures. 5' and 3' sequences were equally represented in the generated EST database. The analysis of sequence redundancy was performed by sequence comparison using local BLASTN through a clustering system running under an alpha version of Biopipeline® and by using the Cap3 contig assembly program [22]. The Biopipeline® clustering revealed a total of 318 non-redundant sequences, meanwhile Cap3 running with an overlap cut-off identity of 95% and a minimum overlap of 25 bases detected a total of 29 contigs composed of two to four sequences and 249 singletons. The observed discrepancy between the unigene set outcomes from both methods is based on the different algorithms used by each program. Manual check of the outcome results confirmed that comparison using Biopipeline® was more efficient in detecting redundancy without losing sensitivity in the detection of gene variants. Thus, further sequence comparisons were performed using the 318 non-redundant sequences as unigen set. Sequence redundancy varied among the different cDNA libraries (Figure 1). The least redundant library was the R4 flower library with a total of 140 unique sequences from 227 analyzed ESTs (62%). In contrast, the most redundant cDNA libraries were: the R1 flower bud (13% of unique sequences) and the root cDNA library (7% of unique sequences). The leaf and stem cDNA libraries exhibited intermediate redundancy levels, with 49 and 28 % of non redundant sequences, respectively. The high level of redundancy in the early flower bud library compared to the late flower bud is likely to be related to differences in this specific subtraction protocol. While the R4 flower library was subtracted with a non-related driver cDNA (leaf cDNA), allowing the detection of transcripts not represented (or represented at a lower level) in the leaf tissue, the R1 flower bud cDNA was arrested with an mRNA population from the same organ but at a different developmental stage. Transcripts from the same organ/tissue share a high number of identical mRNAs and, consequently, a relatively reduced pool of differentially expressed transcripts remains unsubtracted at a specific developmental stage. In the case of the root library, the analysis of redundancy should be treated with caution due to the small number of cDNA molecules that remained unsubtracted after the hybridization step. Thus, studies on predicted functionality were not conducted for this latter cDNA library. The leaf and stem cDNA libraries exhibited higher levels of redundancy compared to the R4 flower cDNA library. The higher redundancies in these two libraries are due to a high representation of photosynthesis related sequences.
Figure 1.
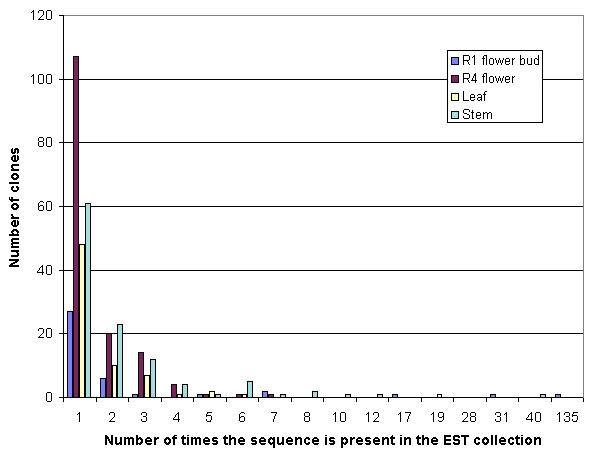
Frequency of redundant clones among ESTs from different organ-specific cDNA libraries.
Analysis of organ-specificity among non-redundant sequences confirmed that a high proportion of the non-redundant sequences in each library corresponded to sequences only detected in that tissue. In the R4 flower and stem cDNA libraries, 93.5% and 98.7% of the analyzed sequences were unique to those libraries, respectively (Table 1). A global analysis including all constructed libraries revealed that 87.8% of the generated non-redundant ESTs were indeed differentially expressed sequences.
EST analysis based on predicted gene function
Sunflower ESTs were grouped into different functional categories according to their predicted gene products based on sequence comparison with the current SWISS-PROT/ TrEMBL (SWALL) data bases. Annotation was performed based on Gene Ontology (GO) [24] terms and functional categories were defined accordingly (Figure 2). This annotation allows the classification of generated ESTs by function [23] with the aim to create universal vocabulary for consensus annotation [24]. A complete list of non-redundant sequences generated here, including BLASTX top hit sequence in SWALL, GO term definition and GO identification number for each sequence is provided on Additional file 1.
Figure 2.
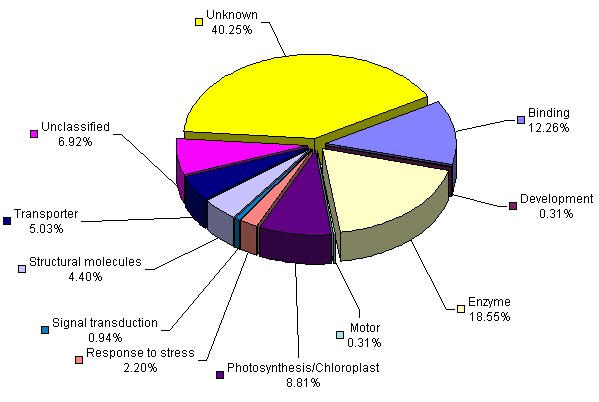
Expression analysis of ESTs from organ-specific cDNA libraries. cDNA clones with significant similarity to protein sequences in SWALL were classified according to Gene Ontology annotation. Sequences with no hits to known protein sequences from BLASTX comparison were classified as unknown. ESTs with significant similarity according to BLASTX comparison but with no GO term definition associated to them were referred as unclassified. Functional analysis includes all non-redundant generated ESTs.
A total of 190 sequences (60 %), out of 318 non-redundant ESTs, showed significant similarity to known gene sequences in the database with a stringency level (E value) of 10-3 and a score value higher than 80. No significant differences in average insert length in both were detected between the sequences that match previous entries on GenBank and those that did not show similarities. These results indicate that the lengths of the sequences reported in this study are good enough to retrieve significant hits in GenBank database. Out of the remaining 128 sequences (40 %) that exhibited no significant similarity to known genes, 90 sequences (70%) exhibited significant homology to ESTs with unknown function on public databases while the remaining sequences representing 32 % are new reported sequences. The Compositae is represented in the GenBank by 113,149 entries of which 44,961 correspond to recently deposit sunflower ESTs and the rest to lettuce (Lactuca sativa; Composite Genome Initiative, CGI, http://cgpdb.ucdavis.edu/database/cgpdb.php. Out of these 44,961 sunflower ESTs, 15,248 are unique sequences, and only circa 2,061 are functionally annotated sequences (TIGR Gene Indices, http://www.tigr.org/tdb/tgi/hagi. In spite of this extensive amount of available information, sequence comparison of the 318 non-redundant sequences generated in this study against 37,208 unique H. annuus and L. sativa sequences (HaGI and LsGI http://www.tigr.org/tdb/tgi/lsgi, TIGR) showed that 197 (59.9%) did not exhibit significant similarity to previously reported sunflower ESTs whilst 228 (69.3%) did not match L. sativa ESTs. The important level of homology found with other plant ESTs that do not belong to the Compositae family indicate that this fact was not due to highly variable or non-coding sequences present at the 3' end of the mRNA. Since the ESTs in this study are derived from polyA RNA and thus enriched in 3' end sequences of the mRNAs, while the ESTs recently deposited at the CGI are enriched in the 5' end of the mRNA, a comparative study of outcome BLASTX was performed in order to determine if the 197 newly detected sunflower genes were indeed represented at GenBank by previously deposited sunflower ESTs from different gene regions. This analysis revealed that most of these sequences share annotation but do not share identities at a nucleotide level, thus some of them are likely to be variants of gene families.
The "unclassified" class correspond to sequences that showed significant similarity to SWALL sequences using BLATX search but they do not have an associated GO term. Most of these sequences correspond to hypothetical proteins with unknown function. The relative abundance of EST categories varied according to the analyzed library (Figure 3). ESTs showing no significant similarity (unknown) represented 56% of the analyzed sequences in the R4 flower cDNA library, while this category was considerably lower in the other libraries, ranging from 16 to 47%. Previous studies reported similar values of predicted novel genes isolated from different normalized cDNA libraries. Asamizu et al. [15] reported that 45% of non-redundant ESTs generated from different plant tissues including aboveground organs, flower buds, roots and liquid-culture seedlings were predicted to be novel genes. In a drought-stressed normalized cDNA library from rice seedlings, up to 28.2% of the non-redundant sequences were novel [17].
Figure 3.
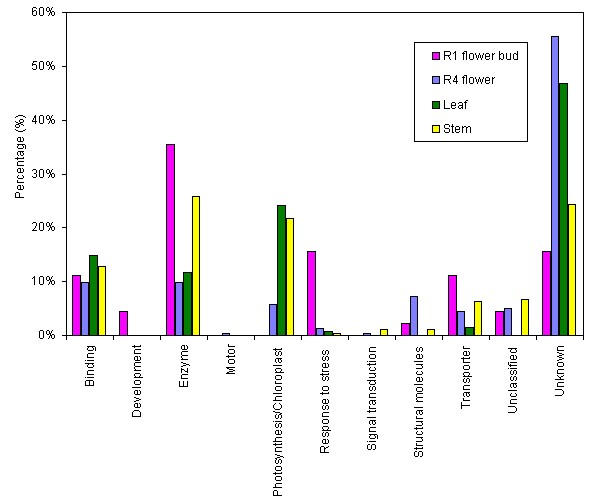
Comparison of ESTs classified by predicted function among four organ-specific cDNA libraries. Functional classification of all generated ESTs was done as described in Figure 2. Percentage of ESTs included in each functional class is compared among four differential cDNA libraries.
"Structural proteins" and "motor" as well as sequences related to cell growth and metabolisms, here included in the "enzyme" class, showed a low level of representation compared to the corresponding values obtained by non-normalized cDNA libraries [25,26]. A similar under representation of ESTs from the cell metabolisms category was reported for other normalized cDNA libraries [16]. This result shows that the normalization step that took place in the construction of the cDNA libraries was efficient in diminishing the level of highly abundant transcripts equally represented in the different analyzed tissues.
As expected, ESTs related to the "photosynthesis/chloroplast" class were highly abundant in the leaf (24%) and stem (22%) libraries while these sequences were absent in the R1 flower bud and very low represented (6%) in the R4 flower cDNA library. Conversely, the leaf and stem libraries showed a low representation of ESTs homologous to stress related sequences, which barely reached 1%. The proportion of "response to stress" sequences showed a higher representation in the R1 flower bud library (16%). Besides the sequences classified as "response to stress" class according to GO terms, there are some other ESTs included in other categories such as "enzyme", "transporter" and "binding" that have been associated to biotic and/or abiotic stress in previous studies [27-32]. Table 2 includes a complete list of 33 non-redundant differential ESTs related to defence and stress response according to GO terms and literature references. Interestingly, agronomical important sequences related to response and/or defence to pathogens such as glucanases (BU671807), germin-like proteins (BU671889) and polygalacturonase inhibitor proteins (BU671906) are new reported ESTs sequences for sunflower as they are not represented in the current EST database. Most of these defence related transcripts were differentially detected in the R1 flower bud library without exposure to any external stimuli, thus reinforcing the importance of designing highly specific organ/developing stage cDNA libraries for detection of low abundant transcripts. This observation confirmed previous reports of higher level of defence related transcripts in developing flowers [33-36]. The "enzyme" class is highly represented in the stem (30%) and R1 flower bud (36%) libraries compared to the leaf and R4 flower library. This class includes a significant number of defence related enzymes differentially detected in the R1 flower bud and stem cDNA libraries. Within this group, those ESTs with significant similarity to pathogen defence-related genes like those coding for germin-like proteins, lipid transfer proteins, polygalacturonase inhibitor factors, protease inhibitors, as well as those genes related to abiotic stress responses like fructosyl transferase, salt-stress induced tonoplast, aquaporin protein, dehydrin protein were mostly detected as unique or low copy number sequences. On the other hand, the more abundant stress related protein genes like glucanases, catalases, peroxidases, jasmonate-induced proteins, thaumatin-like proteins, heat shock proteins were detected more frequently in most of the constructed cDNA libraries. These results are consistent with a previous report on the identification of defence-related genes by suppression subtractive hybridization in rice [27]. In that study the authors compared this strategy with a differential screening performed on a non-differential cDNA library. They found that the suppressed subtracted hybridization allowed the detection of medium-low abundant genes such as protein kinases and transcription factors whilst the differential screening technique detected mostly abundant transcripts such as PR genes.
Table 2.
Differential ESTs related to response to biotic and/or abiotic stress
| ANa | GO IDb | GO functional definitionb | ANc | BLASTX hitd | E value |
| BU671794 | GO:0000238 | Phosphoethanolamine N methytransferase | Q944HO | Putative phosphoethanolamine N methytrans | 2.00E-26 |
| BU671801 | GO:0004332 | Fructose biphosphate aldolase | Q9SXX5 | Plastidic aldolase | 2.00E-91 |
| BU671803 | GO:0042027 | Cyclophilin-type peptidy-prolyl cis-trans isomerase | Q9M530 | Cyclophilin (EC 5.2.1.8) (Peptidyl-prolyl cis-trans isomerase) (PPIase) | 3.00E-18 |
| BU671805 | GO:0008246 | Electron transfer flavoprotein | Q39640 | Glycolate oxidase | 2E-94 |
| BU671807 | GO:0004553 | Hydrolase | Q9M473 | Putative-beta 1,3-glucanase | 5.00E-38 |
| BU671832 | GO:0003773 | Heat shock protein | Q02028 | Stromal 70 kDa heat shock-related protein | 3.00E-27 |
| BU671840 | GO:0004096 | Catalase | P45739 | Catalase | 1.00E-95 |
| BU671841 | GO:0004332 | Fructose biphodphata aldolase | P93565 | homologous to plastidic aldolases | 3.00E-21 |
| BU671845 | GO:0000287 | Magnesium binding | Q93WE2 | Magnesium chelatase subunit | 4.00E-36 |
| BU671864 | GO:0004332 | Fructose-bisphosphate aldolase | Q9SXX5 | Plastidic aldolase | 1.00E-54 |
| BU671866 | GO:0009058 | Biosynthesis | Q39049 | Magnesium chelatase subunit. | 4E-16 |
| BU671867 | GO:0030145 | Magnesium binding | Q943W1 | Oxygen-evolving enhancer protein 1 | 2.00E-92 |
| BU671886 | GO:0004299 | Proteasome endopeptidase | O23708 | Proteasome subunit alpha type 2 | 2.00E-17 |
| BU671887 | GO:0005215 | Transporter | Q9ZR68 | Aquaporin 1 | 2.00E-84 |
| BU671888 | GO:0004553 | Hydrolase, hydrolyzing O-glycosyl compounds | Q9M453 | Putative beta-1,3-glucanase. | 3.00E-58 |
| BU671889 | GO:0030145 | Manganese binding | O48999 | Germin-like protein 3 | 2.00E-35 |
| BU671904 | GO:0004564 | Beta-fructofuranosidase | O81985 | 1,2-beta-fructan 1F-fructosyltransferase | 2.00E-51 |
| BU671906 | GO:0005489 | Electron transport | Q94L58 | Polygalacturonase inhibitor protein | 3.00E-39 |
| BU671909 | GO:0016068 | Immediate hypersensitivity response | Q93YX9 | Lipid transfer protein | 1.00E-37 |
| BU671910 | GO:0016068 | Immediate hypersensitivity response | Q9M6B8 | Lipid transfer protein | 2.00E-15 |
| BU671924 | GO:0004197 | Cysteine-type endopeptidase | Q8VWS1 | Putative cysteine proteinase | 4.00E-42 |
| BU671928 | GO:0003755 | Peptidyl-prolyl cis-trans isomerase | Q8L5T1 | Peptidylprolyl isomerase (Cyclophilin) | 3.00E-18 |
| BU671929 | GO:0004222 | Metalloendopeptidase | O22941 | Putative zinc protease | 1.00E-61 |
| BU671944 | GO:0005509 | Calcium ion binding | O49301 | T26J12.7 protein. | 7.00E-13 |
| BU671955 | GO:0009607 | Response to biotic stimulus | P13046 | Pathogenesis-related protein R major form | 3.00E-34 |
| BU671960 | GO:0004766 | Spermidine synthase | 48658 | Spermidine synthase 1 (EC 2.5.1.16) | 1.00E-21 |
| BU671972 | GO:0008168 | Methyltransferase | Q9LW67 | Ankyrin-like protein | 7.00E-33 |
| BU671989 | GO:0004601 | Peroxidase | O64970 | Cationic peroxidase 2 | 1.00E-40 |
| BU671977 | GO:0005489 | Electron transporter | O04002 | Chloroplast drought-induced stress protein of 32 kDa | 6.00E-41 |
| BU672016 | GO:0004867 | Serine protease inhibitor | Q8LNY0 | Protease inhibitor 2 | 2.00E-13 |
| BU672055 | GO:0004869 | Cysteine protease inhibitor | Q9MB08 | Multicystatin. | 2.00E-12 |
| BU672102 | GO:0005489 | Electron transporter | O04002 | Chloroplast drought-induced stress protein of 32 kDa | 2,00E-56 |
| BU672106 | GO:0004124 | Cysteine synthase | Q8Y0X6 | Probable cysteine synthase B (CSASE B) protein (EC 4.2.99.8). | 9.00E-21 |
a GenBank accession number of ESTs from the present study. b Identification number and functional definition according to GO annotation c The GenBank accession number of most similar sequence to the sunflower EST. d Similarity search was conducted using BLASTX program. EST putative function was assigned according to the highest similar sequence on GenBank.
In the present study, the "binding" class is equally represented in all the analyzed libraries, although this class includes sequence with putative involment in diverse processes such as transcription and translation factors, ATPase and cation binding proteins. Within this group low abundant transcripts like those coding for transcription factors and homeotic factors were specially detected in the R4 flower cDNA library. ESTs with homology to genes coding for signalling enzymes as MAP protein kinase and serine/threonin phosphatase were only detected in the stem cDNA library (Figure 2b). The functional category of "transporter" is represented by sequences with similarity to carrier protein genes as ATP-binding cassette (ABC) and electron transporters that were mainly detected in the R4 flower library. ESTs with similarity to homeobox genes here included in "development" were only detected in the early flower bud cDNA library. The homeobox sequences isolated in this work did not show similarity to previously reported sunflower homeobox genes [37-39]. Preliminary results showed that some of the agronomical interesting sequences, including those putatively related to response to biotic and abiotic stress, revealed polymorphisms when used as genetic markers in the analysis of genetically segregant populations derived from the crossing of parental lines with contrasting biotic and abiotic stress resistance behaviour (not shown).
The application of suppressed subtracted hybridization technology for the detection of differential ESTs allowed the identification of novel sequences in sunflower from a relative small number of analyzed sequences in spite of the large number of ESTs that have been recently release. Particularly interesting was the detection of a significant number of ESTs related to response to both abiotic and biotic stresses, as well as low abundance transcripts with high similarity to homeobox genes, transcription factors and signalling component genes that were not represented in the sunflower EST division at the GenBank. The R4 flower cDNA library was the library that provided the largest number of novel genes in sunflower, whilst the R1 flower bud library was particularly enriched in defence related genes. The detection of these novel sequences could contribute to the development of EST-based markers for important agronomic traits such as resistance to pathogens and tolerance to different environmental stresses such as extreme temperatures and drought, which are aspects crucial for sunflower crop improvement in many of the cultivated areas in the world.
Conclusions
The application of suppressed subtracted hybridization technology enabled the isolation of a significant number of organ-specific sunflower ESTs and allowed the identification of novel sequences from a relative small number of analyzed sequences. Redundancy level and percent of novel sequence detection varied among differential libraries reinforcing the importance of a careful selection of both target and driver transcript population according to project aims. In this work the R4 flower cDNA library provided the largest number of novel genes in sunflower, whilst the R1 flower bud library was particularly enriched in defence related genes. Some of the novel sequences reputed here share annotation but do not share identities at a nucleotide level with sunflower ESTs on public databases and thus, they are likely to be variants of gene families. We report for the first time in sunflower a significant number of novel sequences related to responses to abiotic and biotic stresses as well as low abundant transcripts with high similarity to homeobox genes, transcription factors and signalling components.
Methods
Plant material
Sunflower seedlings (public inbred line RHA89) were grown under controlled green house conditions (20–24°C and 16 h light/ 8 h dark cycle), and then transplanted to the field during the crop season to develop mature plants. Leaves, stems, and capitulum buds from 1 to 2 cm of diameter (early flower buds) and 3 to 4 cm of diameter (late flower buds) were harvested from two months old plants and immediately frozen in liquid nitrogen. Roots were harvested from 15 day old plants grown in sand under green house conditions. All samples were stored frozen at -80°C until processed.
Total and poly (A+) RNA isolation
Total RNA was extracted from approximately 2 g of tissue using TRIzol® reagent following manufacturer recommendations (InVitrogen, USA). Poly (A+) RNA was isolated from 200–500 μg of total RNA using NucleoTrap® System (Promega, USA). RNA integrity was analyzed by checking its electrophoretic mobility on 1.5 % agarose gels in ME buffer (400 mM MOPS, 100 mM Na acetate, 10 mM EDTA pH 8.0, in diethyl-pyrocarbonate treated water). mRNA quantification was performed by UV absorbance at 260 nm (GenQuant pro, Amersham-Pharmacia, UK).
Construction of cDNA libraries
Differential cDNA libraries were constructed from different tissues including leaves, stems, roots and flower buds and from different developmental stages (e.g. R1 and R4 according to the description of sunflower growth stages by Schneiter and Miller [40]) using PCR-Select cDNA Subtraction Kit® (Clontech, USA). Firstly, cDNA was synthesized from 0.5–2.0 μg of poly (A+) RNA from the two types of tissues being compared. The tester (target tissue) and driver (reference tissue) cDNAs were then digested with RsaI, that yields blunt end fragments of approximately 400 bp length in average. We defined different driver populations for the different specific libraries, depending on specific interests. Leaf cDNA collection was arrested against a late flower bud cDNA population. Stem early flower bud and root cDNA collections were arrested against a leaf cDNA population.
Both, tester and driver, cDNA populations were processed following manufacturer instructions, with some modifications. The tester cDNA was subdivided into two halves, and each half was ligated to different cDNA adaptors. Two hybridization rounds were performed with an excess of driver cDNA. Hybridization conditions were performed as recommended by the manufacturer. The resulted products were subjected to two cycles of PCR with adaptor targeted primers to amplify the desired differentially expressed sequences. Amplifications were performed on a PT-100 DNA thermocycler (MJ Research, USA). First PCR master mix contained 10x PCR reaction buffer, 0.2 mM dNTPs, 0.4 μM PCR primer 1 and Advantage cDNA polymerase (Clontech, USA). PCR was performed under the following conditions: 94°C (30 sec) denaturing step followed by 27 cycles each consisting of a denaturation step at 94°C (30 sec), an annealing step at 66°C (30 sec) and an elongation step at 72°C (10 min). The second PCR master mix contained 10x PCR reaction buffer, 0.2 mM dNTPs, 0.4 μM nested PCR primer 1, 0.4 μM nested PCR primer 2 and Advantage cDNA polymerase, PCR was run through 12 cycles each consisting of a denaturing step at 94°C (30 sec), an annealing step at 66° (30 sec) and an elongation step at 72°C (1.5 min).
cDNA molecules were size-selected and fractions larger than 250 bp were cloned non-directionally into the pGem-T-Easy Vector® (Promega, USA). Ligation was performed at 4°C for 48 h and the resulting ligation product was used to transform Escherichia coli (XL1-blue strain) by electroporation (Pulse Controller, BioRad, USA).
Template preparation
cDNA libraries were plated onto solid Luria Bertani (LB) medium containing ampicillin. Recombinant clones were selected by β-galactosidase activity in media containing X-GAL and IPTG. White colonies were randomly picked to 364 well plates containing Freezing Medium (36 mM K2HPO4, 13.2 mM KH2PO4, 1.7 mM sodium citrate, 0.4 mM MgSO4, 6.8 mM (NH4)2 SO4, in LB medium and 4.4 % glycerol), grown overnight and later stored at -70°C. Recombinant plasmids were isolated using REAL 96 prep kit (Qiagen, Germany) as recommended by the supplier. Insert sizes of individual recombinant clones were examined by electrophoresis of EcoRI digestion products on 1.2 % agarose gels in TAE buffer [41].
Sequencing and sequence analysis
Recombinant plasmids were single-pass sequenced from the T7 universal primer site at sequencing facilities (Laboratorio de Alta Complejidad, IMyZA – CICVyA – INTA Castelar, Argentina; Centro de Biologia Molecular e Engenharia Genética – CBMEG Universidade Estadual de Campinas, Sao Paulo, Brazil and/or Department of Plant Pathology, Kansas University). Reverse sequencing was performed from the SP6 primer site, only when the forward sequences failed or were uninformative due to a short length. The generated EST sequences were stored in a relational database in which both 5' and 3' sequences were equally represented. Vector and uninformative sequences were automatically removed using computer program routines. The processed sequence were output to FASTA formatted files and a pile up (Biopipeline®) step routine written by in-house staff (S.L., Bioaxioma S.A.) was applied to detect remaining vector artifacts by comparing against a full vector sequence database. Redundancy was also analyzed by means of a clustering systems running under an alpha version of Biopipeline®. This system displays a graphic matrix which aligns the top scoring hits sequences in a score matrix. Sequences that exhibited more than 80% identity over total large sequence were considered identical or closely related and were assigned to a specific group. Sequence alignment of those highly similar sequences was confirmed by sequence alignment programs (ClustalW [42]). Contig analysis of the grouped ESTs was done using the contig assembly program Cap3 [21].
Sequence similarities searches against different protein databases were conducted using Advanced BLAST program [43]. Default BLAST parameter values were used except for the E value (E = 10-3). The top scoring hits were automatically annotated according to the putative function returned by BLASTX. Gene Ontology (GO) annotation was performed using the GOblet software package [44] and a GO term associated to each sequence showing a significant similarity hit by BLASTX against SWALL search was defined. Sequences comparison against plant division ESTs, HaGI and LsGI were performed locally using BLASTN. These datasets were downloaded from public databases and the "Standalone WWW BLAST Server" from the National Center for Biotechnology Information (NCBI; ftp://ftp.ncbi.nih.gov/blast).
Authors' contribution
PF carried out subtracted cDNA libraries, DNA sequencing, and participated in data analysis, EST annotation and manuscript preparation. SL developed the Biopipeline ® software for sequence comparison analysis, NP directed bioinformatic EST analysis, participated in local and global sequence comparison, HEH conceived the study and coordinated its development, and RH designed the construction of differential EST database, coordinated its analysis and drafted the manuscript. All authors read and approved the final manuscript.
Supplementary Material
A complete list of non-redundant EST, including GenBank accession number (AN) of the generated sequence and its corresponding GO identification number (GO ID), GO function definition inferred from sequence similarity, BLASTX top hit sequence and AN outcome from searches against SWALL is provided.
Acknowledgments
Acknowledgements
We are grateful to Valeria Peralta for the greenhouse work, to Dr. Roberto Perazzo and Lic. Gustavo Guida for his assistance on Biopipeline® step routine development and to Lic. Alejandro D'Angelo for technical support on database programming routines. We thank Dr. Mariana del Vas for critical reading of the manuscript. This research was supported by the ANPCyT/FONCYT; BID 1201 AC/AR PID 024 and by ASAGIR, Argentina. Ing. Agr. P. Fernandez holds a doctoral fellowship from the University of Buenos Aires, Dr. R. Heinz and Dr. N. Paniego are career members of the Consejo Nacional de Investigaciones Científicas y Técnicas (CONICET, Argentina) and Dr. H.E. Hopp is a career member of the Comisión de Investigaciones Científicas de la Provincia de Buenos Aires (CIC) and Professor at the Facultad de Ciencias Exactas y Naturales, University of Buenos Aires (UBA).
Contributor Information
Paula Fernández, Email: pfernandez@cicv.inta.gov.ar.
Norma Paniego, Email: npaniego@castelar.inta.gov.ar.
Sergio Lew, Email: sergio.lew@bioaxioma.com.
H Esteban Hopp, Email: ehopp@cicv.inta.gov.ar.
Ruth A Heinz, Email: rheinz@cicv.inta.gov.ar.
References
- Berry ST, Allen RJ, Barnes SR, Caligari PDS. Molecular-marker analysis of Helianthus annuus L. 1. Restriction fragment length polymorphism between inbred lines of cultivated sunflower. Theor Appl Genet. 1994;89:435–441. doi: 10.1007/BF00225378. [DOI] [PubMed] [Google Scholar]
- Berry ST, Leon AJ, Hanfrey CC, Challis P, Burkholz A, Barnes SJ, Rufener GK, Lee M, Caligari PDS. Molecular-marker analysis of Helianthus annus L. 2. Construction of an RFLP linkage map for cultivated sunflower. Theor Appl Genet. 1995;91:195–199. doi: 10.1007/BF00220877. [DOI] [PubMed] [Google Scholar]
- Gentzbittel L, Zhang YX, Vear F, Griveau B, Nicolas P. RFLP studies of genetic relationship among inbred lines of the cultivated sunflower. Helianthus annuus L.: evidence for distinct restorer and maintainer germplasm pools. Theor Appl Genet. 1994;89:419–425. doi: 10.1007/BF00225376. [DOI] [PubMed] [Google Scholar]
- Gentzbittel L, Vear F, Zhang Y-X, Berville A, Nicolas P. Development of a consensus linkage RFLP map for cultivated sunflower. Theor Appl Genet. 1995;90:1079–1086. doi: 10.1007/BF00222925. [DOI] [PubMed] [Google Scholar]
- Gentzbittel L, Mestries E, Mouzeyrat F, Badaoqui S, Vear F, Tourvieille de Labrouhe D, Nicolas P. A composite map of expressed sequences and phenotypic traits of the sunflower (Helliantus annuus L.) genome . Theor Appl Genet. 1999;99:218–234. doi: 10.1007/s001220051228. [DOI] [Google Scholar]
- Gedil MA, Wye C, Berry S, Segers B, Peleman J, Jones R, Leon A, Slabaugh MB, Knapp SJ. An integrated restriction fragment length polymorphism-amplified fragment length polymorphism linkage map for cultivated sunflower. Genome. 2001;44:213–221. doi: 10.1139/gen-44-2-213. [DOI] [PubMed] [Google Scholar]
- Jan CC, Vick BA, Miller JK, Kahler Al, Butler ETI. Construction of an RFLP linkage map for cultivated sunflower. Theor Appl Genet. 1998;96:15–22. doi: 10.1007/s001220050703. [DOI] [Google Scholar]
- Mokrani L, Gentzbittel l, Azanza F, Fitamant L, Al-Chaarani G, Sarrafi A. Mapping and analysis of quantitative trait loci for grain oil content and agronomic traits using AFLP and SSR in sunflower (Helianthus annuus L.) Theor Appl Genet. 2002;106:149–56. doi: 10.1007/s00122-002-1011-4. [DOI] [PubMed] [Google Scholar]
- Tang S, Yu JK, Slabaugh MB, Shintai DK, Knapp SJ. Simple sequence repeat map of the sunflower genome. Theor Appl Genet. 2002;105:1124–1136. doi: 10.1007/s00122-002-0989-y. [DOI] [PubMed] [Google Scholar]
- Paniego N, Echaide M, Muñoz M, Fernández L, Torales S, Faccio P, Fuxan I, Carrera M, Zandomeni R, Suárez EY, Hopp HE. Microsatellite isolation and characterization in sunflower (Helianthus annuus L.) Genome. 2002;45:34–43. doi: 10.1139/g01-120. [DOI] [PubMed] [Google Scholar]
- Cato SA, Gardener RC, Richardson TE. A rapid PCR Method for genetically mapping ESTs. Theor Appl Genet. 2001;102:296–306. doi: 10.1007/s001220051646. [DOI] [Google Scholar]
- Bonaldo MF, Lennon G, Soares MB. Normalization and subtraction: two approaches to facilitate gene discovery. Genome Res. 1996;6:791–806. doi: 10.1101/gr.6.9.791. [DOI] [PubMed] [Google Scholar]
- Patanjali SR, Parimoo S, Weissman SM. Construction of a uniform-abundance (normalized) cDNA library. Proc Natl Acad Sci USA. 1991;88:1943–1947. doi: 10.1073/pnas.88.5.1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soares MB, Bonaldo MF, Jelene P, Su L, Lawton L, Efstratiadis A. Construction and characterization of a normalized cDNA library. Proc Natl Acad Sci USA. 1991;27:9228–32. doi: 10.1073/pnas.91.20.9228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asamizu E, Nakamura Y, Sato S, Tabata S. A large scale analysis of cDNA in Arabidopsis thaliana : generation of 12028 non-redundant expressed sequence tags from normalized and size-selected cDNA library. DNA Res. 2002;7:175–180. doi: 10.1093/dnares/7.3.175. [DOI] [PubMed] [Google Scholar]
- Ali S, Holloway B, Taylor WC. Normalization of cereal endosperm EST libraries for structural and functional genomic analysis. Plant Mol Biol Rep. 2002;18:123–132. [Google Scholar]
- Reddy AR, Ramakrishna AC, Sekhar AC, Nagablushana I, Ravindra Babu P, Bonaldo MF, Soares MB, Bennetzen JL. Novel genes are enriched in normalized cDNA libraries from drought-stressed seedlings of rice (Oryza sativa L. subsp. Indica cv. Nagina 22) Genome. 2002;45:204–211. doi: 10.1139/g01-114. [DOI] [PubMed] [Google Scholar]
- Diatchenko L, Lau YF, Campbell AP, Chenchick A, Moqaddam F, Huang B, Lukyanov S, Konstantin L., Gurskaya N, Sverdlov E, Siebert PD. Suppression subtractive hybridization: A method for generating differentially regulated or tissue-specific cDNA probes and libraries. Proc Natl Sci USA. 1996;93:6025–6030. doi: 10.1073/pnas.93.12.6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D., Buttler H, Cherry JM, Davis AP, Dolinsky K, Dwight SS, Eppig JT, et al. Gene ontology: tool for the unification of biology. Nature Genet. 2000;25:25–29. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Endo M, Kokubun Y, Higaashitani A, Tabata S, Watanabe M. Analysis of expressed sequence tags of flower buds in Lotus japonicum. DNA Res. 2000;7:213–126. doi: 10.1093/dnares/7.3.213. [DOI] [PubMed] [Google Scholar]
- Takemura M, Fujishige K, Hyodo H, Ohashi Y, Kami C, Nishii A, Ohyama K, Kohchi T. Systematic isolation of genes expressed at low levels in inflorescence apices of Arabidopsis thaliana. DNA Res. 1999;6:275–82. doi: 10.1093/dnares/6.5.275. [DOI] [PubMed] [Google Scholar]
- Huang X, Adams MD, Zhou H, Kerlavage AR. A tool for analyzing and annotating genomic sequences. Genomics. 1997;46:37–45. doi: 10.1006/geno.1997.4984. [DOI] [PubMed] [Google Scholar]
- Ouzounis CA, Coulson RMR, Enright AJ, Kunin V, Pereira-Leal JB. Classification schemes for protein structure and function. Nature Reviews. 2003;4:509–519. doi: 10.1038/nrg1113. [DOI] [PubMed] [Google Scholar]
- Camon E, Barrell D, Brooksbank C, Magrane M, Apweiler R. The Gene Ontology Annotation (GOA) project-application of GO in Swiss-Prot, TrEMBL and InterPro. Comp and Funct Genom. 2003;4:71–74. doi: 10.1002/cfg.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Arabidopsis Genome Initiative Analysis of the genome sequences of the flowering plant Arabidopsis thaliana. Nature. 2002;408:796–815. doi: 10.1038/35048692. [DOI] [PubMed] [Google Scholar]
- Carson D, Botha FC. Preliminary analysis of expressed sequence tags for sugarcane. Crop Sci. 2000;40:1769–1779. [Google Scholar]
- Xiong L, Lee M-W, Yang Y. Identification of defense-related rice genes by suppression subtracted hybridization and differential screening. Mol Plant-Microbe Interact. 2001;14:685–692. doi: 10.1094/MPMI.2001.14.5.685. [DOI] [PubMed] [Google Scholar]
- Urdangarin MC, Norero NS, Broekaert WF, de la Canal L. A defensin gene expressed in sunflower inflorescence. Plant Physiol Biochem. 2000;38:253–258. doi: 10.1016/S0981-9428(00)00737-3. [DOI] [Google Scholar]
- Broekaert WF, Cammue BPA, De Bolle MFC, Thevissen K, De Samblanx GW, Osborn RW. Antimicrobial peptides from plants. Crit Rev Plant Sci. 1997;16:267–323. [Google Scholar]
- Bernier F, Berna A. Germins and germin-like proteins: Plant do-all proteins. But what do they do exactly? Plant Physiol Biochem. 2001;39:545–554. doi: 10.1016/S0981-9428(01)01285-2. [DOI] [Google Scholar]
- Mauch F, Mauch-Mani B, Boller T. Antifungal hydrolases in pea tissue II. Inhibition of fungal growth by combinations of -1,3-glucanase. Plant Physiol. 1988;88:936–942. doi: 10.1104/pp.88.3.936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vigers AJ, Roberts WK, Selitrennikoff CP. A new family of plant antifungal proteins. Mol Plant-Microbe Interact. 1991;4:315–323. doi: 10.1094/mpmi-4-315. [DOI] [PubMed] [Google Scholar]
- Lotan T, Ori N, Fluhr R. Pathogenesis-related proteins are developmentally regulated in tobacco flowers. Plant Cell. 1989;1:881–887. doi: 10.1105/tpc.1.9.881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale AD, Wahleithner JA., Lund M, Bonnett HT, Kelly A, Meeks-Wagner DR, Peacock WJ, Dennis ES. Chitinase, -1,3-glucanase, osmotin andextensin are expressed in tobacco explants during flower formation. Plant Cell. 1990;2:673–684. doi: 10.1105/tpc.2.7.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Q, Kawata EE, Morse MJ, Wu HM, Cheung AY. A flower-specific cDNA encoding a novel thionin in tobacco. Mol Gen Genet. 1992;234:89–96. doi: 10.1007/BF00272349. [DOI] [PubMed] [Google Scholar]
- Atkinson AH, Heath RL, Simpson RJ, Clarke AE, Anderson MA. Proteinase inhibitors in Nicotiana alata stigmas are derived from a precursor protein which is processed into five homologous inhibitors. Plant Cell. 1993;5:203–213. doi: 10.1105/tpc.5.2.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan RL, Gonzales DH. A cDNA encoding an HD-Zip protein from sunflower. Plant Physiol. 1994;106:1687–1688. doi: 10.1104/pp.106.4.1687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gago GM, Almoguera C, Jordano J, Gonzalez DH, Chan RL. Hahb-4, a homeobox-leucine zipper gene potentially involved in abscisic acid-dependent responses to water stress in sunflower. Plant Cell Environ. 2002;25:633–640. doi: 10.1046/j.1365-3040.2002.00853.x. [DOI] [Google Scholar]
- Valle EM, Gonzales DH, Gago G, Chan RL. Isolation and expression pattern of hahrI, a homeobox-containing cDNA from Helianthus annuus. Gene. 1997;196:61–68. doi: 10.1016/S0378-1119(97)00193-5. [DOI] [PubMed] [Google Scholar]
- Schneiter AA, Miller JF. Description of Sunflower Growth Stages. Crop Science. 1981;11:635–638. [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2. Cold Spring Harbor: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–80. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Bio. 1990;215:208–218. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Henning S, Groth D, Lehrach H. Automated gene Ontology annotation for anonymous sequence data. Nucleic Acids Res. 2003;31:3712–3715. doi: 10.1093/nar/gkg582. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
A complete list of non-redundant EST, including GenBank accession number (AN) of the generated sequence and its corresponding GO identification number (GO ID), GO function definition inferred from sequence similarity, BLASTX top hit sequence and AN outcome from searches against SWALL is provided.