

Abstract
A new efficient synthesis of 6-methyl-6-vinylfulvene was developed, starting from the 6-(2-hydroxyethyl)-6-methylfulvene. With larger quantities of the title compound in hand, its photooxygenation with singlet oxygen was studied. Cyclization of the cyclopropanone intermediate to both vinyl moieties in the unsaturated system was observed, whereas the saturated endoperoxide gave mostly the cyclopentenone derivative. m-CPBA attacks exclusively the endocyclic double bonds and gives the 3-cyclopentenones via the unstable epoxides.
Although the synthesis of fulvenes has been reported over 100 years ago by Thiele,1 fulvene synthesis have been the subject of a large number of studies since these cross-conjugated compounds have proven not only of theoretical interest, but also served as important substrates in organometallic chemistry, synthetic organic chemistry and organic peroxide chemistry.2 Moreover, their unique electronic and spectroscopic properties as well as cycloaddition chemistry have provided impetus for developing new syntheses for this class of compounds since there does not seem to exist a universal method for the preparation of fulvenes of all substitution patterns. Thiele’s synthesis of condensing a carbonyl compound with cyclopentadiene in the presence of a base, in most cases sodium ethoxide or methoxide gives good results with simple carbonyl compounds, whereas Little’s variant employing pyrrolidine as base in methanol solution has proven to be the method of choice for simple aliphatic ketones and aromatic aldehydes.3 More recently we reported a survey of fulvene syntheses in terms of application of each method to a particular type of substrate and reported and improved method for the synthesis several of 6-vinylfulvenes as well as those carrying functional groups tethered to the exocyclic double bond by one or several carbons.4 Neuenschwander and his coworkers’ contributions to the syntheses, reactivity and physical as well as spectroscopic properties of fulvenes have been extraordinary;5 they reported for the first time the syntheses of 6-vinylfulvenes by the Thiele (NaOEt base) method at low temperatures, albeit in poor yields: 6-methyl-6-vinylfulvene, for instance was obtained only in 0.5% yield.6 Under Little’s conditions (pyrrolidine base), only the Diels-Alder product between 3-butenone (methylvinylketone, MVK) and cyclopentadiene was isolated.7 In connection with our studies on the thermal rearrangements, cycloadditions and singlet oxygenations, we required 6-methyl-6-vinylfulvene in larger quantities. Herein we report an exceptionally convenient and high yield synthesis of the title compound and also disclose its reactivity toward singlet oxygen (1O2) as well as oxygen atom donors.
The synthesis of 6-methyl-6-vinylfulvene commences with the synthesis of fulvene 2 we had previously reported.3 During the chromatographic purification we also isolated a small amount of a non-polar yellow component that proved to be the bisfulvenylether 3. Its formation could stem from a dehydration on the silicagel column to the vinyl group and acid-catalyzed ether formation with alcochol 2.
Next, alcohol 2 was converted to the corresponding mesylate with methane sulfonyl chloride in CH2Cl2 at 0 °C in the presence of triethylamine,8 to give the mesylate that without further purification was treated with 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) in CH2Cl2 at 0 °C. From this reaction, 6-methyl-6-vinylfulvene (4) was isolated in 97% yield over two steps! (62% from cyclopentadiene).
With compound 4 now available in large quantities in a three-step procedure, we decided to investigate its cycloadditions. Our first objective was to explore its reactivity toward oxidizing agents, in particular singlet oxygen (1O2).9 6,6-susbtituted fulvenes had been reported by Uda et al.11 as well as Ohloff et al.12 to undergo 1,4-addition onto singlet oxygen at low temperatures, the resulting unsatable endoperoxides rearranged to 2(3H)-oxepinones at room temperature via postulated allene oxide-cyclopropanone intermediates. More recently, we have been able to isolate the saturated fulvene endoperoxide by in situ diazene reduction13 and show that allene oxide and cyclopropanones are indeed intermediates in the thermal isomerizations of fulvene endoperoxides, by direct isolation and characterization of a stable allene oxide, as well as intra- and intermolecular trapping/cycloaddition reactions.14 We also reported on base-catalyzed rearrangements of the saturated endoperoxides including those from 6-vinylfulvenes. The question was, whether the extra vinyl group at the 6-position of the fulvene would participate in the thermal isomerization of the unsaturated fulvene endoperoxide.
Upon singlet oxygenation of 4 at -78 °C and gradual warming of the photolysate to room temperature, a product mixture was obtained, the components of each were cleanly separated from one another by repeated chromatography on silica gel (pet.ether/ethyl acetate, first 1:1, then 10:1) and were shown by means of NMR and IR spectroscopy as well as HRMS to be 5, 6 (only (E)- isomer was formed),15 and 7 in order of their elution from the column.
The formation of 5 was not unexpected in light of Uda’s and Ohloff’s results, though it was formed only as a very minor product in this case. The formation of 7 also has precedent: 6,6-dimethylfulvene gives upon photooxygenation at 15-20 °C the 5,5-dimethyl analog of 7.16 However, compound 6 or an analog thereof had never been reported to stem from isomerization of a fulvene endoperoxide. Obviously, products, 6 and 7 arise from intramolecular trapping of the cyclopropanone intermediate 9 by either vinyl group at C2 and C3, respectively. Compound 10 apparently undergoes an acid-catalyzed double bond isomerization to 6 during the chromatography.
Next, we studied the synthesis, thermal rearrangement as well as reagent catalyzed transformations of the saturated endoperoxide 11 that was obtained from 8 by diazene reduction, generated in situ from potassium azodicarboxylate and acetic acid at -78 °C (the actual diazene formation and reduction sets in at ca. -30 °C during the gradual warm up to 0 °C).17 Endoperoxide 11 was refluxed in CH2Cl2 for 1 hour, and the mixture separated by column chromatography on silica gel (pet. Ether/EtOAc 1:1) to give 10 and 11 in yields of 56% and 4%, respectively.
Whereas compound 12 was formed by an intramolecular vinylcyclopropanone-cyclopentenone cyclization,14c the bicyclic acetal 13 stems from an intramolecular 1,3-dipolar cyclaoaddition of the cyclopropanone intermediate onto the formyl group (the exocyclic double bond configuration in 13, which was formed as a single isomer, was tentatively assigned).14a Cyclopentenone 12 was the sole product from the CoTPP18 catalyzed isomerization of 11 (83% yield).
Finally, the reactivity of fulvene 4 toward m-chloroperbenzoic acid (m-CPBA) was tested. It was known that fulvenes undergo epoxidation with nucleophilic oxygen atom donors such as hydroperoxide or t-butylhydroperoxide ions at the exocyclic double bond due to the electrophilic nature of the C6 carbon19 whereas the electrophilic epoxidizing agent dimethyldioxirane (DMD) epoxidizes the endocyclic double bond to give short- lived monoepoxy intermediates that swiftly undergo ring opening and isomerization to 3-cyclopenten-1-ones.20 To the best of our knowledge, the epoxidation of fulvenes with m-CPBA had not previously been reported. Upon reaction of a slight excess of m-CPBA with 4, one obtained a 1.6:1 mixture of the two isomeric 3-cyclopenten-1-ones 14 and 15 in 40% isolated yield. The inseparable mixture was purified by column chromatography on silica gel, and the individual signals in the 1H NMR spectrum unambiguously assigned to each isomer by means of extensive 2D NMR experiments.
These results are analogous to those obtained by Adam et al. on the DMD epoxidation of 6-substituted pentafulvenes, except we have not been able to isolate any bisepoxides, even with excess m-CPBA. Apparently, the unstable epoxide intermediates undergo ring opening under the slightly acidic conditions before they can be epoxidized a second time.
In summary, we have developed an efficient synthesis of 6-methyl-6-vinylfulvene (4) in an overall yield of 62% from cyclopentadiene and studied its oxidation chemistry. The singlet oxygenations as well as the thermal and reagent catalyzed decompositions of the endoperoxides derived thereof proved quite interesting in that the cyclopropanone intermediate was trapped intramolecularly by the vinyl group(s) in the unsaturated and saturated systems, leading to functionalized cyclopentenones 6, 7 and 12, respectively.
Treatment of 4 with m-CPBA proceeded cleanly resulting in the epoxidation of the endocyclic double bond, leading to both possible epoxides that underwent acid-catalyzed isomerization to the corresponding 3-cyclopentenones 14 and 15. We are currently insvestigating the cycloadditions of 4 and its thermal electrocyclization to dihydropentalenes and will report our results from these reactions shortly.
Supplementary Material
Scheme 1.
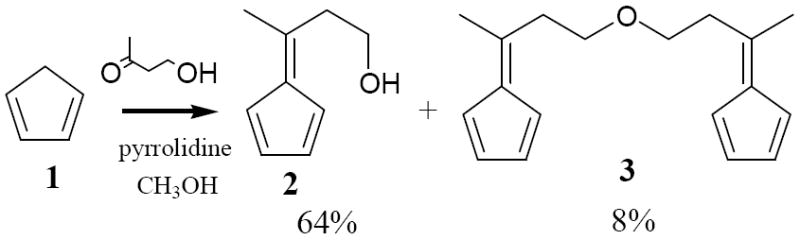
Synthesis of 6-(2-hydroxyethyl)-6-methylfulvene
Scheme 2.
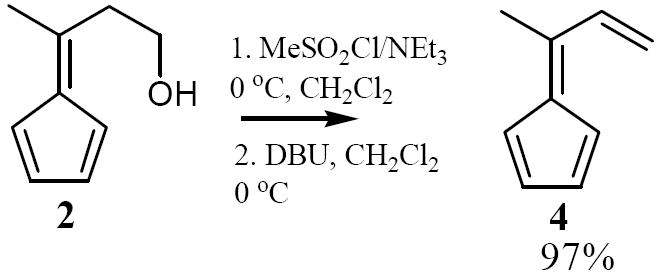
Synthesis of 6-methyl-6-vinylfulvene (4) from 2
Scheme 3.
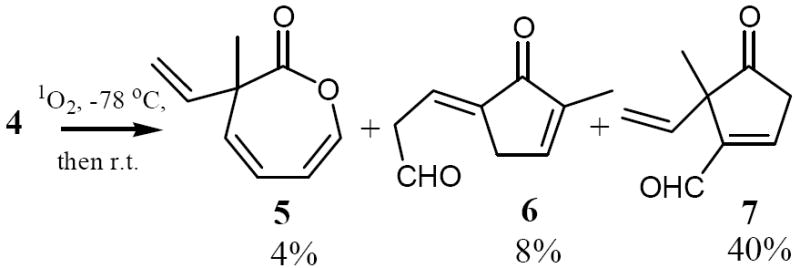
Photooxygenation of 4
Scheme 4.
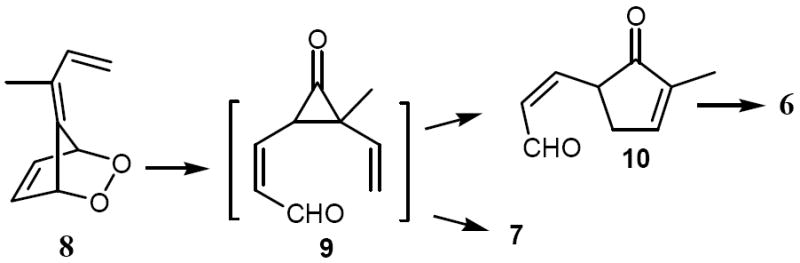
Vinylcyclopropane-cyclopentenone cyclization modes
Scheme 5.
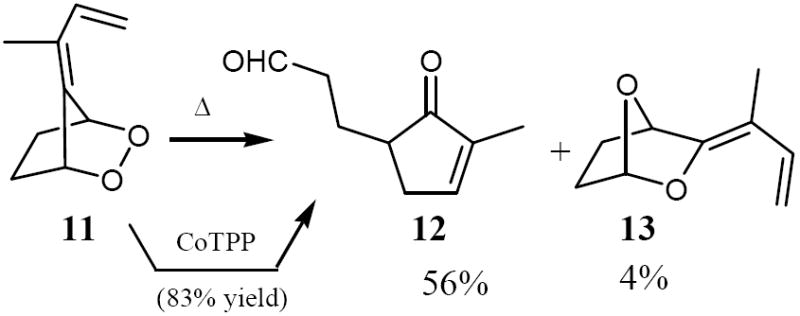
Synthesis and thermolysis of endoperoxide 11
Scheme 6.
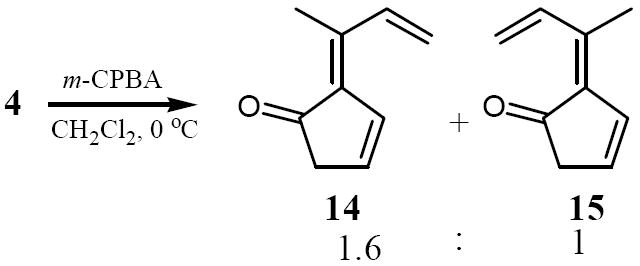
Epoxidation of 4; formation of 3-cyclopenten-1-ones 14 and 15
Acknowledgments
This work was supported by funds from the National Institutes of Health, MBRS-SCORE Program-NIGMS (Grant No. GM52588. We also acknowledge funding from the National Science Foundation (Grants No. DBI-0521342and DUE-9451624)for the purchase of 500 MHz and 300 MHz NMR spectrometers.
Footnotes
Supplementary Material
Experimental procedures and characterization of the products by 1H and 13C NMR, IR, HRMS data are available. Supplementary data associated with this article can be found, in the online version, at
References
- 1.Thiele J. Chem Ber. 1900;33:666. [Google Scholar]
- 2.For reviews, see: Yates P. Advances in Alicyclic Chemistry. Vol. 2. Academic Press; New York: 1968. Fulvenes; pp. 59–184.Bergman ED. Chem Rev. 1968;68:41–84.Day JH. Chem Rev. 1953;53:167–189.Zeller KP. Pentafulvenes. Methoden der Organischen Chemie. 1985;5(2C):504–684.
- 3.Stone KJ, Little RD. J Org Chem. 1984;49:1849. [Google Scholar]
- 4.Erden I, Xu F-P, Sadoun A, Smith W, Sheff G, Ossun M. J Org Chem. 1995;60:813–820. [Google Scholar]
- 5.Neuenschwander M. Fulvenes. In: Patai S, editor. The Chemistry of Double-Bonded Functional Groups, Supplement A. John Wiley & Sons; New York: 1989. pp. 1131–1268. [Google Scholar]
- 6.a Neuenschwander M, Meuche D, Schaltegger H. Helv Chim Acta. 1964;47:1022–1032. [Google Scholar]; b Neuenschwander M, Meuche D, Schaltegger H. Helv Chim Acta. 1963;46:1760. [Google Scholar]
- 7.Griesbeck A. J Org Chem. 1989;54:4981–82. [Google Scholar]
- 8.Compound 3 undergoes Diels-Alder reaction with dimethyl acetylenedicarboxylate in refluxing benzene to give a monoadduct; 1H NMR (300 MHz, CDCl3): δ 6.9 (narrow m, 2H), 6.5 (narrow m, 4H), 4.4 (narrow m, 2H), 3.8 (s, 6H), 3.55 (t, J= 7.0 Hz, 2H), 3.45 (t, J= 5.5 Hz, 2H), 2.8 (t, J= 7.0 Hz, 2H), 2.25 (s, 3H), 2.15 (m, 2H), 1.5 (s, 3H); 13C NMR (75 MHz, CDCl3): δ 165.4, 164.3, 152.4, 151.9, 150.7, 144.3, 142.7, 121.3, 121.0, 101.4, 70.7, 69.8, 54.1, 53.9, 52.7, 37.7, 33.6, 22.2, 17.3 ppm; FT-IR: υ 3072.4, 2950, 2856, 1720, 1638, 1434, 1368, 1319, 1283, 1254, 1217, 1168, 1107, 1037, 919, 768, 735 cm-1; HRMS calcd for C24H28O5 396.1937, found 396.1937.
- 9.Crossland RK, Servis KL. J Org Chem. 1970;35:3195–6. [Google Scholar]
- 10.Wasserman HH, Murray RW. Singlet Oxygen. Academic Press; New York: 1979. [Google Scholar]
- 11.Harada N, Suzuki S, Uda H, Ueno H. J Am Chem Soc. 1971;94:1777–78. [Google Scholar]
- 12.Skorianetz W, Schulte-Elte KH, Ohloff G. Angew Chem. 1972;84:311–12. [Google Scholar]
- 13.Adam W, Erden I. Angew Chem Int Ed Engl. 1978;17:210. [Google Scholar]
- 14.a Erden I, Amputch M. Tetrahedron Lett. 1987;28:3779–82. [Google Scholar]; b Erden I, Drummond J, Alstad R, Xu F. Tetrahedron Lett. 1993;34:1255–58. [Google Scholar]; c Erden I, Drummond J, Alstad R, Xu F. J Org Chem. 1993;58:3611–12. [Google Scholar]; Erden I, Xu F, Cao W. Angew Chem Int Ed Engl. 1997;36:1516–18. [Google Scholar]
- The (E)-stereochemistry of the proton on the exocyclic double bond in 6 is based on the chemical shift of the signal at 6.8 ppm; the corresponding (Z)-olefinic proton has been reported to absorb in a similar compound at δ ~5.7 ppm; see: Noyori R, Ohnishi Y, Kato M. J Am Chem Soc. 1972;94:5105–6.Dubois E, Dubois M. C R Acad Sci, Ser C. 1963;256:715.
- 16.Skorianetz W, Schulte-Elte KH, Ohloff G. Helv Chim Acta. 1971;54:1913–22. [Google Scholar]
- 17.a Adam W, Eggelte HJ. J Org Chem. 1977;42:3987–8. doi: 10.1021/jo00444a055. [DOI] [PubMed] [Google Scholar]; b Coughlin DJ, Salomon RG. J Am Chem Soc. 1977;99:655–657. doi: 10.1021/ja00444a079. [DOI] [PubMed] [Google Scholar]; c Coughlin DJ, Brown RS, Salomon RG. J Am Chem Soc. 1979;101:1533–1539. [Google Scholar]
- 18.Boyd JD, Foote CS, Imagawa DK. J Am Chem Soc. 1980;102:3641–2.Sutbeyaz Y, Secen H, Balci M. J Org Chem. 1988;53:2312–17. doi: 10.1021/jo962092+. and references cited therein.
- 19.a Alder K, Flock FH, Lessenick H. Chem Ber. 1957;90:1709. [Google Scholar]; b Froese RDJ, Organ MJ, Goddard JD, Stack TDP, Trost BM. J Am Chem Soc. 1995;117:10931–38. [Google Scholar]
- 20.Adam W, Hadjiarapoglou L, Meffert A. Tetrahedron Lett. 1991;32:6697–6700. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.